
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNuclear quantum dynamics on the ground electronic state of neutral silver dimer 107Ag109Ag probed by femtosecond NeNePo spectroscopy†
Jiaye
Jin
 *,
Max
Grellmann
and
Knut R.
Asmis
*
*,
Max
Grellmann
and
Knut R.
Asmis
*
Wilhelm-Ostwald-Institut für Physikalische und Theoretisch Chemie, Universität Leipzig, Linnéstr. 2, 04103 Leipzig, Germany. E-mail: Jiaye.Jin@uni-leipzig.de; Knut.Asmis@uni-leipzig.de
First published on 29th August 2023
Abstract
The nuclear quantum dynamics on the ground electronic state of the neutral silver dimer 107Ag109Ag are studied by femtosecond (fs) pump–probe spectroscopy using the ‘negative ion – to neutral – to positive ion’ (NeNePo) excitation scheme. A vibrational wave packet is prepared on the X1Σ+g state of Ag2via photodetachment of mass-selected, cryogenically cooled Ag2− using a first ultrafast pump laser pulse. The temporal evolution of the wave packet is then probed by an ultrafast probe pulse via resonant multiphoton ionization to Ag2+. Frequency analysis of the fs-NeNePo spectra obtained for a single isotopologue and pump–probe delay times up to 60 ps yields the harmonic (ωe = 192.2 cm−1), quadratic anharmonic (ωexe = 0.637 cm−1) and cubic anharmonic (ωeye = 3 × 10−4 cm−1) constants for the X1Σ+g state of neutral Ag2. The fs-NeNePo spectra obtained at different pump wavelengths provide insight into the excitation mechanism. At a pump wavelength of 510 nm instead of 1010 nm, resonant excitation of a short-lived electronically excited state of the anion followed by autodetachment results in population of higher-energy vibrational levels of the neutral ground state. In contrast, at 1140 nm dynamics with a slightly shorter beating period and different relative phase are observed. The present study demonstrates that isotopologue-specific fs-NeNePo spectroscopy provides accurate vibrational constants of mass-selected neutral clusters in their electronic ground state in the terahertz spectral region, which remains difficult to obtain directly in the frequency domain with any other type of spectroscopy of comparable sensitivity.
1. Introduction
Metal clusters possess remarkable photoelectric and catalytic properties, making them subject of current research.1,2 Studies on metal clusters isolated in the gas phase are particularly important. They do not only provide detailed information on their intrinsic properties, like geometric and electronic structure, molecular vibrations, bonding energies and time scales for internal energy redistribution in the absence of a perturbing environment, but also represent ideal model systems for benchmarking modern quantum chemistry methods.3–5The geometric structure of clusters in the gas phase can be probed by various spectroscopic techniques, of which infrared action spectroscopy is probably one of the most powerful and generally applicable methods.6–8 However, the fundamental vibrational frequencies of small metal clusters lie in the far-IR region and therefore such experiments in the frequency domain are typically limited to free-electron-laser facilities.7 A complementary approach to obtain vibrational information is femtosecond (fs) pump–probe spectroscopy.9–11 Here, ultrafast laser pulses are used to generate a vibrational wave packet and probe its time evolution. The obtained transient traces provide fruitful information for investigating coherent nuclear motion on a particular potential energy surface (PES) as well as energy flow over multiple PESs on a femtosecond to picosecond (ps) time scale.
Femtosecond pump–probe experiments have been extensively applied to study excited electronic states of metal clusters in the gas phase.4,9,12 From such experiments, in which the transient signal is typically monitored by detecting either photoabsorption, photoemission, photoelectron emission or fragment-ion formation, information on low-energy vibrational frequencies and even anharmonicities can be obtained.13,14 Characterizing vibrational coherences on the ground state PES of neutral molecules is more challenging, but can be achieved, for example, by impulsive stimulated Raman scattering,15 pulse interference,16 and dump-pump excitation schemes.17,18 However, experiments on neutral clusters typically suffer from the lack of mass selection prior to laser irradiation. The negative-neutral-positive (NeNePo) excitation scheme overcomes this limitation.19 Mass-selected anions are excited with a first femtosecond laser pulse centred at a suitable wavelength, leading to (single photon) photodetachment of an electron and the preparation of a wave packet on the electronic ground state of the neutral species. The nuclear dynamics are then probed by a second, time-delayed femtosecond laser pulse, which ionizes the neutral species, typically in a resonant, multiphoton process. The mass-selected cation yield as a function of the delay time between pump and probe pulse yields the fs-NeNePo spectrum. Fs-NeNePo spectroscopy has been mainly applied to study the wave-packet dynamics of neutral metal as well as metal-containing clusters, namely, Agn (n = 2, 3, 4, 5 and 9),19–25 Ag2Au,26 and Cu(H2O)1,2.27,28
Here we combine two-color fs-NeNePo spectroscopy with a cryogenic ion trap tandem mass spectrometer29 to characterize the wave-packet dynamics on the electronic ground state of the silver dimer 107Ag109Ag, isotopologue-specifically for the first time. Previous, lower frequency resolution single-color NeNePo experiments revealed quantum beats with a period of 180 fs up to delay times of 2.5 ps, corresponding to a vibrational frequency of roughly 185 cm−1.20,30,31 In the present study, we probe a single isotopologue at considerably longer delay times, up to 60 ps, observe multiple recurrences of the vibrational wave packet involving the 22 lowest vibrational levels and hence obtain more detailed vibrational information with high frequency resolution (0.5 cm−1), including higher-order vibrational constants, together with new insights into the photoexcitation mechanism.
