
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
On the antiaromatic–aromatic–antiaromatic transition of the stacked cyclobutadiene dimer†
Mesías
Orozco-Ic
 *ab and
Dage
Sundholm
*a
*ab and
Dage
Sundholm
*a
aDepartment of Chemistry, Faculty of Science, University of Helsinki, P.O. Box 55, A. I. Virtasen aukio 1, FIN-00014 Helsinki, Finland. E-mail: mesias.orozcoic@helsinki.fi; dage.sundholm@helsinki.fi
bDonostia International Physics Center (DIPC), 20018 Donostia, Euskadi, Spain
First published on 17th April 2023
Abstract
We have studied the changes in the aromatic nature of two cyclobutadiene (C4H4) molecules on decreasing the intermolecular distance and approaching the cubane structure in a face-to-face fashion. The analysis based on the calculations of the magnetically induced current density and the induced magnetic field shows that the aromaticity of the two C4H4 rings changes from a strongly antiaromatic character at long distances to an aromatic transition state of stacked C4H4 rings at intermediate internuclear distances when approaching the antiaromatic state of cubane.
1 Introduction
Non-covalent π–π stacking interactions of aromatic rings play a crucial role in stabilizing biological systems.1–3 However, chemical concepts like π–π stacking and aromaticity are difficult to measure. Therefore, they are mainly characterized by using computational approaches.3–6 The molecular magnetic response is one of the commonly used methods to identify the aromatic nature of molecules.7–11 In the presence of an external magnetic field, aromatic molecules sustain a diatropic ring current flowing in the classical direction that induces a secondary magnetic field in the opposite direction to the applied external one.12–14 Antiaromatic molecules sustain a paratropic ring current flowing in the non-classical direction. The paratropic ring current induces a secondary magnetic field strengthening the applied external magnetic field. Non-covalent π–π stacking can also affect the aromaticity or vice versa.15–18 Ring currents usually flow along covalent bonds around molecular rings.19–21 However, there are also examples of molecules with stacked rings exhibiting through-space current densities.16,22–26An interesting property of π stacked molecules is the dependence of the tropicity of the ring current on the distance between the antiaromatic rings. The ring current is paratropic representing antiaromaticity when the face-to-face stacked molecules weakly interact far away from each other and diatropic indicating aromaticity when they strongly interact at a shorter distance between the molecular rings.15,16,27,28 Bean and Fowler found that at short interring distances the rotational transitions between the occupied and virtual frontier orbitals of the dimer are forbidden, whereas the translationally allowed transitions dominate resulting in a diatropic ring current.16 There is also a significant current density between the rings suggesting a through-space delocalization of the π electrons.15,16
The distance dependence of the aromaticity of the C4H4 dimer is even more complicated. The global minimum of the C4H4 dimer is cubane (C8H8), which is a cage structure belonging to the Oh point group with single C–C bonds between C4H4 rings. Cubane has single sp3 hybridized bonds between the C atoms and pyramidal C–H bonds.29 In the presence of an external magnetic field, cubane exhibits a paramagnetic response and sustains a paratropic ring current like C4H4.30,31
Li and Houk studied the dimerization of cyclobutadiene in detail.32 They noted that the face-to-face stacked structure is a transition state with two degenerate imaginary vibrational modes.32 Later, Alonso et al. showed that there is a change in the degree of antiaromaticity along the face-to-face reaction coordinate.33 They employed the harmonic oscillator model of aromaticity (HOMA)34 to determine the degree of aromaticity. For this purpose, they also calculated the fluctuation aromatic (FLU)35 and nucleus-independent chemical shift (NICS)36 indices. The three approaches yielded somewhat inconsistent interpretations. The HOMA and FLU calculations showed that the rings are aromatic when they are between 3.4 Å and 1.9 Å apart, while the NICS values suggested that the dimer is antiaromatic in this interval.33 However, these calculations were performed for structures belonging to the D4h point group, which prevents analyzing changes originating from the transition from the rectangular (D2h) structure of the non-interacting C4H4 molecules to the octahedral (Oh) structure of cubane.33
It is known that calculations of single NICS values can lead to misinterpretations of the aromatic character of three-dimensional molecules and the aromatic character of molecules containing stacked rings.18,37–39 Although a change in the degree of aromaticity of the C4H4 dimer in the face-to-face stacking process has been suggested, it has not been elucidated to what extent it happens and at what distance between the C4H4 rings it occurs.
