
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Laplace inverted pulsed EPR relaxation to study contact between active material and carbon black in Li–organic battery cathodes†
Davis Thomas
Daniel
 *ab,
Conrad
Szczuka
a,
Peter
Jakes
a,
Rüdiger-A.
Eichel
ac and
Josef
Granwehr
ab
*ab,
Conrad
Szczuka
a,
Peter
Jakes
a,
Rüdiger-A.
Eichel
ac and
Josef
Granwehr
ab
aInstitute of Energy and Climate Research (IEK-9), Forschungszentrum Jülich, Jülich, 52425, Germany. E-mail: d.daniel@fz-juelich.de
bInstitute of Technical and Macromolecular Chemistry, RWTH Aachen University, Aachen 52056, Germany
cInstitute of Physical Chemistry, RWTH Aachen University, Aachen 52056, Germany
First published on 24th April 2023
Abstract
The addition of conductive additives during electrode fabrication is standard practice to mitigate a low intrinsic electronic conductivity of most cathode materials used in Li-ion batteries. To ensure an optimal conduction pathway, these conductive additives, which generally consist of carbon particles, need to be in good contact with the active compounds. Herein, we demonstrate how a combination of pulsed electron paramagnetic resonance (EPR) relaxometry and inverse Laplace transform (ILT) can be used to study such contact. The investigated system consists of PTMA (poly(2,2,6,6-tetramethylpiperidinyloxy-4-ylmethacrylate)) monomer radicals, which is a commonly used redox unit in organic radical batteries (ORB), mixed at different ratios with Super P carbon black (CB) as the conductive additive. Inversion recovery data were acquired to determine longitudinal (T1) relaxation time constant distributions. It was observed that not only the position and relative amplitude, but also the number of relaxation modes varies as the composition of PTMA monomer and CB is changed, thereby justifying the use of ILT instead of fitting with a predetermined number of components. A hypothesis for the origin of different relaxation modes was devised. It suggests that the electrode composition may locally affect the quality of electronic contact between the active material and carbon black.
1 Introduction
Improving the energy density of Li-ion batteries is of vital importance for meeting next-generation energy storage needs.1,2 A commonly proposed solution is to reduce the amount of inactive components such as conductive additives or binders in the battery.3 Conductive additives are necessary to improve the bulk electronic conductivity of electrodes, since most of the electrode materials utilised in Li-ion batteries exhibit low intrinsic electronic conductivity.4 However, since the conductive additives do not contribute to the capacity of the battery in most cases, the amount of additive in the electrode needs to be optimised. This aspect is particularly crucial for advancing sustainable battery technologies, such as organic radical polymer batteries (ORB),5 where conductive additives are used in significant amounts.6,7 ORB cathodes with up to 70 wt% of conductive additive have been reported.8 For instance, PTMA (poly(2,2,6,6-tetramethylpiperidinyloxy-4-ylmethacrylate)), a commonly employed radical polymer, exhibits limited electrical conductivity and requires up to 50 wt% of conductive additive for optimal performance.9,10Electrochemical performance of ORBs utilising PTMA as the active material is significantly affected by the type and amount of conductive additive used,11–13 highlighting the need for optimisation of electrode composition. The electron transfer mechanism in PTMA based ORBs involves a two step process – electron transfers between the redox active units on the polymer backbone, and electron transfer through the conductive additive.14 A continuous conduction pathway between active material, conductive additive, and current collector is necessary for electrochemical accessibility and high electronic conductivity. In particular, interfacial contacting between redox active units and carbon additives may be a conductivity bottleneck11,15 that has been shown to influence the overall battery performance.16,17 For ORB active materials that feature an insulating polymer backbone, such as PTMA, a disruption in the contact at the interface between current collector and conductive additive can be caused by the polymer backbone itself, leading to low intrinsic conductivity.14,18 Investigating the electronic contact between the active material and the conductive additive could reveal conductivity bottlenecks and aid in optimising the electrode composition. Therefore, a generally applicable technique to study such contact in composite electrode materials is desirable.
Electron paramagnetic resonance (EPR) is a spectroscopic technique to investigate the environment and dynamics of unpaired electrons in materials. Since the redox states in batteries often involve unpaired electrons, EPR can be utilised to investigate these systems. With respect to battery materials, continuous wave (CW) EPR is often used to confirm the presence of unpaired electrons and for quantification of radical density.6,19,20 Moreover, pulsed EPR techniques,21–24in operando CW EPR25 and EPR imaging26,27 for battery research have also been demonstrated. PTMA based ORBs consist of nitroxide radicals as the redox active unit, which are well-characterised using EPR28 at low concentrations. EPR spectroscopic investigations of PTMA mainly utilise CW EPR techniques, as densely packed nitroxide radicals make the application of advanced EPR techniques challenging. Due to spin exchange interaction between the radicals,29 the EPR line broadens initially and starts narrowing after exceeding the slow exchange limit, leading to a loss in resolution of 14N hyperfine features. High spin concentration and spatially close nitroxide radicals on the polymer chain also lead to shortening of the relaxation times, making the system less amenable to pulsed EPR techniques.
For ORBs with nitroxide radicals as redox units, in situ30 and in operando22 CW EPR have shown the presence of electrochemically inactive radicals on the cathode film. The presence of such inactive redox centres, which do not undergo a change in their redox states while charging or discharging, inhibits the maximum attainable capacity of the ORB. The formation of inactive redox centres in the cathode film could be a result of disruption in contact between the conductive additive and the active material during battery cycling,31 disabling the isolated radicals from participating in the electrochemical reaction. In this case, insight into the distribution of these inactive radical centres and their interaction with the conductive additive could help in identifying important capacity-loss pathways.
The distribution of paramagnetic centres (aggregated or sparsely distributed) and their interaction with the environment can be accessed through their spin relaxation behaviour.32,33 Measurement of the spin–lattice relaxation time constant, T1, is mainly done using inversion or saturation recovery techniques. In saturation recovery, the populations of all EPR levels are equalised with a long microwave excitation, followed by a detection sequence that measures the recovery of equilibrium magnetisation. Inversion recovery uses a π pulse to invert the longitudinal magnetisation, and after a variable delay a spin echo sequence is used to monitor its recovery towards equilibrium.
In addition to true T1 relaxation, which involves an exchange of energy between the spin system and its environment (lattice), apparent relaxation may also contribute to relaxation measurements. In an inversion recovery experiment of samples with a broad EPR spectrum, the bandwidth of the microwave pulse may only suffice to excite a small fraction of spins. Processes such as molecular motions, electron spin exchange and spin flip-flops of nearby spins can transfer magnetisation to spins which are not directly excited, leading to apparent relaxation through spectral diffusion. Spectral diffusion is known to contribute primarily to inversion recovery, but saturation recovery is only unaffected when using saturation sequences with a duration on the order of T1.34 In this work, inversion recovery, which offers a higher sensitivity than saturation recovery, is used to obtain T1 relaxation curves.
