
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceExciton delocalization in a fully synthetic DNA-templated bacteriochlorin dimer†
Olga A.
Mass
 *a,
Devan R.
Watt
a,
Lance K.
Patten
a,
Ryan D.
Pensack
a,
Jeunghoon
Lee
ac,
Daniel B.
Turner
a,
Bernard
Yurke
ab and
William B.
Knowlton
ab
*a,
Devan R.
Watt
a,
Lance K.
Patten
a,
Ryan D.
Pensack
a,
Jeunghoon
Lee
ac,
Daniel B.
Turner
a,
Bernard
Yurke
ab and
William B.
Knowlton
ab
aMicron School of Materials Science & Engineering, Boise State University, Boise, Idaho 83725, USA. E-mail: olgamass@boisestate.edu
bDepartment of Electrical & Computer Engineering, Boise State University, Boise, Idaho 83725, USA
cDepartment of Chemistry and Biochemistry, Boise State University, Boise, Idaho 83725, USA
First published on 16th October 2023
Abstract
A bacteriochlorophyll a (Bchla) dimer is a basic functional unit in the LH1 and LH2 photosynthetic pigment–protein antenna complexes of purple bacteria, where an ordered, close arrangement of Bchla pigments—secured by noncovalent bonding to a protein template—enables exciton delocalization at room temperature. Stable and tunable synthetic analogs of this key photosynthetic subunit could lead to facile engineering of exciton-based systems such as in artificial photosynthesis, organic optoelectronics, and molecular quantum computing. Here, using a combination of synthesis and theory, we demonstrate that exciton delocalization can be achieved in a dimer of a synthetic bacteriochlorin (BC) featuring stability, high structural modularity, and spectral properties advantageous for exciton-based devices. The BC dimer was covalently templated by DNA, a stable and highly programmable scaffold. To achieve exciton delocalization in the absence of pigment–protein interactions critical for the Bchla dimer, we relied on the strong transition dipole moment in BC enabled by two auxochromes along the Qy transition, and omitting the central metal and isocyclic ring. The spectral properties of the synthetic “free” BC closely resembled those of Bchla in an organic solvent. Applying spectroscopic modeling, the exciton delocalization in the DNA-templated BC dimer was evaluated by extracting the excitonic hopping parameter, J to be 214 cm−1 (26.6 meV). For comparison, the same method applied to the natural protein-templated Bchla dimer yielded J of 286 cm−1 (35.5 meV). The smaller value of J in the BC dimer likely arose from the partial bacteriochlorin intercalation and the difference in medium effect between DNA and protein.
Introduction
A dimer of photosynthetic pigment bacteriochlorophyll a (Bchla) is the basic functional unit of the light-harvesting antenna system in purple bacteria.1,2 Namely, 16 dimers of Bchla form a B875 ring in the core antenna LH1, and 8–9 dimers form a B850 ring in the peripheral antenna LH2. The close positioning of the pigments in the Bchla dimer with a center-to-center distance of about 9 Å results in the collective sharing of the excited states.2 In such collective sharing, the excited state of a dimer (a molecular exciton or Frenkel exciton3) can be viewed as a quasiparticle that resides on one neighboring pigment or another according to the quantum-mechanical principle of superposition. In other words, the exciton is delocalized between the two pigments. The exciton delocalization extended to other dimers in each antenna ring enables excitation energy transfer to the reaction center with a high quantum efficiency.2,4–6 The quantum state of exciton delocalization that a photosynthetic antenna maintains at ambient temperatures and in often harsh environmental conditions is highly desirable for engineering excitonic systems7–9 in artificial photosynthesis,10,11 organic optoelectronics,12–14 and room temperature quantum computing.15–18 Hence, it is important to access stable and tunable synthetic analogs of Bchla dimer exhibiting exciton delocalization at room temperature.The structural basis of Bchla is a tetrahydroporphyrin, a 4e−/4H+ reduced porphyrin with alternating pyrrole and pyrroline rings known as bacteriochlorin macrocycle. The steady-state absorption spectrum of the bacteriochlorin chromophore consists of four characteristic porphyrin bands: the fundamental Soret bands By and Bx, and the Qx and Qy bands. In bacteriochlorin, the Qy band responsible for the light absorption to produce the lowest singlet excited state is significantly more intense (ε ∼100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cm−1 M−1) and redshifted than that in the porphyrin due to the reduction of the porphyrin opposing pyrrole rings. Such long-wavelength absorption can enable pure exciton states not mixed with the transitions of the UV region, which—in addition to the exceptional narrowness and minimal vibronic structure of the bacteriochlorin Qy band—are spectral features strongly advantageous for engineering exciton circuits.15
000 cm−1 M−1) and redshifted than that in the porphyrin due to the reduction of the porphyrin opposing pyrrole rings. Such long-wavelength absorption can enable pure exciton states not mixed with the transitions of the UV region, which—in addition to the exceptional narrowness and minimal vibronic structure of the bacteriochlorin Qy band—are spectral features strongly advantageous for engineering exciton circuits.15
According to the molecular exciton model developed by Kasha,3 exciton delocalization is primarily enabled by the excitonic Coulombic coupling of the dye transition dipole moments (TDMs). The intermolecular chromophore distance strongly influences the strength of the excitonic coupling. An intermolecular distance that is too short can result in concentration quenching.19 In contrast, a distance that is too long prevents excitonic coupling; transfer of the excitation energy then occurs via the Föster mechanism between an acceptor and donor chromophore, as seen between the antenna rings B800 to B850 or B850 to B875.5 In the photosynthetic antenna of purple bacteria, the ideal intermolecular distance of 0.4–1.0 nm and orientation of Bchla is maintained by a protein scaffold. In both LH1 and LH2, a Bchla dimer is templated by an α/β apoprotein subunit; though, the protein subunit of LH2 contains an additional distant Bchla monomer.2 In addition to securing relative positions of Bchla dyes, specific non-covalent interactions between amino acids and Bchla are thought to enhance its TDM, and red-shift its site energy.20,21
The examples of bacteriochlorin dimer exhibiting signatures of excitonic coupling and delocalization are limited to a few reported systems created for fundamental studies of photosynthesis and utilizing natural Bchla or its semisynthetic derivatives. Namely, in the absence of a scaffold, a dimer of isolated Bchla was formed via self-aggregation in solution,22,23 and by tethering Bchla dyes via a non-conjugating linker.24 In addition, a Bchla dimer was non-covalently templated by natural α/β apoprotein (reconstructed 820B complex),25 and by synthetic proteins.26,27 The limitation of the self-aggregation method is the presence of a large monomer population or high-order aggregates; while linking two dyes or utilizing non-covalent protein templating is limited to small size aggregates of 1–2 dyes. Using proteins as scaffolds can also be challenging due to their strong propensity to aggregate in solution, conformational sensitivity to modifications, and complex design rules based on 24 amino acid building blocks. Another challenge associated with the above systems is the instability of natural and semisynthetic Bchla due to its favorable conversion into a porphyrin and limited synthetic tunability owing to a full set of substituents around the Bchla macrocycle. These studies highlighted the need for (1) stable and tunable bacteriochlorin, and (2) a covalent templating method with a stable scaffold that can yield precise control over the number of dyes in small and high-order dye aggregates.
DNA is a versatile scaffold owing to its predictable and robust self-assembly and stability,28,29 and simple design rules of only 4 nucleotides encoding the structure of the DNA. Having a hydrophilic negatively charged backbone, DNA is stable in an aqueous solution. The structure of the DNA is robust to local modifications. Further, a dye can be tethered to a specific site of DNA via a variety of covalent linkers, thus enabling control over the number of dyes and their positions relative to the DNA.30 These advantages of dye covalent templating via DNA enabled exciton delocalization in aggregates of such dye families as azo dyes,31–33 merocyanines,34 polymethine dyes,35–43 squaraines,44–48 and perylenes.49 While several studies have shown that aggregates of porphyrins,50–54 the most related dye family to bacteriochlorins, can be covalently templated via DNA, the extent of exciton delocalization was not assessed.
Here, we explored if a fully synthetic DNA-templated bacteriochlorin dimer can exhibit exciton delocalization. We employed a specific type of branched DNA structure, a 4-arm DNA construct known as a Holliday Junction (HJ) that was previously shown to template a variety of dyes via a post-modification of the HJ core36,43,47 and was shown to be particularly advantageous in solubilizing hydrophobic molecules.47 For the dye, we synthesized a novel analog of Bchla using a de novo bacteriochlorin methodology developed by Lindsey.55,56 Bacteriochlorins available through this methodology are designed with geminal dimethyl groups in pyrroline rings to make them far more chemically and thermally stable than natural Bchla. The diversity of de novo bacteriochlorin synthetic modifications enabled us to create bacteriochlorin (BC) with a large TDM comparable with that of Bchla, as well as equip BC with a covalent linker for DNA tethering (Fig. 1). To assess the suitability of DNA to template a bacteriochlorin dimer exhibiting exciton delocalization, we calculated the strength of the excitonic coupling in terms of the excitonic hopping parameter, J, in the DNA-templated bacteriochlorin dimer and the natural Bchla dimer templated by α/β apoprotein in solution known as 820B complex. Semiempirical modeling of steady-state spectra based on the theoretical approach of Kühn, Renger, and May57 determined J values of 26.6 and 35.5 meV in bacteriochlorin and Bchla dimers, respectively, as well as their distinct dimer geometries. While BC exhibited TDM sufficiently high to achieve exciton delocalization in the aqueous DNA environment and the absence of dye–protein interactions, bacteriochlorin dimerization was likely disrupted by partial intercalation into DNA. Collectively, these results indicate that DNA nanotechnology can be a facile means to achieve exciton delocalization in synthetic bacteriochlorin aggregates by selecting complementary bacteriochlorin chemical structures and DNA templates.