Qualitative and quantitative analyses of global and local aromaticity can be addressed by calculating and visualizing the magnetic response and the magnetically induced current density of the studied molecule.19,40 In this work, we analyze the changes in the aromatic character by calculating the magnetic response for a number of steps of the process in which two face-to-face oriented C4H4 rings approach each other from far away until they reach the cubane structure (see Fig. 1). Our study based on the calculations of the magnetically induced current density8,41,42 and the induced magnetic field43–45 reveals that the C4H4 rings change their strongly antiaromatic nature to an aromatic transition state to again reach the antiaromatic state of cubane.
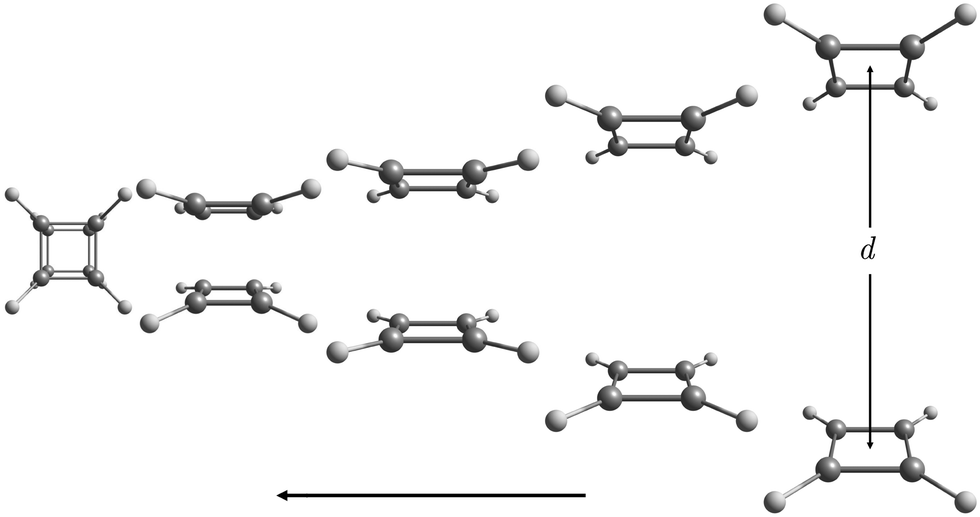 | ||
| Fig. 1 The picture shows different steps in the process of bringing two C4H4 rings together until they join forming the cubane structure. The molecular structures are optimized for fixed interring distances d in the range of 15 ≥ d ≥ 2.96a0 (bohr). | ||
2 Computational methods
The molecular structures were optimized at the density functional theory (DFT) level using the B3LYP functional46 and the semi-empirical dispersion interaction (D3(BJ))47 in combination with basis sets of the triple-ζ polarization quality (def2-TZVP).48 The molecular structures were constrained to the D2h point group. For given distances between the C4H4 rings, the rest of the structural degrees of freedom was fully optimized. The molecular structures were optimized for interring distances d in the range of d ∈ [2.96,15]a0 (bohr) in a step of 0.1a0. Single-point calculations at the second-order Møller–Plesset perturbation theory (MP2)49 and the coupled-cluster single, double, and perturbative triple (CCSD(T))50 levels were performed for each d value. The Cartesian coordinates can be found in the ESI.† Vibrational analyses were performed on the optimized structures with d = 7.522, 4.435, and 2.961a0 (3.996, 2.347, and 1.566 Å). The optimized structures belong to the D2h, D4h and Oh point groups, respectively.Re-optimizations at the spin-component-scaled MP2 (SCS-MP2) level51 using the def2-TZVP basis sets48 were performed at these minima (saddle points). The interring distance was not optimized. Single-point MP2 and CCSD(T) energy calculations using these geometries were repeated using quadruple-ζ polarization quality basis sets (def2-QZVP).48
The magnetic response was addressed by calculating the magnetically induced current density8,41,42 (Jind) and the induced magnetic field43–45 (Bind) using the GIMIC8,41,42 and Aromagnetic52 programs, respectively. The electronic structure calculations were carried out using the Turbomole program.53,54 The magnetic response calculations were performed at the DFT level using the BHandHLYP functional55 and the def2-TZVP basis sets48 because the BHandHLYP functional yielded a good agreement with reference data for magnetic properties calculated at the CCSD(T) level.56 We assume that the strength of the induced magnetic field depends linearly on the strength of the external one. In the presentation of the induced magnetic field, we therefore formally apply an external field of 1 T parallel (|Bext| = 1 T) to the z axis, which coincides with the highest symmetry axis of the studied molecular systems. The analysis of Bind can then be reduced for the discussion of its scalar z component (Bindz), which is the dominant one.43–45 The ring-current strengths (Iind) of the C4H4 molecules were calculated by integrating the Jind flux through a plane which intersects one of the C–C bonds.8,41,42 Changes in the current–density flux can be obtained by calculating profiles of the current–density strength along one coordinate of the integration plane.8,41,42 The profile curve is the derivative of Iind (dIind/dz) with respect to z-coordinate. The profile curves along the z-axis show how Iind changes on decreasing the interring distance.