Different paramagnetic species exhibit varied relaxation behaviour.35 While nitroxide radicals represent a spin system with localised unpaired electrons, conductive additives such as carbon blacks may have contributions from both localised paramagnetic defect centres and mobile conduction electrons,36,37 allowing the use of EPR techniques for their characterisation.36–41 Conduction electrons and nitroxide radicals can be distinguished according to their line shape,42,43 but also differ in their relaxation characteristics. In case of conduction electrons, T1 ≈ T2 is often found.41,43,44 For immobilised nitroxide radicals, T1 ≫ T2 is commonly observed.45,46 In mixed electronic systems, such as the ones investigated in this work, exchange interactions between localised and delocalised electrons serve as an efficient relaxation pathway for the localised electrons.47–50 Nitroxide radicals that interact with faster relaxing electron spins in their vicinity have been reported to undergo similar relaxation enhancement through exchange or dipolar interactions,51,52 enabling the determination of parameters such as inter-spin distances.53 The interaction strength of a nitroxide radical with a carbon black conduction electron, therefore, can be inferred from the relaxation enhancement of the nitroxide radical.
The microscopically heterogeneous nature of nitroxide based active material and carbon black composite samples, with potentially varying grades of contact quality, may cause a distribution of relaxation time constants rather than a single relaxation time. In order to obtain relaxation time distributions, inversion algorithms are suitable, especially in cases where the number of relaxation components is unknown. In order to stabilise the solution of such an inversion, regularisation techniques such as Tikhonov regularisation can be employed.
While Tikhonov regularisation is widely used in EPR for the calculation of distance distributions,54 it has not found widespread application for the analysis of relaxation data. Since most time-domain EPR relaxation studies are conducted on solid samples, negative contributions in the distribution cannot be excluded a priori.55,56 Therefore, the commonly used non-negativity constraint to stabilise the ill-posed inversion with exponential kernel57,58 may lead to inaccurate or unstable results. Employing a conventional fitting technique using, for example, a mono-exponential, stretched exponential, or multi-exponential model, proves more robust and simpler to implement.59 However, a combination of a uniform penalty regularisation, which is less prone to wildly oscillating solutions of inversions with exponential kernel than Tikhonov regularisation in standard form,60 and a zero-crossing penalty proved to be a suitable alternative.61 Thereby, uncertainty of the results is represented as linewidth.62 The aim of the inversion is to find the broadest possible relaxation modes that fit the experimental data without showing non-random, systematic residuals.63
Herein, we demonstrate the use of pulsed EPR relaxometry in combination with Laplace inversion, or inverse Laplace transform (ILT), to study the quality of electronic contact in a composite electrode system. The composite system consists of PTMA monomer, shown in Fig. 1, as the active material, and Super P carbon black (CB) as the conductive additive. Super P mainly consists of spherical carbon particles that form a conductive network within the composite electrode.64,65 The PTMA monomer to CB weight ratio (PMCR) was varied systematically in the composite samples. Continuous-wave (CW) EPR was used to identify the number and distribution of paramagnetic centres, and pulsed EPR was employed to probe the relaxation characteristics of the active material. Laplace inversion of the relaxation data was used to afford relaxation distributions and establish correlations between the composition and the quality of PTMA monomer–CB contact.
 | ||
| Fig. 1 Chemical structure of PTMA monomer. | ||
2 Experimental
2.1 Sample preparation
Composite samples of PTMA (poly(2,2,6,6-tetramethylpiperidinyloxy-4-ylmethacrylate)) monomer (Sigma Aldrich, Germany) and Super P carbon black (Alfa Aesar, Germany) were prepared by adding nitroxide solutions in toluene to the Super P powder. The amount of solution added to CB was kept constant at 100 μL for all samples. The nitroxide concentration was adjusted to obtain the desired weight ratios. The samples were then dried at 60 °C for two weeks in an oven under air, followed by crushing the samples using a mortar and pestle to obtain a fine powder. The powder samples were transferred to 2 mm outer diameter EPR tubes and filled to a height of 30 mm. The ‘wet’ composite samples were obtained by adding 50 μL of toluene to dried composite samples within the EPR tube. The EPR tube was left undisturbed until the solvent fully permeated and soaked the sample.2.2 EPR spectroscopy
