
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The role of covalency in enhancing stability of Eu and Am complexes: a DFT comparison of BTP and BTPhen†
Izaak
Fryer-Kanssen
a,
Thomas
Malcomson
 *b,
Jonathan
Austin
c and
Andrew
Kerridge
*a
*b,
Jonathan
Austin
c and
Andrew
Kerridge
*a
aDepartment of Chemistry, Lancaster University, Bailrigg, Lancaster LA1 4YB, UK. E-mail: a.kerridge@lancaster.ac.uk
bDepartment of Chemistry, School of Natural Sciences, University of Manchester, Oxford Road, Manchester, M13 9PL, UK. E-mail: thomas.malcomson@manchester.ac.uk
cNational Nuclear Laboratory, Chadwick House, Birchwood Park, Warrington, WA3 6AE, UK
First published on 7th June 2023
Abstract
We compare the stabilities and bonding nature of [Eu/Am(BTPhen)2(NO3)]2+ complexes to those previously reported for [Eu/Am(BTP)3]3+, and investigate whether more accurately reflecting the reaction conditions of the separation process by considering [Eu/Am(NO3)3(H2O)x] (x = 3, 4) complexes instead of aquo complexes increases the selectivity of the separation ligands BTP and BTPhen for Am over Eu. The geometric and electronic structures of [Eu/Am(BTPhen)2(NO3)]2+ and [Eu/Am(NO3)3(H2O)x] (x = 3, 4) have been evaluated using density functional theory (DFT) and used as the basis for analysis of the electron density through the application of the quantum theory of atoms in molecules (QTAIM). Increased covalent bond character for the Am complexes of BTPhen over Eu analogues was found, with this increase more pronounced than that found in BTP complexes. BHLYP-derived exchange reaction energies were evaluated using the hydrated nitrates as a reference and a favourability for actinide complexation by both BTP and BTPhen was found, with the BTPhen ligand found to be more selective, with relative stability ≈0.17 eV greater than BTP.
1 Introduction
The processing of spent nuclear fuel has been an area of great focus since the inception of nuclear energy. While initial processing had militaristic aims, focus has shifted in later decades to minimising the environmental impact of spent nuclear fuel storage and maximising fuel resources. This focus remains today, with particular regard to the development of next-generation nuclear reactors. PUREX (Plutonium Uranium Redox EXtraction) remains the process by which uranium and plutonium is recovered from spent fuel, producing a raffinate which contains lanthanides (Ln) and the minor actinides (MA, typically considered to comprise Np, Am, Cm), the latter of which are responsible for most of the residual radioactivity. Removal of these minor actinides can reduce the radiotoxicity of the eventual waste product derived from PUREX raffinate, reducing the environmental impact of any subsequent geological disposal. The challenge, however, lies in the separation of the minor actinides from the lanthanides, the chemistry of the latter strongly resembling that of AmIII and CmIII.Despite this challenge, which is further exacerbated by the requirement that any process must stand up to highly acidic conditions with significant radioactivity, several ligands for Am/Eu separation have been identified and successfully tested, such as the N-donor ligand ‘trio’ developed for the European Selective ActiNide EXtraction (SANEX) process: the bis-triazinylpyridines (BTPs, 1), a family of ligands with high AmIII/EuIII separation factors, with the drawback of problematic stripping of the ligands due to their strong binding with the actinide;1–6 bis-triazinylbipyridines (BTBPs, 2), which show good selectivity and increased back-extraction capability in comparison to the BTPs, with the drawback of slow kinetics without the use of a phase-transfer agent;4–7 and the bis-triazinylphenanthrolines (BTPhens, 3), which have much faster reaction kinetics than the BTBPs, owing to the cis-locked nature of the 1,10-phenanthroline moiety which replaces the 2,2,-bipyridine of the BTBPs, as well as high selectivity and efficiency (Fig. 1).6,8,9
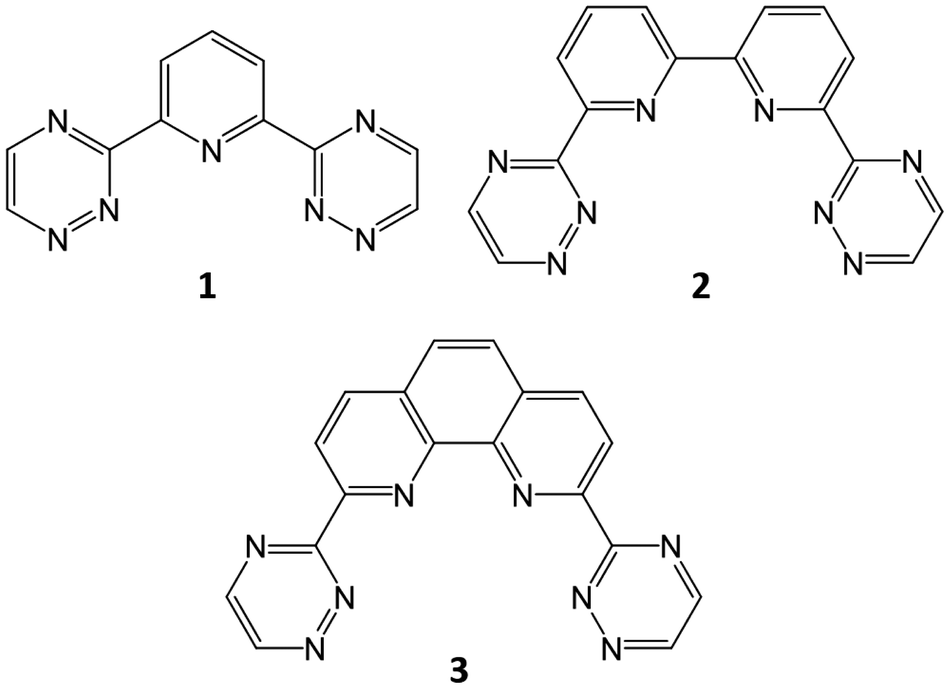 | ||
| Fig. 1 N-donor ligands for An/Ln separation: BTP (1); BTBP (2); and BTPhen (3). BTP and BTPhen (1 and 3) are considered in this study. | ||
The tridentate BTP ligand forms symmetrical 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ligand:metal complexes with the Ln/An centre with an overall charge of 3+. The existing literature shows that a wide range of computational and analytical methods have been used to investigate the differences in structure between selected LnIII and AnIII complexes. A combined effort of electrospray mass spectroscopy,11,12 time-resolved laser-induced fluorescence spectroscopy (TRLFS),13–19 X-ray photoelectron spectroscopy (XPS) and extended X-ray absorption fine structure spectroscopy (EXAFS)13,16,20–24 with complementary DFT studies16,21,25,26 have probed the structure of [Ln/An(BTP)n]3+ (n = 1–3), with a general focus on the complexes of Ln = Eu, Gd and An = U–Cm due to their relevance to the separation process. A trend of decreasing metal–ligand bond length with decreasing LnIII ionic radius was observed spectroscopically across the lanthanides,13,16,20–24 in contrast to the bond lengths of the actinide complexes, which were observed to be largely independent of AnIII ionic radius.13,21,27,28
:1 ligand:metal complexes with the Ln/An centre with an overall charge of 3+. The existing literature shows that a wide range of computational and analytical methods have been used to investigate the differences in structure between selected LnIII and AnIII complexes. A combined effort of electrospray mass spectroscopy,11,12 time-resolved laser-induced fluorescence spectroscopy (TRLFS),13–19 X-ray photoelectron spectroscopy (XPS) and extended X-ray absorption fine structure spectroscopy (EXAFS)13,16,20–24 with complementary DFT studies16,21,25,26 have probed the structure of [Ln/An(BTP)n]3+ (n = 1–3), with a general focus on the complexes of Ln = Eu, Gd and An = U–Cm due to their relevance to the separation process. A trend of decreasing metal–ligand bond length with decreasing LnIII ionic radius was observed spectroscopically across the lanthanides,13,16,20–24 in contrast to the bond lengths of the actinide complexes, which were observed to be largely independent of AnIII ionic radius.13,21,27,28