3 Results and discussion
The interaction energy between the C4H4 rings as a function of the interring distance is shown in Fig. 2. Similar potential energy curves are obtained at different levels of theory. The potential energy curve can be divided into three regions. In the first region (denoted as R1), there is a weak interaction between the C4H4 rings due to van der Waals attraction (d ≥ 7a0). In the second region (denoted as R2), where the C4H4 rings interact more strongly, the energy is ca. 30 kcal mol−1 below the energy at the van der Waals minimum calculated at the CCSD(T) level. The narrow region at short distances (R3) is where the C4H4 rings bind covalently forming cubane. The global minimum (cubane) is at d = 2.961a0 (see Table 1). However, the minimum of the potential energy curve at d = 4.435a0 in the R2 region is actually a saddle point with two imaginary frequencies. The van der Waals minimum at d = 7.522a0 is also a saddle point with four imaginary frequencies (see Table 1). The same energetic behavior is confirmed by MP2 and CCSD(T) calculations using the def2-QZVP basis sets and the molecular structures optimized at the SCS-MP2/def2-TZVP level (see Table 1).48,51 | ||
| Fig. 2 The interaction energy (Eint) between the C4H4 rings as a function of the interring distance calculated at different levels of theory using the def2-TZVP basis set. The vertical dashed lines divide the potential energy curve into the three regions. | ||
| d (a0) | B3LYP-D3(BJ) | MP2 | CCSD(T) | MP2a | CCSD(T)a | Imaginary frequencies | NICS(0) | B ind z (0) | I ind |
|---|---|---|---|---|---|---|---|---|---|
| a Calculated using the def2-QZVP basis sets and the molecular structure optimized at the SCS-MP2/def2-TZVP level. | |||||||||
| 7.0 | 61.1 | 75.5 | 65.5 | 73.9 | 64.4 | 58.9i, 44.2i, 34.5i, 33.6i | 28.2 | 113.6 | −19.9 |
| 4.4 | 28.2 | 16.3 | 35.6 | 14.4 | 34.3 | 447.8i, 447.8i | −32.0 | 17.4 | 4.7 |
| 2.96 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | — | 15.3 | 59.1 | −4.6 |
The calculated magnetic response elucidates that the three minima correspond to the transitions from two antiaromatic rings in the R1 region, to an aromatic transition state in R2, and cubane in R3, which is dominated by paratropic ring currents corresponding to antiaromaticity. The change in the tropicity along the face-to-face reaction coordinate is identified by computing the ring-current strengths for each d value as shown in Fig. 3. Along this reaction coordinate, the ring-current strength of each C4H4 ring goes from being strongly paratropic of ca. −20 nA T−1 to diatropic of ca. 5 nA T−1, and revert to a paratropic ring current of ca. −5 nA T−1 when forming cubane.