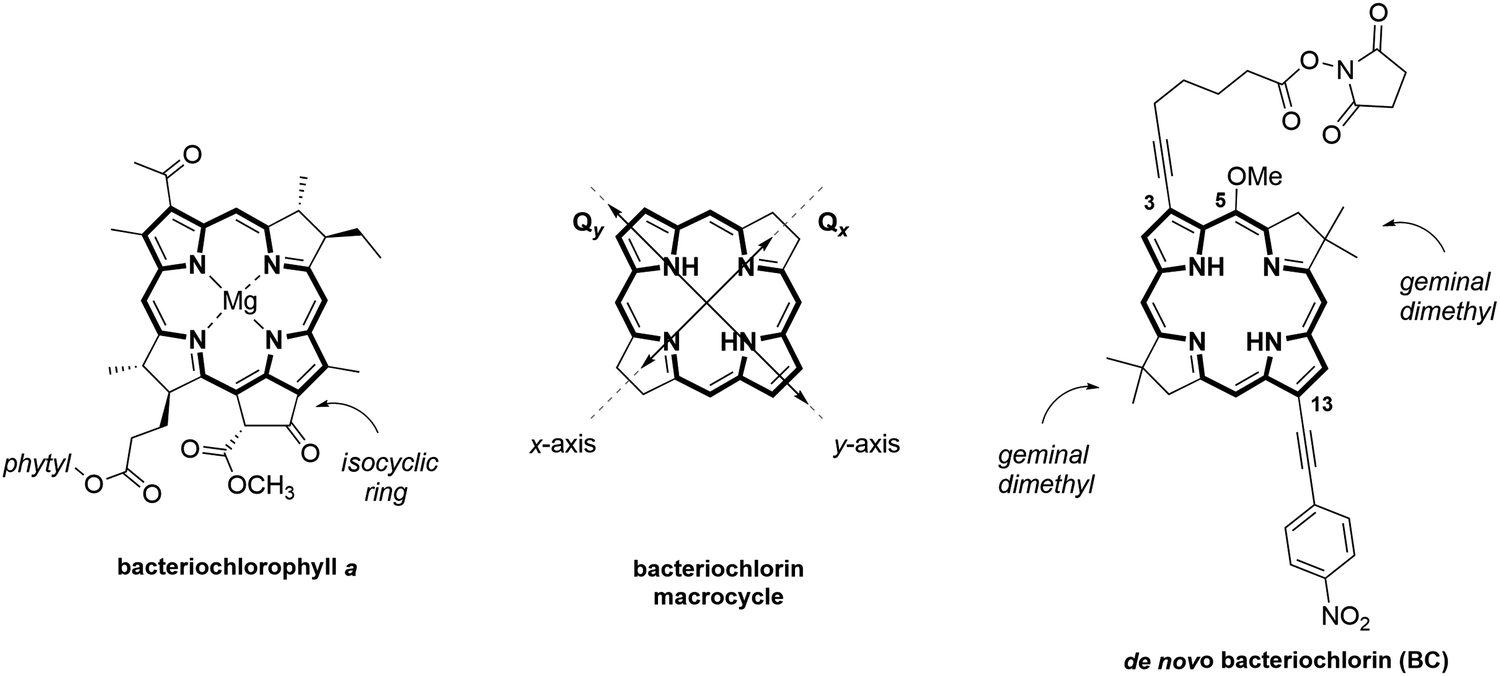 | ||
| Fig. 1 Chemical structures of (left) natural bacteriochlorophyll a, (center) bacteriochlorin macrocycle depicting Qx and Qy transitions polarized along molecular x- and y-axis, respectively, and (right) synthetic bacteriochlorin BC. | ||
Results
Dye design
To create a DNA-templated dimer for this study, we chose the synthetic bacteriochlorin analog of natural Bchla depicted in Fig. 1 as BC. The synthetic BC contains a bacteriochlorin macrocycle of Bchla but exhibits greater thermal and chemical stability due to the presence of geminal dimethyl groups that prevent dehydrogenation into a porphyrin. Established de novo methodology for synthetic bacteriochlorins offers diverse synthetic modularity of their chemical and photophysical properties.55,58,59 The BC side substituents were selected to increase the Qy transition dipole moment magnitude, which is critical for stronger excitonic coupling.60 Survey studies demonstrated that auxochromes—functional groups that alter the spectral properties of a chromophore—positioned along the Qy TDM axis (attachment sites 2-, 3- and 12-, 13-) have the strongest influence on bacteriochlorin spectral properties. Specifically, ethynyl-type groups caused a significant enhancement of Qy magnitude.61 Based on these observations, we designed BC to carry 13-(4-nitrophenyl)ethynyl and 3-ethynyl auxochromes and N-hydroxysuccinimide ester (NHS) linker for DNA tethering. The presence of the isocyclic ring and the central metal were shown to cause Qy to redshift, but had a small effect on Qy magnitude,62 and were therefore omitted in the BC structural design. The methoxy group at the 5-position was installed to provide the means for the asymmetric chemical derivatization of the macrocycle.63Chemical synthesis
The synthesis of the target NHS-bacteriochlorin BC for tethering to DNA relied on the selective derivatization of the 3,13-dibromo-5-methoxybacteriochlorin building block (Fig. 2). First, β-bromodihydrodipyrrin-acetal 1 was prepared according to the established streamlined multistep synthesis.64 Self-condensation of 1 into 2 in the presence of TMSOTf/2,6-DTBP was carried out according to the reported procedure55 with several modifications. In particular, reducing the amount of TMSOTf from 5 to 3 molar equivalents, added in increments over 1 h, afforded 2 with 92% purity in 54% yield. Monitoring the reaction progress by thin layer chromatography indicated reaction completion in 3 h. We found that high purity of 1 is critical for achieving a high yield of its self-condensation. Next, bacteriochlorin 2 was subjected to two subsequent Sonogashira reactions under the conditions for asymmetric disubstitution previously developed by Yu and Ptaszek, taking advantage of the 5-methoxy group sterically hindering the 3-position.63 Compound 2 was treated with 1-ethynyl-4-nitrobenzene in the presence of Pd(PPh3)4 and K2CO3 in DMF to afford 3 functionalized with 13-(4-nitrophenyl)ethynyl group in 28% yield; small amounts of the disubstituted product and unreacted starting material were observed. Treatment of 3 with 6-heptynoic acid NHS ester 4, catalyzed by (PPh3)2PdCl2 in Et3N/DMF, afforded the desired NHS ester BC in 18% yield. Newly reported compounds 3 and BC were characterized by 1H NMR, 13C NMR, ESI-MS, steady-state absorption, and fluorescence spectroscopies.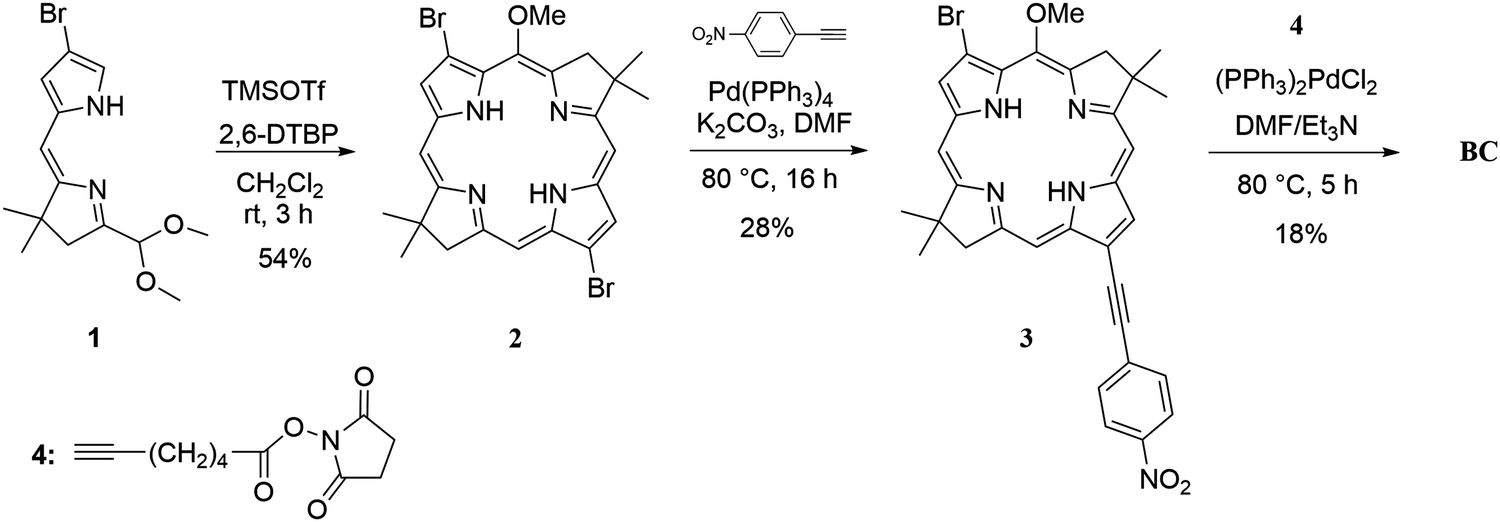 | ||
| Fig. 2 Synthetic route to the de novo NHS ester bacteriochlorin BC. | ||
DNA templating
To template a BC dimer covalently to DNA, we chose a four-armed HJ construct. Previously, we showed that conformational isomerization of HJ between two isoforms65 could result in a racemic mixture of two dimer enantiomorphs tethered to DNA via covalent linkers of equal length.66 In that study, we also employed covalent linkers of unequal length to cause a conformational bias toward one HJ isoform and, hence, one optical isomer of the dimer.66 Here, we implemented an alternative approach to avoid HJ conformational isomerization by spanning two HJ arms via a short chain of 14 unpaired thymidines. This approach was employed previously to constrain an unlabeled HJ.67,68 The HJ was assembled using three oligo strands: A, C, and BD containing the thymidine chain. Oligo strands C and BD were internally modified with the amino thymidine T* for dye tethering. The BC in the form of NHS esters were tethered to BD and C oligo strands (Fig. 3a), purified by high-performance liquid chromatography, and characterized by ESI-MS at Integrated DNA Technologies (Section S2, ESI†). To create HJ constructs, oligo strands A, C, and BD were combined in equimolar amounts in aqueous buffer 1× TBE 15 mM MgCl2 to the final concentration of 1.5 μM or 4.0 μM. Due to the sensitivity of bacteriochlorins to high temperatures, we avoided a standard DNA anneal procedure, which would have required heating the samples to 94 °C followed by cooling. Instead, to hybridize oligos into HJs, the samples were incubated at room temperature for 2 h. In this manner, we created the BC dimer and a control monomer construct (Fig. 3b). The formation of BC–HJ constructs was confirmed by non-denaturing polyacrylamide gel electrophoresis (Section S3, ESI†). | ||
| Fig. 3 Molecular designs of bacteriochlorin–DNA constructs. (a) an oligo post-modification with BC depicting chemical structures of the thymidine amino modifier (T*) and the resulting structure of BC tethered to the thymidine in an oligo strand. (b) Schematics of BC monomer and BC dimer. Each green diamond represents BC. The HJ strands are labeled A, BD, and C. Strand BD contains a chain of 14 unpaired thymidines. | ||
Optical properties
Spectral properties of “free” BC (i.e., not covalently attached to DNA) were characterized by measuring absorption and fluorescence in toluene at room temperature. Free BC exhibited an absorption spectrum characteristic for bacteriochlorins featuring four absorption bands: By(0,0) Soret at 355 nm, Bx(0,0) Soret at 375 nm, Qx(0,0) at 530 nm, and Qy (0,0) at 757 nm (Fig. 4 and Table 1). For comparison, the synthetic precursors 3,13-dibromo bacteriochlorin 2 without ethynyl groups and bacteriochlorin 3, containing one ethynyl group at the 13-position, absorbed at 723 and 749 nm, respectively (Fig. S10, ESI†). The serial redshift of the Qy band upon ethynyl mono and disubstitution indicated that both ethynyl groups were strongly conjugated with the macrocycle in BC. A weak vibronic progression was observed to higher energy in the absorption spectrum of BC. As typical for synthetic bacteriochlorins, the Qy band was exceptionally narrow with a full width at half-maximum (fwhm) of 23 nm (402 cm−1). To evaluate the molar extinction of the long-wavelength absorption band in BC, we relied on the established approach61 to calculate the peak-intensity ratio and integrated manifold ratio of Qy and Soret (By and Bx), then compared the ratios to those of Bchla (Table 1). The absorption manifolds of BC and Bchla were similar, indicating that the molar extinction of Qy in BC should be at least as large as the reported extinction of Bchla of 92000 cm−1 M−1.69 In addition, we measured the absorption of “free” BC in a protic solvent such as ethanol for later comparison with the absorption of BC covalently attached to DNA in an aqueous environment (Fig. 4 and Table 1). From toluene to ethanol, BC exhibited a small blueshift and a small decrease in Qy intensity relative to the Soret manifold. The observed effect of the protic solvent on BC absorption was in good agreement with a previous report on structurally similar synthetic bacteriohlorins.70 Lastly, the BC fluorescence peaked at 761 nm in toluene (756 nm in ethanol), yielding a Stokes shift of 3 nm (4 nm in ethanol) (Fig. S11, ESI†). In ethanol, the BC fluorescence scaled by absorptance decreased by 20% compared to that in toluene.