Additionally, there are many examples of computational investigations into the selective binding of these ligands, both for the BTP ligand and other nitrogen donor ligands, such as BTBP (6,6′-bis(1,2,4-triazin-3-yl)-2,2′-bipyridine) and BTPhen (2,9-bis(1,2,4-triazin-3-yl)-1,10-phenanthroline).26,29–41 Focal to many of these studies are the differences in energies of the LnIII and AnIII complexes. Despite the large separation factors exhibited by these ligands, these energetic differences amount to only hundredths to tenths of an electronvolt;31,32,34,38 for instance, Lan et al.31–33 report that for the reaction M(NO3)3 (H2O)4 + L → M(L)(NO3)3 + 4H2O, (L = BTBPs) the formation of M(L)(NO3)3 is favoured energetically when M = Am compared to M = Eu by 0.13 eV in the DFT-calculated Gibbs free energy for L = BTBP and 0.07 eV for L = CyMe4-BTBP, obtained using the B3LYP xc-functional.31 For BTP, Trumm et al.34 report the formation of the Cm complex to be 2.3 kcal mol−1 (≈0.1 eV) more favourable than Gd in the gas-phase, calculated at the MP2 level on DFT structures optimised using the BHLYP xc-functional.
The high AmIII/EuIII separation factors exhibited by these three families of ligands is thought to be due to an enhanced covalent interaction in the actinides due to the increased radial extent, and hence chemical availability, of the 5f orbitals, compared to the more core-like 4f orbitals of the lanthanides. In support of this, recent studies have shown growing evidence of correlations between covalent bond character and bond stability.10,42–45 Our previous work employing Bader's quantum theory of atoms in molecules (QTAIM)46 applied to density functional theory (DFT) derived electron densities has demonstrated an increase in the covalent character of the metal–ligand bonds of [An(H2O)9]3+ and [An(BTP)3 ]3+ complexes (An = Am, Cm) compared to their Ln analogues (Ln = Eu, Gd), which was more pronounced in the BTP complexes than in the aquo complexes.10 Additionally, [An(H2O)9]3+ + [Ln(BTP)3]3+ → [Ln(H2O)9]3+ + [An(BTP)3]3+ exchange reaction energies were shown to favour An complexation by BTP for the Eu ↔ Am and Gd ↔ Cm reactions. Together, this selectivity of the BTP ligand and increase in covalent bonding character for Am over Eu and Cm over Gd implies a small but significant electronic contribution to An–BTP bond stability and the selectivity found experimentally.
Here, we present the results of DFT-based quantum chemical simulations and subsequent QTAIM analysis of [Eu/Am(BTPhen)2(NO3)]2+, in order to provide insight into the origin of the experimentally observed selectivity of the BTPhen ligand for the actinide through the evaluation of exchange reaction energies and the quantification of covalent character in equivalent Am–N and Eu–N bonds. We continue to present an analysis into the stability of [Eu/Am(BTP)3]3+ complexes relative to the corresponding nitrate complexes.
Additionally, we investigate whether the properties of the [Eu/Am(BTPhen)2(NO3)]2+ are greater in magnitude than those previously reported for the BTP ligand.10 We also present the results of the same simulation and analysis applied to the [Eu/Am(NO3)3(H2O)x] (x = 3, 4) complexes, in an effort to better replicate the conditions of the separation process.
2 Methods
All calculations were performed using version 6.6 of the TURBOMOLE quantum chemistry code47 using scalar-relativistic DFT. Three xc-functionals were employed: BLYP,48,49 B3LYP50,51 and BHLYP.52 BLYP is a functional based on the generalised gradient approximation (GGA), whereas B3LYP and BHLYP are hybrid GGA functionals, incorporating 20% and 50% of exact exchange, respectively. All optimisations were performed using the def-SVP (Eu and Am) and def2-SVP (H, C, N and O) basis sets of polarised double-ζ quality,53 which is referred to from here onwards as def(2)-SVP. Single-point-energy calculations were performed at these optimised geometries using the def(2)-TZVP basis sets of polarised triple-ζ quality.53 All calculations were performed in the presence of a water-like continuum solvent defined using the COSMO model54 with the default radii rO = 1.72 Å, rC = 2.00 Å, rN = 1.83 Å, rH = 1.30 Å, rEu = 2.22 Å, and rAm = 2.22 Å. Scalar relativistic effects were accounted for by replacing Eu and Am core electrons with the small-core pseudopotentials of Dolg and co-workers:55–57 28 and 60 core electrons were replaced, respectively. Topological and integrated properties of the electron density were investigated with version 14 of the AIMAll code.58During the analysis of the topological and integrative properties (Tables 3 and 4), ΔAm/Eu is taken as the percentage by which a given metric is greater for Am over Eu, N(Ω) = integrated electron density in atomic basin Ω, q(Ω) = total charge of basin Ω, λ(Ω) = localization index of basin Ω, ![[small rho, Greek, macron]](https://www.rsc.org/images/entities/i_char_e0d1.gif) BCP = mean magnitude of the electron density at the M–N BCPs,
BCP = mean magnitude of the electron density at the M–N BCPs, ![[small delta, Greek, macron]](https://www.rsc.org/images/entities/i_char_e0c5.gif) (Ω1,Ω2) = mean delocalisation index between atomic basins Ω1 and Ω2 and
(Ω1,Ω2) = mean delocalisation index between atomic basins Ω1 and Ω2 and 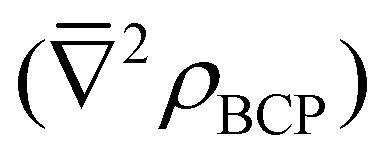 = mean Laplacian of ρ at the M–N BCPs. B3LYP-derived values are given in parentheses. All values are in atomic units (a.u.).
= mean Laplacian of ρ at the M–N BCPs. B3LYP-derived values are given in parentheses. All values are in atomic units (a.u.).