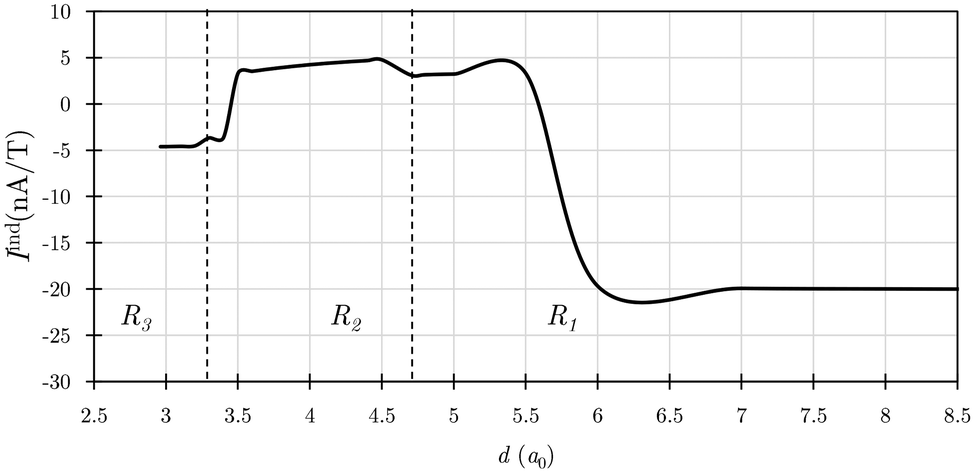 | ||
| Fig. 3 The ring-current strength (Iind) as a function of the interring distance (d) calculated at the BHandHLYP/def2-TZVP level. The vertical dashed lines divide the d range into three regions. | ||
At large distances (d > 7a0), the interaction energy between the rings is small and dominated by the van der Waals interaction. In the R1 region, the C4H4 rings are rectangular belonging to the D2h point group. We study the magnetic response for the structure at d = 7a0, which is near the van der Waals minimum representing the molecular structure in the R1 region. The optimized structure at the van der Waals minimum is a saddle point with four imaginary frequencies (see Table 1).
From the magnetic point of view, the C4H4 rings show independent paratropic ring currents and large deshielded Bindz cones that overlap between the rings. Animations of the magnetically induced current density are shown in the ESI.†
The NICS(0) and Bindz (0) values are strongly positive (Table 1), which is practically the magnetic response of a single C4H4 molecule.37 The antiaromatic character also explains why the R1 structures have a rather small gap of 3.6 eV between the highest-occupied molecular orbital (HOMO) and the lowest-unoccupied molecular orbital (LUMO). The HOMO–LUMO gap for the dimer in R1 is very similar to the one for cyclobutadiene. The aromatic transition state in R2 has a significantly larger HOMO–LUMO gap of 4.96 eV than the one for cyclobutadiene. Cubane has by far the largest HOMO–LUMO gap of 8.23 eV. The HOMO–LUMO gap as a function of the interring distance d is shown in Fig. 4.
 | ||
| Fig. 4 The HOMO–LUMO gap as a function of the interring distance calculated at the B3LYP-D3(BJ)/def2-TZVP level. | ||
The C–C bonds of the C4H4 rings of the optimized molecular structure at d = 4.435a0 are equally long. Thus, the structure in R2 belongs to the D4h point group. The potential energy curve has sharp peaks at the boundaries between the three regions because there are different occupied frontier orbitals in the adjacent regions (see Fig. 2). For a given d value, the occupation of the frontier orbitals suddenly changes in the self-consistent-field optimization of the orbitals leading to a lower energy.
The highest occupied frontier orbitals of the molecular structure in R2 have ungerade parity and the lowest unoccupied frontier orbitals have even parity. Since translation has ungerade parity and rotation has even parity, the formal excitation from the occupied orbitals to the unoccupied ones is translationally allowed but rotationally forbidden, which leads to the aromatic nature of the transition state.16
The HOMO of the molecular structure in R1 belongs to the B3g of the D2h point group and LUMO+1 has also gerade parity implying that the formal excitation from the HOMO to LUMO+1 is rotationally allowed leading to the antiaromatic cyclobutadiene dimer at long interring distances.16 Thus, at the borderline between R1 and R2, the frontier B3u and B3g orbitals flip from occupied to unoccupied and vice versa.
The HOMO, HOMO−1, LUMO and LUMO+1 of cubane belong to the T2u, T2g, T1u and A1g irreducible representations of the Oh point group, respectively. The HOMO−1 to LUMO transition is rotationally allowed leading to antiaromaticity. The HOMO to LUMO+1 transition is translationally allowed and would contribute to the aromatic nature of cubane. Since the HOMO and HOMO−1 as well as the LUMO and LUMO+1 are almost degenerate, the contributions are expected to be of the same size with the opposite sign leading to a rather weak ring current. The maximum in the potential energy curve at the borderline between R2 and R3 is also due to the flipping of the occupied and virtual frontier orbitals.
The diatropic ring-current is strongest for the saddle point at d = 4.435a0 (see Fig. 3 and 6). Following the imaginary frequencies of the saddle point at d = 4.435a0 and relaxing the symmetry constraints lead to a syn-type structure where two C–C bonds are formed between the layers, which Li and Houk also reported.32 They pointed out that the saddle point is stabilized due to a reduction in the antiaromaticity. At the CCSD(T) level, the transition state is energetically ca. 36 kcal mol−1 above cubane, which is the global minimum.
