
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Unveiling the double triplet nature of the 2Ag state in conjugated stilbenoid compounds to achieve efficient singlet fission†
Letizia
Mencaroni
 a,
Martina
Alebardi
a,
Fausto
Elisei
a,
Irena
Škorić
b,
Anna
Spalletti
a and
Benedetta
Carlotti
*a
a,
Martina
Alebardi
a,
Fausto
Elisei
a,
Irena
Škorić
b,
Anna
Spalletti
a and
Benedetta
Carlotti
*a
aDepartment of Chemistry, Biology and Biotechnology, University of Perugia, via elce di sotto 8, 06123 Perugia, Italy. E-mail: benedetta.carlotti@unipg.it
bDepartment of Organic Chemistry, Faculty of Chemical Engineering and Technology, University of Zagreb, 10000 Zagreb, Croatia
First published on 17th July 2023
Abstract
In this investigation, the excited-state evolution in a series of all-trans stilbenoid compounds, displaying a low-lying dark singlet state of 2Ag-like symmetry nearly degenerate with the bright 1Bu state, was unveiled by employing advanced ultrafast spectroscopies while probing the effect of solvent polarizability. Together with the dual emission, femtosecond transient absorption and broadband fluorescence up-conversion disclosed the double nature of the 2Ag-like state showing both singlet features, a lifetime typical of a singlet and the ability to emit, and a triplet character, exhibiting a triplet-like absorption spectrum. The ultrafast formation (in hundreds of femtoseconds) from the non-relaxed upper singlet state led to the identification of 2Ag as the correlated triplet pair of singlet fission. The spectral difference obtained by comparison of transient absorption peaks of the 2Ag (1TT) and the triplet states was found to be in remarkable agreement with the observed triplet yield and the 1(TT) separation rate constant. Indeed, this spectral shift provided an experimental method to gain qualitative insight into the ease of separation of the 1(TT) and the relative SF efficiency. The highly conjugated polyene-like structures enable the ultrafast formation of the double triplet, but then the large binding energy prevents the triplet separation and thus the effective completion of singlet fission. Even though thermodynamically feasible for all the investigated stilbenoids according to TD-DFT calculations, singlet fission resulted to occur efficiently in the case of 1-(pyridyl-4-ylethenyl)-4-(p-nitrostyryl)benzene and nitro-styrylfuran with the triplet yield reaching 120% and 140%, respectively, triggered by their greatly enhanced intramolecular charge transfer character relative to the other compounds in the series.
Introduction
Singlet fission (SF), the down-conversion of a singlet excited state in two independent triplet excitons, has aroused great interest as it promises to boost the solar cell efficiency beyond the Shockley–Queisser limit.1,2 Despite the tremendous research effort, the number of SF-capable materials is still limited to a few classes of chromophores, namely acenes,3–6 perylene/terrylenedimides,7 diphenylisobenzofurans,8 and diketopyrrole–pyrroles.9 More recently, much of this effort has been directed to homo or hetero-dimers of the prototypical SF systems10,11 and donor–acceptor (DA)-type conjugated oligomers/polymers.12–16 This is because the intramolecular SF is more attractive as it allows for better control of its efficiency, being insensitive to the intermolecular packing, and facilitates the processing techniques when dealing with photovoltaic applications.Particularly, the most important intermediate species in the SF process is a transient multiexciton (ME) state, having a triplet nature but overall singlet spin multiplicity. Thus, it is usually called the “correlated triplet pair state” (1TT) or the biexciton state and is able to split into two independent triplets.17 In former studies concerning some carotenoids,18 polymers,19–23 and small molecules, such as quinoidal thiophenes,24,25 the lowest singlet excited state with Ag symmetry, similar to that present in C2h-symmetry molecules such as α,ω-diphenylpolyenes,26 has been found to coincide with the 1(TT) state.17,27,28 The 2Ag state in fact is a biexciton state which can be described by various configurations, including a pair of interacting triplet states. The electronic structure of the polyene-like molecules then shows the upper singlet state, the bright mono-excited S2 state of 1Bu symmetry, as the one reached by photoexcitation according to the selection rules. However, the role of the intermediate 2Ag state in intramolecular SF has sometimes proven to be controversial.29 In fact, when dealing with short-chain polymers, the triplets in the 2Ag state are so strongly coupled (such that it is possible to define a binding energy) that they undergo fast geminate recombination or annihilation making 2Ag behave like an ordinary singlet state. In this case, the S2 → S1→ S0 decay is better represented by ordinary internal conversion processes.30 For larger polyenes instead, the possibility of the two triplet excitons residing far away in the molecular structure reduces the coupling, favouring the separation and thus opening the second step of the singlet fission process (S1 or 1(TT)→ 2T1). However, Campos et al.31 have demonstrated that lengthening the polymer chain is not sufficient to open SF, suggesting that the chromophores should be separated by appropriate covalent bridges.32 Zhu et al.20 pointed out that an “ideal” mechanism for efficient SF would be the strong S1/CT/1(TT) coupling, in order to promote the ultrafast intramolecular formation of the ME state, and thanks to properly tuned intermolecular electronic interactions, ensuring the separation of the triplet pair between neighbouring molecules in this manner. The mediation of the CT was already proven to favour the SF phenomenon33 aiding the triplet pair decorrelation.29 Nevertheless, as already observed for other push–pull SF candidates,13,34 the intramolecular SF process is highly sensitive to the degree of charge transfer character.14,35–37 Even if in polymers with too little charge transfer character, the biexciton singlet state decays without forming triplets, it is also known that the much-stabilized CT state becomes a trap state being detrimental to SF.20,34,38
In light of expanding the pool of SF capable materials, the interest in the photobehavior of a series of poorly-fluorescent, highly-polarizable conjugated all-trans geometrical isomers of stilbenoid compounds (Chart 1) was re-awaken as they show a low-lying dark singlet state of 2Ag-like symmetry, nearly degenerate with the bright upper 1Bu-like state. The synthesis and photophysical characterization of the investigated stilbenoid compounds have been previously addressed.39–43 Both steady-state absorption and fluorimetric analysis jointly with the temperature effect revealed for the thiophene and the naphthyl containing compounds a low emission capability due to the presence of a low-lying singlet state of Ag symmetry. The reactive decay was also carefully studied pointing out almost negligible photoisomerization yield. Therefore, the main excited state deactivation pathways are expected to be non-radiative and non-reactive. The push–pull PN and DF compounds, characterized by the highly dipolar structure due to the presence of the electron-deficient NO2 unit, have also been included in the series as showing similar experimental evidence (negligible emission and photoreaction) in non-polar solvents.44–46
 | ||
| Chart 1 Investigated molecules. | ||
Herein, steady-state, fast and ultrafast spectroscopic techniques have been applied to the compounds in Chart 1 aiming at verifying the possible involvement of singlet fission, among the other competitive deactivation pathways, in their excited-state deactivation dynamics. The impact of different solvent polarizability, which allows tuning the energy gap between the two singlet excited states of different nature, was carefully evaluated through femtosecond transient absorption and broad-band fluorescence upconversion techniques. In fact, polarizability is expected to affect the ionic Bu state much better than the covalent 2Ag state, resulting in a progressive stabilization of the former upon increasing the refractive index of the medium. The controversial role of the dark 2Ag state in triggering SF for this series of stilbenoid compounds will be finally discussed.