 | ||
| Fig. 4 Acquired steady-state absorption spectrum of “free” BC in toluene (solid dark green) and in ethanol (solid light green) and a reference spectrum of “free” Bchla (dotted green) in toluene obtained from ref. 69. The spectra are normalized at the Qy maximum. | ||
| “Free” dye | Solvent | λ By(0,0), Bx(0,0)/nm | λ Qx(0,0)/nm | λ Qy(0,0)/nm | ε Qy/cm−1 M−1 | fwhm Qy/cm−1 (nm) | I Qy/IByc | I Qy/IBxd |
|
|---|---|---|---|---|---|---|---|---|---|
| a At room temperature. b Absorption data from ref. 69 (in toluene). c Ratio of the intensities of the By(0,0) and Qy(0,0) bands. d Ratio of the intensities of the Bx(0,0) and Qy(0,0) bands. e Ratio of the integrated intensities of the B (By and Bx; 320–420 nm) and Qy [Qy(0,0) and vibrational progression including Qy(1,0); 660–850 nm] bands. | |||||||||
| BC | Ethanol | 353, 369 | 522 | 750 | — | 466 (26) | 1.12 | 1.03 | 0.64 |
| BC | Toluene | 355, 375 | 530 | 757 | — | 402 (23) | 1.58 | 1.44 | 0.75 |
| Bchla | Toluene | 361, 396 | 580 | 780 | 92000 |
579 (35) | 1.30 | 2.04 | 1.03 |

The BC monomer covalently templated by HJ was characterized by steady-state absorption, fluorescence, and circular dichroism (CD) spectroscopies. To evaluate the solvating environment of the BC covalently attached to DNA, the spectral properties of the monomer (i.e., a single dye attached to the strand BD in HJ) were compared to those of “free” BC in a nonpolar solvent such as toluene and in a polar protic solvent such as ethanol (Fig. 4 and 5a). The monomer exhibited the Soret maxima (By and Bx) at 356 and 375 nm, Qx at 533 nm and Qy at 767 nm. Relative to the “free” BC in toluene and ethanol, the monomer's Soret had minor redshifts with the reversed intensity ratio of By and Bx. In contrast, the Q bands exhibited more noticeable redshifts, with the Qx red-shifted by 3 nm and 11 nm, and the Qy red-shifted by 10 nm and 17 nm relative to toluene and ethanol, respectively. A significant redshift of the Q bands relative to the “free” BC in ethanol suggested the monomer being exposed to a nonpolar DNA groove solvation environment rather than an external aqueous environment. Another characteristic of the BC–DNA interaction was obtained from the spectral bandwidth. The Qy bandwidth of the monomer [fwhm = 461 cm−1 (27 nm)] was comparable with that of “free” BC in ethanol and toluene, indicating that such events as macrocycle distortion or multiple modes of binding that would typically result in absorption broadening relative to the “free dye” did not take place in the BC monomer construct. Finally, the hypochromicity was evaluated by calculating monomer Qy molar extinction. The DNA absorption peak appeared in the monomer spectrum at 260 nm (Fig. S12, ESI†). Based on the DNA theoretical extinction coefficient, the Qy molar extinction (εQy) of the monomer was calculated to be 104811 cm−1 M−1. The value of εQy of the BC monomer is higher than that of free Bchla in toluene,69 which suggests no hyperchromicity effect in an aqueous DNA environment.
 | ||
| Fig. 5 (a) Acquired steady-state absorption of BC monomer and dimer tethered to HJ. The sample concentration for absorption was 1.5 μM in 1× TBE 15 mM MgCl2. Acquired steady-state circular dichroism of (b) BC monomer, (c) BC dimer tethered to HJ, and (d) magnified Qy region. The sample concentration for CD was 4.0 μM in 1× TBE 15 mM MgCl2. The spectra were recorded at room temperature. | ||
To gain more insight into the interactions of BC with DNA, we characterized the BC monomer by CD spectroscopy (Fig. 5b). The CD of the monomer exhibited a profound DNA bisignate couplet at 244 and 280 nm indicative of a well-formed B-DNA duplex. In the bacteriochlorin region, a weak negative feature (17 M−1 cm−1) was observed in the vicinity of the Soret Bx region at 376 nm, while no signal was observed in the Qx and Qy regions. Since BC is achiral, the observed negative signal was attributed to the induced CD (ICD) due to the coupling between the DNA bases and a bacteriochlorin Bx Soret transition. The position of a ligand relative to DNA can be gauged from the sign and magnitude of the ICD.71 A small positive ICD is typical for the intercalated ligands with the TDM oriented perpendicular to the base pair axis, while a small negative ICD occurs when the ligand TDM is parallel to the long base pair axis. On the other hand, a strong positive CD signal is associated with a ligand bound to the minor groove, thus having its TDM positioned 45° to the bases. Therefore, the small negative ICD in the vicinity of Bx suggested that bacteriochlorin could be oriented such that its x-molecular axis is parallel to the base pair long axis. Since bacteriochlorin molecular geometry is unfavorable for intercalation due to sterically bulky geminal dimethyl groups in the opposing pyrroline rings, the bacteriochlorin could be partially intercalated via its nitrophenyl substituent. This assumption was further supported by the reported strong intercalating propensity of nitrophenyl ligands.72 The absence of the observed coupling between the Qy transition and DNA was expected given the drastic difference in the Qy and DNA transition energies. Lastly, the monomer did not exhibit fluorescence even at a high sample concentration of 4 μM (Fig. S11, ESI†). Only fluorescence traces from “free” BC were observed in the monomer sample. Fluorescence quenching of monomer could be explained with the partial bacteriochlorin intercalation or photoinduced electron transfer to DNA.73,74
Next, we performed steady-state spectroscopy to characterize the DNA-templated BC dimer. In the absorption spectrum, BC dimer exhibited the following changes relative to the monomer spectrum: Qy blueshift from 767 nm to 764 nm, and Qx and Soret redshifts of 2 nm (Fig. 5a and Fig. S12, ESI†). The perturbations in the dimer absorption relative to that of the monomer seemed to indicate a weak excitonic coupling between the chromophores in face-to-face or H-aggregate orientation. No fluorescence was observed in the BC dimer (Fig. S11, ESI†), indicating excited-state quenching often observed in H-aggregates.75 An additional signature of excitonic coupling in the BC dimer was evident from its CD spectrum (Fig. 5c). The dimer exhibited three CD features in the dye region not observed in the CD spectrum of the monomer. First, the dimer showed a negative band at 356 nm corresponding to the By Soret absorption maximum. In addition, the dimer spectrum exhibited a negative band at 538 nm in the Qx region, and a weak bisignate couplet with the intersection point at 767 nm corresponding to the monomer Qy absorption maximum (Fig. 5d). The bisignate couplet was characterized by Cotton effects at 755 and 781 nm of equal magnitude and opposite sign indicative of the exciton-coupled dimer. Due to the small signal magnitude and its proximity to the noisy range of the 900 nm detector limit, we carried out several CD measurements optimizing the instrument settings; the Qy bisignate couplet was consistently observed under different settings (Fig. S13, ESI†). While the dimer Soret and Qx spectral features could arise from both BC excitonic coupling and electronic interactions with DNA bases, the spectral changes in the vicinity of Qy were attributed solely to excitonic coupling and delocalization in the BC dimer. The Davydov splitting of 441 cm−1 (a difference between Cotton effects of the bisignate couplet) afforded spectral excitonic coupling strength of 220.5 cm−1 (27.4 meV).