Integration of the magnetically induced current density (Jind) passing through a plane that cuts the C–C bond of one of the C4H4 rings confirms that there is a diatropic ring current (Iind) of 4.7 nA T−1 that flows around each ring (see Fig. 5 and animations in the ESI†). The profiles of the ring-current strength in Fig. 6 show that the ring current at an interring distance of 7a0 consists of two almost independent paratropic ring currents in the C4H4 rings. The double peak shows that the ring current is sustained in the π orbitals of the C4H4 rings. Cubane with an interring distance of 2.961a0 sustains a paratropic ring current in the σ orbitals at the C4H4 plane. The transition state at d = 4.435a0 sustains a diatropic ring current in the σ orbitals of the two C4H4 rings. However, it also sustains a strong diatropic ring current between the C4H4 rings. Despite this, the Bindz plots exhibit deshielding cones that are smaller than those in the R1 region suggesting a lower degree of antiaromaticity. However, the calculation of the induced magnetic field does not directly reveal that the transition state is aromatic. Thus, interpretations of the aromatic/antiaromatic character based on NICS(0) and Bindz (0) calculations are not completely unproblematic.
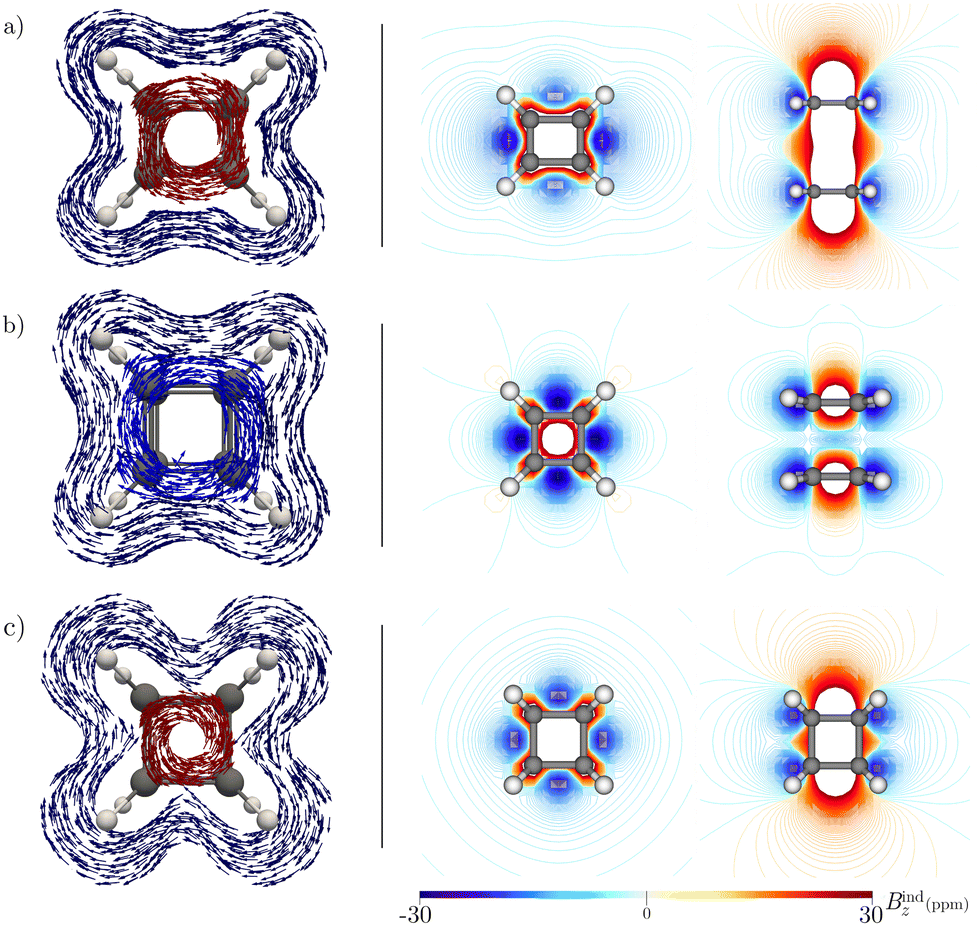 | ||
| Fig. 5 The pictures in left panel show the Jind vector maps when (a) d = 7.0, (b) d = 4.4, and (c) d = 2.96a0. The diatropic and paratropic contributions to the current density in the C4H4 plane are shown in blue and red, respectively. The pictures in the right panel show the Bindz isolines calculated in the vertical symmetry plane and in the horizontal symmetry plane of the stacked C4H4 rings. The calculations were performed at the BHandHLYP/def2-TZVP level. | ||
 | ||
| Fig. 6 The ring-current strength profiles (dIind/dz) along the z-axis are calculated for the two C4H4 rings with distances of d = 7.0, 4.4, and 2.96a0 using an integration plane that intersects both rings. The horizontal dashed lines denote the positions of the C4H4 rings. Integration of the dIind/dz curves yields twice the ring-current strengths in Table 1. | ||
In the R3 region, the energy decreases significantly due to the formation of interring carbon bonds between the C4H4 rings. Finally, the cubane structure is obtained at d = 2.961a0 (see Fig. 2), where the aromatic nature obtained in R2 switches to antiaromaticity. The paratropic ring current flows mainly in the planes of the four-membered rings that are perpendicular to the direction of the external magnetic field (see Fig. 5 and 6). The diatropic ring-current is the strongest for the saddle point at d = 4.435a0 (see Fig. 3 and 6).