2. Methods
The experiments were performed using a home-built tandem mass spectrometer with an integrated and temperature-controllable radio-frequency linear quadrupole ion trap and ultrashort laser pulses produced from an amplified Ti:sapphire femtosecond laser system.2.1 Tandem mass spectrometer
The experimental setup (shown in Fig. 1) is similar to that used in previous NeNePo studies with the exception of a newly installed cryogenic quadrupole ion trap.19–21,24,26 Silver cluster anions are produced by aggregation using a silver target (99.99%, Kurt J. Lesker) as the anode of a DC magnetron sputtering source (TORUS, Kurt J. Lesker) enclosed in a liquid nitrogen cooled source chamber. The plasma is produced and stabilized by applying a continuous argon (99.999%, Air Liquide) and helium (99.999%, Air Liquide) gas flow. The beam of neutral and ionic particles expands through a 7 mm diameter nozzle and subsequently through a 2 mm diameter skimmer, which is held at a sufficiently positive potential in order to repel cationic species. The beam of anions is collimated in a He-filled radio frequency (RF) decapole ion guide (Q0 in Fig. 1), then deflected into 90° by the quadrupole ion deflector D0 into the first RF quadrupole mass filter Q1. Silver dimer anions, corresponding to the isotopologue 107Ag109Ag−, are mass-selected by the first quadrupole mass filter, deflected into 90° by the deflector D1 and continuously accumulated in a helium-filled linear quadrupole ion trap Q2 (0.20 mbar partial pressure). Q2 is directly attached to the second stage of a closed-cycle He-cryostat (Sumitomo SRDK-408E2, F50H compressor). The trap temperature is controlled using a Cernox sensor and a heating cartridge connected to a Lake Shore Model 335 temperature controller. Collisions with the He buffer gas thermalize the trapped anions close to the trap temperature, which is held at 20 K.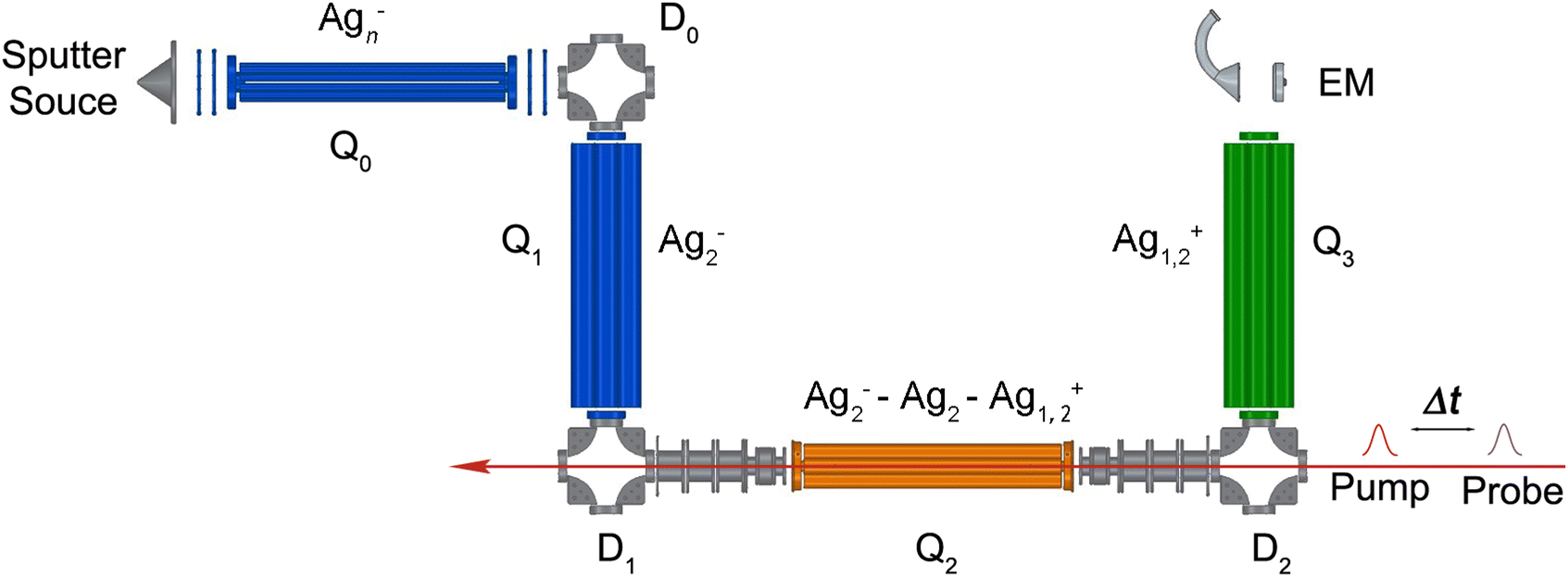 | ||
| Fig. 1 Experimental scheme of the linear quadrupole ion-trap tandem mass spectrometer (see text for details). D0–2: electrostatic 90° quadrupole deflectors; Q0: radiofrequency decapole ion guide; Q1,3: quadrupole mass filters; Q2: cryogenically cooled, radio-frequency linear quadrupole ion trap; EM: channel electron multiplier detector with conversion dynode. The pump and probe femtosecond laser pulses propagate along the ion-trap axis. | ||
The pump and probe femtosecond laser pulses are applied along the ion-trap axis and focused near the center of the ion trap. The trapped anions can absorb photons, leading to photodetachment of electrons and subsequent photoionization of neutral clusters. Positively charged ions are no longer confined along the trap axis and are expelled from the trap. Cations leaving the trap via the exit lens are deflected into 90° (D2) and focused into a second mass-selecting quadrupole mass filter (Q3). The yield of mass-selected cations is measured by a subsequent off-axis electron multiplier detector (EM). NeNePo spectra are obtained by monitoring the mass-selected cation counts for 2000 ms as function of the delay time between two femtosecond laser pulses with a step interval of 20 fs.
2.2 Laser system
The ultrafast laser setup is shown in Fig. 2. A chirped-pulse Ti:sapphire regenerative-amplifier system (Solstice Ace, Spectra Physics) is used to generate a linearly polarized femtosecond laser pulse centred at 800 nm, with a duration of approximately 35 fs, at a repetition rate of 1 kHz and with pulse energy of 7 mJ. The beam is then split into two arms (a pump and a probe arm) using a 50–50 beam splitter. In the pump arm, the photodetachment pulse, tunable in wavelength between 510–1140 nm (1.1–2.4 eV), is generated using an ultrafast optical parametric oscillator/amplifier laser system (OPA, TOPAS Prime, Light Conversion) combined with a wavelength extender (NIRUVIS, Light Conversion). In the probe arm, the photoionization probe pulse (402 nm, 3.1 eV) is obtained by frequency doubling of the 800 nm pulse in a harmonic generation unit (S/T-HG, Spectra-Physics) and sent through a prism-pair compressor to minimize group-velocity dispersion. The probe pulse is then directed onto a linear translation delay stage (DLS225, Newport), which produces a variable delay time relative to the pump pulse. The minimum delay-time step on the delay stage is 1 fs. The relative light polarization of the probe pulse to the pump pulse is controlled by a MgF2λ/2 waveplate (EKSMA Optics). The duration of the pulses is measured using an autocorrelator (Mini TPA, A.P.E.). Typically, the duration of the pump pulse is approximately 35–50 fs full width at half maximum (FWHM) (sech2), while the duration of the probe pulse is about 30–40 fs (sech2). The frequency spectra of the pulses are characterized by a compact CCD spectrometer (CCS200, Thorlabs). Two pulses are collinearly combined and propagate along the ion-trap axis through a thin CaF2 lens (f = +1000 mm), resulting in a 0.35 mm diameter focal point of the 402 nm radiation. The time zero (delay time = 0 fs) between the two laser pulses is determined by measuring the current (via a picoammeter, Keithley Model 6485) on a retractable copper plate mounted in the vacuum on which the two pulses interfere.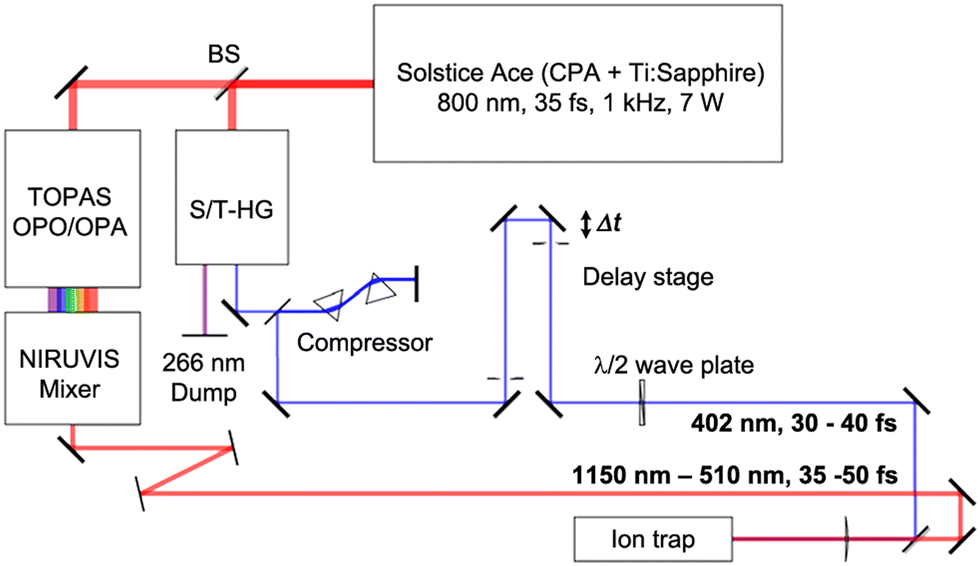 | ||
| Fig. 2 Schematic of the laser setup (see text for details) used for generating fs pump pulses (red path) for photdetachment and temporally delayed probe pulses (blue path) for photoionization. | ||
2.3 Frequency analysis
Frequency analysis of the oscillatory part for NeNePo spectra is performed by using a fast Fourier transform (FFT) or a short time windowed Fourier transform (STFT) with a suitable window interval. First, the oscillatory part of the spectrum is extracted by removing the background using the symmetric least-square method.32,33 A Kaiser window function (α = 4.5) is used to balance the main-lobe width and side-lobe level. Zero values are padded to the oscillatory part until 23 times the data length, in order to minimize the influence of a non-integral cut of the period. The period and the initial phase are determined by fitting the oscillatory components to a sinusoidal function containing an exponential damping term, f(t) = Ae−t/τ![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) sin(2π/T + ϕ), where A is the oscillation amplitude, τ is the dephasing life time, T is the period of the oscillation, and ϕ is the initial phase of the oscillation.