2.1 Structural characterisation
We have previously reported the structures of [Ln(H2O)9]3+ and [Ln(BTP)3]3+ for Ln = Ce–Lu obtained using the BLYP, B3LYP and BHLYP xc-functionals.10 For [Ln(H2O)9]3+, the B3LYP and BHLYP functionals gave Ln–O bond lengths typically ≈0.01–0.03 Å shorter than those obtained with BLYP, with a more pronounced difference in the middle of the series (Gd–Dy); additionally, BHLYP-derived [Eu(BTP)3]3+ bond lengths lay closer to EXAFS literature values than those derived using B3LYP. Similar functional differences were observed for Ln–N bond lengths in [Ln(BTP)3]3+, with BLYP significantly overestimating bond lengths for the middle of the series. High expectation values of 〈S2〉 (in comparison to formal values) for these poor-quality BLYP structures was indicative of significant spin contamination, which the inclusion of exact exchange was found to significantly reduce.10 These overestimated expectation values, along with the poor performance of the BLYP functional when compared to experimentally determined structural data, indicated that the origin of this poor performance lay in the description of the electronic structure of the Ln ion, leading us to focus only on the electronic structures of the B3LYP and BHLYP-optimised complexes. As such, throughout this paper, we report only results obtained with the B3LYP and BHLYP functionals; due to the performance when compared to similar experimental systems10,31,32,34,38 and the production of better S2 values, the primary focus of the discussion will be on BHLYP values.For [Eu/Am(BTPhen)2(NO3)]2+ and [Eu/Am(NO3)3(H2O)x] (x = 3, 4) geometries were obtained through optimisation with B3LYP and BHLYP. Additionally, in our previous study, bond lengths were found to be typically ≈0.05 Å shorter in the presence of a continuum aqueous solvent than in the gas phase, and in better agreement with experimental values. For this reason, we also employed the COSMO solvent model when optimising the present complexes.
3 Results & discussion
3.1 Structure – [Eu/Am(BTPhen)2(NO3)]2+
Unlike the 3:1 complexes formed by the tridentate BTP ligands, the larger tetradentate BTPhen ligand forms 2:1 complexes with Eu and An ions.8 The [Eu/Am(BTPhen)2]3+ complex has a coordination number of only 8 and can accommodate solvent molecules to achieve a coordination number of 9 or 10.8 As the ions to be separated by the SANEX process are maintained in a nitric acid solution, we have here investigated the [Eu/Am(BTPhen)2(NO3)]2+ complex, which incorporates a bidentate nitrate ligand, as reported for the [Ln(CyMe4-BTPhen)2(NO3)]2+ complexes isolated and characterised by Lewis et al. (Ln = Eu) and Whittaker et al. (Ln = Pr, Eu, Tb, Yb) (Fig. 2).8,59 Average optimised M–N and M–Onitrate bond lengths for [Eu/Am(BTPhen)2(NO3)]2+ are reported in Table 1.
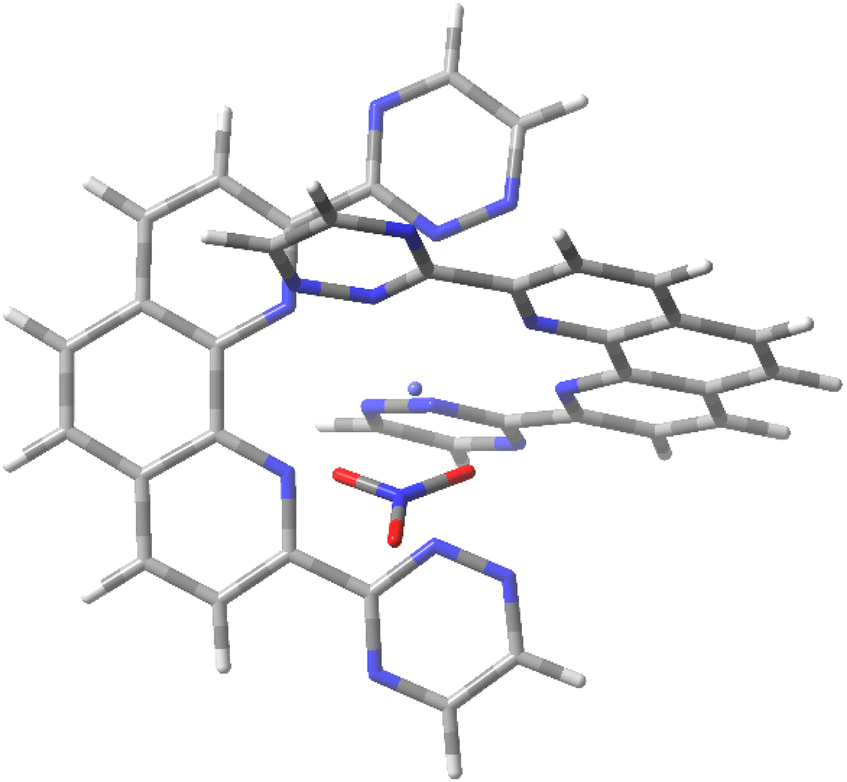 | ||
| Fig. 2 BHLYP/def(2)-SVP optimised geometries of [Am(BTPhen)2(NO3)]2+. | ||
| Complex |
![[R with combining macron]](https://www.rsc.org/images/entities/i_char_0052_0304.gif) (MN) (MN) |
(MON) |
|---|---|---|
| [Eu(BTP)3]3+ | 2.57 | |
| (2.59) | ||
| [Am(BTP)3]3+ | 2.60 | |
| (2.61) | ||
| [Eu(BTPhen)2NO3]2+ | 2.61 | 2.55 |
| (2.63) | (2.55) | |
| [Am(BTPhen)2NO3]2+ | 2.63 | 2.60 |
| (2.65) | (2.58) |
The BHLYP- and B3LYP-calculated Eu–N bond lengths in BTPhen are ≈0.03–0.05 Å longer than the mean [Eu(CyMe4-BTPhen)2(NO3)]2+ Eu–N bond lengths (2.587, 2.582 Å) reported by Lewis et al.8 Additionally, it would appear that B3LYP is overestimating M–N and underestimating M–O compared to BHLYP.
As in the BTP complexes, the BHLYP- and B3LYP-calculated bond lengths for the BTPhen complexes are similar, with at most a 0.02 Å difference. The Am–N bond lengths are ≈0.02 Å longer than the Eu–N bond lengths in the BTPhen complexes, consistent with the ≈0.03 Å Eu/Am–N bond length difference in the BTP complexes and Eu/Am–N bond length differences seen in other theoretical studies, for example that of Trumm et al. in 2015 in which the bond lengths for thirteen Gd complexes were all 0.02–0.05 Å shorter than their Cm analogues.60 Additionally, while the M–N bond lengths in the BTPhen complexes are ≈0.03–0.04 Å longer than in the BTP complexes, the Eu–ON bond lengths are 0.03–0.05 Å shorter than the corresponding Am–ON bond lengths in the BTPhen complexes.