Integration of Jind for cubane yields a ring-current strength of −4.62 nA T−1 in each ring. The Bindz calculations confirm that cubane is deshielded suggesting antiaromaticity. Havenith et al. explained that the paratropicity of cubane is due to collective contributions from the four-membered rings.31 Although cubane has been considered to be super-antiaromatic due to the large positive NICS(0) value of 15.3 ppm,30 we show here that this is not the case. When the two C4H4 rings approach each other, the C4H4 dimer reaches an aromatic transition state with different occupied frontier orbitals than at long distances between the rings. Cubane is antiaromatic sustaining paratropic ring currents in the σ orbitals of the strained sp3 bonds. The large Bindz value of −58.9 ppm in the middle of a C4H4 ring of cubane is due to the paratropic ring current in the ring plane.
4 Conclusions
We have studied how the aromatic character changes when two cyclobutadiene rings approach each other in the face-to-face orientation from far away as separate molecules to the cubane structure. The magnetically induced current density and the ring-current strength have been calculated at different distances between the C4H4 rings. The induced magnetic field in the vicinity of the molecule has also been calculated in each step along the reaction coordinate. The calculations show that the C4H4 rings are antiaromatic at long distances and that cubane is also dominated by paratropic ring currents, whereas the transition state at intermediate interring distances is aromatic sustaining diatropic ring currents in the two rings and between them. Calculations at the B3LYP-D3(BJ), MP2, and CCSD(T) levels show that the interaction energy between the C4H4 rings increases as they get closer. The face-to-face reaction coordinate can be divided into three regions with significantly different tropicities of the magnetically induced current density leading to significant differences in the induced magnetic field in the vicinity of the molecule. The first region of the reaction coordinate represents the regime where the C4H4 rings interact weakly. Their magnetic response is similar to two non-interacting antiaromatic cyclobutadiene molecules. The van der Waals minimum (saddle point) is obtained at an interring distance of 7.522a0. The second region in the range of d = [3.4,4.7]a0 corresponds to a transition state with strongly interacting aromatic π-stacked C4H4 rings belonging to the D4h point group. The interring distance of the aromatic transition state is 4.435a0. Finally, for short interring distances that are less than 3.4a0, interring carbon bonds with a length of 2.961a0 are formed yielding the cubane structure belonging to the Oh point group. The strongly strained cubane structure is known to be antiaromatic even though it has sp3 hybridized molecular orbitals forming single C–C bonds.Author contributions
D. S. conceived the idea of the project. M. O.-I. and D. S. performed the calculations. The authors discussed the results and contributed to the final manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Academy of Finland through the project number 340583, the Magnus Ehrnrooth Foundation, the Waldemar von Frenckell's foundation, and the Swedish Cultural Foundation in Finland. We acknowledge computational resources from the CSC – IT Center for Science, Finland.Notes and references
- M. L. Waters, Curr. Opin. Chem. Biol., 2002, 6, 736–741 CrossRef CAS PubMed.
- T. Chen, M. Li and J. Liu, Cryst. Growth Des., 2018, 18, 2765–2783 CrossRef CAS.
- C. R. Martineza and B. L. Iverson, Chem. Sci., 2012, 3, 2191–2201 RSC.