sin(2π/T + ϕ), where A is the oscillation amplitude, τ is the dephasing life time, T is the period of the oscillation, and ϕ is the initial phase of the oscillation.
2.4 Excitation scheme
The excitation scheme used in the present experiments is shown in Fig. 3. The first linear polarized femtosecond laser pulse (pump pulse), centred at a wavelength of 510 nm (bandwidth: 9.6 nm, pulse energy: 1 μJ) and with a pulse duration of 45 fs, photodetaches an electron from Ag2−, launching a vibrational wave packet exclusively on the neutral electronic ground state of Ag2 (X1Σ+g).34–36 The corresponding photon energy (2.43 eV) substantially exceeds the electron affinity (EA) of Ag2− (1.1 eV),34–36 but remains below the first triplet excited electronic state of Ag2 (a3Σ+u), predicted 1.6 eV above the X1Σ+g state.20,37 The evolution of the nuclear motion is then probed using a second femtosecond laser pulse (probe pulse, centred at 402 nm, 40 fs, 5.0 nm FWHM, 1.5 μJ), linearly polarized at the magic angle to the pump pulse, in order to avoid rotation dynamics, which resonantly ionizes neutral Ag2 by 1 + 2 multiphoton ionization via the strong A1Σ+u ← X1Σ+g transition.30,38 Cations are automatically expelled from the ion trap and are subsequently mass-selectively detected as a function of the delay time. The fs-NeNePo spectrum is frequency-analysed using fast Fourier transformation with a resolution down to about 0.5 cm−1 (delay time ≈ 60 ps).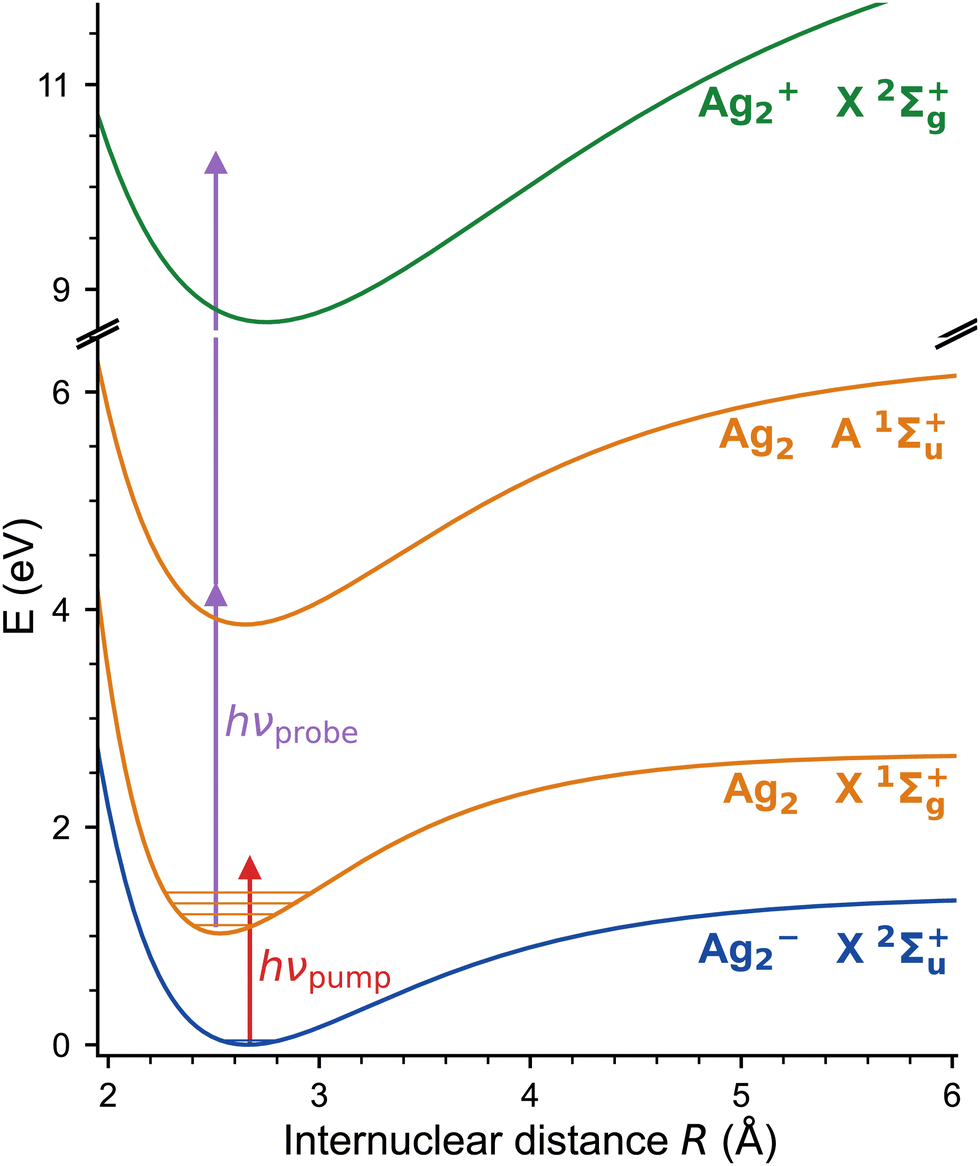 | ||
| Fig. 3 NeNePo pump–probe excitation scheme for the silver dimer over three charge states (blue: anion, orange: neutral, green: cation). Mass-selected Ag2− anions X2Σ+u are photodetached by the first ultrafast laser pulse (red arrow) to the electronic ground state of neutral Ag2 X1Σ+g. The wave-packet dynamics are probed by a second, delayed ultrafast laser pulse (purple arrows) via 1 + 2 resonance-enhanced multiphoton ionization. The potential energy curves correspond to the Morse potential obtained from the published molecular constants.30,34–38 | ||
3. Results
The obtained fs-NeNePo spectrum is shown in Fig. 4a over the complete range of delay times (−3 ps to 61 ps) and with an interval of 20 fs. The other panels (Fig. 4b–d) contain magnified sections of the transient trace. For negative delay times, i.e., when the probe pulse arrives prior to the pump pulse, the fs-NeNePo spectrum (see Fig. 4b) shows a low and constant signal of Ag2+ cations, indicating that cation formation is inefficient and without clear time-dependence. A sharp rise