3.2 Structure – [Eu/Am(NO3)3(H2O)x] (x = 3, 4)
In our previous study, we compared the BTP and aqueous complexes of Eu and Am. However, in the SANEX process, the ions must be extracted from the nitric acid environment of the PUREX raffinate. As such, we have optimised structures of [Eu/Am(NO3)3(H2O)x] (x = 3, 4) complexes, to investigate whether the separation ligands exhibit greater An affinity when the ions are initially bound by nitrate ligands. In these complexes the nitrate ligands were modelled in a bidentate coordination mode, which is more favourable than the monodentate mode.61 Water was added in order to complete the first coordination sphere, with hydration numbers of 3 and 4 chosen to achieve coordination numbers of 9 and 10, matching the coordination numbers of the BTP and BTPhen complexes, respectively (Fig. 3). Average optimised M–ON and M–OW bond lengths for the [Eu/Am(NO3)3(H2O)x] (x = 3, 4) complexes are reported in Table 2. | ||
| Fig. 3 BHLYP/def(2)-SVP optimised geometries of [Am(NO3)3(H2O)x] (x = 3 (A), 4 (B)). | ||
| Complex |
(MON) |
(MOW) |
|---|---|---|
| [Eu(NO3)3(H2O)3] | 2.47 | 2.41 |
| (2.48) | (2.43) | |
| [Am(NO3)3(H2O)3] | 2.51 | 2.46 |
| (2.51) | (2.48) | |
| [Eu(NO3)3(H2O)4] | 2.51 | 2.47 |
| (2.51) | (2.50) | |
| [Am(NO3)3(H2O)4] | 2.54 | 2.52 |
| (2.54) | (2.54) |
As in the BTP and BTPhen complexes, B3LYP-calculated bond lengths are similar to those obtained with BHLYP, with at most 0.03 Å difference. In both the x = 3 and x = 4 complexes, the average M–OW bond lengths are shorter than the M–ON bond lengths, by ≈0.03–0.05 Å when x = 3 and up to 0.04 Å when x = 4. Bond lengths are consistently longer by ≈0.03–0.07 Å when x = 4 than when x = 3, likely due to additional steric crowding caused by the additional bound water. The bond lengths in the Eu complexes are ≈0.03–0.05 Å shorter than their Am counterparts.
3.3 Eu vs. Am bonding in BTP and BTPhen complexes
To investigate the bonding character in the [Eu/Am(BTPhen)2 (NO3)]2+ and [Eu/Am(NO3)3(H2O)x] (x = 3, 4) complexes, we have again employed the QTAIM, which we have previously used to investigate the bonding character in the [Eu/Am(H2O)9]3+ and [Eu/Am(BTP)3]3+ complexes.10 These calculations were based on single-point-energy calculations using a SARC all-electron basis sets of polarised triple-zeta quality62–65 along with the second-order Douglas–Kroll–Hess Hamiltonian in order to account for scalar relativistic effects; however, recently added functionality in our chosen analysis software has allowed us to instead use densities generated using the def(2)-TZVP basis sets, employing effective core potentials (ECP) (Tables 3 and 4), which we also employ when evaluating our reaction energies. For consistency we have therefore repeated the analysis of the [Eu/Am(H2O)9]3+ and [Eu/Am(BTP)3]3+ complexes with these densities. Comparison of QTAIM values calculated utilising an ECP with those determined using an all electron basis set10 produces only small variations for the [Eu/Am(H2O)9]3+ and [Eu/Am(BTP)3]3+ complexes; this similarity lends credence to both the appropriateness of the def(2)-TZVP//ECP model chemistry as well as to the robust transferability of the QTAIM analysis.| Complex | q(M) |
![[q with combining macron]](https://www.rsc.org/images/entities/i_char_0071_0304.gif) (N) (N) |
N(M) − λ(M) |
(M,N) |
BCP(M–N) |
|
|---|---|---|---|---|---|---|
| [Eu(BTP)3]3+ | 2.29 | −0.99 | 1.02 | 0.204 | 0.0422 | 0.131 |
| (2.18) | (−0.84) | (1.12) | (0.221) | (0.0416) | (0.122) | |
| [Am(BTP)3]3+ | 2.25 | −0.98 | 1.21 | 0.240 | 0.0451 | 0.137 |
| (2.13) | (−0.84) | (1.36) | (0.264) | (0.0453) | (0.128) | |
| Δ Am/Eu | 18% | 17% | 7% | |||
| (21%) | (19%) | (9%) | ||||
| [Eu(BTPhen)2(NO3)]2+ | 2.31 | −1.07 | 1.00 | 0.187 | 0.0386 | 0.121 |
| (2.19) | (−0.91) | (1.12) | (0.201) | (0.0377) | (0.112) | |
| [Am(BTPhen)2(NO3)]2+ | 2.27 | −1.06 | 1.18 | 0.222 | 0.0417 | 0.128 |
| (2.15) | (−0.91) | (1.33) | (0.240) | (0.0414) | (0.120) | |
| Δ Am/Eu | 18% | 19% | 8% | |||
| (19%) | (20%) | (10%) |

QTAIM partitions a molecular system into a contiguous space-filling set of atomic volumes bound by a zero flux surface which satisfies the condition ∇ρ(r)·n(r) = 0, where ρ(r) is the magnitude of the electron density at a point r and n(r) is a vector normal to the surface at that point. Molecular structure is revealed by critical points (CPs) in the electron density, i.e. points in space at which the first derivatives of ρ vanish. Two types of CPs are nuclear critical points (NCPs), which are local maxima of ρ(r) at the atomic nuclei, and bond critical points (BCPs), which lie at the points at which a minimum in ρ(r) between nuclei coincides with the interatomic surface defined by the zero-flux condition. Early characterisation of chemical bonding in QTAIM focused on the concept by which a bond path can be defined by two NCPs with a BCP situated on the line of minimum ρ(r), and the properties of the electron density at the BCP can be used to characterise the bonding interaction; however, in recent years a number of molecules have been presented in which a BCP and, as a result, a bond path is missing between two bonded atoms, leading to the need for refinement in QTAIM bond characterisation.66–68 Three QTAIM metrics are typically used for this characterisation:69 the magnitude of the electron density, ρBCP, the Laplacian of the electron density, ∇2ρBCP, and the energy density at the BCP, HBCP. For a covalent interaction, a rule of thumb is that ρBCP is large and positive (>0.2 a.u.), while ∇2ρBCP and HBCP are negative.
To enhance our ability to accurately characterise both existing bonds and their character, and to supplement this topological analysis, we also consider several integrated properties of the electron density: the atomic charge (q(M)), the localisation (λ(M)) and delocalisation (δ(M,N)) indices70 due to their relation to the interatomic exchange–correlation energy, and the difference between the total electron density in the atomic basin and the localisation index (N(M) − λ(M)). Additionally, we have discounted HBCP; as we have previously argued, HBCP can be considered effectively equal to 0 in these complexes.10 Reported in Table 3 are the topological and integrated properties of [Eu/Am(BTPhen)2(NO3)]2+ as well as those derived from the repeated analysis of [Eu/Am(BTP)3]3+ discussed above.
There is a slight excess of 0.04 (0.05) a.u. in the atomic charge on the metal centres of the Eu complexes compared to the Am complexes with both B3LYP and BHLYP; however, as we have previously argued, the localisation indices are more informative than q(M) with regard to bonding.71–73 The difference between the total electron density in the atomic basin and the localisation index, N(M) − λ(M), provides the number of non-localised electrons in the atomic basin. Δ(N(M) − λ(M))vAm/Eu, the percentage by which N(M) − λ(M) is greater for the Am complexes over that of the Eu complexes, is significant in both the BTP and BTPhen complexes – 18% (21%) and 18% (19%), respectively.