- C. A. Hunter, K. R. Lawson, J. Perkins and C. J. Urch, J. Chem. Soc., Perkin Trans. 2, 2012, 651–669 Search PubMed.
- J. W. G. Bloom and S. E. Wheeler, Angew. Chem., Int. Ed., 2011, 50, 7847–7849 CrossRef CAS PubMed.
- G. Merino, M. Solà, I. Fernández, C. Foroutan-Nejad, P. Lazzeretti, G. Frenking, H. L. Anderson, D. Sundholm, F. P. Cossío, M. A. Petrukhina, J. Wu, J. I. Wu and A. Restrepo, Chem. Sci., 2023 10.1039/D2SC04998H.
- M. Solà, A. I. Boldyrev, M. K. Cyrañski, T. M. Krygowski and G. Merino, Aromaticity and Antiaromaticity: Concepts and Applications, John Wiley & Sons, Ltd, 2022 Search PubMed.
- D. Sundholm, H. Fliegl and R. J. F. Berger, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2016, 6, 639–678 CAS.
- J. A. N. F. Gomes and R. B. Mallion, Chem. Rev., 2001, 101, 1349–1384 CrossRef CAS PubMed.
- P. Lazzeretti, Prog. Nucl. Magn. Reson. Spectrosc., 2000, 36, 1–88 CrossRef CAS.
- A. Balaban, P. von Ragué Schleyer and H. S. Rzepa, Chem. Rev., 2005, 105, 3436–3447 CrossRef CAS PubMed.
- N. F. Ramsey, Phys. Rev., 1950, 78, 699–703 CrossRef CAS.
- J. A. Pople, Mol. Phys., 1958, 1, 175–180 CrossRef CAS.
- R. McWeeny, Mol. Phys., 1958, 1, 311–321 CrossRef CAS.
- C. Corminboeuf, P. von, R. Schleyer and P. Warner, Org. Lett., 2007, 9, 3263–3266 CrossRef CAS PubMed.
- D. E. Bean and P. W. Fowler, Org. Lett., 2008, 10, 5573–5576 CrossRef CAS PubMed.
- D. Sundholm, M. Rauhalahti, N. Özcan, R. Mera-Adasme, A. L. J. Kussmann and C. Ochsenfeld, J. Chem. Theory Comput., 2017, 13, 1952–1962 CrossRef CAS PubMed.
- M. Orozco-Ic, J. Barroso, N. D. Charistos, A. Muñoz-Castro and G. Merino, Chem. – Eur. J., 2019, 26, 326–330 CrossRef PubMed.
- D. Sundholm, M. Dimitrova and R. J. F. Berger, Chem. Commun., 2021, 57, 12362–12378 RSC.
- E. Paenurk and R. Gershoni-Poranne, Phys. Chem. Chem. Phys., 2022, 24, 8631–8644 RSC.
- Y.-C. Lin and D. Sundholm, J. Chem. Theory Comput., 2006, 2, 761–764 CrossRef CAS PubMed.
- L. N. Wirz, M. Dimitrova, H. Fliegl and D. Sundholm, J. Phys. Chem. Lett., 2018, 9, 1627–1632 CrossRef CAS PubMed.
- V. Vijay, M. Madhu, R. Ramakrishnan, A. Benny and M. Hariharan, Chem. Commun., 2020, 56, 225–228 RSC.
- M. Orozco-Ic, R. R. Valiev and D. Sundholm, Phys. Chem. Chem. Phys., 2022, 24, 6404–6409 RSC.
- K. Wypych, M. Dimitrova, D. Sundholm and M. Pawlicki, Org. Lett., 2022, 24, 4876–4880 CrossRef CAS PubMed.
- L.-X. Bai, M. Orozco-Ic, X. Zarate, D. Sundholm, S. Pan, J.-C. Guo and G. Merino, Molecules, 2022, 27, 7407 CrossRef CAS PubMed.
- M. Orozco-Ic, A. Restrepo, A. Muñoz-Castro and G. Merino, J. Chem. Phys., 2019, 151, 014102 CrossRef PubMed.
- R. Nozawa, J. Kim, J. Oh, A. Lamping, Y. Wang, S. Shimizu, I. Hisaki, T. Kowalczyk, H. Fliegl, D. Kim and H. Shinokubo, Nat. Commun., 2019, 10, 3576 CrossRef PubMed.
- P. E. Eaton and T. W. Cole, J. Am. Chem. Soc., 1964, 86, 3157–3158 CrossRef CAS.