X-Band CW EPR spectra were recorded using a Bruker Elexsys E540 EPR spectrometer operating at 9.33 GHz. The spectra were recorded at room temperature as first derivatives of the absorption function, with a field modulation amplitude of 0.05 mT, modulation frequency of 100 kHz, and microwave power of 0.6325 mW. All pulsed EPR experiments were conducted on a Bruker ELEXSYS E580 X-band spectrometer with a Bruker EN 4118-X-MD4 pulse ENDOR resonator. The temperature was maintained at cryogenic levels with a helium cryostat (Oxford Instruments, CF935). For experiments at cryogenic temperatures, the samples were flash frozen using liquid nitrogen and then inserted into the cryostat. The π/2 and π pulse lengths were set to 16 ns and 32 ns respectively. Field swept echo detected EPR spectra were acquired using a standard two-pulse Hahn echo sequence with an inter-pulse delay τ of 200 ns. For T1 measurements, an inversion recovery sequence (π–tR–π/2–τ–π–τ–echo) with a 4-step phase cycle66 was used. τ was kept at 200 ns and the initial delay tR after inversion was set to 400 ns, which was incremented linearly in steps of 1.5 μs to obtain an inversion recovery time trace with 512 points.2.3 Laplace inversion
T 1 relaxation time distributions were obtained using Laplace inversion with an exponential kernel, where the measured signal sk of the kth recovery data point was represented as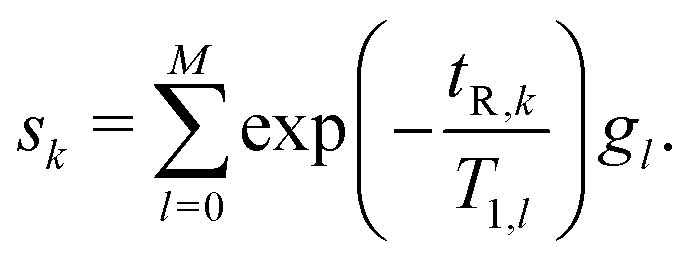 | (1) |
3 Results and discussion
3.1 CW EPR
Fig. 2a shows the CW EPR spectra for a series of PTMA monomer–CB composite samples. The ratios on the right edge of each spectrum denote the PTMA monomer to CB weight ratio (PMCR). Hereafter, the composite samples are referred by their respective PMCR. The topmost spectrum is the one of neat CB (marked as Super P). The CB spectrum is characterised by a symmetrical Lorentzian line with g = 2.0023 and a peak-to-peak linewidth of ΔB = 0.54 mT. The line narrows to 0.35 mT upon treatment with solvent or evacuation. Exchange interaction of CB electrons and paramagnetic molecular oxygen causes the EPR line of CB to broaden, and treatment with solvent displaces the paramagnetic oxygen from CB surfaces, resulting in a narrower line. Similar behaviour of the EPR line has been reported for various carbon blacks.36,40,68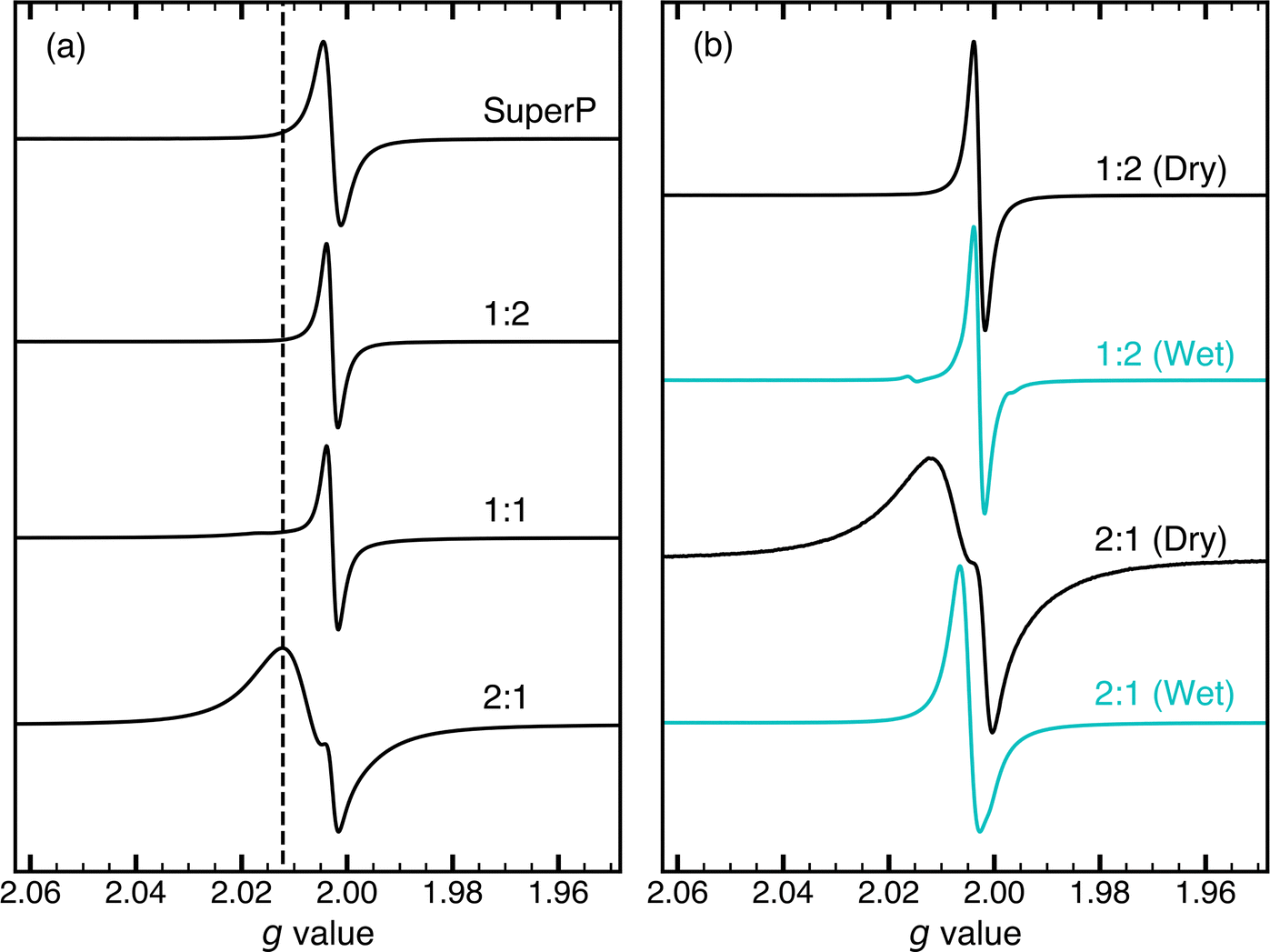 | ||
| Fig. 2 X-Band (9.33 GHz) continuous wave EPR spectra of composite samples acquired at 295 K with a modulation amplitude of 0.05 mT and modulation frequency of 100 kHz. Ratios on the right edge of each spectrum denotes the PMCR. (a) EPR spectra of composite samples with varying PMCR compared to neat CB (Super P). The dashed line denotes the contribution from the nitroxide species. (b) Comparison of EPR spectra of wet (cyan) and dry (black) composite samples. The characteristic three line EPR spectrum of the nitroxide species is observed (feature at g = 2.015). | ||
In comparison to neat CB, the spectra of composite samples showed a narrower linewidth of ΔB = 0.35 mT for the CB component, indicating that the CB conduction electrons do not undergo exchange with paramagnetic oxygen in these samples. This suggests that CB interfaces in the composite samples are not exposed to molecular oxygen, possibly due to residual solvent or being coated by the PTMA monomer. For the nitroxide component, a broad EPR line was observed in 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, owing to spin exchange interactions between nitroxide radicals. This is indicative of densely packed nitroxide radicals. The intensity of this broad nitroxide component decreases as the relative amount of PTMA monomer decreases, and it is resolved to a lesser extent in 1:1. For composite samples with a higher relative amount of CB than 1:1, the nitroxide component was not resolved. For PMCR of 1:6, 1:20 and 1:30, the spectral features were similar to a PMCR of 1:2, and only the contribution from CB was resolved.
:1, owing to spin exchange interactions between nitroxide radicals. This is indicative of densely packed nitroxide radicals. The intensity of this broad nitroxide component decreases as the relative amount of PTMA monomer decreases, and it is resolved to a lesser extent in 1:1. For composite samples with a higher relative amount of CB than 1:1, the nitroxide component was not resolved. For PMCR of 1:6, 1:20 and 1:30, the spectral features were similar to a PMCR of 1:2, and only the contribution from CB was resolved.