in cation signal is observed within 180 fs around time zero, followed by clear oscillatory features. The first maximum of the Ag2+ signal appears at a delay time of 180 fs, longer than the width of the cross-correlation signal of the two femtosecond pulses and hence reflecting the motion of the wave packet. The oscillatory component of the Ag2+ signal has a period of about 180 fs and is attributed to vibrational wave-packet dynamics on the ground state potential, consistent with the previous, single-color fs-NeNePo study using 400 nm laser pulses.20 The fs-NeNePo spectrum obtained monitoring the monomer 107Ag+ exhibits a first maximum at 220 fs (see Fig. S1 in ESI†) followed by the same period of about 180 fs, indicating a different Frank–Condon window for the formation of Ag+ fragment. The oscillatory part of the Ag2+ signal fades out after about ≈4 ps, but reappears for delay times of 27 ps (Fig. 4c) and 54 ps (Fig. 4d) with periods of 180 to 200 fs. These wave packet recurrences are clear signatures of the anharmonicity of the potential.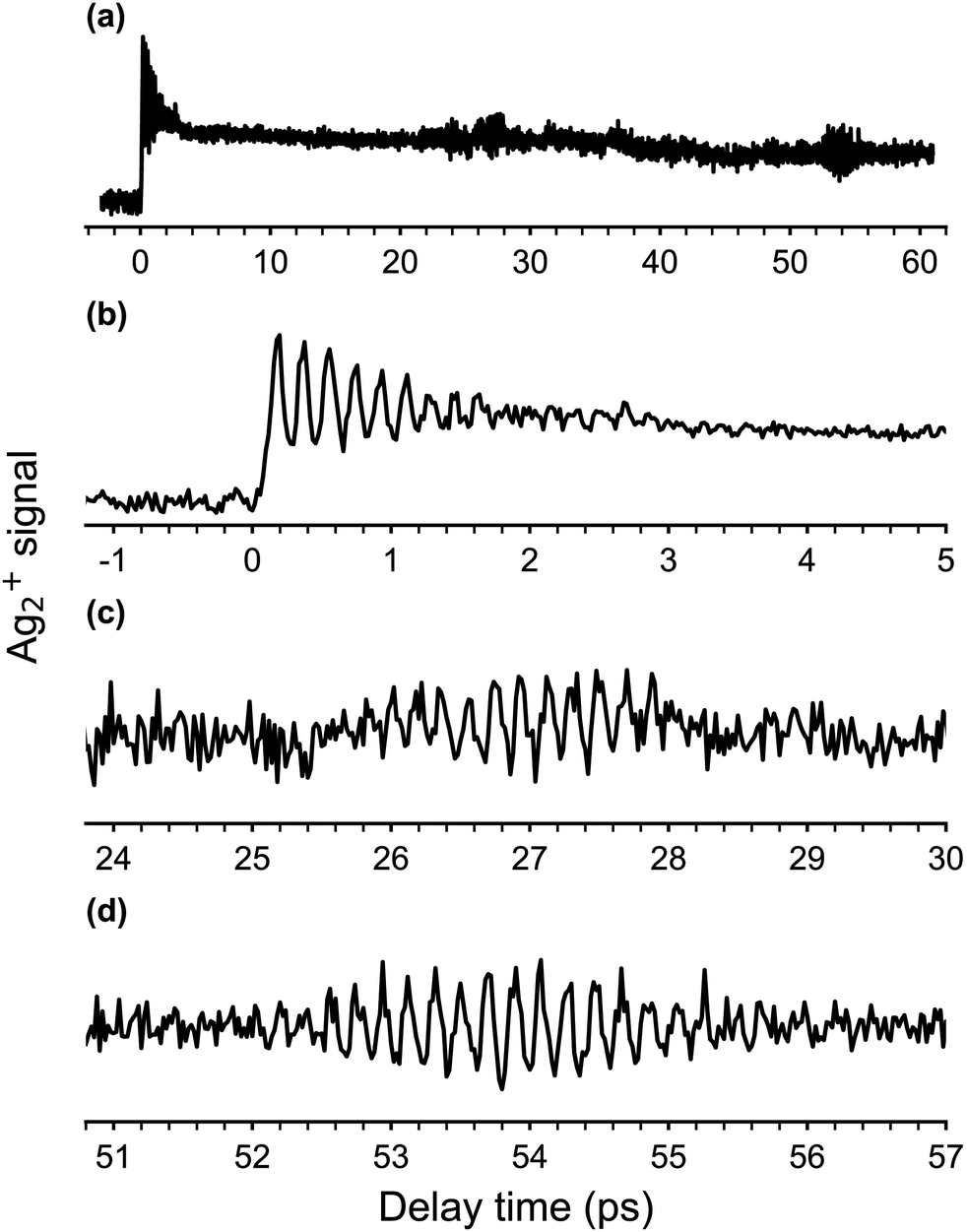 | ||
| Fig. 4 (a) Two-color fs-NeNePo spectrum (λpump = 510 nm, λprobe = 402 nm, magic angle polarization) for the Ag2−/Ag2/Ag2+ obtain by monitoring the 107,109Ag2+ cation signal over a delay time range from −3 to 61 ps. (b)–(d) Magnified section of the fs-NeNePo spectrum shown in (a) for delay times of (b) −1 ps to 5 ps, (c) 24 ps to 30 ps (first recurrence) and (d) 51 ps to 57 ps (second recurrence). | ||
We also measured fs-NeNePo spectra of the pump pulse centred at 1010 nm (1.23 eV), i.e., with photon energies close to the vertical detachment energy (VDE) of Ag2−. These transient traces are shown in Fig. 5b in the delay time range from −0.1 ps to 2 ps. We observe a similar signal to that obtained at 510 nm (Fig. 5a), also at longer delay times (see Fig. S2, ESI†). A pump pulse wavelength of 1140 nm (1.09 eV), corresponding to an (average) photon energy slightly below the EA, results in a qualitatively different fs-NeNePo spectrum (see Fig. 5c). The first maximum appears at a similar delay time (with respect to Fig. 5a), but is markedly smaller in intensity than the second maximum. This second maximum appears around 320 fs, roughly 50 fs earlier than in the spectrum obtained at 510 nm in Fig. 5a, and the beating signal has a minimally smaller period of 182 fs. Wave-packet recurrences are not observed, probably due to the lower oscillatory cation signal as a result of the smaller photodetachment cross section close to threshold.