Results
Spectral and fluorescence properties
The effect of solvent polarizability was studied to probe the changes in the evolution of the excited state with varying relative positions of the upper S2 (1Bu) and S1 (2Ag) states by spanning the refractive index (n) from 1.3030 (Hx/PFHx) to 1.7410 (DIM). Fig. 1 depicts the polarizability effect on the absorption and emission spectra in the representative case of 2,5-(2TE)2T. The structured absorption profile (vibronic progression ∼1280 cm−1) red-shifts ca. 30 nm when going from the lowest polarizable medium (Hx/PFHx, 5/5 v/v, mixture) to the highest polarizable solvent, DIM. The bathochromic shift of the spectrum is attributed to the progressive stabilization of the allowed 1Bu state with increasing n. When it comes to the steady-state fluorescence spectra, they are clearly due to the superimposition of two emissions and the effect of polarizability better resides on different spectral shapes. In fact, the position and relative intensity of the structured emission bands (vibrational progression, Δν = 1370 cm−1 in Hx/PFHx) are clearly modified by the polarizability, with the simultaneous narrowing of the emission spectrum when approaching a more polarizable environment (FWHM = 4900 and 3350 cm−1 in Hx/PFHx and DIM, respectively). In the less polarizable media two distinct contributions to the fluorescence of 2,5-(2TE)2T can be distinguished (Fig. 1): the hypsochromic 1Bu state, thermally populated by the lowest 2Ag, having the 0–0 vibronic peak as a shoulder at 446 nm in Hx/PFHx which shifts to 460 nm in Tol and 471 nm in DMSO, upon increasing the solvent polarizability; and the bathochromic 2Ag state, centred below 500 nm, less sensitive to the solvent. Particularly, the emission spectrum recorded in DIM appears to be the most diverse in the series. In fact, the high refractive index is responsible for the inversion of the states in this medium and, thus, the emission profile can be entirely assigned to the stabilized 1Bu state in line with previous findings.47 Similar spectral features were highlighted in other naphthyl and thiophene containing compounds (Fig. S1–S4, ESI†), whereas PN and DF show bell-shaped emission spectra due to the ICT nature of its singlet excited state (Fig. S4, ESI†).44–46 | ||
| Fig. 1 Normalized absorption (left) and emission (right) spectra of 2,5-(2TE)2T in solvents of different polarizabilities. | ||
Additional evidence for the presence of the low-lying forbidden Ag-like state for all the investigated compounds arouses when taking into account the fluorescence quantum yields and relative kinetic constants which both reveal a trend to increase with n (see Table 1 and Table S5, ESI†). As for 2,5-(2TE)2T, the 107 s−1 radiative rate constant values measured in solvents of low polarizability are ascribed to the forbidden nature of the lowest excited singlet state (2Ag), whereas kF gains almost one order of magnitude when passing from IP to BrN (kF = 1.1 × 108 s−1). This trend reproduces the progressive mix of the fully-allowed, emissive 1Bu and the dark, forbidden 2Ag due to the stabilization of the former. The closer the two singlet excited states, the stronger the vibronic coupling,48 resulting in the increased oscillator strength of the 2Ag (S1) → 1Ag (S0) transition, leading to larger emission capability (ϕF) and probability (kF). Analogous outcomes were found for the other stilbenoid compounds under study, with the most remarkable solvent effect observed in the case of DF. In fact, as for the highly-dipolar furan derivative, the ϕF resulted to be negligible in solvents of low polarizability (ϕF ∼ 10−5, kF ∼ 106 s−1) while being sizable in BrN (ϕF = 0.23, kF = 9.2 × 108 s−1) and An (ϕF = 0.09, kF = 2.8 × 108 s−1).
| n | ϕ F | τ F/ns | k F/s−1 | |
|---|---|---|---|---|
| a From ref. 40. b From fs-TA measurements. c From ref. 46. | ||||
| 1NPH | ||||
| IP | 1.3540 | 0.13a | 11.3a | 1.2 × 107 |
MeCH/3-MP 9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 :1 |
1.4184 | 0.18a | 11.0a | 1.6 × 107 |
| Tol | 1.49693 | 0.34a | 9.8a | 3.5 × 107 |
| CS2 | 1.6277 | 0.02a | 0.4a | 5.2 × 107 |
| 2,5-(2TE)2T | ||||
| Hx/PFHx (5/5) | 1.3030 | 0.060 | 2.1 | 2.9 × 107 |
| IP | 1.3540 | 0.025 | 1.8 | 1.4 × 107 |
| Tol | 1.49693 | 0.071 | 1.8 | 3.9 × 107 |
| BrN | 1.6570 | 0.16 | 1.5 | 1.1 × 108 |
| PN | ||||
| Tol | 1.49693 | 0.0022c | 0.365b | 6.0 × 106 |
| Tol/An 7:3 |
1.50293 | 0.009c | 0.38b | 2.4 × 107 |
| Tol/An 5:5 |
1.50695 | 0.015c | 0.40b | 3.8 × 107 |
| Tol/An 3:7 |
1.51098 | 0.030c | 0.20b | 1.5 × 108 |
| An | 1.5170 | 0.067c | 0.36b | 1.9 × 108 |
| DF | ||||
| MeCH/3-MP 9:1 |
1.4184 | 4 × 10−5 | 0.033b | 1.3 × 106 |
| DHN | 1.474 | 5 × 10−5 | 0.058b | 8.3 × 105 |
| Tol | 1.49693 | 6 × 10−4 | 0.089b | 6.7 × 106 |
| Tol/An 9:1 |
1.49894 | 2.5 × 10−3 | 0.049b | 5.1 × 107 |
| Tol/An 7:3 |
1.50293 | 5.2 × 10−3 | 0.068b | 8.1 × 107 |
| Tol/An 5:5 |
1.50695 | 0.011 | 0.080b | 1.4 × 108 |
| An | 1.5170 | 0.090 | 0.32b | 2.8 × 108 |
| BrN | 1.6570 | 0.23 | 0.25b | 9.2 × 108 |
Triplet properties
The low fluorescence efficiency measured for the investigated compounds implies the involvement of non-radiative deactivation pathways in their excited-state dynamics. Nanosecond laser flash photolysis was thus applied to investigate and quantify the contribution of the triplet excited state formation. The triplet properties (triplet absorption spectrum, triplet quantum yield and lifetime) were investigated in air-equilibrated and nitrogen-purged Tol solutions (see Fig. 2 and Table 2) as well as in various solvents of different polarizabilities (see Fig. S6, S7 and Tables S6, S7, ESI†). The triplet transient absorption spectra were found to reproduce those previously published,39,42,45–47,49 obtained through sensitization experiments. | ||
| Fig. 2 Triplet transient absorption spectra obtained in nitrogen-purged Tol solutions of 1NPH, 2,5-(2TE)2T, PN and DF45 (λexc = 355 nm). | ||
| Sample | C/10−5 M | λ T/nm | τ T/μs | ϕ T | k T/107 s−1 |
|---|---|---|---|---|---|
| 1NPH | 2.0 | 490 | 14.5 | 0.02 ± 0.01 | 0.20 |
| 2,5-(2TE)2T | 2.0 | 540 | 6.29 | 0.14 ± 0.02 | 7.8 |
| PN | 0.9 | 515 | 0.84 | 0.80 ± 0.10 | 220 |
| 3.0 | 1.04 ± 0.15 | ||||
| 5.0 | 1.16 ± 0.17 | ||||
| DF | 2.0 | 570 | 1.30 | 0.97 ± 0.15 | 1090 |
| 7.0 | 1.27 ± 0.19 | ||||
| 10 | 1.4 ± 0.2 |
Looking at Fig. 2, it is apparent that the triplet quantum yields for the four reported compounds 1NPH, 2,5-(2TE)2T, PN and DF are substantially different, considering the signal-to-noise ratio of the spectra. In the case of 2,5-(2TE)2T, the triplet formation efficiency (ϕT) was found to be 14% in Tol. Interestingly, the solvent refractive index influenced the triplet yield: for 2,5-(2TE)2TϕT is measured to be 9% in the Hx/PFHx mixture and doubled in BrN (see Table S6, ESI†). A slight increase of the relative kinetic constants (kT = ϕT/τF ∼107 s−1) was also observed upon increasing the solvent polarizability. As for the noisy ns-TA spectra of 1NPH, a lower triplet yield of ca. 2% was obtained in Tol, together with a slower triplet formation time (kT ∼105 s−1) with respect to 2,5-(2TE)2T. The concentration effect in the limited investigated range (from 2 × 10−5 to 1 × 10−4 M) did not produce any impact on the triplet production of 2,5-(2TE)2T and 1NPH, in any of the investigated solvents.