Modeling
To quantify the strength of excitonic coupling responsible for exciton delocalization spectroscopically observed in the BC dimer, we applied semiempirical modeling42,48 based on the theoretical approach of Kühn, Renger, and May (Section S8, ESI†).57 For closely positioned and collectively excited chromophores, the excitonic coupling is primarily based on dipole–dipole Coulomb interaction of the TDMs. According to Kasha's molecular exciton model,3 the strength of the Coulomb interaction (or hopping interaction) can be expressed with the exciton hopping parameter J. To determine J in the bacteriochlorin dimer, we simultaneously modeled BC dimer experimental absorption and CD spectra utilizing a Holstein-like76 Frenkel Hamiltonian60 (eqn (S1), ESI†). In brief, the modeling generated various theoretical dimer configurations and calculated J as Coulomb dipole–dipole coupling for each dimer configuration using the extended dipole approximation77 (eqn (S4), ESI†). Placing two point charges of the opposite sign along the Qy transition accounted for the J dependence on the TDM center-to-center distance, angle, and displacement. The TDM magnitude of the DNA-templated BC monomer was determined from its experimental absorption spectrum to be 6.9 D (eqn (S5), ESI†). To account for the dye–DNA interactions and medium effect, the experimental TDM value of the monomer and solvent refractive index were incorporated in the numerical coefficient J0 (eqn (S7), ESI†). Next, the J values were used to populate the Holstein-like augmented Frenkel Hamiltonian consisting of the electronic and vibronic components and generate theoretical absorption and CD spectra (eqn (S10) and (S11), ESI†). The theoretical spectra were compared with the experimental absorption and CD spectra and assigned a fitness score. The search for the best-fit spectra and the associated J value continued by iterations in TDM orientations guided by stochastic gradient descent. To ensure that the found dimer configuration was the unique solution to fit the experimental spectra, we modeled each system twice, starting with two extreme initial dimer configurations, face-to-face and end-to-end.KRM modeling, as described above, was performed for two representative cases of the bacteriochlorin–HJ system. In Case 1, we assumed a homogeneous dimer system where bacteriochlorin dyes were fully available to form the dimer (Fig. 6a). In Case 2, we assumed a heterogeneous system of a dimer and two monomers, where a population of the intercalated dyes was not sterically available to form a dimer, thus resulting in excess of “optical” monomers (i.e., distant dyes in the dimer HJ construct) and a dimer population (Fig. 6b). When modeling Case 2, the “optical” monomers were kept sufficiently distant from the dimer and each other to prevent their coupling. The modeling of Case 2 was performed as 50% dimer and 50% monomer; other dimer–monomer ratios could not be examined due to the limitation of the KRM modeling code.
 | ||
| Fig. 6 Two representative cases of bacteriochlorin-HJ system and their KRM modeling. (a) Case 1: a homogeneous system of one dimer population; (b) Case 2: a heterogeneous system of a dimer and monomer populations. (c) and (d) Acquired steady-state absorption and circular dichroism spectra (solid lines) of BC monomer and BC dimer and theoretical KRM fit (dash line) for (c) Case 1, and (d) Case 2. To account for only 50% of the bacteriochlorin molecules responsible for the observed CD in Case 2, the experimental spectrum magnitude was increased by 2. Vertical bars represent energy eigenstates with heights relative to their transition amplitudes. The bar segments show the relative electronic (blue) versus vibronic (orange) contribution to each transition. | ||
The theoretical spectra that afforded the best fit for the BC dimer systems 1 and 2 are shown in Fig. 6c and d. The J values were 18.5 meV (149 cm−1) and 26.6 meV (214 cm−1), for Cases 1 and 2, respectively. While modeled spectra for both cases exhibited good agreement with the experimental results (Table S5, ESI†), the extracted value of J for Case 2 was in closer agreement with the spectral coupling strength of 27.4 meV from Davydov splitting, and so Case 2 appeared to be a more realistic description of the bacteriochlorin-HJ system. The KRM-derived J of 26.6 meV could be underestimated if the fraction of the intercalated dye population was higher than the modeled 50%.
As a first step to compare the DNA-templated BC dimer and the protein-templated Bchla dimer, we applied the same spectroscopic modeling approach to calculate J in the natural Bchla dimer from the 820B complex. For this modeling, we used experimental absorption and CD spectra of 820B reported by Parkes-Loach et al. (Fig. S14 and S15, ESI†).25 Isolated from the LH1 antenna of Rhodospirillum rubrum, 820B consisted of two Bchla molecules templated by α/β apoprotein, and lacked the carotenoid and ubiquinone components.25 Because the absorption of the reference Bchla monomer templated by apoprotein was unavailable, we used the reported absorption of “free” Bchla (i.e., 777B) in an aqueous buffer containing methanol from ref. 25 (Fig. S14, ESI†). The absorption maxima of “free” Bchla in an aqueous buffer and 820B dimer were 777 and 820 nm, respectively. The energy difference between “free” Bchla and 820B dimer is likely due to a combination of excitonic coupling and protein-Bchla interactions. To account for the protein-Bchla interactions in the KRM modeling, we included the offset energy of 50 meV (403 cm−1). Applying a TDM of 7.5 D calculated from the spectrum of 777B, experimental absorption and CD spectra of 820B dimer were fitted with the theoretical curves (Fig. 7). The KRM modeling determined J in isolated 820B dimer to be 35.5 meV (286 cm−1) which was in close agreement with the strength of excitonic coupling of about 300 cm−1 calculated by other methods.78–80 To further characterize the spectral properties of the BC and Bchla dimers, we compared the origin of excitonic transitions obtained from the computed eigenvalues and eigenvectors vectors of the Holstein-like Hamiltonian.76 The KRM modeling indicated that the three lowest energy transitions of the BC dimer (Case 2) were mostly of electronic origin, i.e., associated with the 0–0 transition. At higher energy (and shorter wavelength), the KRM modeling derived an additional transition of vibronic origin associated with 0–n transition due to weak vibronic coupling. The Bchla dimer was characterized by a similar nature of excitonic transitions (Fig. 7). Overall, both BC and Bchla dimers showed that most of their oscillator strength is of the electronic transitions, which is advantageous in diminishing exciton decoherence originated from the exciton–vibron coupling.15
 | ||
| Fig. 7 Steady-state absorption and circular dichroism spectra (solid lines) of 777B “free” dye in phosphate buffer containing methanol and n-octyl β-D-glucopyranoside, and 820B dimer templated by apoprotein at 10 μM in phosphate buffer containing n-octyl β-D-glucopyranoside from ref. 25, and theoretical KRM fit (dash line). Vertical bars represent energy eigenstates with heights relative to their transition amplitudes. The bar segments show the relative electronic (blue) versus vibronic (orange) contribution to each transition. | ||
As a second step of comparison, we analyzed the geometries of the DNA-templated BC dimer (Case 2) and protein-templated Bchla dimer. The mutual TDM orientations that afforded the best spectral fit were extracted from the vector spherical and Cartesian coordinates (Fig. 8 and Table S6, ESI†). The dimer geometries were described in terms of center-to-center distance R, and oblique angle α to characterize a deviation from a parallel TDM orientation. In addition, slip angles θs were extracted to characterize a displacement of one TDM relative to another and, hence, an aggregate type (i.e., J-dimers exhibit θs < 54.7° while H-dimers exhibit θs > 54.7°). In the Bchla dimer, TDMs adopted a head-to-tail orientation (J-aggregate) evident in the sharp slip angles of 35° and 7° with a center-to-center distance of 10.1 Å and a 28° obliquity. The TDM orientation in Bchla dimer derived by KRM from the spectral data of reconstructed 820B complex in solution25 was in close agreement with that of the Bchla dimer unit in the crystal structure of LH1-R1 Thermochromatium tepidum at a resolution of 1.9 Å.81 On the other hand, in BC dimer, the TDMs adopted a face-to-face orientation (H-aggregate) evident in the slip angles of 97° and 68° with R of 6.7 Å and an oblique angle of 30°. To visualize prospective molecular dimer geometries, the theoretical Qy polarized along the molecular y-axes were aligned with the extracted TDM vectors; dye macrocycles were oriented arbitrarily face-to-face (Fig. 8).