The broadening of the nitroxide EPR line in 1:1 and the absence of a resolved nitroxide component for samples with a higher relative amount of CB could be a result of relaxation rate enhancement of nitroxides in the immediate vicinity of CB. The linewidth in CW EPR spectra is dependent on relaxation, where a short spin–spin relaxation time T2 leads to EPR line broadening. Stochastic collisions between paramagnetic centres is known to cause relaxation enhancement through spin exchange interaction.34,46 In the composite samples, relaxation enhancement of the nitroxide radicals could be afforded through frequent encounters with mobile conduction electrons of CB, shortening the longitudinal relaxation time T1 due to exchange interactions.49,53 In polymer–CB composites, electron conduction has been explained by tunnelling with potential-barrier modulation at low temperature and, possibly, thermal activation over the potential barrier at high temperature.69 Both mechanisms critically depend on a close spatial vicinity of the two types of paramagnetic centres. This indicates a preferential surface coverage of the CB particles with PTMA monomer rather than the formation of PTMA monomer clusters in the 1:1 sample and samples with higher relative amount of CB. In contrast, in the 2:1 sample, some fraction of densely packed nitroxides seem to be present that are sufficiently isolated from the CB, such that the relaxation is slower and the nitroxide component can be resolved. In a different context of a Gd3+-doped mixed LiYbxY1−xF4 single crystal with varying ratios of diamagnetic and paramagnetic Yb3+ centres in the host crystal, a comparable line broadening beyond detectability had been explained by the formation of an exchange percolation network through the crystal by the paramagnetic centres.70
In order to confirm that the absence of the nitroxide component was not a result of a chemical change of the radical centre due to the heat treatment (see Sample preparation) or the addition of CB, solvent was re-introduced to the dry composite samples. Upon addition of solvent, ‘wet’ 2:1 (see Fig. 2b) showed a narrower linewidth of the nitroxide EPR component, consistent with a reduced contact between the nitroxide radicals and the CB in the presence of a solvent. In 1:1 and in samples with a higher relative amount of CB (see wet 1:2 spectrum in Fig. 2b), a characteristic three-line spectrum of nitroxide was observed. The results indicate that the nitroxide radical centres remain chemically intact in the composite sample, but the relaxation properties are considerably altered by the CB.
3.2 Pulsed EPR
Pulsed EPR was employed to characterise the relaxation behaviour of the PTMA monomer in the presence of CB. For all the composite samples investigated, a spin echo from the nitroxide component was obtained only below 60 K. No spin echo was obtained at the same experimental conditions for the neat CB sample. Field-swept echo detected EPR spectra for composite samples recorded at 50 K were typical of immobilised nitroxide radicals71 (Fig. 3). For the same τ delay, the relative intensity of the central mI = 0 transition compared to mI = 1 and mI = −1 transitions was found to decrease as the amount of nitroxide in the composite sample was increased. This effect is attributed to instantaneous diffusion,34,72–74 which is proportional to spin concentration and is indicative of a network of dipole–dipole coupled spins.34 Since dipole–dipole coupling is distance dependent, the spectral shape of echo detected spectra is dependent on the packing density of the nitroxide radicals in the composite sample.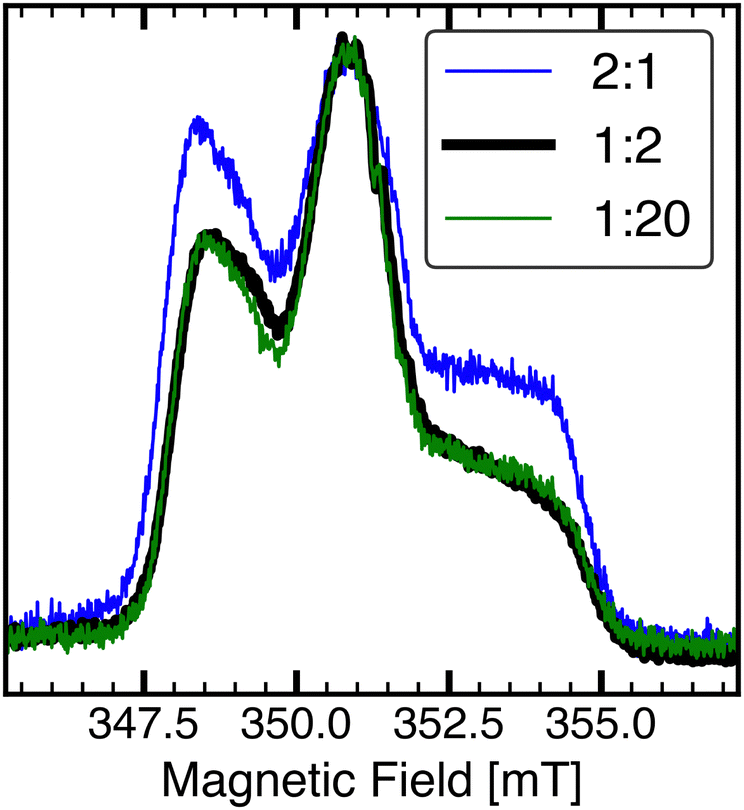 | ||
| Fig. 3 Field-swept echo-detected X-band EPR spectra of PTMA monomer/Super P composite samples at 50 K. The π/2 and π pulse lengths were set to 16 ns and 32 ns, respectively. Echo-detected EPR spectra with τ = 200ns of samples with PTMA monomer-to-Super P ratio of 2:1 (blue), 1:2 (black), and 1:20 (green). Spectra are normalised to the amplitude of the central line. The spectral shape was similar for the 1:2 and 1:20 samples. | ||
The echo detected spectral shape for 2:1 is consistent with a dense packing of nitroxide radicals, while the nitroxides are spatially more distant in 1:2. For a PMCR of 1:6, 1:20 and 1:30, the spectral features were similar to that of 1:2.
A distinction between different nitroxide components as a result of a variation of spin exchange with CB was not apparent from the EPR spectrum directly. Comparing 1:2 and 1:20 in Fig. 3, the spectral shape of the nitroxide was preserved. Since in the strong exchange limit a nitroxide radical in the vicinity of a faster relaxing paramagnetic species will have a relaxation rate approaching that of the fast-relaxing species,53 the sudden observation of an echo from nitroxide radical even down to a PMCR of 1:30, while the CB species is still not observable, may hint at a reduced exchange interaction between CB conduction electrons and nitroxide radicals. Such an explanation is consistent with a reduced electron tunnelling rate at lower temperature.69 Nonetheless, exchange is not expected to be quenched completely, hence T1 relaxation may be a sensitive probe for the contact between CB and nitroxides.
Inversion recovery experiments for measuring T1 were conducted at the field position of maximum intensity. T1 anisotropy was studied for the 2:1 sample, using Laplace inversion along the recovery dimension and regularisation along both dimensions.75 Only a minor field dependence of T1 was observed (see Fig. S3, ESI†). Fig. 4 shows the inversion recovery time traces for composite samples with varying PMCR. T1 increases as the composition is changed from 2:1 to 1:2 and starts decreasing after 1:2. In 2:1, due to the dense packing of nitroxide radicals, the dominant contribution to relaxation is expected to be spin interactions between the nitroxide radicals, which account for the majority of the lattice. In addition, the relaxation curve might contain contributions from spectral diffusion processes,34 which are prominent in aggregated spin systems with small inter-spin distances such as in 2:1. Spectral diffusion tends to shorten apparent spin–lattice relaxation times.53,72,76 As the nitroxides get more dispersed through the carbon lattice in 1:2, the aforementioned contributions decrease and T1 increases from 2:1 to 1:2. The increase in T1 from 2:1 to 1:2 indicates that at high PTMA monomer content, the majority of nitroxide radicals exhibit relaxation that is influenced by nitroxide–nitroxide contact. Decreasing the number of nitroxide–nitroxide close contacts or increasing the distance slows the relaxation down. For 1:6, where CB accounts for most of the lattice, a further increase in dispersion of nitroxide radicals does not lead to a slower relaxation, suggesting that the nitroxide radicals are well-separated and a different relaxation mechanism dominates. The faster relaxation observed in 1:6 compared to 1:2 indicates a more efficient energy transfer between spin system and the lattice. From the perspective of CB, the conductive network is dependent on the inter-particle contact,31,77 which improves as the PTMA monomer is more dispersed, allowing the formation of more continuous conduction pathways and a better dissipation of energy through the lattice. As the PTMA monomer–CB contact improves further in samples with PMCR below 1:6, T1 decreases further.