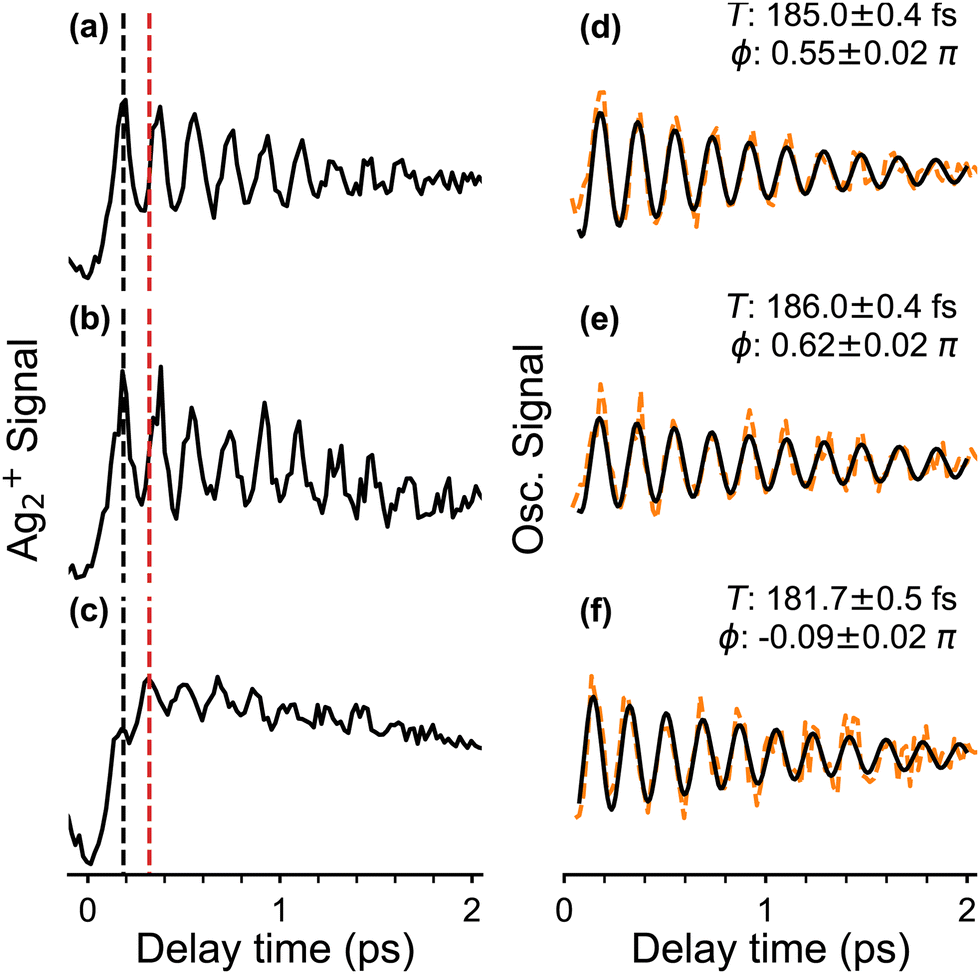 | ||
| Fig. 5 Ag2+ cation signal (a)–(c) and oscillatory components (d)–(f) for delay times from −0.1 to 2.0 ps. Three different pump wavelengths λpump are applied (a) and (d) λpump = 510 nm, (b) and (e) λpump = 1010 nm and (c) and (f) λpump = 1140 nm. Black and red dashed lines in (a)–(c) mark the delay time of different maxima in the fs-NeNePo spectra. A sinusoidal oscillator containing an exponential damping is assumed for fitting the oscillatory curves. See Section 2.3 for details. The fitting period (in fs) and the initial phase are indicated. | ||
The initial phase (right panel in Fig. 5) of the oscillatory part contributing to the NeNePo spectra is obtained at different pump laser wavelengths is determined by fitting to a sinusoidal function with an exponential damping term. The two NeNePo spectra obtained from pump wavelengths of 510 nm and 1010 nm (Fig. 5a and b) are characterized to have a similar beating period as well as a similar oscillation phase (Fig. 5d and e). In contrast, at 1140 nm (Fig. 5f) the beating frequency is slightly shorter (182 fs vs. ≥185 fs) and also the phase is substantially different (−0.1π rad vs. 0.5π rad). This indicates that energetically higher lying vibrational levels of the neutral Ag2 ground state are less efficiently populated at photodetachment energies close to threshold, effectively changing the launching point for the wave packet. Furthermore, interference effects resulting from two-photon photodetachment may also contribute to the observed phase of fs-NeNePo spectrum at 1140 nm.39
Frequency domain spectra are obtained from the transient traces using the fast Fourier transform (FFT). For pump wavelengths of 510 nm and 1010 nm, NeNePo spectra with delay times up to 61 ps were obtained and the corresponding frequency spectra are presented in Fig. 6. (See Fig. S3, ESI† for a comparison of the FFT spectra obtained at early delay times for all three pump wavelengths discussed above.). The FFT spectrum of the 510 nm/402 nm fs-NeNePo spectrum (in Fig. 6a) reveals more than 20 roughly equidistant lines in the spectral range corresponding to the wavenumbers from 160 cm−1 to 191 cm−1 with a spacing of 1.2 cm−1. We assign these frequency bands (from right to left) to the vibrational interval ΔGν+1/2 = G(ν + 1) − G(ν) of adjacent vibrational eigenstates with increasing vibrational quantum number ν of the X1Σ+g state of neutral Ag2. The highest energy frequency band at 190.9 cm−1 is assigned to ΔG1/2 (v = 0), in agreement with ω0 derived from the previously published molecular constants of 107Ag109Ag (ωe = 192.4 cm−1 and ωexe = 0.643 cm−1).38,40 Note, the absolute assignment of the populated vibrational levels based on the FFT spectrum alone (see Fig. S4, ESI† for a magnified section of the FFT spectrum around 191 cm−1) is not straightforward, but the assignment of relative vibrational quantum numbers is. An assignment to vibrational coherence from the electronic ground state of the anion (X2Σ+u), which exhibits a vibrational frequency of 145 cm−1, can be ruled out.34
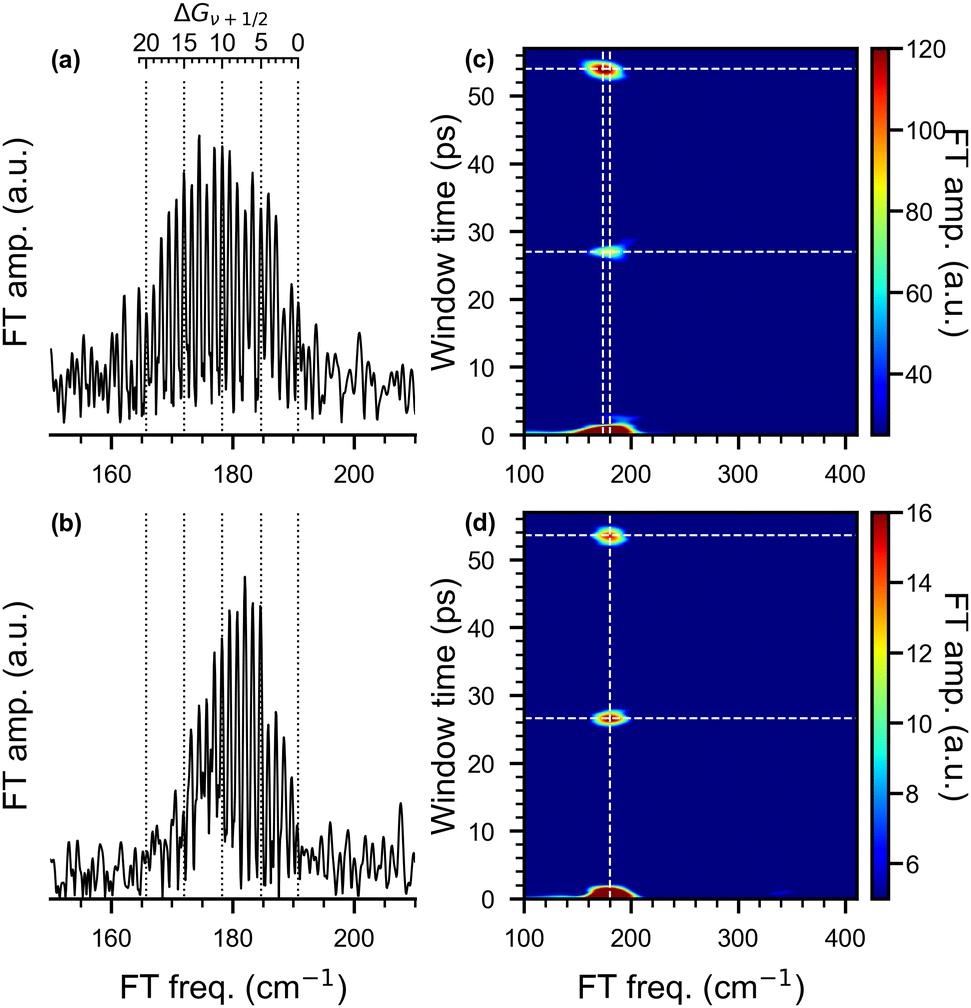 | ||
| Fig. 6 Fast Fourier transform (FFT) analysis (left column) of the oscillatory trace obtained from the fs-NeNePo spectrum in 0 ps to 60 ps delay time and the wavelengths of pump pulse, (a) λpump = 510 nm and (b) λpump = 1010 nm. The bands are assigned to the vibrational interval ΔGν+1/2 indicated by the vibrational quantum number ν. Short time Fourier transform (STFT) maps (right column) obtained by sliding a 2 ps time window along the delay time for pump pulse wavelengths of (c) λpump = 510 nm, (d) λpump = 1010 nm. | ||
Interestingly, the FFT spectrum obtained for a pump pulse wavelength of 1010 nm (see Fig. 6b) reveals only a subset of the peaks observed in Fig. 6a. It shows the same bands at higher frequencies, but no clearly resolved signal below 173 cm−1. The peak at 173 cm−1 corresponds to ΔG29/2(ν = 14) and hence the excited vibrational levels are close to those previously reported for (nanosecond) anion photoelectron spectra.34 Hence, photodetachment involving 1010 nm photons yields the population of the vibration levels of the neutral ground state expected from Franck–Condon considerations, while the broader FFT spectrum observed for 510 nm resolves vibration levels up to ΔG43/2(ν = 21). Such a dependence of the vibrational dynamics on the pump pulse wavelength is unexpected. This suggests that the preparation of a superposition of vibrational eigenstates on the electronic ground-state PES of neutral Ag2 is not exclusively determined by the Franck–Condon factors for photodetachment from the electronic ground state of the anion. There must be a different mechanism involved in the photodetachment process (see below).