 | ||
| Fig. 8 KRM-derived TMD vector orientations and dimer geometries of isolated Bchla dimer (820B complex) templated by α/β apoprotein, and BC dimer templated by DNA HJ. (a) and (b) Three-dimensional TMD vector plots with TMD plane projections XY, XZ, and YZ. (c)–(h) Dimer geometries with the geometric parameters: center-to-center distance R in Å, and oblique angle α in ° shown in the oblique view, and slip angles θs in ° shown in the side view. The TDM vectors were aligned with the dye Qy transitions; the dye core rotations were arbitrarily chosen. | ||
Discussion
Motivated by reports of exciton delocalization in natural light-harvesting aggregates observed at room temperature,82–85 we explored the potential to create a similar system using more robust engineering materials: stable and tunable synthetic bacteriochlorin and self-programmable DNA. Namely, by calculating the excitonic hopping parameter J, we explored if a DNA-templated dimer of Bchla synthetic analog can exhibit exciton delocalization suitable for building excitonic devices. The J value represents the strength of dipole–dipole Coulomb coupling, which is the main contribution to the excitonic coupling responsible for exciton delocalization between closely positioned chromophores. In fact, J is proportional to the product of TDMs of closely positioned dyes (eqn (S4), ESI†). The magnitude of the TDM primarily stems from the dye core structure but can be enhanced (or reduced) by side substituents and the environment. In natural and synthetic bacteriochlorins, placing auxochromes on opposing pyrrole rings was shown to have the strongest influence on the Qy TDM, while the addition of the isocyclic ring and central metal had a less pronounced effect.61 Though synthetic means of installing an isocyclic ring and zinc central metal have been demonstrated,86,87 we were able to use 3,13-disubstitution alone to achieve a large TDM in the BC. Ethynyl-type groups were chosen to enable a robust conjugation with the macrocycle. The resulting synthetic bacteriochlorin BC exhibited comparable TDM to natural Bchla, evident from a comparison of their integrated absorption manifolds (Table 1) and featured a narrower Qy band with a smaller vibronic progression relative to the natural Bchla.Proceeding with covalent templating BCvia DNA, we found that bacteriochlorin was likely in proximity to nucleobases rather than externally bound to DNA based on the Qy bathochromic shift relative to the “free” dye in ethanol. Further, bacteriochlorin might undergo DNA intercalation based on the weak negative feature in the Bx Soret region of the monomer CD spectrum. We assumed that BC was partially intercalated via its nitrophenyl substituent so the Bx transition was parallel to the long base pair axis. This assumption was further supported by the molecular geometry of BC having bulky geminal dimethyl groups unfavorable for the macrocycle stacking between the base pairs. On the other hand, no hypochromicity nor ICD was observed for the Qy transition in the monomer spectrum, indicating the electronic interactions between Qy and DNA bases did not take place, likely due to a large difference in their transition energies. Apparently, the BC dimer construct exhibited signatures of the excitonic coupling and delocalization evident in the small Qy absorption blueshift and the symmetric bisignate CD couplet. Indeed, a dimer could form between two intercalated dyes. Alternatively, the intercalation could prevent full dye dimerization if several favorable but distant intercalation sites were available on the DNA template. Lacking the direct means to assess the effect of intercalation, we modeled two representative cases: in Case 1, we assumed all dyes covalently attached to DNA were available to form a dimer; in Case 2, 50% of the dyes were sterically unavailable for dimerization, while another 50% of the dyes formed a dimer. The excitonic hopping parameter J was calculated to be 18.5 meV and 26.6 meV for Cases 1 and 2, respectively. The J values derived for Case 2 agreed with the measured excitonic coupling of 27.4 meV from Davydov splitting.
To directly compare exciton delocalization in de novoBC dimer templated by DNA and Bchla dimer templated by protein, we used the same modeling approach to calculate excitonic hopping parameter J in isolated 820B complex, which consisted of Bchla dimer templated by α/β apoprotein.25 The J of 820B, found to be 35.5 meV (286 cm−1), was in close agreement with the previous calculations by others of the excitonic coupling in the Bchla dimer unit of LH1 and LH2 antennas.79,80,82 While both free Bchla and BC exhibited comparable TDM magnitudes in the organic solvent, the J in DNA-templated de novoBC dimer was about 10 meV (30%) smaller than that in the 820B dimer templated by apoprotein in solution. There are several possible explanations for this difference. First, if monomer intercalation occurred, it would disturb efficient dimerization, which would cause an underestimation of J in BC dimer. Second, the 30% difference between J in Bchla dimer and BC dimer corresponded to the 1.2× difference in the TDM and could be attributed to the medium effects and local polarization of Bchla by the protein, which do not occur in DNA. Another difference between the DNA-templated BC and protein-templated Bchla was found in the dimer geometries. The KRM-derived geometry of 820B agreed with the geometry of the Bchla dimer unit in the LH1 antenna crystal structure at 1.9 Å resolution.81 The dyes in 820B were found to be oriented head-to-tail (i.e., a J-aggregate) with a center-to-center distance of 10.1 Å with the 28° oblique angle (Fig. 8). In contrast, the dyes in DNA-templated BC dimer adopted an H-aggregate orientation with a center-to-center distance of 6.7 Å and 30° oblique angle. We attributed the difference in the aggregate geometry primarily to the polarity of the medium. In an overall hydrophobic protein environment, the position of each Bchla is secured via van der Waals interactions with surrounding amino acids, while in an aqueous DNA environment, the orientation of the dyes in the aggregate is governed by minimizing solvent-exposed dye surface as a manifestation of the hydrophobic effect. Therefore, to access oblique or head-to-tail TDM orientations in DNA-templated bacteriochlorin aggregates, future structural designs should be focused on mitigating face-to-face bacteriochlorin alignment utilizing steric substituents or coordination chemistry.
Conclusion
We created a fully synthetic DNA-templated BC dimer exhibiting exciton delocalization and featuring stability and functional modularity. Incorporating an NHS linker enabled BC covalent attachment to the DNA HJ with spanned arms. A complement of two non-natural side substituents (not present in Bchla) installed in BC afforded spectral properties of Bchla and a sufficient TDM to achieve exciton delocalization in the aqueous DNA environment and the absence of pigment–protein interactions. Therefore, the synthetic BC enabled us to utilize the unique spectral properties of bacteriochlorins: exceptionally narrow NIR absorption with minimum vibronic contribution, in highly programmable DNA nanotechnology29 to engineer exciton-based materials. Future structural considerations should be focused on the careful selection of bacteriochlorin substituents and complementary DNA templates guided by systematic studies of bacteriochlorin-DNA non-covalent binding modes. A key goal would be to produce a synthetic bacteriochlorin dimer J-aggregate, in which the TDMs align with head-to-tail orientation. As bacteriochlorin dimers (both natural and synthetic) exhibit excitonic coupling in the weak coupling regime, increasing the number of dyes in the aggregates will be required to achieve high J.Experimental methods
Chemical synthesis
:1 dichloromethane/hexanes). The reaction mixture was diluted with dichloromethane, washed (saturated aqueous NaHCO3, water, and brine), dried (Na2SO4), and concentrated using a rotary evaporator. The crude was purified by column chromatography [silica, dichloromethane/hexanes 1:3 then 1:1) to afford 2 as a dark green solid (92.0 mg, 92 wt% purity, 54%). 1H NMR (CDCl3, 600 MHz) δ, ppm: −1.98 (brs, 1H), −1.76 (brs, 1H), 1.93 (s, 12H), 4.34 (s, 3H), 4.40 (s, 4H), 8.51 (s, 1H), 8.52 (s, 1H), 8.70, (s, 1H), 8.74 (s, 1H), 8.75 (s, 1H). λabs (CH2Cl2)/nm 349, 370, 504, 723.
:1 then 3:1) to afford 3 as a red solid (8 mg, 28%). 1H NMR (CDCl3, 600 MHz) δ, ppm: −1.94 (brs, 1H), −1.64 (brs, 1H), 1.95 (s, 6H), 1.96 (s, 6H), 4.35 (s, 3H), 4.43 (s, 2H), 4.44 (s, 2H), 8.00 (d, J = 8.7 Hz, 2H), 8.37 (d, J = 8.7 Hz, 2H), 8.51 (s, 1H), 8.62 (s, 1H), 8.75 (d, J = 2.4 Hz, 1H), 8.85 (d, J = 2.0 Hz, 1H), 8.93 (s, 1H); λabs (CH2Cl2)/nm 350, 365, 521, 749. 13C NMR (CDCl3, 600 MHz) δ, ppm: 31.1, 31.2, 45.5, 46.3, 48.1, 51.5, 64.9, 90.8, 94.6, 96.47, 96.51, 98.5, 107.2, 114.2, 124.08, 124.10, 124.9, 125.8, 127.7, 130.9, 132.5, 134.8, 135.4, 135.8, 137.9, 147.2, 156.6, 160.9, 169.6, 171.5. HRMS (ESI/Q-TOF) m/z: [M+] calcd for C33H30BrN5O3 623.1532; found 623.1538.
:1). The product was washed with anhydrous methanol (2 mL) to afford BC as a red solid (3 mg, 18%). 1H NMR (CDCl3, 600 MHz) δ, ppm: −1.97 (brs, 1H), −1.60 (brs, 1H), 1.94 (s, 12H), 2.01–2.08 (m, 2H), 2.12–2.27 (m, 2H), 2.82–2.92 (m, 8H), 4.41 (s, 3H), 4.43 (s, 4H), 8.00 (d, J = 8.2 Hz, 2H), 8.37 (d, J = 8.2 Hz, 2H), 8.51 (s, 1H), 8.59 (s, 1H), 8.74 (d, J = 1.9 Hz, 1H), 8.82 (d, J = 1.8 Hz, 1H), 8.90 (s, 1H); 13C NMR (CDCl3, 600 MHz) δ, ppm: 20.2, 24.3, 25.8, 28.3, 30.9, 31.1, 31.2, 45.4, 46.2, 48.3, 51.5, 64.9, 91.1, 94.5, 96.4, 96.8, 98.3, 113.6, 115.2, 124.1, 124.2, 126.7, 131.1, 132.1, 132.5, 134.6, 135.7, 135.8, 137.6, 147.2, 156.9, 160.3, 168.7, 169.3, 171.6. λabs (toluene)/nm 355, 375, 530, 757. HRMS (ESI/Q-TOF) m/z: [M+] calcd for C44H42N6O7 766.3115; found 766.3094.