For both the BTP and BTPhen complexes, the small values of BCP and the positive values of their Laplacian indicate that the metal–ligand interaction is predominantly ionic, as would be expected. While M–N BCP values of the BTPhen complexes are lower than those of the BTP complexes, the lengths of the M–N bonds are longer in the BTPhen complexes. Instead we can consider ΔAm/Eu, the percentage by which the Am BCP values are larger than their Eu counterparts. Despite the Eu–N bond lengths being shorter than the Am–N bond lengths, ΔAm/Eu is 7% (9%) for the BTP complexes, while ΔAm/Eu for the BTPhen complexes is slightly larger at 8% (10%). In the same way, we can consider ΔAm/Eu, the percentage by which (Am, N) is greater than (Eu, N), which is 17% (19%) for the BTP complexes and 19% (20%) for the BTPhen complexes.
Altogether, the QTAIM analysis of the BTP and BTPhen complexes show a slightly greater difference in covalency for the BTPhen complexes than seen in the BTP complexes. While these percentages are small, we have previously reported a 4% increase in the BHLYP-derived value of BCP for An (An = Am, Cm) which is ≈3 standard deviations larger than the mean Ln (Ln = Ce–Lu) value (corresponding B3LYP-derived BCP values are 6% larger and 4% standard deviations from the mean Ln value).10
3.4 Eu vs. Am bonding in aquo and nitrate complexes
We have previously argued that if covalent stabilisation of the An–N bond plays a role in the actinide selectivity of BTP, and hence BTPhen, complexes, then the difference in covalent character between the An–N and Ln–N bonds should be expected to be more pronounced than in the M–O bonds of the aquo complexes.10 QTAIM analysis of the nona-aquo complexes of Eu and Am confirmed that this was the case, with Δ(N(M) − λ(M))Am/Eu, ΔAm/Eu and ΔAm/Eu values which were all lower than their equivalents in the BTP complexes.10 Here, we extend this argument to the [Eu/Am(NO3)3(H2O)x] (x = 3, 4) complexes of Eu and Am, the topological and integrated properties for which are tabulated in Table 4, as well as the repeated analysis of [Eu/Am(H2O)9]3+.
| Complex | q(M) |
(ON) |
(OW) |
N(M) − λ(M) |
(M,O) |
BCP
|
|
|---|---|---|---|---|---|---|---|
| [Eu(H2O)9]3+ | 2.46 | −1.27 | 0.91 | 0.196 | 0.0422 | 0.187 | |
| (2.37) | (−1.20) | (1.00) | (0.213) | (0.0440) | (0.177) | ||
| [Am(H2O)9]3+ | 2.45 | −1.27 | 1.05 | 0.225 | 0.0455 | 0.188 | |
| (2.36) | (−1.20) | (1.14) | (0.244) | (0.0455) | (0.178) | ||
| Δ Am/Eu | 15% | 15% | 3% | ||||
| (15%) | (14%) | (4%) | |||||
| [Eu(H2O)3(NO3)3] | 2.40 | −0.67 | −1.27 | 0.98 | 0.205 | 0.0470 | 0.185 |
| (2.27) | (−0.62) | (−1.20) | (1.09) | (0.227) | (0.0465) | (0.172) | |
| [Am(H2O)3(NO3)3] | 2.38 | −0.67 | −1.27 | 1.12 | 0.234 | 0.0484 | 0.185 |
| (2.25) | (−0.62) | (−1.20) | (1.25) | (0.259) | (0.0485) | (0.173) | |
| Δ Am/Eu | 14% | 14% | 3% | ||||
| (14%) | (14%) | (4%) | |||||
| [Eu(H2O)3(NO3)4] | 2.41 | −0.67 | −1.26 | 0.96 | 0.182 | 0.0421 | 0.167 |
| (2.28) | (−0.62) | (−1.20) | (1.08) | (0.213) | (0.0440) | (0.177) | |
| [Am(H2O)3(NO3)4] | 2.38 | −0.67 | −1.26 | 1.11 | 0.210 | 0.0438 | 0.170 |
| (2.26) | (−0.62) | (−1.19) | (1.24) | (0.244) | (0.0455) | (0.178) | |
| Δ Am/Eu | 16% | 15% | 4% | ||||
| (15%) | (15%) | (6%) |

While N(M) − λ(M), ΔAm/Eu, BCP and 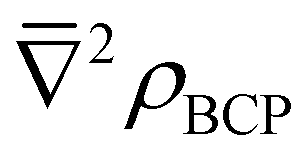 values vary in magnitude slightly between complexes, they are broadly similar, and the small values of BCP and positive
values vary in magnitude slightly between complexes, they are broadly similar, and the small values of BCP and positive 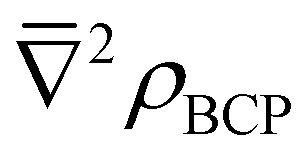 again indicate an ionic metal–ligand interaction. Looking once more at the percentage differences, Δ(N(M) − λ(M))Am/Eu values are 14–16% (14–15%) for the nona-aquo and hydrated nitrate complexes, compared to the 18% increase (19–21%) in the BTP and BTPhen complexes; ΔAm/Eu values are 3–4% (4–6%) compared to 7–8% (9–10%) and ΔAm/Eu values are 14–15% (14–15%) compared to 17–19% (19–20%). Altogether, a trend in the Δ(N(M) − λ(M))Am/Eu values emerges, as follows:
again indicate an ionic metal–ligand interaction. Looking once more at the percentage differences, Δ(N(M) − λ(M))Am/Eu values are 14–16% (14–15%) for the nona-aquo and hydrated nitrate complexes, compared to the 18% increase (19–21%) in the BTP and BTPhen complexes; ΔAm/Eu values are 3–4% (4–6%) compared to 7–8% (9–10%) and ΔAm/Eu values are 14–15% (14–15%) compared to 17–19% (19–20%). Altogether, a trend in the Δ(N(M) − λ(M))Am/Eu values emerges, as follows:
| [Eu/Am(BTPhen)2(NO3)]2+ > [Eu/Am(BTP)3]3+ > [Eu/Am(NO3)3(H2O)4] > [Eu/Am(H2O)9]3+ > [Eu/Am(NO3)3(H2O)3] | (1) |
3.5 Stabilities of BTP and BTPhen complexes relative to aquo and hydrated trinitrate complexes
BHLYP/def(2)-TZVP single-point energy calculations were performed at BHLYP/def(2)-SVP optimised geometries. Results from these calculations were used to calculate self-consistent-field (SCF) energies of the exchange reactions (1–7), tabulated in Table 5. To investigate the stability of the BTP complexes of Am over Eu analogues, we have previously considered the exchange reaction (1) (Table 5). To more accurately reflect the conditions of the separation process, here we also consider the exchange reactions (2) and (3), which replace the nona-aquo complexes in eqn (1) with M[(H2O)3(NO3)x] (x = 3, 4) complexes. Further, to investigate the stability of the BTPhen complexes of Am over Eu, we also consider the exchange reactions (4–6), as well as an exchange reaction between the BTP and BTPhen complexes, reaction (7).| Reaction | E r (eV) | |
|---|---|---|
| (1) | [Eu(BTP)3]3+ + [Am(H2O)9]3+ → [Eu(H2O)9]3+ + [Am(BTP)3]3+ | −0.01 |
| (−0.05) | ||
| (2) | [Eu(BTP)3]3+ + [Am(NO3)3(H2O)3] → [Eu(NO3)3(H2O)3] + [Am(BTP)3]3+ | −0.09 |
| (−0.04) | ||
| (3) | [Eu(BTP)3]3+ + [Am(NO3)3(H2O)4] → [Eu(NO3)3(H2O)4] + [Am(BTP)3]3+ | −0.03 |
| (0.02) | ||
| (4) | [Eu(BTPhen)2NO3]2+ + [Am(H2O)9]3+ → [Eu(H2O)9]3+ + [Am(BTPhen)2NO3]2+ | −0.19 |
| (−0.22) | ||
| (5) | [Eu(BTPhen)2NO3]2+ + [Am(NO3)3(H2O)3] → [Eu(NO3)3(H2O)3] + [Am(BTPhen)2NO3]2+ | −0.26 |
| (−0.21) | ||