In neat contrast, for DF a triplet yield approaching unity in dilute solution (97% at 2 × 10−5 M), and surprisingly reaching 140% in the most concentrated solution under study (1 × 10−4 M), was measured in Tol (see Table 2). Analogously, a sizable triplet yield was measured for PN in dilute solution (80% at 9 × 10−6 M) that increases to 120% at 5 × 10−5 M. The ultrafast formation of their triplet state (kT ∼109/1010 s−1), together with the exceptionally high triplet production efficiency exceeding 100%, suggest that a peculiar multiple exciton generation process might be operative for these nitro-derivatives. Similarly to fluorescence, the triplet properties of DF and PN were proven to be drastically affected by the solvent polarity: a sharp drop in the ϕT values was observed in the polar solvent due to the competition with both fluorescence and internal conversion from the lowest ICT singlet state.45,46
Ultrafast dynamics
In order to trace down the excited-states evolution, femtosecond transient absorption measurements were carried out by exciting each sample at 400 nm in solvents of different polarizabilities. Femtosecond transient absorption (fs-TA) results obtained for 2,5-(2TE)2T, PN and DF in Tol are shown as representative cases in Fig. 3.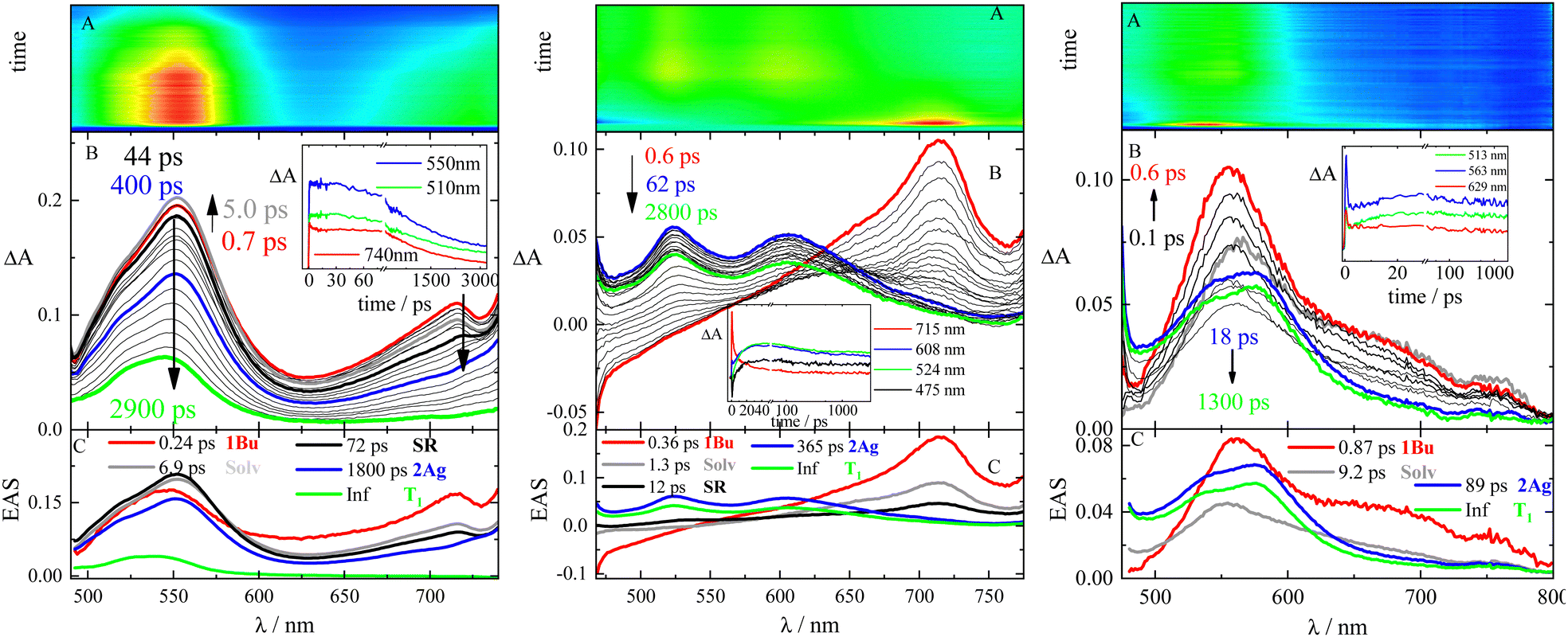 | ||
| Fig. 3 Fs-TA measurements of compounds 2,5-(2TE)2T (left), PN (central) and DF (right) in Tol (λpump = 400 nm). Panel A: experimental 3D matrix; Panel B: representative spectra at different delay times and representative kinetics (inset) at different wavelengths; and Panel C: EAS (evolution associated spectra) obtained by global analysis. | ||
The temporal evolution of the TA spectra for 2,5-(2TE)2T in Tol is shown in panel B of Fig. 3 (left). At first, the representative spectra observed at early delays after excitation were characterized by two positive excited state absorption (ESA) bands at ca. 550 and 720 nm. The global fit of the data revealed the presence of five exponential components whose evolution associated spectra (EAS), obtained by global analysis, are reported in panel C of Fig. 3 (left). The following transient species were unveiled: (i) the first ultrafast transient with ca. 250 fs lifetime (red spectrum) was assigned to S2, 1Bu state; probably mixed with 2Ag, (ii) the grey and black transients were interpreted as the solvation dynamic/vibrational cooling (6.9 ps) and structural relaxation (72 ps) processes, respectively; (iii) the blue profile, peaking at 550 nm, with a lifetime of 1800 ps, perfectly matching the τF obtained by TC-SPC, was identified as the dark S1, the relaxed 2Ag state; (iv) the longer-lived transient, with the maximum ESA at 540 nm, was referred to the first triplet excited state (T1) considering the infinite (Inf) lifetime and the perfect overlap to the ns-TA triplet spectrum. A deeper analysis of fs-TA results also disclosed some unconventional kinetics in the photobehavior of 2,5-(2TE)2T in Tol. In fact, the 2Ag state is likely populated after photoexcitation from the non-relaxed 1Bu state within the first 250 fs. This could be inferred by the presence of the ESA band of the 2Ag state (540 nm) in the 1Bu red spectrum (540 nm and 725 nm) already at 700 fs after photoexcitation. Between 700 fs (red profile) and 5 ps (grey profile) after photoexcitation, the decrease of the 720 nm-ESA signal is concomitant with the growth of the 550 nm-band. Thereafter, the TA signal slowly decays evolving firstly into the blue spectrum and later toward the slightly blue-shifted green ESA band. Analogous excited-state evolution was detected for the other stilbenoid compounds under investigation (see Table 3 and Fig. S8–S10 and Tables S8–S13, ESI†). The solvent polarizability slightly affects the photobehavior with small changes in the transient lifetimes and the relative intensity of the T1 signal.
| 1NPH | 2,5-(2TE)2T | PN | DF | Assignment |
|---|---|---|---|---|
| 0.25 | 0.24 | 0.36 | 0.86 | Solv/1Bu |
| 6.9 | 1.3 | 9.2 | Solv | |
| 70 | 72 | 12 | VC/SR | |
| 9800 | 1800 | 365 | 89 | 2Ag |
| Inf | Inf | Inf | Inf | T1 |
As for compounds PN and DF, the assignment of the transient species previously reported45,46 was reconsidered here. EAS spectra of DF in Tol are shown in panel C of Fig. 3 (right). The ultrafast red profile (τ = 0.87 ps) peaking at 560 nm, with a broad shoulder at 680–700 nm, was proven to be the bright upper singlet state of 1Bu symmetry. The latter, after vibrational cooling (τ = 9.2 ps), evolves toward the blue profile, having a lifetime of 89 ps. The blue spectrum decays leading to significant population of the green T1 excited state, which then slowly decays far beyond the time windows of 3.2 ns of the experimental set up. A faster and more efficient production of T1 was thus observed for DF relative to the other stilbenoids in the series. The blue transient, previously interpreted as an upper peculiar T1′ state,45 has now been assigned to the doubly-excited singlet state of 2Ag-like symmetry.