The STFT maps obtained with a time window of 2 ps, shown on the right of in Fig. 6, reveal quantum beats resulting from the anharmonic nature of the potential energy curve in a frequency- and time-resolved way. For a pump wavelength of 510 nm, the broad band extending from 160–195 cm−1 in the FFT spectrum (Fig. 6a) appears slightly shifted in the wavenumber and with varying intensity at window times of 27.0 and 54.0 ps in the STFT map (Fig. 6c). Wave-packet recurrences are observed at multiples of 27 ps, which is consist with the spacing between adjacent vibrational intervals (≈1.24 cm−1). The shift of the center frequency indicates that the low-frequency components contribute more to the recurrence features than the high-frequency ones. For a pump wavelength of 1010 nm, the STFT map (Fig. 6d) also shows wave-packet recurrences, but at slightly shorter window times of 26.5 and 53.5 ps, consist with interval spacings of 1.26 and 1.24 cm−1. The larger interval spacing and shorter recurrence time hints at the contribution of more high-frequency components in this spectrum. The central frequencies of all these recurrence features are all located at around 180 cm−1.
Based on the experimental vibrational intervals ΔGν+1/2 (see Table S1, ESI†), we determined vibrational constants of the silver dimer isotoplogue 107Ag109Ag by fitting the experimental values to a Dunham expression (see SI for details). The so obtained harmonic (ωe), quadratic anharmonic (ωexe) and cubic anharmonic (ωeye) terms are compared to previously published values in Table 1. Our vibrational constants show excellent agreement with previously determined values using Fourier transform absorption spectroscopy of the A1Σ+u ← X1Σ+g transitions for hot silver dimer produced in an oven.40 Fig. S5 and S6 (ESI†) displays the corresponding Birge–Sponer plot (ΔGν+1/2vs. v) and the curve obtained from the molecular constants in ref. 38 and 40.
4. Discussion
In order to assess the effect of isotopologue-selective measurements on the wave-packet dynamics, we simulated the oscillatory features of the fs-NeNePo spectra. Effects related to isotopologue-dependent wave-packet dynamics have been reported previously, for example, for the potassium as well as the bromine dimer.41–44 To this end, we considered the wave-packet dynamics for the three silver dimer isotopologues 107,107Ag2, 107,109Ag2 and 109,109Ag2. The relative shift in energy of the vibrational levels for two isotopologues expressed in terms of the vibrational constants is given by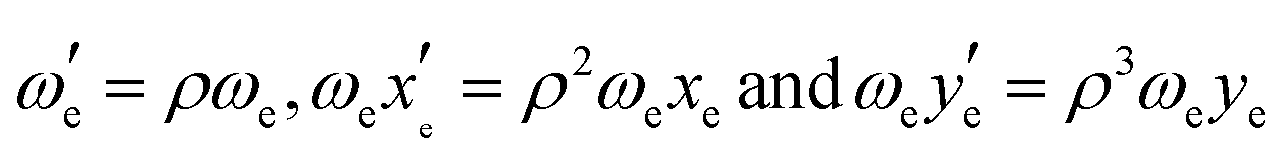 with
with 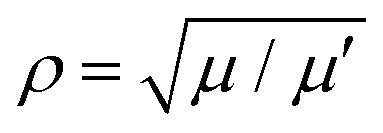 , where μ is the reduced mass of the isotopologue and μ′ is for the lighter isotopologue. The simulations are shown in Fig. S7 and S8 (ESI†). Isotopologue-dependent effects are difficult to observe at small delay times of less than 2 ps (left column in Fig. S8, ESI†), due to the relatively small shift of the vibrational periods of the silver dimer isotopologues. The differences are more apparent at longer delay times, i.e. at the recurrence times around 26 ps (center column) and 52 ps (right column). Without isotopologue selection, the first recurrence at 26 ps appears weaker (compared to that at 52 ps), owing to the out-of-phase oscillation of the 107,109Ag2 isotopologue signal with respect to the other two minor isotopologues.
, where μ is the reduced mass of the isotopologue and μ′ is for the lighter isotopologue. The simulations are shown in Fig. S7 and S8 (ESI†). Isotopologue-dependent effects are difficult to observe at small delay times of less than 2 ps (left column in Fig. S8, ESI†), due to the relatively small shift of the vibrational periods of the silver dimer isotopologues. The differences are more apparent at longer delay times, i.e. at the recurrence times around 26 ps (center column) and 52 ps (right column). Without isotopologue selection, the first recurrence at 26 ps appears weaker (compared to that at 52 ps), owing to the out-of-phase oscillation of the 107,109Ag2 isotopologue signal with respect to the other two minor isotopologues.
Interestingly, we do not observe fractional recurrences of the vibrational wave packets, which have been studied in detail for other systems, like the alkali metal and halogen dimers.43,45–47 In the reported studies, the wave packet breaks up into sets of sub-wave-packets at times t/Trev = p/q, where p/q is an irreducible fraction of integers. Experimental evidence of the fractional recurrence for the wavepacket of the primary frequency ω is that the revival of the high order q/p·ω frequency components appears at p/q·Trev time in the STFT map. But this is not found for the silver dimer, as shown in the Fig. 6, probably due to the low signal-noise ratio of the oscillatory cation signal. The simulated fractional revival (see Fig. S7, ESI†) is much weaker than the first and second full recurrences.