500 M−1 cm−1. The bacteriochlorin-HJ samples were prepared by combining equimolar amounts of three oligomers in 1× TBE, 15 mM MgCl2 pH 8 buffer solution to the final DNA concentrations of 1.5 μM and 4.0 μM for steady-state absorption and CD measurements, respectively. All DNA samples were allowed to hybridize by incubation for 2 h at room temperature.
Optical characterization
UV-vis spectra were recorded in duplicates at room temperature on a single-beam Cary 5000 UV-vis-NIR spectrophotometer (Agilent Technologies) in a quartz cuvette with a 1 cm path length (Starna). Absorption spectra were monitored over a 230–900 nm wavelength range. CD measurements were performed on a JACSO-J1500 spectropolarimeter in a 1 cm path-length quartz cuvette (Jasco). Spectra were recorded over the 230–900 nm wavelength range at a speed of 200 nm min−1, 2 s data integration time, 1 nm bandwidth, and 9 accumulations.Author contributions
Conceptualization, OAM; methodology, OAM, BY, WBK; validation, OAM, DRW, LKP; formal analysis, OAM, DRW, LKP; investigation, OAM, DRW, LKP; data curation, OAM, BY, WBK; writing—original draft preparation, OAM, DRW; writing—review and editing, DBT, OAM, BY, RDP, WBK, LKP, JL, DRW; visualization, OAM, DRW, LKP; supervision, OAM, JL, WBK; funding acquisition, OAM, RDP, BY, JL, WBK. All authors have read and agreed to the published version of the manuscript.Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
This research was supported wholly by the U.S. Department of Energy (DOE), Office of Basic Energy Sciences, Materials Sciences and Engineering Division and DOE's Established Program to Stimulate Competitive Research (EPSCoR) program under Award No. DE-SC0020089. The ESI/Q-TOF of bacteriochlorin molecules were obtained at the Biomolecular Research Center at Boise State, which is supported in part by the Institutional Development Awards (IDeA) from the National Institute of General Medical Sciences (Award no. P20GM103408) and the National Institutes of Health (Award no. P20GM109095), the National Science Foundation (Award no. 0619793 and 0923535), the MJ Murdock Charitable Trust, and the Idaho State Board of Education. We thank Dr Shibani Basu for the assistance with recording a circular dichroism spectrum of BC dimer HJ construct. We thank Dr Natalya Hallstrom for the assistance with PAGE electrophoresis of bacteriochlorin-DNA constructs.References
- R. J. Cogdell, N. W. Isaacs, T. D. Howard, K. McLuskey, N. J. Fraser and S. M. Prince, How Photosynthetic Bacteria Harvest Solar Energy, J. Bacteriol., 1999, 181, 3869–3879 CrossRef CAS PubMed.
- X. Hu, A. Damjanovic, T. Ritz and K. Schulten, Architecture and mechanism of the light-harvesting apparatus of purple bacteria, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 5935–5941 CrossRef CAS PubMed.
- M. Kasha, Energy Transfer Mechanisms and the Molecular Exciton Model for Molecular Aggregates, Radiat. Res., 1963, 20, 55–70 CrossRef CAS PubMed.
- M. Chachisvilis, O. Kuhn, T. Pullerits and V. Sundstrom, Excitons in photosynthetic purple bacteria: Wavelike motion or incoherent hopping?, J. Phys. Chem. B, 1997, 101, 7275–7283 CrossRef CAS.
- T. Mirkovic, E. E. Ostroumov, J. M. Anna, R. Van Grondelle, Govindjee and G. D. Scholes, Light Absorption and Energy Transfer in the Antenna Complexes of Photosynthetic Organisms, Chem. Rev., 2017, 117, 249–293 CrossRef CAS PubMed.
- M. Dahlbom, T. Pullerits, S. Mukamel and V. Sundström, Exciton Delocalization in the B850 Light-Harvesting Complex: Comparison of Different Measures, J. Phys. Chem. B, 2001, 105, 5515–5524 CrossRef CAS.
- G. D. Scholes and G. Rumbles, Excitons in nanoscale systems, Nat. Mater., 2006, 5, 683–696 CrossRef CAS PubMed.
- G. D. Scholes, G. R. Fleming, A. Olaya-Castro and R. van Grondelle, Lessons from nature about solar light harvesting, Nat. Chem., 2011, 3, 763–774 CrossRef CAS PubMed.
- G. D. Scholes, Quantum Biology Coherence in photosynthesis, Nat. Phys., 2011, 7, 448–449 Search PubMed.
- M. R. Wasielewski, Self-Assembly Strategies for Integrating Light Harvesting and Charge Separation in Artificial Photosynthetic Systems, Acc. Chem. Res., 2009, 42, 1910–1921 CrossRef CAS PubMed.
- F. Garo and R. Haner, A DNA-based light-harvesting antenna, Angew. Chem., Int. Ed., 2012, 51, 916–919 CrossRef CAS PubMed.
- X. Wang, R. Sha, W. B. Knowlton, N. C. Seeman, J. W. Canary and B. Yurke, Exciton Delocalization in a DNA-Templated Organic Semiconductor Dimer Assembly, ACS Nano, 2022, 16, 1301–1307 CrossRef CAS PubMed.
- D. L. Kellis, S. M. Rehn, B. L. Cannon, P. H. Davis, E. Graugnard, J. Lee, B. Yurke and W. B. Knowlton, DNA-mediated excitonic upconversion FRET switching, New J. Phys., 2015, 17, 115007 CrossRef.
- D. L. Kellis, C. Sarter, B. L. Cannon, P. H. Davis, E. Graugnard, J. Lee, R. D. Pensack, T. Kolmar, A. Jäschke, B. Yurke and W. B. Knowlton, An All-Optical Excitonic Switch Operated in the Liquid and Solid Phases, ACS Nano, 2019, 13, 2986–2994 CrossRef CAS PubMed.
- B. Yurke, R. Elliott and A. Sup, Implementation of a Frenkel exciton-based controlled phase shifter, Phys. Rev. A, 2023, 107, 012603 CrossRef CAS.
- M. R. Wasielewski, M. D. E. Forbes, N. L. Frank, K. Kowalski, G. D. Scholes, J. Yuen-Zhou, M. A. Baldo, D. E. Freedman, R. H. Goldsmith, T. Goodson, M. L. Kirk, J. K. McCusker, J. P. Ogilvie, D. A. Shultz, S. Stoll and K. B. Whaley, Exploiting chemistry and molecular systems for quantum information science, Nat. Rev. Chem., 2020, 4, 490–504 CrossRef CAS PubMed.
- B. Yurke, in Visions of DNA nanotechnology at 40 for the next 40 – a tribute to Nadrian C. Seeman, ed. N. Jonoska and E. Winfree, Springer, Singapore, 2023, pp. 125–169 Search PubMed.
- M. A. Castellanos, A. Dodin and A. P. Willard, On the design of molecular excitonic circuits for quantum computing: The universal quantum gates, Phys. Chem. Chem. Phys., 2020, 22, 3048–3057 RSC.
- G. S. Beddard and G. Porter, Concentration quenching in chlorophyll, Nature, 1976, 260, 366–367 CrossRef CAS.
- R. J. Cogdell, T. D. Howard, N. W. Isaacs, K. McLuskey and A. T. Gardiner, Structural factors which control the position of the Qy absorption band of bacteriochlorophyll a in purple bacterial antenna complexes, Photosynth. Res., 2002, 74, 135–141 CrossRef CAS PubMed.
- E. Wientjes, G. Roest and R. Croce, From red to blue to far-red in Lhca4: How does the protein modulate the spectral properties of the pigments?, Biochim. Biophys. Acta, Bioenerg., 2012, 1817, 711–717 CrossRef CAS PubMed.
- K. Sauer, J. R. L. Smith and A. J. Schultz, The Dimerization of Chlorophyll a, Chlorophyll b, and Bacteriochlorophyll in Solution1, J. Am. Chem. Soc., 1966, 88, 2681–2688 CrossRef CAS.
- J. Linnanto, J. A. I. Oksanen and J. E. I. Korppi-Tommola, Exciton interactions in self-organised bacteriochlorophyll a – aggregates, Phys. Chem. Chem. Phys., 2002, 4, 3061–3070 RSC.
- M. R. Wasielewski, U. H. Smith, B. T. Cope and J. J. Katz, A synthetic biomimetic model of special pair bacteriochlorophyll a, J. Am. Chem. Soc., 1977, 99, 4173 CrossRef CAS PubMed.
- P. S. Parkes-Loach, J. R. Sprinkle and P. A. Loach, Reconstitution of the B873 light-harvesting complex of Rhodospirillum rubrum from the separately isolated. alpha.- and .beta. -polypeptides and bacteriochlorophyll a, Biochemistry, 1988, 27, 2718–2727 CrossRef CAS PubMed.
- I. Cohen-Ofri, M. van Gastel, J. Grzyb, A. Brandis, I. Pinkas, W. Lubitz and D. Noy, Zinc-Bacteriochlorophyllide Dimers in de Novo Designed Four-Helix Bundle Proteins. A Model System for Natural Light Energy Harvesting and Dissipation, J. Am. Chem. Soc., 2011, 133, 9526–9535 CrossRef CAS PubMed.