The 2Ag lifetime for DF showed a trend of increasing upon enhancing the solvent polarizability when solvents of very low polarity (dielectric constant ca. 2) are considered (e.g.: τ2Ag = 33 ps in MeCH/3-MP; τ2Ag = 58 ps in DHN; τ2Ag = 89 ps in Tol; see Table S13, ESI†). However, the ultrafast dynamics for compounds PN and DF substantially differs when a slightly more polar environment is taken into account. In this respect, the highly dipolar PN and DF showed a unique behavior among the investigated stilbenoid compounds. In fact, an ICT singlet state appears mixed with the 2Ag already in Tol/An mixtures (Fig. 4). The ICT then becomes a distinct transient species in more polar solvents (An, dielectric constant ca. 4), thus opening a new decay pathway competitive to 1Bu → 2Ag conversion and leading to the disappearance of 2Ag (see Fig. 4 and Tables S12, S13, ESI†).
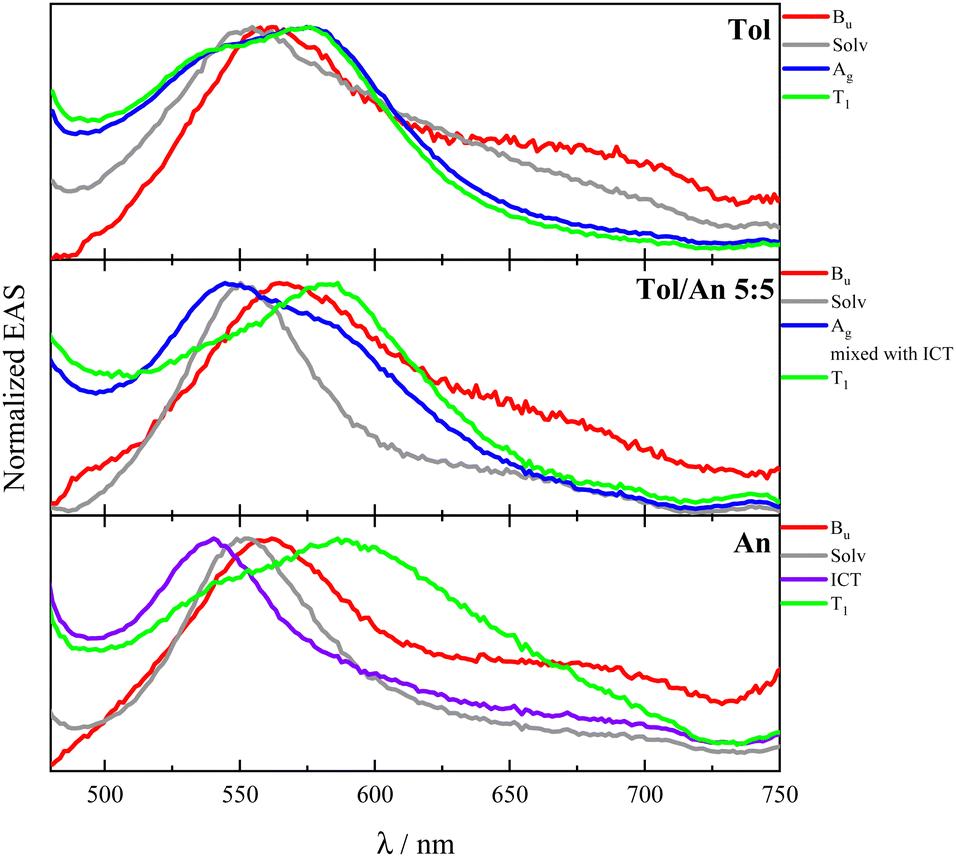 | ||
| Fig. 4 Fs-TA results for DF in solvents of different polarizabilities: normalized evolution associated spectra (EAS) obtained by global analysis. | ||
Fluorescence upconversion experiments with femtosecond temporal resolution (fs-FUC) were performed to gain a deeper insight into the emissive excited-state evolution as a function of solvent polarizability. Fig. 5 portrays the normalized decay associated spectra (DAS) obtained by the best fit of the results for compound 2,5-(2TE)2T. In agreement with the results of the complementary pump-probe investigation, four emissive transients were identified with the missing transient being the dark T1. The red profile (τ < 700 fs) is representative of the 1Bu excited state, followed by the grey and black profiles related to the solvation/structural relaxation processes, whereas the red-shifted, long-lived (1–2 ns) emission was assigned to the relaxed forbidden 2Ag state. In line with the steady-state fluorimetric results discussed above, the fs-FUC measurements clearly unveiled the dual fluorescence from both the bright 1Bu and the dark 2Ag state, particularly in less polarizable media. As a result, the wide red spectra in Hx/PFHx and IP, spanning from 450 to 700 nm, in Fig. 5 consists actually of the combined emission contributions from both 1Bu (λMAX = 495 nm) and 2Ag (λMAX = 575 nm), confirming the ultrafast formation of 2Ag within the laser pulse. When moving toward more polarizable media, the 1Bu emission undergoes a marked red-shift (λMAX = 520 nm in BrN), simultaneously leading to the increase in the 2Ag emission intensity, that roughly remains in the same spectral position (575 nm). In the higher refractive index regime, the two contributions to the fluorescence are more challenging to separate due to the progressive mixing of the two states. The reduced energy gap between the two singlets enables their stronger vibronic coupling.48 As a consequence, the oscillator strength of 2Ag increases, borrowed by the closer 1Bu, and this justifies the gain of one order of magnitude in the emission quantum yields and the kinetic constants when passing from IP to BrN, discussed so far. In conclusion, the double emission present in the ultrafast red transient shown in Fig. 5 highlights the 2Ag state to be populated immediately after photoexcitation possibly by a coherent process from the non-relaxed Bu state, in agreement with the ultrafast S2 → S1 conversion observed through the fs-TA measurements (Fig. 3).29 The fluorescence upconversion results also show the emitting capability of the 2Ag transient for all other investigated compounds (as detailed in the ESI†), including PN and DF for which this species is clearly characterized by a triplet-like ESA.