- D. Moran, M. Manoharan, T. Heine and P. von R. Schleyer, Org. Lett., 2003, 5, 23–26 CrossRef CAS PubMed.
- R. W. A. Havenith, P. W. Fowler and E. Steiner, Chem. Phys. Lett., 2003, 371, 276–283 CrossRef CAS.
- Y. Li and K. N. Houk, J. Am. Chem. Soc., 1996, 118, 880–885 CrossRef CAS.
- M. Alonso, J. Poater and M. Solà, Struct. Chem., 2007, 18, 773–783 CrossRef CAS.
- J. Kruszewski and T. Krygowski, Struct. Chem., 1972, 13, 3839–3842 Search PubMed.
- E. Matito, M. Solà, P. Salvadora and M. Duran, Faraday Discuss., 2007, 135, 325–345 RSC.
- P. von, R. Schleyer, C. Maerker, A. Dransfeld, H. Jiao and N. J. R. van E. Hommes, J. Am. Chem. Soc., 1996, 118, 6317–6318 CrossRef PubMed.
- G. Monaco and R. Zanasi, J. Phys. Chem. Lett., 2017, 8, 4673–4678 CrossRef CAS PubMed.
- M. Orozco-Ic, M. Dimitrova, J. Barroso, D. Sundholm and G. Merino, J. Phys. Chem. A, 2021, 125, 5753–5764 CrossRef CAS PubMed.
- J. Poater, J. M. Bofill, P. Alemany and M. Solà, J. Org. Chem., 2006, 71, 1700–1702 CrossRef CAS PubMed.
- G. Merino, A. Vela and T. Heine, Chem. Rev., 2005, 105, 3812–3841 CrossRef CAS PubMed.
- J. Jusélius, D. Sundholm and J. Gauss, J. Chem. Phys., 2004, 121, 3952–3963 CrossRef PubMed.
- H. Fliegl, S. Taubert, O. Lehtonen and D. Sundholm, Phys. Chem. Chem. Phys., 2011, 13, 20500–20518 RSC.
- G. Merino, T. Heine and G. Seifert, Chem. – Eur. J., 2004, 10, 4367–4371 CrossRef CAS PubMed.
- T. Heine, R. Islas and G. Merino, J. Comput. Chem., 2007, 28, 302–309 CrossRef CAS PubMed.
- R. Islas, T. Heine and G. Merino, Acc. Chem. Res., 2012, 45, 215–228 CrossRef CAS PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648 CrossRef CAS.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- C. Hättig, A. Hellweg and A. Köhn, Phys. Chem. Chem. Phys., 2006, 8, 1159–1169 RSC.
- D. P. Tew, W. Klopper, C. Neiss and C. Hättig, Phys. Chem. Chem. Phys., 2007, 9, 1921–1930 RSC.
- S. Grimme, J. Chem. Phys., 2003, 118, 9095–9102 CrossRef CAS.
- M. Orozco-Ic, J. L. Cabellos and G. Merino, Aromagnetic, 2016, Cinvestav–Mexico Search PubMed.
- R. Ahlrichs, M. Bär, M. Häser, H. Horn and C. Kölmel, Chem. Phys. Lett., 1989, 162, 165–169 CrossRef CAS.
- S. G. Balasubramani, G. P. Chen, S. Coriani, M. Diedenhofen, M. S. Frank, Y. J. Franzke, F. Furche, R. Grotjahn, M. E. Harding, C. Hättig, A. Hellweg, B. Helmich-Paris, C. Holzer, U. Huniar, M. Kaupp, A. Marefat Khah, S. Karbalaei Khani, T. Müller, F. Mack, B. D. Nguyen, S. M. Parker, E. Perlt, D. Rappoport, K. Reiter, S. Roy, M. Rückert, G. Schmitz, M. Sierka, E. Tapavicza, D. P. Tew, C. van Wüllen, V. K. Voora, F. Weigend, A. Wodynski and J. M. Yu, J. Chem. Phys., 2020, 152, 184107 CrossRef CAS PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 1372–1377 CrossRef CAS.
- S. Lehtola, M. Dimitrova, H. Fliegl and D. Sundholm, J. Chem. Theory Comput., 2021, 17, 1457–1468 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Cartesian coordinates of the optimized molecular structures and animations of the current densities. See DOI: https://doi.org/10.1039/d3cp01350b |
| This journal is © the Owner Societies 2023 |