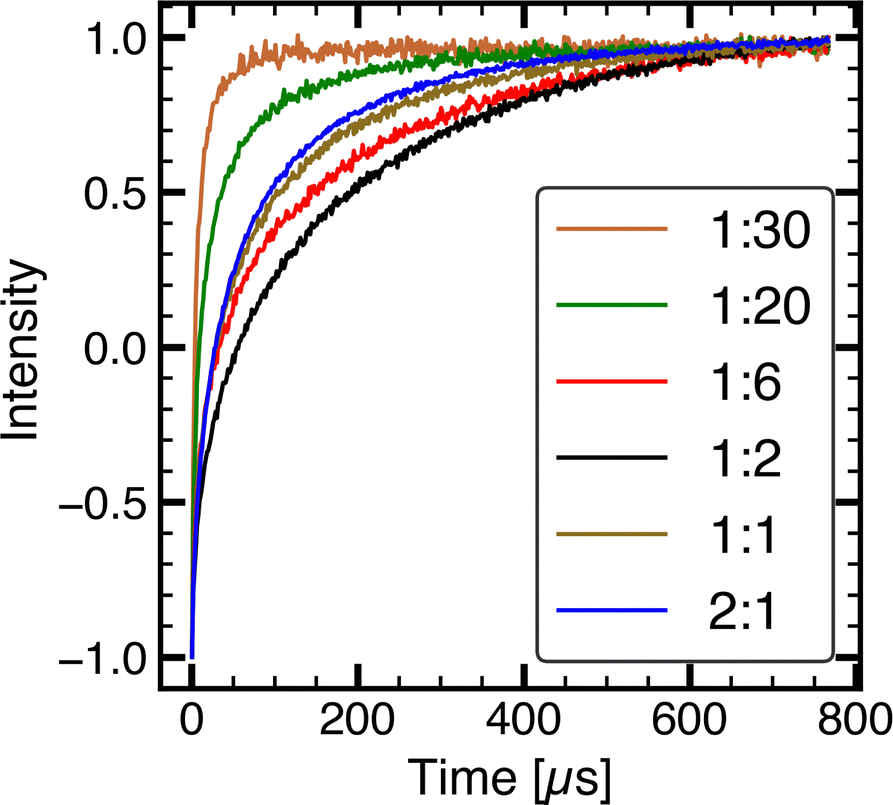 | ||
| Fig. 4 Comparison of inversion recovery time traces for varying ratios of PTMA monomer to Super P, acquired at 50 K. The 1:30 composite sample shows the fastest T1 relaxation. While field-swept echo-detected EPR spectra of 1:2 and 1:20 were similar, T1 relaxation shows a significant difference. For comparing relaxation curves from different composite samples, inversion recovery data were scaled between −1 and 1. | ||
The change in relaxation trend from 1:2 to 1:6 shows that the dependence of relaxation on sample composition is not a simple monotonic correlation with the amount of CB. If the distribution of PTMA monomer within the CB lattice is non-uniform, the presence of distinct PTMA monomer populations differing in their contact quality with CB within the same composite sample is also possible. As evident when comparing relaxation curves of 2:1 and 1:30, relaxation of the nitroxide–nitroxide contact dominated system is significantly different from a system where nitroxide–CB contact is dominant. Therefore, a variation in the quality of PTMA monomer–CB contact may become apparent as a distribution in relaxation times.
3.3 Laplace inversion
In order to obtain relaxation time distributions from time domain inversion recovery data, ILT was conducted. The possibility of multiple relaxation environments in the investigated system makes ILT an apt tool to be used here, as relaxation time distributions can be obtained without assuming the number of relaxation components. Fig. 5 shows the T1 relaxation time distributions obtained using ILT with an exponential kernel. The T1 distributions did not show any physically implausible features or systematic artefacts, and the residuals from fits were representative of random noise devoid of any apparent systematic features (see Fig. S1 in the ESI†). The absence of non-random features in the residuals indicates that the chosen kernel is compatible with the data, and that Laplace inversion is able to accurately extract all the features contained in the data from the inversion recovery experiment. Exchange features, such as contributions with a negative sign, were not apparent in the distributions, albeit exchange cannot be excluded either.55 While the use of a non-negativity constraint is not justified a priori56 in case of solid-state samples where exchange or cross-relaxation may be expected, the investigated system showed no significant differences when a non-negativity penalty was used (see Fig. S4, ESI†). | ||
| Fig. 5 Relaxation time distributions (black) for varying PMCR. Inversion recovery experiments were conducted at 50 K. T1 data were inverted using an exponential kernel to obtain the distributions. Labels on the right edge denote the PTMA monomer-to-Super P weight ratio. The distributions were fit (orange) to Gaussian functions in log space. Refer to text for the discussion on individual components 1 (blue), 1′ (light blue), 2 (brown), 2′ (green) and 3 (red). | ||
The distributions indicated at least two distinct relaxation components for each composite sample except maybe for 1:30, where the obtained distribution was asymmetric yet not obviously multi-component. For all the other samples, T1 of the two components differed by at least an order of magnitude, ranging from 70 μs to 380 μs for the slow relaxing component and from 1 μs to 20 μs for the fast relaxing component. In order to derive mean relaxation time constants and amplitudes (Fig. 6), the distributions were fit using Gaussian functions in log space. Integrals from the Gaussian fits were utilised for determining the relative weight of different relaxation components (Fig. 6a). Composite samples with PMCR of 2:1, 1:1, 1:20 and 1:30 could be satisfactorily fit with a two-Gaussian model, while a three-Gaussian model was required for 1:2 and 1:6. Nonetheless, these fits only represent an effective approximation and care must be taken when attempting a physical interpretation.