 | ||
| Fig. 5 Fs-FUC results of 2,5-(2TE)2T in solvents of different polarizabilities: decay associated spectra (DAS) obtained by global analysis. | ||
Discussion
Taking into consideration the comparison of the normalized ESA spectra obtained in the femtosecond and nanosecond regime reported in Fig. 6 and Fig. S14, ESI,† a certain similarity between the 2Ag and the T1 ESA was noticed. In particular, the 2Ag absorption band perfectly matches the spectrum referred to the first triplet excited state in the case of DF and closely resembles those of PN and 2,5-(2TE)2T. Therefore, the low-lying 2Ag state, populated in less than 1 ps, emits with a lifetime which ranges from tens of picoseconds to a few nanoseconds according to the stilbenoid structure, but simultaneously manifests a triplet-like absorption profile. The double (singlet–triplet) nature of this peculiar transient, already observed in other conjugated polyenes, well correlates with the intermediate species in singlet fission: the correlated triplet pair 1(TT).17,27,28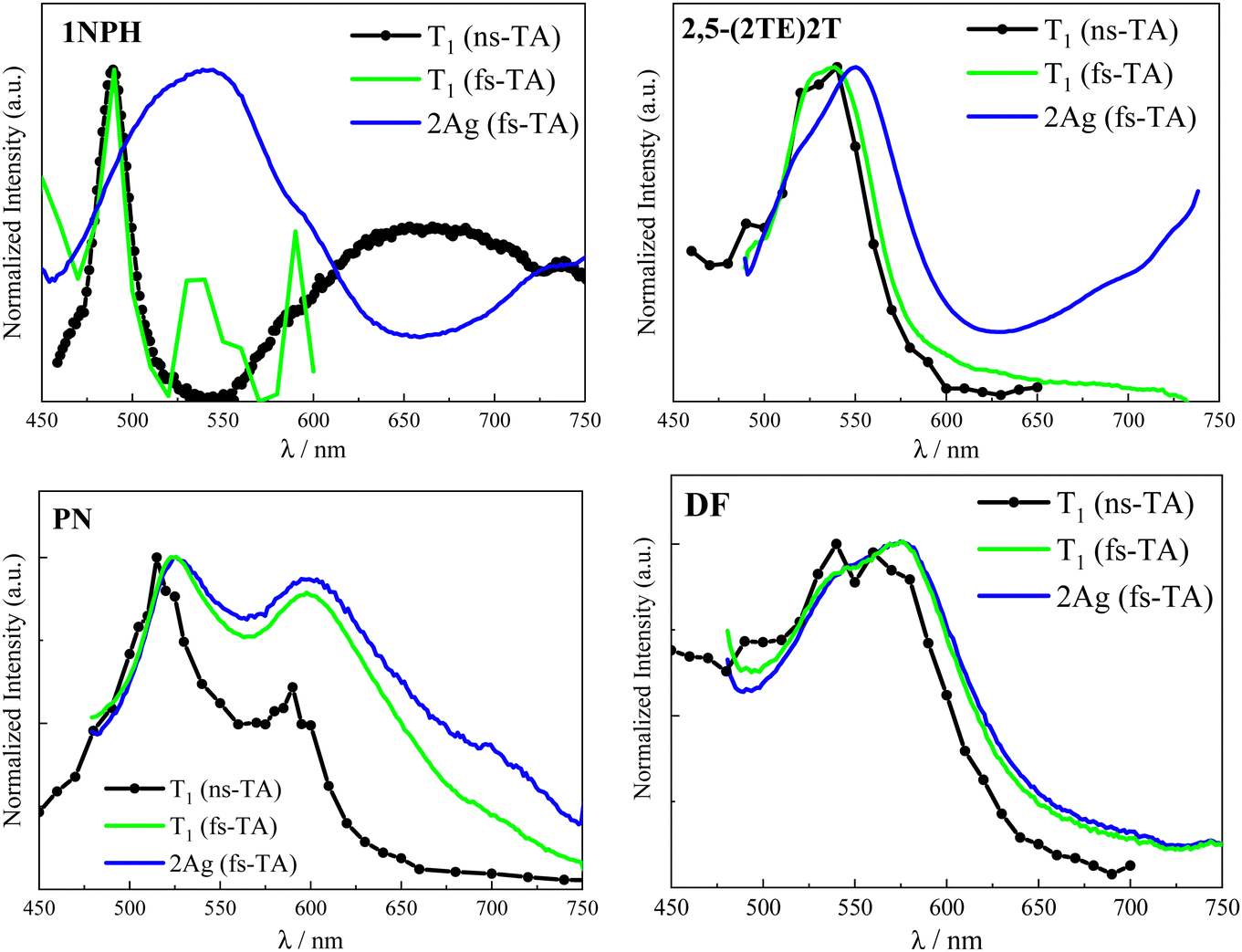 | ||
| Fig. 6 Comparison between T1 ESA spectra as obtained by ns-TA (black, scattered-line) and by fs-TA (green line) experiments and 2Ag ESA spectra as obtained by fs-TA experiments (blue) for compounds 1NPH, 2,5-(2TE)2T, PN and DF in Tol. | ||
Before the SF phenomenon could be invoked to explain the 1Bu → 2Ag evolution, the relative energies of the first excited states need to be considered so that the thermodynamic feasibility of the process can be discussed. Table 4 summarizes the predicted vertical energies as obtained by TD-DFT calculations at the optimized S0 geometry. The chosen level of theory intrinsically limits the predictive power of the quantum mechanical calculations employed herein as DFT only refers to monoelectronic excitations. Hence, any prediction of the 2Ag state fails as it should be described by multireference configurations. However, the TD-DFT results fairly reproduce the experimental absorption band of bright 1Bu, which corresponds to the first accessible excited state via one-photon absorption. Additionally, taking into account that the 2Ag state (likely 1(TT)) is populated by the possible vibrationally-hot, non-relaxed S2 (1Bu) state, the latter should be considered to verify the strict energy level matching conditions of SF: E(S2) ≥ 2 × E(T1). The last column of Table 4 suggests that the SF process in the case of the naphthyl and thiophene containing compounds would be exothermic (see also the ESI†), while in the case of PN and DF energy barriers of ca. 0.30 and 0.36 eV, respectively, should be overcome. When performing the ultrafast spectroscopic experiments, we employ a femtosecond pump laser peaked at 400 nm and characterized by a FWHM of about 50 nm. Considering the energetics of the Bu → Ag transition for PN and DF, as predicted by the calculations (Table 4), it is possible that the small activation energy required to populate the correlated triplet pair is provided by the thermal energy, also considering the eventual heating effect resulting from the laser irradiation of the sample, or that the Ag formation occurs from a vibrationally-hot non-relaxed S2 state (see Fig. 7).
 | ||
| Fig. 7 Sketch of the crossing of Bu and Ag potential curves, showing the ultrafast formation of Ag in 1NPH (left) and DF (right) facilitating the SF process. Transient absorption signals in Tol are also shown: the Ag (1TT) → Sn′ transition (blue arrow) differs from the T1 → Tn transition (green arrow) in the case of 1NPH, where the Ag state is highly stabilized, while a 1(T1T1) → 1(T1Tn′) transition very similar to the T1 → Tn transition (green) better describes the transient absorption signal of Ag for compound DF. | ||
A further experimental attempt to determine the triplet energy in order to verify the possible involvement of SF was devoted to the study of the luminescence spectra of the samples in a glass matrix (MCH/3MP 9/1 v/v) at 77 K. Unfortunately, no phosphorescence signal was detected for the naphthyl and thiophene containing compounds, likely due to either the low triplet quantum yields or to the low sensitivity of the InGaAs detector for their phosphorescence expected in the near-infrared spectral range. On the other hand, phosphorescence was revealed in the visible for DF, peaked at 670 nm (1.85 eV) in excellent agreement with the computationally predicted triplet energy (Table 4) and showing a 2.9 ms decay lifetime (Fig. S16 and S17, ESI†).
Quantum mechanical calculation also allowed to discard the hypothesis of an ultrafast ISC taking place in these stilbenoid systems. As a matter of fact, for the thienyl derivatives 2,5-(2TE)2T, D2TO and 2,5-(PhBu)2T, (n,π*) states are expected to lie at high energy and thus they do not play a role in their excited state deactivation (see ref. 43 and Tables S15–S17, ESI†). Actually, as for PN and DF, the quantum-mechanical calculations predicted triplet states of an n,π* character, energetically close to the π,π* 1Bu45,46 that might justify fast intersystem crossing rates in agreement with the El-Sayed rules.50 However, this interpretation would not explain the totality of our experimental results. In contrast to the conventional scenario (ultrafast ISC), the proposed mechanism (ultrafast SF) seems to take into account all our experimental evidence.