The mechanism behind populating more than twenty vibrational levels (see Fig. 6a) is not directly obvious. For lower photodetachment energies, the lowest energy band in the anion photoelectron spectrum of Ag2− reveals resolved vibrational levels up to an electron binding energy of 1.2 eV, corresponding to the population of up to ν = 8 of neutral Ag2.34 However, in later studies that use higher photodetachment energies this band shows a longer, unresolved tail extending up to 1.5 eV, corresponding to the population of vibrational levels up to v ≈ 21.36 Moreover, these latter spectra also show a weak,35,36 unassigned feature at even higher energies around 1.65 eV, clearly below first electronic state of neutral Ag2, but asymptotically approaching the bond dissociation limit (1.83 ± 0.2 eV) of Ag2− for Ag(5d104s1) + Ag(5d104s2) fragmentation.48 This suggests that there may be an additional photodetachment channel involving an electronic excited state of the anion. Depending on the pump wavelength this excited state can be resonantly excited, followed by fast autodetachment and efficient population of higher vibrational levels of the neutral electronic ground state. We can only speculate on the nature of the involved electronically excited state of the anion. It could be a 2Σ+g state, populated by promoting an electron from a non-bonding σu molecular orbital to a bonding σg molecular orbital. Its atomic asymptote corresponds to Ag(5d104s1) + Ag(5d104s14p1) fragments. Although this dissociation channel lies 3.7 eV higher than that of the ground state (Ag(5d104s1) + Ag(5d104s2)),49 stronger bonding in the excited state will result in a considerably lower 2Σ+g ← 2Σ+u excitation energy.
Additional information can be obtained from the time domain data. The phase of the oscillatory part of the NeNePo signal contains information on when ionization occurs relative to the initial formation of the wave packet.13,50–52 A value of the phase close to 0 π or 2 π, as is observed for a pump wavelength of 1140 nm, suggests that ionization from the neutral state occurs close to the equilibrium bond length of the neutral. In contrast, a phase of π/2, obtained for pump wavelengths of 510 nm and 1010 nm, indicates ionization at one of the turning points of the wave-packet dynamics.
It should be noted that two Franck–Condon windows, one for photodetachment and one for photoionization, determine the oscillatory cation signal. The predicted potential energy curve for the A1Σ+u state of Ag2 results in a nearly symmetric Franck–Condon envelope for the A1Σ+u ← X1Σ+g transitions for low-lying vibrational levels of the X1Σ+g state.53 However, the higher-lying vibrational levels of X1Σ+g have larger absorption coefficients to the higher-lying vibrational levels of A1Σ+u. This indicates that the X1Σ+g state has better ionization efficiency at the outer (longer bond lengths) than the inner turning point for high-lying vibrational levels. Therefore, the oscillatory signal observed with pump wavelengths of 510 nm and 1010 involve ionization at the outer turning point with an initial phase of π/2. This also holds, when the electronically excited state of Ag2− preferentially auto-detaches an electron to produce high-lying vibrational levels.
Note, the strong electronic field resulting from the ultrashort laser pulses, typically with peak intensities in (or higher than) the terawatt cm−2 range, can induce energy level shifts as a result of the optical Stark effect.54–56 The peak intensity of the pump and the probe pulses in the present experiments are more than an order of magnitude smaller (10–40 petawatt cm−2) and light-induced Stark shifting of the quantum levels of the neutral Ag2 is not expected.
5. Conclusions
The present analysis of the fs-NeNePo spectra demonstrates that characterizing the nuclear wave-packet dynamics in neutral silver dimers with ∼50 fs duration laser pulses over a period of 60 ps in the time domain yields high resolution vibrational information. In general, beating frequencies corresponding to wavenumbers below ∼400 cm−1 can be resolved and determined with a resolution down to 0.5 cm−1. In addition, the observation of the wave-packet recurrence of a single isotopologue at longer delay times yields direct information on the anharmonic nature of the PES. Hence, combining ultrafast laser systems with the fs-NeNePo excitation scheme does not only allow studying the ultrafast nuclear quantum dynamics of mass-selected neutral clusters in the gas phase, but also introduces a complementary strategy for measuring vibrational spectra in the terahertz regime. In addition, valuable information on the photoexcitation mechanism can be extracted from such experiments.Author contributions
J. J. and M. G. performed the experimental works. J. J. curated and analysed the data. J. J. and M.G. prepared the figures. J. J., M. G. and K. R. A. wrote and revised the manuscript. J. J. and K. R. A. designed the study, coordinated the project, supervised the research and contributed to the data interpretations.Conflicts of interest
There are no conflicts of interests.Acknowledgements
This work was funded by the Deutsche Forschungsgemeinschaft DFG as part of the major instrumentation grant INST 268/381-1. K. R. A. thanks Ludger Wöste for his continuing support and Daniel Neumark (UC Berkeley) for fruitful discussions. J. J. acknowledges the Alexander von Humboldt-Stiftung for a postdoctoral fellowship and thanks Bernd von Issendorff for helpful discussions on the sputter source. M. G. gratefully acknowledges a doctoral scholarship from the Studienstiftung des deutschen Volkes. We thank the precision mechanics workshop of the physical departments at Freie Universität Berlin und Universität Leipzig as well as Marko Kutschbauch and all building technicians at Universität Leipzig for technical and machinery works. We also acknowledge Jürgen Jäschke, Marcel Jorewitz and Alexandra Giermann for technical support within the laboratory.References
- D. Grandjean, E. Coutiño-Gonzalez, N. T. Cuong, E. Fron, W. Baekelant, S. Aghakhani, P. Schlexer, F. D'Acapito, D. Banerjee, M. B. J. Roeffaers, M. T. Nguyen, J. Hofkens and P. Lievens, Science, 2018, 361(6403), 686 CrossRef CAS PubMed.
- X. Zhang, M. Zhang, Y. Deng, M. Xu, L. Artiglia, W. Wen, R. Gao, B. Chen, S. Yao, X. Zhang, M. Peng, J. Yan, A. Li, Z. Jiang, X. Gao, S. Cao, C. Yang, A. J. Kropf, J. Shi, J. Xie, M. Bi, J. A. van Bokhoven, Y.-W. Li, X. Wen, M. Flytzani-Stephanopoulos, C. Shi, W. Zhou and D. Ma, Nature, 2021, 589(7842), 396 CrossRef CAS PubMed.
- S. Link and M. A. El-Sayed, Annu. Rev. Phys. Chem., 2003, 54, 331 CrossRef CAS PubMed.
- T. E. Dermota, Q. Zhong and A. W. Castleman, Chem. Rev., 2004, 104(4), 1861 CrossRef CAS PubMed.
- Z. Luo, A. W. Castleman and S. N. Khanna, Chem. Rev., 2016, 116(23), 14456 CrossRef CAS PubMed.
- M. A. Duncan, Int. J. Mass Spectrom., 2000, 200(1–3), 545 CrossRef CAS.
- K. R. Asmis, A. Fielicke, G. von Helden and G. Meijer, The Chemical Physics of Solid Surfaces: Vibrational spectroscopy of gas-phase clusters and complexes, in, Atomic clusters: From gas phase to deposited, The chemical physics of solid surfaces, ed. D. Woodruff, Elsevier, Amsterdam, Boston, 1st edn, 2007, vol. 12, pp. 327–75 Search PubMed.
- J. Roithová, Chem. Soc. Rev., 2012, 41(2), 547 RSC.
- P. M. Kraus, M. Zürch, S. K. Cushing, D. M. Neumark and S. R. Leone, Nat. Rev. Chem., 2018, 2(6), 82 CrossRef CAS.
- M. Maiuri, M. Garavelli and G. Cerullo, J. Am. Chem. Soc., 2020, 142(1), 3 CrossRef CAS PubMed.
- S. Rafiq, N. P. Weingartz, S. Kromer, F. N. Castellano and L. X. Chen, Nature, 2023, 620(7975), 776 CrossRef CAS PubMed.
- S. V. Kruppa, F. Bäppler, C. Holzer, W. Klopper, R. Diller and C. Riehn, J. Phys. Chem. Lett., 2018, 9(4), 804 CrossRef CAS PubMed.
- M. Gruebele and A. H. Zewail, J. Chem. Phys., 1993, 98(2), 883 CrossRef CAS.
- A. Stolow and J. G. Underwood, Adv. Chem. Phys., 2008, 139, 497 CrossRef CAS.