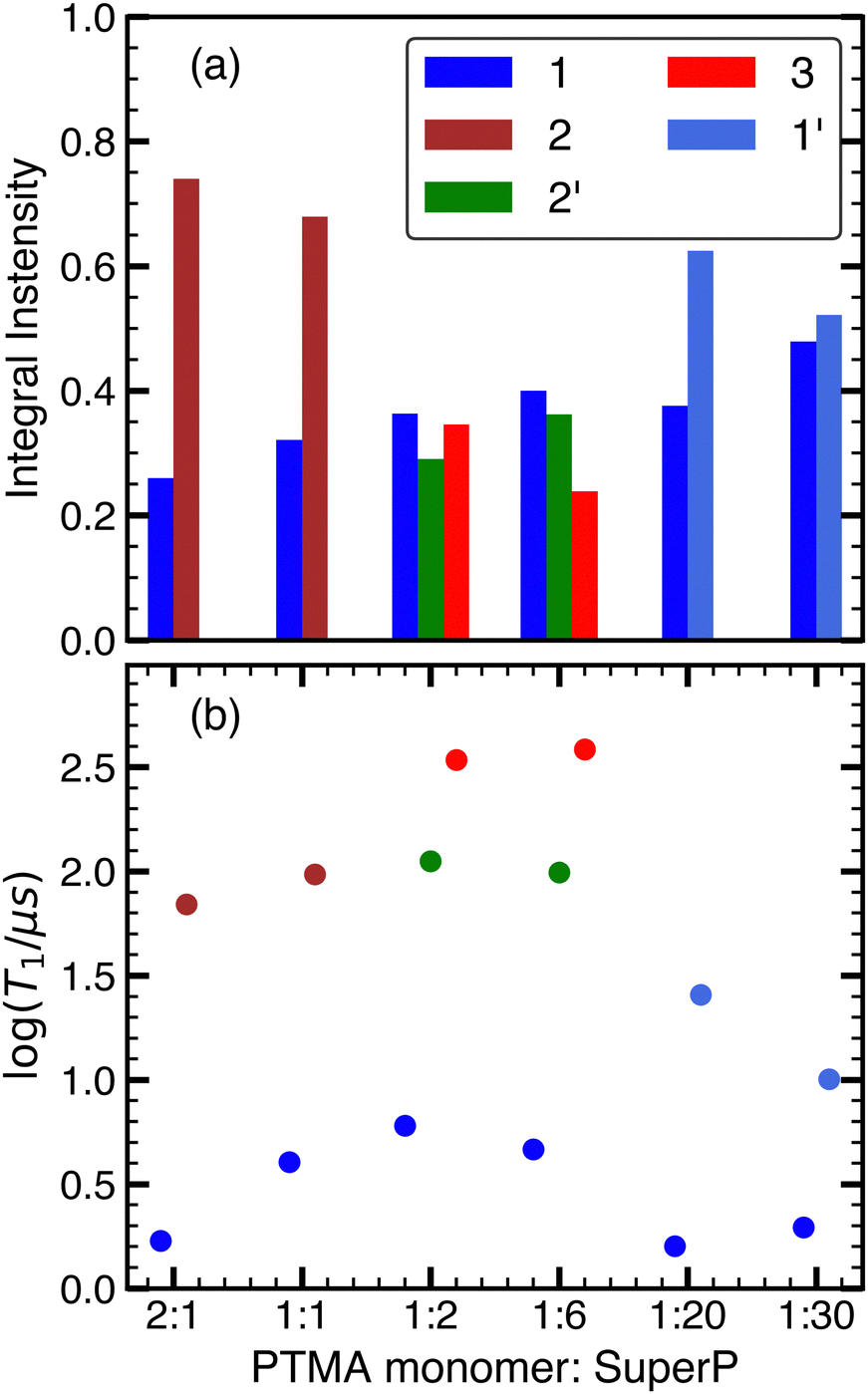 | ||
| Fig. 6 T 1 values and integrals obtained by fitting relaxation time distributions with Gaussian functions in log space. The categorisation into different components, indicated by colours and numbers in the legend, is empirical; it is used to address the contributions in the main text. (a) Integrals of relaxation components shown in Fig. 5. (b) T1 values of relaxation components obtained from the mode of Gaussian fits shown in Fig. 5. | ||
The qualitative relaxation trend observed from the inversion recovery curves were largely followed by both (or all three) components, with an initial increase in T1 from 2:1 to 1:2, followed by a plateau until 1:6 and then a decrease down to 1:30.
In changing the composition from 2:1 to 1:30, the lattice changes from a nitroxide dominated environment in 2:1 (CB-in-PTMA monomer, where the PTMA monomer forms a percolation network) to a CB dominated environment in 1:30 (PTMA monomer-in-CB, with Super P forming a percolation network). In-between, a mixture of PTMA monomer and CB is formed, where both species form local clusters of variable size. This segregation into three different types of composition appears also reproduced in the relaxation time distributions.
At a high CB content of 1:30 and 1:20, with good contact between PTMA monomer and CB as discussed above, PTMA appears to occupy two different environments, labelled 1 and 1′ in Fig. 5 and 6, with different relaxation properties. Possible environments may be in-pore and ex-pore CB surfaces, another option could be graphitic and amorphous surface regions.75 However, the qualitative nature of the T1 distribution analysis with Gaussian modes becomes apparent when comparing the result for the 1:20 sample at 50 K and at 30 K (Fig. 7). At 30 K, the 1:20 distribution appears to contain three overlapping, but distinguishable relaxation modes. Even at this reduced temperature, fast relaxation components with T1 < 20 μs are visible, consistent with an intimate contact between PTMA monomer and CB.
 | ||
| Fig. 7 Comparison of relaxation time distributions for PTMA monomer to Super P ratios of 1:2 and 1:20. Inversion recovery experiments were conducted at 30 K and T1 data was inverted using an exponential kernel to obtain the distributions. | ||
As the PMCR increases to 1:6, the two fast relaxing components at 50 K cannot be clearly distinguished anymore, and they are fitted by a single, broadened Gaussian mode centred at ≈4 μs (labelled 1 in Fig. 5 and 6). This may hint towards a differentiation between graphitic and amorphous regions by PTMA, which is expected to blur as the CB surface gets fully covered with PTMA monomer. At the same time, two considerably more slowly relaxing components (3 and 2′) with T1 values ranging from 95 to 400 μs appear. Notice that only in the intermediate PMCR range two Gaussian components are necessary to obtain an adequate fit of the slowly relaxing part of the T1 distribution, yet the distribution itself does not show distinguishable features. However, the 1:2 sample at 30 K (Fig. 7) does show distinguishable modes, hence a different physical origin for the three Gaussian components appears plausible. A possible explanation could be the formation of a second PTMA monomer layer without direct contact to the CB. Exchange with CB would be indirect, hence reduced, via the directly adsorbed layer. On the other hand, in the absence of extended nitroxide clusters showing bulk-like dipole–dipole interaction, which are amplified by spatial correlation of PTMA monomer orientations, also relaxation mediated by interactions with other nitroxides is reduced. The formation of such an intermediate layer on top of adsorbed surface PTMA monomers, but without full contact to bulk-like extended nitroxide clusters, could be analogous to structures found for ionic liquids on CB surfaces.75 Such a layer may show significantly different physical properties compared to adsorbed nitroxides as well as bulk-like nitroxide clusters.