Even though the ultrafast formation of the 2Ag state is occurring in all the stilbenoid compounds under study, the behaviour of the molecules in the series is found to be different: either the 2Ag state shows a pure singlet character (as proposed in the case of the naphthyl-derivative) or leads to the independent triplet separation with small (for the thienyl derivatives) or great efficiency (PN and DF). The reason for the ineffective triplet production might rely on the fact that the 2Ag triplet pair is so strongly coupled that the separation would require overcoming a huge energy barrier; hence, the independent triplets are trapped in the tightly bound 1(TT). As stated above, the estimation of the binding energy of the correlated triplet pair requires higher levels of theory that are, for instance, CASPT2 or RAS-SF methods.51 In the absence of more sophisticated quantum mechanical calculations, we searched for an experimental strategy to gain qualitative insight into the strength of inter-triplet interactions within the biexciton. Interesting experimental information on the 1(TT) coupling strength has surprisingly come from the comparison of the T1 and 2Ag ESA bands (Fig. 6 and Fig. S14, ESI†). As a matter of fact, a direct correlation could be drawn: the more their spectral profiles overlap, the more triplet character the 2Ag state retains, the higher the triplet yield that was measured (see Table 5 where the comparison was quantified as ΔET1−Ag, the difference between the T1 and 2Ag spectral maxima). In contrast, the worse the overlapping, the more 2Ag behaves as a pure singlet excited state, and the lower the triplet production, rendering the discrimination between the intramolecular SF and the ordinary ultrafast internal conversion non-trivial. For example, the almost perfect overlap between the 2Ag ESA band to the green T1 in the case of DF (Fig. 6, right panel), for which ΔE ≈ 0.0037 eV, leads to ϕT = 0.97 in dilute solution. For PN, a slightly higher ΔE value of 0.013 eV implies a lower but still substantial ϕT value of 80%. As for the case of 2,5-(2TE)2T, the spectral superimposition resulted in a ΔE ≈ 0.046 eV, ca. 20 times larger than DF, with an overall triplet quantum yield of 14%. Nicely, the ΔE in the case of 2,5-(2TE)2T matches the estimated energy barrier (3.8 kcal mol−1) for a thermally activated, non-radiative and non-reactive relaxation process previously observed.39 When it comes to 1NPH, the energy difference between the ESA maxima is enlarged by another order of magnitude (ΔE ≈ 0.243 eV), with a consequent reduction of the ϕT value, down to 2%. This may be either due to a pure singlet character of the 2Ag state in the case of 1NPH or to the fact that the two triplets are highly interacting and unable to separate into independent triplets. The intermediate behaviors of 2,5-(2TE)2T and PN give strength to the new experimental methodology proposed here, which provides qualitative insight into the inter-triplet binding energy. For these compounds, the similarity between the two spectra is pronounced, even though the small shift between their relative peaks (11 and 3 nm, respectively) suggests a stronger inter-triplet interaction relative to DF in agreement with their lower triplet yield. Furthermore, an additional interrelation can be inferred between the spectral overlay (ΔE) and the deactivation rate constant of the double triplet (kTT calculated as 1/τ2Ag and reported in Table 5): the larger the ΔE value, the more strongly the triplets interact, the smaller the kTT value. Moreover, a good linear correlation has been found for the plot of kTT against the reciprocal of ΔE (R2 = 0.998), see Fig. S15 ESI.† Strong triplet–triplet interaction within 1(TT) may imply the stabilization of the 2Ag state compared to S2, 3(TT) and 5(TT), leading to smaller kinetic constants, like those typical of the decay of a forbidden singlet excited state. In contrast, weaker T–T interactions, which do not cause further stabilization of 2Ag, would allow for a close proximity of the latter to the 3(TT) and above all to the 5(TT) state (crucial for the triplet separation step),52–55 thus opening new pathways to fast independent triplet population. These results are illustrated in Fig. 7 where the two-limiting cases of 1NPH (2Ag constituted by tightly-bound triplets behaving as an ordinary singlet) and DF (2Ag constituted by weakly-bound triplets behaving as the correlated triplet pair, leading to efficient SF) are shown. Fig. 7 also depicts in the case of 1NPH that only the v = 0 and v = 1 states can be likely reached by photoexcitation with the spectrally broad femtosecond pump pulse (375–425 nm). However, as 2Ag is much stabilized relative to 1Bu, the population of the former by 1Bu is still possible. On the other hand, in the case of DF, for which a more bathochromic absorption spectrum was observed, the broad laser excitation is able to access hot vibrational states of 1Bu allowing for the coherent population of the 2Ag state, even if located at higher energy, overcoming the predicted energetic barrier.
| λ T1/nm | λ 2Ag/nm | Δλ/nm | ΔE/cm−1 | ΔE/eV | τ 2Ag/ps | k TT/108 s−1 | ϕ T | |
|---|---|---|---|---|---|---|---|---|
| 1NPH | 490 | 540 | 50 | 1890 | 0.234 | 9800 ± 900 | 1.01 | 0.02 ± 0.01 |
| 2,5-(2TE)2T | 539 | 550 | 11 | 371 | 0.046 | 1800 ± 200 | 5.56 | 0.14 ± 0.02 |
| PN | 523 | 526 | 3 | 109 | 0.013 | 365 ± 35 | 27.4 | 0.80 ± 0.10 |
| DF | 575 | 576 | 1 | 30 | 0.0037 | 89 ± 9 | 112 | 0.97 ± 0.15 |
Our time resolved spectroscopic results show that the compounds bearing the longest polyene chain undergo internal conversion as the only efficient relaxation pathway,56 indicating that the lengthening of the structure is not always a winning strategy for achieving SF.31 On the other hand, not only ultrafast, possibly coherent, 1Bu → 2Ag conversion but also efficient independent triplet separation from the double triplet state of 2Ag nature is enabled in highly dipolar 1-(pyridyl-4-ylethenyl)-4-(p-nitrostyryl)benzene and nitro-styrylfuran. These findings prove that the mediation of virtual ICT is crucial to activating complete and efficient SF in the case of conjugated stilbenoid compounds.57
The SF rate (Bu → Ag conversion) evaluated through the ultrafast spectroscopic data is found to be higher than the diffusional rate governing intermolecular interactions between distinct molecules in solution. Hence, the ultrafast triplet formation kinetics may be related to intramolecular processes occurring at short time delays after excitation, rather than slower diffusion-controlled bimolecular encounters. An intramolecular production of the correlated triplet pair would also be consistent with the absence of significant concentration effects on the steady-state absorption spectra (see Fig. S5, ESI† for the representative case of DF), implying the formation of aggregates to be excluded. However, concentration effects were observed on ϕT measured via ns-TA for DF and PN. The presence of a second nearby chromophore may indeed favor the independent triplet separation, accomplishing the second step of SF and leading to the observed increase of the triplet yield with concentration. Therefore, taken together, our experimental results would be in line with ultrafast INTRAmolecular double triplet formation followed by an INTERmolecular independent triplet separation.20,30,34,58
Conclusions
In this study, the nature of the S2 (1Bu) →S1 (2Ag) conversion in a series of six conjugated stilbenoid compounds was unveiled, by employing state-of-the-art ultrafast spectroscopy techniques while probing the effect of solvent polarizability. Steady-state and time-resolved emission measurements revealed dual emission in less polarizable media and progressive mixing of the bright S2 (1Bu) and dark S1 (2Ag) states as the refractive index of the medium increases, due to the preferential stabilization of the ionic 1Bu state. The closer the two states, the higher the 2Ag emission probability thanks to the stronger vibronic coupling. Femtosecond transient absorption and fluorescence up-conversion disclosed the double nature of the 2Ag state showing not only singlet features that are the lifetime typical of a singlet state (hundreds of ps up to few nanoseconds) and ability to emit, but also a triplet character, indicated by its triplet-like absorption spectrum. These spectroscopic results provided deeper insight into the formation mechanism of the peculiar 2Ag state, likely occurring from a non-relaxed singlet with 1Bu symmetry. The ultrafast appearance time of this triplet-like species hinted at the S2 → S1 decay as the first step of SF, that is, the formation of the correlated triplet pair. TD-DFT calculations revealed that SF would be thermodynamically feasible in these molecules, occurring from the non-relaxed S2 state, possibly according to the coherent mechanism.59–62 Moreover, the difference between the T1 and 2Ag spectral maxima, ΔET1–Ag was found to be in remarkable agreement with the observed triplet yield and 2Ag deactivation rate constant. Hence, this work succeeded in providing a simple experimental method, based only on the spectral comparison, to gain qualitative insight into the ease of separation of the 1(TT), which then enables efficient SF. In fact, when dealing with the thiophene and naphthyl containing compounds low triplet quantum yields were obtained, possibly due to the large binding energy between the coupled triplets which traps the triplet excitons within the 2Ag state (large energy differences between the T1 and 2Ag spectral maxima, ΔET1–Ag). Hence, even though the highly conjugated polyene-like structures enable the ultrafast formation of the correlated triplet pair, the large binding energy prevents the efficient completion of SF. Surprisingly, in the presence of small ΔET1–Ag, a triplet yield of 140% and 120% was found in concentrated solutions of the nitro-styrylfuran (DF) and nitro-pyridine derivative (PN), respectively, pointing to the involvement of a multiple exciton generation phenomenon as SF in the excited-state evolution. Different from all the other molecules of the series, DF and PN show a remarkable ICT character provided by the highly dipolar structure, thanks to the presence of the nitro group, being one of the strongest electron-acceptors and the electron-rich furan or pyridine units at opposite sites. The mediation of a virtual CT state in the case of DF and PN may favour the coherent SF phenomenon aiding also the triplet pair decorrelation29,33,57 and allowing for the efficient achievement of the second step of the process. Ultimately, this study constitutes a step forward in understanding the role of the Ag state in the excited-state evolution of highly conjugated stilbenoid compounds, and aims at guiding the design of new small polyene-like molecules capable of SF. A successful strategy to implement efficient SF could be based on tuning the charge-transfer character, by functionalising the molecular structure with appropriate substituents, in order to achieve the best trade-off between conjugation (ultrafast population of 2Ag) and decorrelation of triplet pairs (separation of independent triplets with high yield).Author contributions
L. M.: data curation, investigation, methodology, writing–original draft, and writing–review & editing; M. A.: data curation, investigation, methodology, and writing–review & editing; F. E.: investigation, funding acquisition, and writing–review & editing; I. S.: investigation, resources, and writing–review & editing; A. S.: conceptualization, data curation, supervision, and writing–review & editing; B. C.: investigation, supervision, writing–original draft, and writing–review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work has been funded by the European Union – NextGeneration EU under the Italian Ministry of University and Research (MUR) National Innovation Ecosystem grant ECS00000041 – VITALITY. The authors acknowledge the University of Perugia and the MUR for support within the project Vitality.Notes and references
- M. B. Smith and J. Michl, Annu. Rev. Phys. Chem., 2013, 64, 361–386 CrossRef CAS PubMed.