- K. R. Reddy, J. Jiang, M. Krayer, M. A. Harris, J. W. Springer, E. Yang, J. Jiao, D. M. Niedzwiedzki, D. Pandithavidana, P. S. Parkes-Loach, C. Kirmaier, P. A. Loach, D. F. Bocian, D. Holten and J. S. Lindsey, Palette of lipophilic bioconjugatable bacteriochlorins for construction of biohybrid light-harvesting architectures, Chem. Sci., 2013, 4, 2036–2053 RSC.
- N. C. Seeman, Structural DNA nanotechnology: an overview, Methods Mol. Biol., 2005, 303, 143–166 CAS.
- N. C. Seeman, DNA in a material world, Nature, 2003, 421, 427–431 CrossRef PubMed.
- V. L. Malinovskii, D. Wenger and R. Haner, Nucleic acid-guided assembly of aromatic chromophores, Chem. Soc. Rev., 2010, 39, 410–422 RSC.
- H. Kashida, H. Asanuma and M. Komiyama, Alternating hetero H aggregation of different dyes by interstrand stacking from two DNA-dye conjugates, Angew. Chem., Int. Ed., 2004, 43, 6522–6525 CrossRef CAS PubMed.
- H. Kashida, M. Tanaka, S. Baba, T. Sakamoto, G. Kawai, H. Asanuma and M. Komiyama, Covalent incorporation of methyl red dyes into double-stranded DNA for their ordered clustering, Chem. - Eur. J., 2006, 12, 777–784 CrossRef CAS PubMed.
- T. Fujii, H. Kashida and H. Asanuma, Analysis of Coherent Heteroclustering of Different Dyes by Use of Threoninol Nucleotides for Comparison with the Molecular Exciton Theory, Chem. – Eur. J., 2009, 15, 10092–10102 CrossRef CAS PubMed.
- J. Dietzsch, D. Bialas, J. Bandorf, F. Würthner and C. Höbartner, Tuning Exciton Coupling of Merocyanine Nucleoside Dimers by RNA, DNA and GNA Double Helix Conformations, Angew. Chem., Int. Ed., 2022, 61, e202116783 CrossRef CAS PubMed.
- B. L. Cannon, D. L. Kellis, L. K. Patten, P. H. Davis, J. Lee, E. Graugnard, B. Yurke and W. B. Knowlton, Coherent Exciton Delocalization in a Two-State DNA-Templated Dye Aggregate System, J. Phys. Chem. A, 2017, 121, 6905–6916 CrossRef CAS PubMed.
- B. L. Cannon, L. K. Patten, D. L. Kellis, P. H. Davis, J. Lee, E. Graugnard, B. Yurke and W. B. Knowlton, Large Davydov Splitting and Strong Fluorescence Suppression: An Investigation of Exciton Delocalization in DNA-Templated Holliday Junction Dye Aggregates, J. Phys. Chem. A, 2018, 122, 2086–2095 CrossRef CAS PubMed.
- P. D. Cunningham, Y. C. Kim, S. A. Diaz, S. Buckhout-White, D. Mathur, I. L. Medintz and J. S. Melinger, Optical Properties of Vibronically Coupled Cy3 Dimers on DNA Scaffolds, J. Phys. Chem. B, 2018, 122, 5020–5029 CrossRef CAS PubMed.
- S. M. Hart, W. J. Chen, J. L. Banal, W. P. Bricker, A. Dodin, L. Markova, Y. Vyborna, A. P. Willard, R. Häner, M. Bathe and G. S. Schlau-Cohen, Engineering couplings for exciton transport using synthetic DNA scaffolds, Chem, 2021, 7, 752–773 CAS.
- D. Heussman, J. Kittell, L. Kringle, A. Tamimi, P. H. von Hippel and A. H. Marcus, Measuring local conformations and conformational disorder of (Cy3)(2) dimer labeled DNA fork junctions using absorbance, circular dichroism and two-dimensional fluorescence spectroscopy, Faraday Discuss., 2019, 216, 211–235 RSC.
- R. J. Mazuski, S. A. Díaz, R. E. Wood, L. T. Lloyd, W. P. Klein, D. Mathur, J. S. Melinger, G. S. Engel and I. L. Medintz, Ultrafast Excitation Transfer in Cy5 DNA Photonic Wires Displays Dye Conjugation and Excitation Energy Dependency, J. Phys. Chem. Lett., 2020, 4163–4172, DOI:10.1021/acs.jpclett.0c01020,.
- F. Nicoli, M. K. Roos, E. A. Hemmig, M. Di Antonio, R. de Vivie-Riedle and T. Liedl, Proximity-Induced H-Aggregation of Cyanine Dyes on DNA-Duplexes, J. Phys. Chem. A, 2016, 120, 9941–9947 CrossRef CAS PubMed.
- S. K. Roy, O. A. Mass, D. L. Kellis, C. K. Wilson, J. A. Hall, B. Yurke and W. B. Knowlton, Exciton Delocalization and Scaffold Stability in Bridged Nucleotide-Substituted, DNA Duplex-Templated Cyanine Aggregates, J. Phys. Chem. B, 2021, 125, 13670–13684 CrossRef CAS PubMed.
- K. M. Duncan, H. M. Byers, M. E. Houdek, S. K. Roy, A. Biaggne, M. S. Barclay, L. K. Patten, J. S. Huff, D. L. Kellis, C. K. Wilson, J. Lee, P. H. Davis, O. A. Mass, L. Li, D. B. Turner, J. A. Hall, W. B. Knowlton, B. Yurke and R. D. Pensack, Electronic Structure and Excited-State Dynamics of DNA-Templated Monomers and Aggregates of Asymmetric Polymethine Dyes, J. Phys. Chem. A, 2023, 127, 4901–4918 CrossRef CAS PubMed.
- M. S. Barclay, S. K. Roy, J. S. Huff, O. A. Mass, D. B. Turner, C. K. Wilson, D. L. Kellis, E. A. Terpetschnig, J. Lee, P. H. Davis, B. Yurke, W. B. Knowlton and R. D. Pensack, Rotaxane rings promote oblique packing and extended lifetimes in DNA-templated molecular dye aggregates, Commun. Chem., 2021, 4, 19 CrossRef CAS PubMed.
- M. S. Barclay, C. K. Wilson, S. K. Roy, O. A. Mass, O. M. Obukhova, R. P. Svoiakov, A. L. Tatarets, A. U. Chowdhury, J. S. Huff, D. B. Turner, P. H. Davis, E. A. Terpetschnig, B. Yurke, W. B. Knowlton, J. Lee and R. D. Pensack, Oblique Packing and Tunable Excitonic Coupling in DNA-Templated Squaraine Rotaxane Dimer Aggregates, ChemPhotoChem, 2022, 6, e202200039 CrossRef CAS.
- S. Basu, K. Cervantes-Salguero, B. Yurke, W. B. Knowlton, J. Lee and O. A. Mass, Photocrosslinking Probes Proximity of Thymine Modifiers Tethering Excitonically Coupled Dye Aggregates to DNA Holliday Junction, Molecules, 2022, 27, 4006 CrossRef CAS PubMed.
- O. A. Mass, C. K. Wilson, G. Barcenas, E. A. Terpetschnig, O. M. Obukhova, O. S. Kolosova, A. L. Tatarets, L. Li, B. Yurke, W. B. Knowlton, R. D. Pensack and J. Lee, Influence of Hydrophobicity on Excitonic Coupling in DNA-Templated Indolenine Squaraine Dye Aggregates, J. Phys. Chem. C, 2022, 126, 3475–3488 CrossRef CAS PubMed.
- O. A. Mass, C. K. Wilson, S. K. Roy, M. S. Barclay, L. K. Patten, E. A. Terpetschnig, J. Lee, R. D. Pensack, B. Yurke and W. B. Knowlton, Exciton Delocalization in Indolenine Squaraine Aggregates Templated by DNA Holliday Junction Scaffolds, J. Phys. Chem. B, 2020, 124, 9636–9647 CrossRef CAS PubMed.
- K. M. Duncan, D. L. Kellis, J. S. Huff, M. S. Barclay, J. Lee, D. B. Turner, P. H. Davis, B. Yurke, W. B. Knowlton and R. D. Pensack, Symmetry Breaking Charge Transfer in DNA-Templated Perylene Dimer Aggregates, Molecules, 2022, 27, 6612 CrossRef CAS PubMed.
- A. Brewer, G. Siligardi, C. Neylon and E. Stulz, Introducing structural flexibility into porphyrin–DNA zipper arrays, Org. Biomol. Chem., 2011, 9, 777–782 RSC.
- D. G. Singleton, R. Hussain, G. Siligardi, P. Kumar, P. J. Hrdlicka, N. Berova and E. Stulz, Increased duplex stabilization in porphyrin-LNA zipper arrays with structure dependent exciton coupling, Org. Biomol. Chem., 2016, 14, 149–157 RSC.
- M. Vybornyi, A. L. Nussbaumer, S. M. Langenegger and R. Häner, Assembling Multiporphyrin Stacks Inside the DNA Double Helix, Bioconjugate Chem., 2014, 25, 1785–1793 CrossRef CAS PubMed.
- M. Endo, M. Fujitsuka and T. Majima, Diastereochemically Controlled Porphyrin Dimer Formation on a DNA Duplex Scaffold, J. Org. Chem., 2008, 73, 1106–1112 CrossRef CAS PubMed.
- B. Albinsson, J. K. Hannestad and K. Börjesson, Functionalized DNA nanostructures for light harvesting and charge separation, Coord. Chem. Rev., 2012, 256, 2399–2413 CrossRef CAS.
- M. Krayer, M. Ptaszek, H.-J. Kim, K. R. Meneely, D. Fan, K. Secor and J. S. Lindsey, Expanded Scope of Synthetic Bacteriochlorins via Improved Acid Catalysis Conditions and Diverse Dihydrodipyrrin-Acetals, J. Org. Chem., 2010, 75, 1016–1039 CrossRef CAS PubMed.