| (6) | [Eu(BTPhen)2NO3]2+ + [Am(NO3)3(H2O)4] → [Eu(NO3)3(H2O)4] + [Am(BTPhen)2NO3]2+ | −0.21 |
| (−0.14) | ||
| (7) | [Eu(BTP)3]3+ + [Am(BTPhen)2NO3]2+ → [Eu(BTPhen)2NO3]2+ + [Am(BTP)3]3+ | −0.17 |
| (−0.17) | ||
As we have stated previously, the reaction energies required to give significant separation factors are not large; an energy difference of 0.12 eV corresponds to a separation factor of 100, or a 99% separation of species.74 In our previous study we reported Er values of −0.01 eV (−0.05 eV) for reaction (1). Here we report that when [Eu/Am(NO3)3(H2O)x] replaces the aqueous complexes in reaction (1) (reactions (2) and (3)), the reaction energy is shifted in favour of the formation of the actinide BTP complex, to −0.09 eV when x = 3 and −0.03 eV when x = 4, although it is worth noting that the B3LYP-derived reaction energies shift in the opposite direction, by 0.01 when x = 3 and by 0.07 when x = 4, leading to a positive reaction energy for the latter (0.02 eV). With these opposing trends in mind, consideration should be given to BHLYP which presents more accurate structural metrics than the equivalent B3LYP calculations; when considering reaction energies of the scale shown here, these small geometric deviations can explain the discrepancy in trends observed.
Most notably, when the separation ligand is BTPhen instead of BTP, the reaction energy is shifted in favour of the actinide by 0.17 eV (with both B3LYP and BHLYP), as seen in reaction (7). While the reaction energy for the BTP ligand is evident of weak selectivity, those of the BTPhen ligand are much higher. This is in accordance to experimental results, which show a much larger separation factor for the BTPhen ligand than BTP,75–78 as well as our QTAIM analysis, which shows a slightly greater difference in covalency for the BTPhen complexes than seen in the BTP complexes. The same shifts in reaction energy are observed for when [Eu/Am(NO3)3(H2O)x] replaces the aqueous complexes in reaction (4), although unlike the BTP ligand all B3LYP derived reaction energies are in favour of the actinide. A potential cause for this could be that the shift in favour of the lanthanide is outweighed by the greater selectivity of the BTPhen ligand.
4 Conclusions
In this work, we have investigated the stability and bonding nature of the separation ligands BTP and BTPhen with EuIII and AmIII, performing QTAIM analysis on DFT-optimised electron densities of [Eu/Am(BTPhen)2(NO3)]2+ with BHLYP/def(2)-TZVP//BHLYP/def(2)-SVP and B3LYP/def(2)-TZVP//B3LYP/def(2)-SVP model chemistries. This analysis has revealed an increased covalent bonding character for the Am complexes of BTPhen over Eu which was slightly more than the increase seen for the BTP complexes. Additionally, exchange reaction energies for the BTPhen ligand utilising nona-aquo complexes as a reference show evidence for stronger selectivity of the BTPhen ligand for Am over Eu than the BTP ligand by ≈0.17 eV at the SCF level.The values presented within this work suggest that while differences in covalent character may present a partial explanation for the trends in selectivity, it cannot account for the full picture; instead, it is suggested that structural differences also represent a significant factor. There is a 0.04 Å increase in the Eu–N bond of [Eu(BTPhen)2(NO3)]2+ when compared to the equivalent BTP complex while Am–N bond lengths for the same complexes only show a 0.03 Å increase. In comparison the 0.02 Å difference Eu–N and Am–N bonds of the [Eu/Am(BTPhen)2(NO3)]2+ complexes can be contrasted to a significantly larger 0.05 Å difference in the Eu/Am–ON bonds; these observations suggest that the stronger Eu–nitrate interactions may act to weaken the corresponding Eu–BTPhen interactions, which in turn acts to promote the stability of the Am–BTPhen complex.
As these ligands were designed to operate in ≥1 M nitric acid solutions, we have also performed this same analysis on [Eu/Am(NO3)3(H2O)x] (x = 3, 4) complexes as a better reference than the nona-aquo complexes. QTAIM analysis revealed only slight differences in any increased covalent bonding character for Am over Eu between these hydrated nitrate complexes (for both x = 3 and 4) and the nona-aquo complexes. However, BHLYP-derived exchange reaction energies using the hydrated nitrates as a reference shift in favour of the actinide for both BTP and BTPhen.
Recent work in the field of Ln/An separation via solvent extraction has investigated how functionalisation effects the selectivity of the BTPhen ligand.79–83
We find that the increase in covalent bonding character for the BTPhen complex of Am over that of Eu is slightly greater than that seen for the BTP complexes, reaction energies which show the BTPhen ligand to be more actinide-selective than BTP, and that when the nona-aquo complexes previously used to calculate Eu ↔ Am exchange reaction energies are replaced with hydrated nitrate complexes the reaction energies shift further in favour of the An species.
While this increase in An covalent character from BTP to BTPhen complexes readily explains the differences in the exchange reaction energies (Table 5), that alone fails to fully describe the exceptionally high separation factor observed through experiment, given the small relative difference in exchange energy on the overall thermodynamics of the system. There are a number of possibilities through which the extent of this separation factor may be explained: while having minimal effect on the relative exchange energy, these values fail to take into account the dissociative barriers involved in the removal of each ligand from the relative An and Ln complexes; additionally, while the relative stabilities of each complex are assessed here in their completely coordinated states (M(BTP)33+ and M(BTPhen)2NO32+), the intermediate structures have yet to be investigated in terms of their relative properties. In addition to the findings presented here, filling in a piece of the overall puzzle, the questions raised in turn warrant further, and dedicated investigation to better shed light on the full picture.