- A. J. Baldacchino, M. I. Collins, M. P. Nielsen, T. W. Schmidt, D. R. McCamey and M. J. Y. Tayebjee, Chem. Phys. Rev., 2022, 3, 021304 CrossRef CAS.
- J. J. Burdett, A. M. Müller, D. Gosztola and C. J. Bardeen, J. Chem. Phys., 2010, 133, 144506 CrossRef PubMed.
- A. Rao, M. W. B. Wilson, J. M. Hodgkiss, S. Albert-Seifried, H. Bässler and R. H. Friend, J. Am. Chem. Soc., 2010, 132, 12698–12703 CrossRef CAS PubMed.
- M. W. B. Wilson, A. Rao, K. Johnson, S. Gélinas, R. di Pietro, J. Clark and R. H. Friend, J. Am. Chem. Soc., 2013, 135, 16680–16688 CrossRef CAS PubMed.
- L. Ma, K. Zhang, C. Kloc, H. Sun, M. E. Michel-Beyerle and G. G. Gurzadyan, Phys. Chem. Chem. Phys., 2012, 14, 8307 RSC.
- S. W. Eaton, L. E. Shoer, S. D. Karlen, S. M. Dyar, E. A. Margulies, B. S. Veldkamp, C. Ramanan, D. A. Hartzler, S. Savikhin, T. J. Marks and M. R. Wasielewski, J. Am. Chem. Soc., 2013, 135, 14701–14712 CrossRef CAS PubMed.
- J. C. Johnson, A. J. Nozik and J. Michl, J. Am. Chem. Soc., 2010, 132, 16302–16303 CrossRef CAS PubMed.
- I. Papadopoulos, M. J. Álvaro-Martins, D. Molina, P. M. McCosker, P. A. Keller, T. Clark, Á. Sastre-Santos and D. M. Guldi, Adv. Energy Mater., 2020, 10, 2001496 CrossRef CAS.
- N. V. Korovina, N. F. Pompetti and J. C. Johnson, J. Chem. Phys., 2020, 152, 040904 CrossRef CAS PubMed.
- S. N. Sanders, E. Kumarasamy, A. B. Pun, K. Appavoo, M. L. Steigerwald, L. M. Campos and M. Y. Sfeir, J. Am. Chem. Soc., 2016, 138, 7289–7297 CrossRef CAS PubMed.
- Y. Kasai, Y. Tamai, H. Ohkita, H. Benten and S. Ito, J. Am. Chem. Soc., 2015, 137, 15980–15983 CrossRef CAS PubMed.
- J. Hu, K. Xu, L. Shen, Q. Wu, G. He, J.-Y. Wang, J. Pei, J. Xia and M. Y. Sfeir, Nat. Commun., 2018, 9, 2999 CrossRef PubMed.
- E. Busby, J. Xia, Q. Wu, J. Z. Low, R. Song, J. R. Miller, X.-Y. Zhu, L. M. Campos and M. Y. Sfeir, Nat. Mater., 2015, 14, 426–433 CrossRef CAS PubMed.
- B. Carlotti, I. K. Madu, H. Kim, Z. Cai, H. Jiang, A. K. Muthike, L. Yu, P. M. Zimmerman and T. Goodson, Chem. Sci., 2020, 11, 8757–8770 RSC.
- A. K. Muthike, B. Carlotti, I. K. Madu, H. Jiang, H. Kim, Q. Wu, L. Yu, P. M. Zimmerman and T. Goodson, J. Phys. Chem. B, 2021, 125, 5114–5131 CrossRef CAS PubMed.
- K. Miyata, F. S. Conrad-Burton, F. L. Geyer and X.-Y. Zhu, Chem. Rev., 2019, 119, 4261–4292 CrossRef CAS PubMed.
- C. C. Gradinaru, J. T. M. Kennis, E. Papagiannakis, I. H. M. van Stokkum, R. J. Cogdell, G. R. Fleming, R. A. Niederman and R. van Grondelle, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 2364–2369 CrossRef CAS PubMed.
- S. N. Sanders, E. Kumarasamy, A. B. Pun, M. L. Steigerwald, M. Y. Sfeir and L. M. Campos, Angew. Chem., 2016, 128, 3434–3438 CrossRef.
- M. T. Trinh, Y. Zhong, Q. Chen, T. Schiros, S. Jockusch, M. Y. Sfeir, M. Steigerwald, C. Nuckolls and X. Zhu, J. Phys. Chem. C, 2015, 119, 1312–1319 CrossRef CAS.
- A. J. Musser, M. Al-Hashimi, M. Maiuri, D. Brida, M. Heeney, G. Cerullo, R. H. Friend and J. Clark, J. Am. Chem. Soc., 2013, 135, 12747–12754 CrossRef CAS PubMed.
- J. Ren, Q. Peng, X. Zhang, Y. Yi and Z. Shuai, J. Phys. Chem. Lett., 2017, 8, 2175–2181 CrossRef CAS PubMed.
- M. R. Antognazza, L. Lüer, D. Polli, R. L. Christensen, R. R. Schrock, G. Lanzani and G. Cerullo, Chem. Phys., 2010, 373, 115–121 CrossRef CAS.
- O. Varnavski, N. Abeyasinghe, J. Aragó, J. J. Serrano-Pérez, E. Ortí, J. T. López Navarrete, K. Takimiya, D. Casanova, J. Casado and T. Goodson, J. Phys. Chem. Lett., 2015, 6, 1375–1384 CrossRef CAS PubMed.
- A. D. Chien, A. R. Molina, N. Abeyasinghe, O. P. Varnavski, T. Goodson and P. M. Zimmerman, J. Phys. Chem. C, 2015, 119, 28258–28268 CrossRef CAS.
- B. S. Hudson and B. E. Kohler, J. Chem. Phys., 1973, 59, 4984–5002 CrossRef CAS.
- A. J. Musser and J. Clark, Annu. Rev. Phys. Chem., 2019, 70, 323–351 CrossRef CAS PubMed.