- H.-J. Kim and J. S. Lindsey, De Novo Synthesis of Stable Tetrahydroporphyrinic Macrocycles: Bacteriochlorins and a Tetradehydrocorrin, J. Org. Chem., 2005, 70, 5475–5486 CrossRef CAS PubMed.
- O. Kühn, T. Renger and V. May, Theory of exciton-vibrational dynamics in molecular dimers, Chem. Phys., 1996, 204, 99–114 CrossRef.
- M. Taniguchi, D. L. Cramer, A. D. Bhise, H. L. Kee, D. F. Bocian, D. Holten and J. S. Lindsey, Accessing the near-infrared spectral region with stable, synthetic, wavelength-tunable bacteriochlorins, New J. Chem., 2008, 32, 947–958 RSC.
- H. Jing, S. Liu, J. Jiang, V.-P. Tran, J. Rong, P. Wang and J. S. Lindsey, Meso bromination and derivatization of synthetic bacteriochlorins, New J. Chem., 2022, 46, 5556–5572 RSC.
- D. Abramavicius, B. Palmieri, D. V. Voronine, F. Šanda and S. Mukamel, Coherent Multidimensional Optical Spectroscopy of Excitons in Molecular Aggregates; Quasiparticle versus Supermolecule Perspectives, Chem. Rev., 2009, 109, 2350–2408 CrossRef CAS PubMed.
- E. Yang, C. Kirmaier, M. Krayer, M. Taniguchi, H.-J. Kim, J. R. Diers, D. F. Bocian, J. S. Lindsey and D. Holten, Photophysical Properties and Electronic Structure of Stable, Tunable Synthetic Bacteriochlorins: Extending the Features of Native Photosynthetic Pigments, J. Phys. Chem. B, 2011, 115, 10801–10816 CrossRef CAS PubMed.
- O. Mass, M. Taniguchi, M. Ptaszek, J. W. Springer, K. M. Faries, J. R. Diers, D. F. Bocian, D. Holten and J. S. Lindsey, Structural characteristics that make chlorophylls green: interplay of hydrocarbon skeleton and substituents, New J. Chem., 2011, 35, 76–88 RSC.
- Z. Q. Yu and M. Ptaszek, Multifunctional Bacteriochlorins from Selective Palladium-Coupling Reactions, Org. Lett., 2012, 14, 3708–3711 CrossRef CAS PubMed.
- M. Krayer, T. Balasubramanian, C. Ruzie, M. Ptaszek, D. L. Cramer, M. Taniguchi and J. S. Lindsey, Refined syntheses of hydrodipyrrin precursors to chlorin and bacteriochlorin building blocks, J. Porphyrins phthalocyanines, 2009, 13, 1098–1110 CrossRef CAS.
- C. Hyeon, J. Lee, J. Yoon, S. Hohng and D. Thirumalai, Hidden complexity in the isomerization dynamics of Holliday junctions, Nat. Chem., 2012, 4, 907–914 CrossRef CAS PubMed.
- O. A. Mass, S. Basu, L. K. Patten, E. A. Terpetschnig, A. I. Krivoshey, A. L. Tatarets, R. D. Pensack, B. Yurke, W. B. Knowlton and J. Lee, Exciton Chirality Inversion in Dye Dimers Templated by DNA Holliday Junction, J. Phys. Chem. Lett., 2022, 13, 10688–10696 CrossRef CAS PubMed.
- A. Kimball, Q. Guo, M. Lu, R. P. Cunningham, N. R. Kallenbach, N. C. Seeman and T. D. Tullius, Construction and analysis of parallel and antiparallel Holliday junctions, J. Biol. Chem., 1990, 265, 6544–6547 CrossRef CAS PubMed.
- L. K. Arciszewska, I. Grainge and D. J. Sherratt, Action of site-specific recombinases XerC and XerD on tethered Holliday junctions, EMBO J., 1997, 16, 3731–3743 CrossRef CAS PubMed.
- PhotochemCAD, https://www.photochemcad.com/, (accessed January 18, 2023).
- N. N. Esemoto, Z. Yu, L. Wiratan, A. Satraitis and M. Ptaszek, Bacteriochlorin Dyads as Solvent Polarity Dependent Near-Infrared Fluorophores and Reactive Oxygen Species Photosensitizers, Org. Lett., 2016, 18, 4590–4593 CrossRef CAS PubMed.
- N. C. Garbett, P. A. Ragazzon and J. B. Chaires, Circular dichroism to determine binding mode and affinity of ligand–DNA interactions, Nat. Protoc., 2007, 2, 3166–3172 CrossRef CAS PubMed.
- M. H. Nasir, E. Jabeen, R. Qureshi, F. L. Ansari, A. Shaukat, U. Nasir and A. Ahmed, Investigation of redox mechanism and DNA binding of novel 2-(x-nitrophenyl)-5-nitrobenzimidazole (x = 2, 3 and 4), Biophys. Chem., 2020, 258, 106316 CrossRef CAS PubMed.
- C. A. M. Seidel, A. Schulz and M. H. M. Sauer, Nucleobase-Specific Quenching of Fluorescent Dyes. 1. Nucleobase One-Electron Redox Potentials and Their Correlation with Static and Dynamic Quenching Efficiencies, J. Phys. Chem., 1996, 100, 5541–5553 CrossRef CAS.
- H. Asanuma, T. Fujii, T. Kato and H. Kashida, Coherent interactions of dyes assembled on DNA, J. Photochem. Photobiol., C, 2012, 13, 124–135 CrossRef CAS.
- J. S. Huff, P. H. Davis, A. Christy, D. L. Kellis, N. Kandadai, Z. S. D. Toa, G. D. Scholes, B. Yurke, W. B. Knowlton and R. D. Pensack, DNA-Templated Aggregates of Strongly Coupled Cyanine Dyes: Nonradiative Decay Governs Exciton Lifetimes, J. Phys. Chem. Lett., 2019, 10, 2386–2392 CrossRef CAS PubMed.
- T. Holstein, Studies of polaron motion: Part I. The molecular-crystal model, Ann. Phys., 1959, 8, 325–342 CAS.
- V. Czikklely, H. D. Forsterling and H. Kuhn, Extended dipole model for aggregates of dye molecules, Chem. Phys. Lett., 1970, 6, 207–210 CrossRef.
- K. Sauer, R. J. Cogdell, S. M. Prince, A. Freer, N. W. Isaacs and H. Scheer, Structure-Based Calculations of the Optical Spectra of the LH2 Bacteriochlorophyll-Protein Complex from Rhodopseudomonas acidophila, Photochem. Photobiol., 1996, 64, 564–576 CrossRef CAS.
- G. D. Scholes, I. R. Gould, R. J. Cogdell and G. R. Fleming, Ab Initio Molecular Orbital Calculations of Electronic Couplings in the LH2 Bacterial Light-Harvesting Complex of Rps. Acidophila, J. Phys. Chem. B, 1999, 103, 2543–2553 CrossRef CAS.
- S. Tretiak, C. Middleton, V. Chernyak and S. Mukamel, Bacteriochlorophyll and Carotenoid Excitonic Couplings in the LH2 System of Purple Bacteria, J. Phys. Chem. B, 2000, 104, 9540–9553 CrossRef CAS.
- L.-J. Yu, M. Suga, Z.-Y. Wang-Otomo and J.-R. Shen, Structure of photosynthetic LH1–RC supercomplex at 1.9 Å resolution, Nature, 2018, 556, 209–213 CrossRef CAS PubMed.
- D. C. Arnett, C. C. Moser, P. L. Dutton and N. F. Scherer, The First Events in Photosynthesis: Electronic Coupling and Energy Transfer Dynamics in the Photosynthetic Reaction Center from Rhodobacter sphaeroides, J. Phys. Chem. B, 1999, 103, 2014–2032 CrossRef CAS.
- R. Kumble, S. Palese, R. W. Visschers, P. Leslie Dutton and R. M. Hochstrasser, Ultrafast dynamics within the B820 subunit from the core (LH-1) antenna complex of Rs. rubrum, Chem. Phys. Lett., 1996, 261, 396–404 CrossRef CAS.
- T. Brixner, J. Stenger, H. M. Vaswani, M. Cho, R. E. Blankenship and G. R. Fleming, Two-dimensional spectroscopy of electronic couplings in photosynthesis, Nature, 2005, 434, 625–628 CrossRef CAS PubMed.
- D. B. Turner, K. E. Wilk, P. M. G. Curmi and G. D. Scholes, Comparison of Electronic and Vibrational Coherence Measured by Two-Dimensional Electronic Spectroscopy, J. Phys. Chem. Lett., 2011, 2, 1904–1911 CrossRef CAS.
- M. Krayer, E. Yang, J. R. Diers, D. F. Bocian, D. Holten and J. S. Lindsey, De novo synthesis and photophysical characterization of annulated bacteriochlorins. Mimicking and extending the properties of bacteriochlorophylls, New J. Chem., 2011, 35, 587–601 RSC.
- C.-Y. Chen, E. Sun, D. Fan, M. Taniguchi, B. E. McDowell, E. Yang, J. R. Diers, D. F. Bocian, D. Holten and J. S. Lindsey, Synthesis and Physicochemical Properties of Metallobacteriochlorins, Inorg. Chem., 2012, 51, 9443–9464 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: 1H NMR and 13C NMR spectra, oligo sequences, ESI-MS and electrophoresis of dye-labeled strands, absorption spectra of bacteriochlorin 2 and 3, 230–850 nm absorption spectra of DNA-bacteriochlorin constructs, fluorescence data, and KRM modeling. See DOI: https://doi.org/10.1039/d3cp01634j |
| This journal is © the Owner Societies 2023 |