In parallel, given the results presented in this work, further investigation into the effect of functionalisation of the BTPhen ligands84 has on both exchange reaction and QTAIM based measure, such as the recently investigated substitution at the 5,6- and 4,7-positions of the 1,10-phenanthroline moiety,85,86 is warranted.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the Nuclear Decommissioning Authority for financial support and the National Nuclear Laboratory for industrial supervision. I. F.-K., T. M., and A. K. thank The High End Computing facility at Lancaster University.Notes and references
- M. J. Hudson, C. E. Boucher, D. Braekers, J. F. Desreux, M. G. B. Drew, M. R. S. J. Foreman, L. M. Harwood, C. Hill, C. Madic, F. Marken and T. G. A. Youngs, New J. Chem., 2006, 30, 1171 RSC
.
- Z. Kolarik, U. Müllich and F. Gassner, Solvent Extr. Ion Exch., 1999, 17, 1155 CrossRef CAS
- Z. Kolarik, U. Müllich and F. Gassner, Solvent Extr. Ion Exch., 1999, 17, 23 CrossRef CAS
- P. J. Panak and A. Geist, Chem. Rev., 2013, 113, 1199 CrossRef CAS PubMed
- Z. Kolarik, Chem. Rev., 2008, 108, 4208 CrossRef CAS PubMed
- M. J. Hudson, L. M. Harwood, D. M. Laventine and F. W. Lewis, Inorg. Chem., 2013, 52, 3414 CrossRef CAS PubMed
- A. Geist, C. Hill, G. Modolo, M. R. S. J. Foreman, M. Weigl, K. Gompper and M. J. Hudson, Solvent Extr. Ion Exch., 2006, 24, 463 CrossRef CAS
- F. W. Lewis, L. Harwood, M. J. Hudson, M. G. B. Drew, J. Desreux, G. Vidick, N. Bouslimani, G. Modolo, A. Wilden, M. Sypula, T. Vu and J. Simonin, J. Am. Chem. Soc., 2011, 133, 13093 CrossRef CAS PubMed
- F. W. Lewis, L. M. Harwood, M. J. Hudson, M. B. Drew, V. Hubscher-Bruder, V. Videva, F. Arnaud-Neu, K. Stamberg and S. Vyas, Inorg. Chem., 2013, 52, 4993 CrossRef CAS PubMed
- I. Fryer-Kanssen, J. Austin and A. Kerridge, Inorg. Chem., 2016, 55, 10034 CrossRef CAS PubMed
- M. Steppert, C. Walther, A. Geist and T. Fanghänel, New J. Chem., 2009, 33, 2437 RSC
- S. Colette, B. Amekraz, C. Madic, L. Berthon, G. Cote and C. Moulin, Inorg. Chem., 2002, 41, 7031 CrossRef CAS PubMed
- M. Steppert, C. Walther, A. Geist and T. Fanghänel, New J. Chem., 2009, 33, 2437 RSC
- S. Trumm, P. J. Panak, A. Geist and T. Fanghänel, Eur. J. Inorg. Chem., 2010, 3022 CrossRef CAS
- S. Trumm, G. Lieser, M. R. S. J. Foreman, P. J. Panak, A. Geist and T. Fanghänel, Dalton Trans., 2010, 39, 923 RSC
- M. A. Denecke, P. J. Panak, M. Weigl, B. Schimmelpfennig and A. Geist, Inorg. Chem., 2005, 44, 8418 CrossRef CAS PubMed
- S. Colette, B. Amekraz, C. Madic, L. Berthon, G. Cote and C. Moulin, Inorg. Chem., 2004, 43, 6745 CrossRef CAS PubMed
- C. M. Ruff, U. Müllich, A. Geist and P. J. Panak, Dalton Trans., 2012, 41, 14594 RSC
- B. B. Beele, E. Rüdiger, F. Schwörer, U. Mullich, A. Geist and P. J. Panak, Dalton Trans., 2013, 42, 12139 RSC
- N. L. Banik, M. a Denecke, A. Geist, G. Modolo, P. J. Panak and J. Rothe, Inorg. Chem., 2009, 33, 2437 Search PubMed
- N. L. Banik, B. Schimmelpfennig, C. M. Marquardt, B. Brendebach, A. Geist and M. A. Denecke, Dalton Trans., 2010, 39, 5117 RSC
- J. Berthet, Y. Miquel, P. B. Iveson, M. Nierlich, P. Thuéry, C. Madic and M. Ephritikhine, J. Chem. Soc., Dalton Trans., 2002, 2, 3265 RSC
- M. G. B. Drew, D. Guillaneux, M. J. Hudson, P. B. Iveson, M. L. Russel and C. Madic, Inorg. Chem. Commun., 2001, 4, 12 CrossRef CAS
- P. B. Iveson, C. Riviére, M. Nierlich, P. Thuéry, M. Ephritikhine, D. Guillaneux and C. Madic, Chem. Commun., 2001, 1512 RSC
- F. Gutierrez, C. Rabbe, R. Poteau and J. P. Daudey, J. Phys. Chem. A, 2005, 109, 4325 CrossRef CAS PubMed
- L. Petit, C. Adamo and P. Maldivi, Inorg. Chem., 2006, 45, 8517 CrossRef CAS PubMed
- M. Hülsen, A. Weigand and M. Dolg, Theor. Chem. Acc., 2008, 112, 23 Search PubMed
- A. Moritz, X. Cao and M. Dolg, Theor. Chem. Acc., 2006, 117, 473 Search PubMed
- F. W. Lewis, L. Harwoord, M. J. Hudson, M. G. B. Drew, V. Hubscher-Bruder, V. Videva, F. A. K. Stamberg and S. Vyas, Inorg. Chem., 2013, 52, 4993 CrossRef CAS PubMed
- D. Guillaumont, J. Phys. Chem., 2004, 108, 6893 CrossRef CAS
- J. Lan, W. Shi, L. Yuan, Y. Zhao, J. Li and Z. Chai, Inorg. Chem., 2011, 50, 9230 CrossRef CAS PubMed
- J. Lan, W. Shi, L. Yuan, Y. Feng, Y. Zhao and Z. Chai, J. Phys. Chem. A, 2012, 116, 504 CrossRef CAS PubMed
- J. Lan, W. Shi, L. Yuan, Y. Zhao, J. Li and Z. Chai, Coord. Chem. Rev., 2012, 256, 1406 CrossRef CAS
- M. Trumm, B. Schimmelpfennig and A. Geist, Nukleonika, 2015, 60, 847 CrossRef
- C. de Sahb, L. a Watson, J. Nadas and B. P. Hay, Inorg. Chem., 2013, 52, 10632 CrossRef CAS PubMed
- Y. Yang, S. Hu, Y. Fang, H. Wei, S. Hu, D. Wang, L. Yang, H. Zhang and S. Luo, Polyhedron, 2015, 95, 86 CrossRef CAS
- A. Zaiter, B. Amine, Y. Bouzidi, L. Belkhiri, A. Boucekkine and M. Ephritikhine, Inorg. Chem., 2014, 53, 4687 CrossRef CAS PubMed
- Y. Yang, J. Liu, L. Yang, K. Li, H. Zhang, S. Luo and L. Rao, Dalton Trans., 2015, 44, 8959 RSC