- L. Wang, T.-S. Zhang, L. Fu, S. Xie, Y. Wu, G. Cui, W.-H. Fang, J. Yao and H. Fu, J. Am. Chem. Soc., 2021, 143, 5691–5697 CrossRef CAS PubMed.
- H. Kim and P. M. Zimmerman, Phys. Chem. Chem. Phys., 2018, 20, 30083–30094 RSC.
- L. Wang, S. Bai, Y. Wu, Y. Liu, J. Yao and H. Fu, Angew. Chem., Int. Ed., 2020, 59, 2003–2007 CrossRef CAS PubMed.
- S. N. Sanders, E. Kumarasamy, A. B. Pun, M. L. Steigerwald, M. Y. Sfeir and L. M. Campos, Chem, 2016, 1, 505–511 CAS.
- R. Ringström, F. Edhborg, Z. W. Schroeder, L. Chen, M. J. Ferguson, R. R. Tykwinski and B. Albinsson, Chem. Sci., 2022, 13, 4944–4954 RSC.
- K. Miyata, Y. Kurashige, K. Watanabe, T. Sugimoto, S. Takahashi, S. Tanaka, J. Takeya, T. Yanai and Y. Matsumoto, Nat. Chem., 2017, 9, 983–989 CrossRef CAS PubMed.
- L. Mencaroni, B. Carlotti, F. Elisei, A. Marrocchi and A. Spalletti, Chem. Sci., 2022, 13, 2071–2078 RSC.
- K. Aryanpour, T. Dutta, U. N. V. Huynh, Z. V. Vardeny and S. Mazumdar, Phys. Rev. Lett., 2015, 115, 267401 CrossRef PubMed.
- E. A. Margulies, C. E. Miller, Y. Wu, L. Ma, G. C. Schatz, R. M. Young and M. R. Wasielewski, Nat. Chem., 2016, 8, 1120–1125 CrossRef CAS PubMed.
- R. M. Young and M. R. Wasielewski, Acc. Chem. Res., 2020, 53, 1957–1968 CrossRef CAS PubMed.
- E. Sundin, R. Ringström, F. Johansson, B. Küçüköz, A. Ekebergh, V. Gray, B. Albinsson, J. Mårtensson and M. Abrahamsson, J. Phys. Chem. C, 2020, 124, 20794–20805 CrossRef CAS.
- G. Ginocchietti, G. Galiazzo, U. Mazzucato and A. Spalletti, Photochem. Photobiol. Sci., 2005, 4, 547–553 CrossRef CAS PubMed.
- E. Marri, G. Galiazzo, F. Masetti, U. Mazzucato, C. Zuccaccia and A. Spalletti, J. Photochem. Photobiol., A, 2005, 174, 181–186 CrossRef CAS.
- G. Bartocci, A. Spalletti, R. S. Becker, F. Elisei, S. Floridi and U. Mazzucato, J. Am. Chem. Soc., 1999, 121, 1065–1075 CrossRef CAS.
- E. Marri, G. Galiazzo and A. Spalletti, Photochem. Photobiol. Sci., 2004, 3, 205–210 CrossRef CAS PubMed.
- I. Baraldi, G. Ginocchietti, U. Mazzucato and A. Spalletti, Chem. Phys., 2007, 337, 168–176 CrossRef CAS.
- B. Carlotti, A. Cesaretti, G. Cacioppa, F. Elisei, I. Odak, I. Škorić and A. Spalletti, J. Photochem. Photobiol., A, 2019, 368, 190–199 CrossRef CAS.
- L. Mencaroni, B. Carlotti, A. Cesaretti, F. Elisei, A. Grgičević, I. Škorić and A. Spalletti, Photochem. Photobiol. Sci., 2020, 19, 1665–1676 CrossRef CAS PubMed.
- B. Carlotti, F. Elisei, U. Mazzucato and A. Spalletti, Phys. Chem. Chem. Phys., 2015, 17, 14740–14749 RSC.
- G. Ginocchietti, E. Cecchetto, L. De Cola, U. Mazzucato and A. Spalletti, Chem. Phys., 2008, 352, 28–34 CrossRef CAS.
- H.-D. Zhang, Q. Qiao, R.-X. Xu and Y. Yan, J. Chem. Phys., 2016, 145, 204109 CrossRef PubMed.
- S. Ciorba, G. Galiazzo, U. Mazzucato and A. Spalletti, J. Phys. Chem. A, 2010, 114, 10761–10768 CrossRef CAS PubMed.
- O. Yushchenko, G. Licari, S. Mosquera-Vazquez, N. Sakai, S. Matile and E. Vauthey, J. Phys. Chem. Lett., 2015, 6, 2096–2100 CrossRef CAS PubMed.
- M. R. Momeni, J. Chem. Theory Comput., 2016, 12, 5067–5075 CrossRef CAS PubMed.
- M. J. Y. Tayebjee, S. N. Sanders, E. Kumarasamy, L. M. Campos, M. Y. Sfeir and D. R. McCamey, Nat. Phys., 2017, 13, 182–188 Search PubMed.
- M. Chen, M. D. Krzyaniak, J. N. Nelson, Y. J. Bae, S. M. Harvey, R. D. Schaller, R. M. Young and M. R. Wasielewski, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 8178–8183 CrossRef CAS PubMed.
- B. S. Basel, J. Zirzlmeier, C. Hetzer, B. T. Phelan, M. D. Krzyaniak, S. R. Reddy, P. B. Coto, N. E. Horwitz, R. M. Young, F. J. White, F. Hampel, T. Clark, M. Thoss, R. R. Tykwinski, M. R. Wasielewski and D. M. Guldi, Nat. Commun., 2017, 8, 15171 CrossRef CAS PubMed.
- H. Sakai, R. Inaya, H. Nagashima, S. Nakamura, Y. Kobori, N. V. Tkachenko and T. Hasobe, J. Phys. Chem. Lett., 2018, 9, 3354–3360 CrossRef CAS PubMed.
- S. M. Bachilo, C. W. Spangler and T. Gillbro, Chem. Phys. Lett., 1998, 283, 235–242 CrossRef CAS.
- J. D. Schultz, J. Y. Shin, M. Chen, J. P. O’Connor, R. M. Young, M. A. Ratner and M. R. Wasielewski, J. Am. Chem. Soc., 2021, 143, 2049–2058 CrossRef CAS PubMed.
- M. Chen, A. F. Coleman, R. M. Young and M. R. Wasielewski, J. Phys. Chem. C, 2021, 125, 6999–7009 CrossRef CAS.
- A. M. Alvertis, S. Lukman, T. J. H. Hele, E. G. Fuemmeler, J. Feng, J. Wu, N. C. Greenham, A. W. Chin and A. J. Musser, J. Am. Chem. Soc., 2019, 141, 17558–17570 CrossRef CAS PubMed.
- W.-L. Chan, T. C. Berkelbach, M. R. Provorse, N. R. Monahan, J. R. Tritsch, M. S. Hybertsen, D. R. Reichman, J. Gao and X.-Y. Zhu, Acc. Chem. Res., 2013, 46, 1321–1329 CrossRef CAS PubMed.
- G. D. Scholes, G. R. Fleming, L. X. Chen, A. Aspuru-Guzik, A. Buchleitner, D. F. Coker, G. S. Engel, R. van Grondelle, A. Ishizaki, D. M. Jonas, J. S. Lundeen, J. K. McCusker, S. Mukamel, J. P. Ogilvie, A. Olaya-Castro, M. A. Ratner, F. C. Spano, K. B. Whaley and X. Zhu, Nature, 2017, 543, 647–656 CrossRef CAS PubMed.
- T. W. Kim, P. Kim, A. W. Mills, A. Chakraborty, S. Kromer, A. J. S. Valentine, F. N. Castellano, X. Li and L. X. Chen, J. Phys. Chem. C, 2022, 126, 11487–11497 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Details of the experimental methods; spectral and fluorescence properties; triplet properties; ultrafast spectroscopy; phosphorescence measurements; and quantum-mechanical calculations. See DOI: https://doi.org/10.1039/d3cp02805d |
| This journal is © the Owner Societies 2023 |