At a high PTMA monomer content, relaxation becomes dominated by dipole–dipole interaction between nitroxide radicals that form extended bulk-like clusters. The slowly relaxing components could not be distinguished in samples with high PTMA monomer content (2:1 and 1:1) and are fit using a single Gaussian mode at ≈70 μs, (component 2 in Fig. 5 and 6). At the same time, the adsorbed layer on CB still persists, as evidenced by the sustained presence of a relaxation component with T1 < 10 μs. Since it can be expected that the bulk-like clusters show some local order, more effective paths for spectral spin diffusion may exist, which affect the observed T1 relaxation rates and prevent a classification of the observed changes in relaxation time distributions at high PTMA monomer content.
Nonetheless, the results for the different samples suggest that the relaxation of each species is influenced by multiple relaxation processes. However, as evidenced by the large difference in relaxation time values, the relative weight of their contribution to the relaxation of each component differs. Within a composite sample, PTMA monomers in good contact with CB relax faster and are less sensitive to changes in the extent of nitroxide dispersion or nitroxide–nitroxide contact. In contrast, PTMA monomers that exist in densely packed clusters seem to relax more slowly and are more sensitive to changes in nitroxide dispersion.
Nitroxide–nitroxide contact could also be monitored using spin-echo dephasing rates, Tm, which show an increase as the relative amount of nitroxide in the composite sample is decreased (see Fig. S5, ESI†). However, the contrast was not as pronounced as for T1 and, while a qualitative correlation was observed, the general validity of Tm as an alternative parameter for investigating the proposed interactions would have to be investigated on a sample-by-sample basis due to the presence of considerable ESEEM (electron spin echo envelope modulation) oscillations.78 To prevent overfitting, a weighted ILT using the amplitude of the ESEEM modulations as non-uniform weights was employed.63
A schematic model for the composite system based on the T1 relaxation distributions is shown in Fig. 8. Different relaxation regimes corresponding to different degrees of PTMA monomer–CB contact are identified. The fastest relaxing component is attributed to well dispersed PTMA monomer in good contact with CB (Fig. 8a, denoted with a blue outline) that forms a percolation network. In contrast, Fig. 8c shows extended nitroxide clusters (brown) showing a weak contact with dispersed CB particles. The system shown in Fig. 8c represents CB in a percolation network of PTMA monomer. Fig. 8b shows smaller clusters of both PTMA monomer (red) and CB. The slowest relaxing component is attributed to these smaller clusters of PTMA monomer that are neither part of an extended PTMA monomer cluster nor in direct contact with CB. The fastest relaxing component at intermediate PMCR represents the primary adsorbed layer (blue) of PTMA monomer in direct contact with CB while an additional layer (green) which builds on the primary layer is attributed to PTMA monomer in an indirect contact with CB. A similar analysis based on Tm data was not feasible due to the presence of ESEEM (electron spin echo envelope modulation) oscillations (see discussion in ESI†).
 | ||
| Fig. 8 Proposed model for the PTMA monomer–Super P (black, filled circles) composite systems based on the relaxation distributions obtained using ILT. Different relaxation regimes are identified corresponding to different degrees of PTMA monomer–CB contact. The colours are qualitatively related to the corresponding species in Fig. 5 and 6. (a) Composite sample with high CB content showing PTMA monomer in a percolation network of CB. PTMA monomer in good contact with CB is denoted as a blue surface layer on the carbon particles. (b) Composite samples in an intermediate relaxation regime showing smaller clusters of both CB and PTMA monomer (red) of varying sizes. PTMA monomer in indirect contact with CB is denoted as a green layer on top of the adsorbed primary layer in blue. White pores represent EPR silent voids, which could either be filled with air or with residual solvent. (c) Composite sample with a high PTMA content containing dispersed CB particles in a percolation network of PTMA monomer (brown). Directly adsorbed PTMA monomer in good contact with CB (denoted by the blue layer) only represents a minor fraction of the total PTMA monomer content. | ||
The relaxation time constant distributions followed the same trends at 30 K, and a better resolution of the relaxation components was observed than at 50 K (see Fig. S2, ESI†). An improved resolution in the relaxation time distribution at lower temperature is likely due to different temperature dependencies of the relaxation components and further reinforced by the higher signal-to-noise ratio.
Since the relaxation time modes for the different components overlap, assignment and separation is not unambiguously possible. Even though five different labels were used for the fitted components in Fig. 5, an actual assignment of five components appears unreasonable. A classification into three different components appears more justified, especially when considering the relaxation results at both temperatures. Furthermore, an exact Gaussian shape in log space for each of the modes is rather unlikely, yet such a fit provided a simple basis for the classification and discussion of the relaxation time distributions. It allowed rationalising the origin of different relaxation components, and correlation of the composition with PTMA monomer–CB contact could be established by following apparent changes in the ILT-derived relaxation distributions. Therefore, the presented technique appears suitable as an analytical tool to compare different protocols for sample preparation.
4 Conclusions
The use of pulsed EPR relaxometry combined with inverse Laplace transform (ILT) was demonstrated to characterise the contact between PTMA monomer and Super P carbon black in composite samples with varying PTMA monomer to Super P weight ratios. Inversion recovery experiments revealed that sample composition and accessibility of carbon black towards the PTMA monomer significantly affects the relaxation characteristics of the PTMA monomer. The time domain relaxation data was inverted using ILT to obtain relaxation time distributions, enabling the identification of different degrees of PTMA monomer–carbon black contact. ILT was found to be apt for detecting changes in the relaxation characteristics of nitroxides in the presence of carbon black and at different distributions of nitroxides within the composite sample. Thereby, pulsed EPR relaxometry showed a different contrast than CW EPR, making it a complementary technique to also identify sample changes that do not lead to an apparent variation in the CW EPR spectra. Different relaxation components differing by at least an order of magnitude were evident in the relaxation distributions and could be monitored for a range of PTMA monomer to carbon black weight ratios. Our analysis indicates that the composition of the sample significantly affects the quality of contact between the PTMA monomer and carbon black. Laplace inverted pulsed EPR relaxation may serve as a robust tool to study such contact also in other, similar systems.Author contributions
Davis Thomas Daniel: conceptualisation, methodology, investigation, formal analysis, writing – original draft, visualisation, Conrad Szczuka: writing – review & editing, Peter Jakes: writing – review & editing, supervision, Rüdiger-A. Eichel: writing – review & editing, supervision, Josef Granwehr: conceptualisation, methodology, software, writing – original draft, supervision. All authors have read and agreed to the published version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank Dr. P. Philipp M. Schleker for valuable discussions. Funding by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) within the priority programme “Polymer-Based Batteries” (SPP 2248) – project number 441255373 – is gratefully acknowledged. The development of the ILT code was supported by an EPSRC First Grant (EP/J001112/1).Notes and references
- T. Placke, R. Kloepsch, S. Dühnen and M. Winter, J. Solid State Electrochem., 2017, 21, 1939–1964 CrossRef CAS.
- J. M. Tarascon and M. Armand, Nature, 2001, 414, 359–367 CrossRef CAS.
- E. Foreman, W. Zakri, M. Hossein Sanatimoghaddam, A. Modjtahedi, S. Pathak, A. G. Kashkooli, N. G. Garafolo and S. Farhad, Adv. Sustainable Syst., 2017, 1, 1700061 CrossRef.