- Y.-X. Yan, E. B. Gamble and K. A. Nelson, J. Chem. Phys., 1985, 83(11), 5391 CrossRef CAS.
- J. Zheng, K. Kwak and M. D. Fayer, Acc. Chem. Res., 2007, 40(1), 75 CrossRef CAS PubMed.
- J. C. Vaughan, T. Hornung, K. W. Stone and K. A. Nelson, J. Phys. Chem. A, 2007, 111(23), 4873 CrossRef CAS PubMed.
- R. Pausch, M. Heid, T. Chen, W. Kiefer and H. Schwoerer, J. Chem. Phys., 1999, 110(19), 9560 CrossRef CAS.
- S. Wolf, G. Sommerer, S. Rutz, E. Schreiber, T. Leisner, L. Wöste and R. S. Berry, Phys. Rev. Lett., 1995, 74(21), 4177 CrossRef CAS PubMed.
- L. D. Socaciu-Siebert, J. Hagen, J. Le Roux, D. Popolan, M. Vaida, S. Vajda, T. M. Bernhardt and L. Wöste, Phys. Chem. Chem. Phys., 2005, 7(14), 2706 RSC.
- H. Hess, S. Kwiet, L. Socaciu, S. Wolf, T. Leisner and L. Wöste, Appl. Phys. B, 2000, 71(3), 337 CrossRef CAS.
- D. W. Boo, Y. Ozaki, L. H. Andersen and W. C. Lineberger, J. Phys. Chem. A, 1997, 101(36), 6688 CrossRef.
- T. Leisner, S. Vajda, S. Wolf, L. Wöste and R. S. Berry, J. Chem. Phys., 1999, 111(3), 1017 CrossRef CAS.
- H. Hess, K. R. Asmis, T. Leisner and L. Wöste, Eur. Phys. J. D, 2001, 16(1), 145 CrossRef CAS.
- T. Leisner, S. Rutz, G. Sommerer, S. Vajda, S. Wolf, E. Schreiber and L. Wöste, Spectroscopy on mass-selected neutral clusters: Femtosecond dynamics of the ground state of Agn, AIP Conf. Proc., 1996, 603–609 CrossRef CAS.
- T. M. Bernhardt, J. Hagen, L. D. Socaciu, R. Mitrić, A. Heidenreich, J. Le Roux, D. Popolan, M. Vaida, L. Wöste, V. Bonacić-Koutecký and J. Jortner, Chem. Phys. Chem., 2005, 6(2), 243 CrossRef CAS PubMed.
- F. Muntean, M. S. Taylor, A. B. McCoy and W. C. Lineberger, J. Chem. Phys., 2004, 121(12), 5676 CrossRef CAS PubMed.
- M. S. Taylor, J. Barbera, C.-P. Schulz, F. Muntean, A. B. McCoy and W. C. Lineberger, J. Chem. Phys., 2005, 122(5), 54310 CrossRef PubMed.
- K. R. Asmis, M. Brümmer, C. Kaposta, G. Santambrogio, G. von Helden, G. Meijer, K. Rademann and L. Wöste, Phys. Chem. Chem. Phys., 2002, 4(7), 1101 RSC.
- V. Beutel, H.-G. Krämer, G. L. Bhale, M. Kuhn, K. Weyers and W. Demtröder, J. Chem. Phys., 1993, 98(4), 2699 CrossRef CAS.
- S. Fedrigo, W. Harbich and J. Buttet, J. Chem. Phys., 1993, 99(8), 5712 CrossRef CAS.
- P. H. C. Eilers, Anal. Chem., 2003, 75(14), 3631 CrossRef CAS PubMed.
- H. F. M. Boelens, P. H. C. Eilers and T. Hankemeier, Anal. Chem., 2005, 77(24), 7998 CrossRef CAS PubMed.
- J. Ho, K. M. Ervin and W. C. Lineberger, J. Chem. Phys., 1990, 93(10), 6987 CrossRef CAS.
- H. Handschuh, C.-Y. Cha, P. S. Bechthold, G. Ganteför and W. Eberhardt, J. Chem. Phys., 1995, 102(16), 6406 CrossRef CAS.
- K. Vetter, S. Proch, G. F. Ganteför, S. Behera and P. Jena, Phys. Chem. Chem. Phys., 2013, 15(48), 21007 RSC.
- A. Przystawik, P. Radcliffe, S. Göde, K. H. Meiwes-Broer and J. Tiggesbäumker, J. Phys. B: At., Mol. Opt. Phys., 2006, 39(19), S1183–S1189 CrossRef CAS.
- G. Herzberg and K. P. Huber, Molecular spectra and molecular structure, Springer, Berlin, 2013 Search PubMed.
- J. J. Gerdy, M. Dantus, R. M. Bowman and A. H. Zewail, Chem. Phys. Lett., 1990, 171(1–2), 1 CrossRef CAS.
- B. Kleman and S. Lindkvist, Ark. Fys., 1955, 9(4), 385 CAS.
- S. Rutz, R. de Vivie-Riedle and E. Schreiber, Phys. Rev. A, 1996, 54(1), 306 CrossRef CAS PubMed.
- A. Lindinger, C. Lupulescu, M. Plewicki, F. Vetter, A. Merli, S. M. Weber and L. Wöste, Phys. Rev. Lett., 2004, 93(3), 33001 CrossRef PubMed.
- J. Heufelder, H. Ruppe, S. Rutz, E. Schreiber and L. Wöste, Chem. Phys. Lett., 1997, 269(1–2), 1 CrossRef CAS.
- I. S. Averbukh, M. J. Vrakking, D. M. Villeneuve and A. Stolow, Phys. Rev. Lett., 1996, 77(17), 3518 CrossRef CAS PubMed.
- S. Rutz, S. Greschik, E. Schreiber and L. Wörste, Chem. Phys. Lett., 1996, 257(3–4), 365 CrossRef CAS.
- M. J. Vrakking, D. M. Villeneuve and A. Stolow, Phys. Rev. A, 1996, 54(1), R37–R40 CrossRef CAS PubMed.
- R. Robinett, Phys. Rep., 2004, 392(1–2), 1 CrossRef CAS.
- V. A. Spasov, T. H. Lee, J. P. Maberry and K. M. Ervin, J. Chem. Phys., 1999, 110(11), 5208 CrossRef CAS.
- A. Kramida and Y. Ralchenko, NIST Atomic Spectra Database, NIST Standard Reference Database 78, National Institute of Standards and Technology, 1999.
- T. Ergler, B. Feuerstein, A. Rudenko, K. Zrost, C. D. Schröter, R. Moshammer and J. Ullrich, Phys. Rev. Lett., 2006, 97(10), 103004 CrossRef PubMed.
- L. Fang and G. N. Gibson, Phys. Rev. Lett., 2008, 100(10), 103003 CrossRef CAS PubMed.
- E. R. Hosler and S. R. Leone, Phys. Rev. A, 2013, 88, 2 Search PubMed.
- R. R. Laher, M. A. Khakoo and A. Antić-Jovanović, J. Mol. Spectrosc., 2008, 248(2), 111 CrossRef CAS.
- T. Bayer, M. Wollenhaupt and T. Baumert, J. Phys. B: At., Mol. Opt. Phys., 2008, 41(7), 74007 CrossRef.
- D. Townsend, B. J. Sussman and A. Stolow, J. Phys. Chem. A, 2011, 115(4), 357 CrossRef CAS PubMed.
- I. R. Solá, J. González-Vázquez, R. de Nalda and L. Bañares, Phys. Chem. Chem. Phys., 2015, 17(20), 13183 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3cp02055j |
| This journal is © the Owner Societies 2023 |