- V. S. Bryantsev and B. P. Hay, Dalton Trans., 2015, 44, 7935 RSC
- C. Mari, M. Miguirditchian, D. Guillaumont, A. Tosseng, C. Berthon, P. Guilbaud, M. Duvail, J. Bisson, D. Guillaneux, M. Pipelier and D. Dubreuil, Inorg. Chem., 2011, 50, 6557 CrossRef PubMed
- M. Trumm and B. Schimmelpfennig, Mol. Phys., 2016, 114, 876 CrossRef CAS
- Q. Huang, J. R. Kingham and N. Naltsoyannis, Dalton Trans., 2015, 44, 2554 RSC
- S. D. Woodall, A. N. Swinburne, A. Kerridge, P. D. Pietro, C. Adam, P. Kaden and L. S. Natrajan, Chem. Commun., 2015, 51, 5402 RSC
- P. D. Pietro and A. Kerridge, Inorg. Chem., 2016, 55, 573 CrossRef PubMed
- P. D. Pietro and A. Kerridge, Phys. Chem. Chem. Phys., 2016, 18, 16830 RSC
-
R. F. W. Bader, Atoms in Molecules: A Quantum Theory, Oxford University Press, 1990 Search PubMed
- R. Ahlrichs, M. Bär, M. Häser, H. Horn and C. Kölmel, Chem. Phys. Lett., 1989, 162, 165 CrossRef CAS
- B. Miehlich, A. Savin, H. Stoll and H. Preuss, Chem. Phys. Lett., 1989, 157, 200 CrossRef CAS
- C. Lee, W. Yang and R. G. Parr, Phys. Rev., 1988, 37, 785 CAS
- A. D. Becke, J. Chem. Phys., 1993, 98, 1372 CrossRef CAS
- A. D. Becke, Phys. Rev., 1988, 38, 3098 CAS
- R. Krishnan, J. S. Binkley, R. Seeger and J. A. Pople, J. Chem. Phys., 1980, 72, 650 CrossRef CAS
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297 RSC
- A. Klamt and G. Schüürmann, Perkins Trans., 1993, 2, 799 RSC
- M. Dolg, H. Stoll and H. Preuss, J. Chem. Phys., 1989, 90, 1730 CrossRef CAS
- X. Cao and M. Molg, J. Chem. Phys., 2001, 115, 7348 CrossRef CAS
- W. Küchle, M. Dolg, H. Still and H. Preuss, J. Chem. Phys., 1994, 100, 7535 CrossRef
- K. A. Keith, AIMAll (Version 19.02.13), aim.tkgristmill.com.
- D. M. Whittaker, T. L. Griffiths, M. Helliwell, A. N. Swinburne, L. S. Natrajan, F. W. Lewis, L. M. Harwood, S. A. Parry and C. A. Sharrad, Inorg. Chem., 2013, 52, 3429 CrossRef CAS PubMed
- M. Trumm, B. Schimmelpfennig and A. Geist, Nukleonika, 2015, 60, 847 CrossRef
- J. Xi, J. Lan, G. Lu, Y. Zhao, Z. Chai and W. Shi, Mol. Simul., 2014, 40, 379 CrossRef CAS
- D. A. Pantazis and F. J. Neese, J. Chem. Theory Comput., 2009, 5, 2229 CrossRef CAS PubMed
- D. A. Pantazis, X. Y. Chen, C. R. Landis and F. J. Neese, J. Chem. Theory Comput., 2008, 4, 908 CrossRef CAS PubMed
- M. Bühl, C. Reimann, D. A. Pantazis, T. Bredow and F. J. Neese, J. Chem. Theory Comput., 2008, 4, 1449 CrossRef PubMed
- D. A. Pantazis and F. J. Neese, J. Chem. Theory Comput., 2011, 7, 677 CrossRef CAS
- C. Foroutan-Nejad, S. Shahbazian and R. Marek, Chem. – Eur. J., 2014, 20, 10140 CrossRef CAS PubMed
- C. Foroutan-Nejad, Angew. Chem., Int. Ed., 2020, 59, 20900 CrossRef CAS PubMed
- S. Sowlati-Hashjin, V. Šadek, S. Sadjadi, M. Karttunen, A. Martín-Pendás and C. Foroutan-Nejad, Nat. Commun., 2022, 13, 2069 CrossRef CAS PubMed
- A. Kerridge, Chem. Commun., 2017, 53, 6685 RSC
- C. Foroutan-Nejad, Z. Badri and R. Marek, Phys. Chem. Chem. Phys., 2015, 17, 30670 RSC
- A. Kerridge, RSC. Adv., 2014, 4, 12078 RSC
- P. Di Pietro and A. Kerridge, Inorg. Chem., 2016, 55, 573 CrossRef CAS PubMed
- R. Beekmeyer and A. Kerridge, Inorganics, 2015, 3, 482 CrossRef CAS
-
K. L. Nash, Metal-Ion Separation and Preconcentration, ed. A. H. Bond, M. L. Dietz and R. D. Rogers, ACS Publications, 1999, pp. 52–78 Search PubMed
- H. Wu, Q. Wu, C. Wang, J. Lan, Z. Liu, Z. Chai and W. Shi, Dalton Trans., 2016, 45, 8107 RSC
- P. Zsabka, K. V. Hecke, L. Adriaensen, A. Wilden, G. Modolo, M. Verwerft, K. Binnemans and T. Cardinaels, RSC Adv., 2021, 11, 6014 RSC
- P. Zsabka, K. V. Hecke, A. Wilden, G. Modolo, M. Verwerft, K. Binnemans and T. Cardinaels, Solvent Extr. Ion Exch., 2020, 38, 194 CrossRef CAS
- F. W. Lewis, L. M. Harwood, M. J. H. Adn, A. Afsar, D. M. Laventine, K. Stastna, J. John and P. Distler, Solvent Extr. Ion Exch., 2018, 36, 115 CrossRef CAS
- M. A. Higginson, N. D. Kyle, O. J. Marsden, P. Thompson, F. R. Livens and S. L. Heath, Dalton Trans., 2015, 44, 16547 RSC
- A. Afsar, D. M. Laventine, L. M. Harwood, M. J. Hudson and A. Geist, Chem. Commun., 2013, 49, 8534 RSC
- M. A. Higginson, O. J. Marsden, P. Thompson, F. R. Livens and S. L. Heath, React. Funct. Polym., 2015, 91, 93 CrossRef
- A. Afsar, P. Distler, L. M. Harwood, J. John and J. Westwood, Chem. Commun., 2017, 53, 4010 RSC
- N. J. Williams, J. Dehaudt, V. S. Bryantsev, H. Luo, C. W. Abney and S. Dai, Chem. Commun., 2017, 53, 2744 RSC
-
I. Fryer-Kanssen, Advancing solvent extraction technology for improved management of contaminated liquors, PhD thesis, Lancaster University, 2017 Search PubMed
- A. Afsar, D. M. Laventine, L. M. Harwood, M. J. Hudson and A. Geist, Chem. Commun., 2013, 49, 8534 RSC
- A. C. Edwards, C. Wagner, A. Geist, N. A. Burton, C. A. Sharrad, R. W. Adams, R. G. Pritchard, R. C. Whitehead and L. M. Harwood, Dalton Trans., 2016, 45, 18102 RSC
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3cp01832f |
| This journal is © the Owner Societies 2023 |