- G. Liu, H. Zheng, A. S. Simens, A. M. Minor, X. Song and V. S. Battaglia, J. Electrochem. Soc., 2007, 154, A1129 CrossRef CAS.
- M. D. Hager, B. Esser, X. Feng, W. Schuhmann, P. Theato and U. S. Schubert, Adv. Mater., 2020, 32, 2000587 CrossRef CAS PubMed.
- K. Nakahara, S. Iwasa, M. Satoh, Y. Morioka, J. Iriyama, M. Suguro and E. Hasegawa, Chem. Phys. Lett., 2002, 359, 351–354 CrossRef CAS.
- H. Nishide, S. Iwasa, Y. J. Pu, T. Suga, K. Nakahara and M. Satoh, Electrochim. Acta, 2004, 50, 827–831 CrossRef CAS.
- D. R. Nevers, F. R. Brushett and D. R. Wheeler, J. Power Sources, 2017, 352, 226–244 CrossRef CAS.
- H. Hishide and T. Suga, Electrochem. Soc. Interface, 2005, 14, 32–36 CrossRef.
- T. Janoschka, M. D. Hager and U. S. Schubert, Adv. Mater., 2012, 24, 6397–6409 CrossRef CAS PubMed.
- J. K. Kim, G. Cheruvally, J. H. Ahn, Y. G. Seo, D. S. Choi, S. H. Lee and C. E. Song, J. Ind. Eng. Chem., 2008, 14, 371–376 CrossRef CAS.
- Q. Huang, D. Choi, L. Cosimbescu and J. P. Lemmon, Phys. Chem. Chem. Phys., 2013, 15, 20921–20928 RSC.
- C. M. Liu, J. Chen, F. Q. Wang and B. L. Yi, Russ. J. Electrochem., 2012, 48, 1052–1057 CrossRef CAS.
- S. Yoshihara, H. Isozumi, M. Kasai, H. Yonehara, Y. Ando, K. Oyaizu and H. Nishide, J. Phys. Chem. B, 2010, 114, 8335–8340 CrossRef CAS PubMed.
- M. Park, X. Zhang, M. Chung, G. B. Less and A. M. Sastry, J. Power Sources, 2010, 195, 7904–7929 CrossRef CAS.
- A. Vlad, J. Rolland, G. Hauffman, B. Ernould and J. F. Gohy, ChemSusChem, 2015, 8, 1692–1696 CrossRef CAS PubMed.
- G. Hauffman, Q. Maguin, J. P. Bourgeois, A. Vlad and J. F. Gohy, Macromol. Rapid Commun., 2014, 35, 228–233 CrossRef CAS PubMed.
- C. H. Lin, J. T. Lee, D. R. Yang, H. W. Chen and S. T. Wu, RSC Adv., 2015, 5, 33044–33048 RSC.
- S. Bahceci and B. Esat, J. Power Sources, 2013, 242, 33–40 CrossRef CAS.
- M. Khodeir, B. Ernould, J. Brassinne, S. Ghiassinejad, H. Jia, S. Antoun, C. Friebe, U. S. Schubert, Z. Kochovski, Y. Lu, E. V. Ruymbeke and J. F. Gohy, Soft Matter, 2019, 15, 6418–6426 RSC.
- C. Szczuka, J. Ackermann, P. P. M. Schleker, P. Jakes, R.-A. Eichel and J. Granwehr, Commun. Mater., 2021, 2, 20 CrossRef CAS.
- I. Kulikov, N. A. Panjwani, A. A. Vereshchagin, D. Spallek, D. A. Lukianov, E. V. Alekseeva, O. V. Levin and J. Behrends, Energy Environ. Sci., 2022, 15, 3275–3290 RSC.
- C. Szczuka, P. Jakes, R.-A. Eichel and J. Granwehr, Adv. Energy Sustainability Res., 2021, 2, 2100121 CrossRef CAS.
- C. Szczuka, R.-A. Eichel and J. Granwehr, ACS Appl. Energy Mater., 2022, 5, 449–460 CrossRef CAS.
- A. Niemöller, P. Jakes, S. Eurich, A. Paulus, H. Kungl, R. A. Eichel and J. Granwehr, J. Chem. Phys., 2018, 148, 014705 CrossRef PubMed.
- A. Niemoller, P. Jakes, R. A. Eichel and J. Granwehr, Sci. Rep., 2018, 8, 1–7 Search PubMed.
- M. Sathiya, J. B. Leriche, E. Salager, D. Gourier, J. M. Tarascon and H. Vezin, Nat. Commun., 2015, 6, 6276 CrossRef CAS PubMed.
- E. P. Kirilina, T. P. Prisner, M. Bennati, B. Endeward, S. A. Dzuba, M. R. Fuchs, K. Möbius and A. Schnegg, Magn. Reson. Chem., 2005, 43, S119–S129 CrossRef CAS PubMed.
- Y. Molin, K. Salikhov and K. Zamaraev, Spin Exchange. Principles and Applications in Chemistry and Biology, Springer, US, 1980, p. 244 Search PubMed.
- Q. Huang, E. D. Walter, L. Cosimbescu, D. Choi and J. P. Lemmon, J. Power Sources, 2016, 306, 812–816 CrossRef CAS.
- K. Sheem, Y. H. Lee and H. S. Lim, J. Power Sources, 2006, 158, 1425–1430 CrossRef CAS.
- J.-L. Du, G. R. Eaton and S. S. Eaton, J. Magn. Reson., Ser. A, 1995, 115, 213–221 CrossRef CAS.
- I. V. Koptyug, S. H. Bossmann and N. J. Turro, J. Am. Chem. Soc., 1996, 118, 1435–1445 CrossRef CAS.
- A. Schweiger and G. Jeschke, Principles of Pulse Electron Paramagnetic Spectroscopy, Oxford University Press, 2001, p. 578 Search PubMed.
- S. S. Eaton and G. R. Eaton, Relaxation Times of Organic Radicals and Transition Metal Ions, ed. L. J. Berliner, G. R. Eaton and S. S. Eaton, Springer US, Boston, MA, 2000, pp. 29–154 Search PubMed.
- R. L. Collins, M. D. Bell and G. Kraus, J. Appl. Phys., 1959, 30, 56–62 CrossRef CAS.
- G. Wagoner, Phys. Rev., 1960, 118, 647–653 CrossRef CAS.
- C. Brosseau, P. Molinié, F. Boulic and F. Carmona, J. Appl. Phys., 2001, 89, 8297–8310 CrossRef CAS.
- A. Watanabe, H. Ishikawa, K. Mori and O. Ito, Carbon, 1989, 27, 863–867 CrossRef CAS.
- J. F. Baugher and B. Ellis, J. Colloid Interface Sci., 1972, 38, 658–659 CrossRef CAS.
- L. S. Singer and G. Wagoner, J. Chem. Phys., 1962, 37, 1812–1817 CrossRef CAS.
- G. Feher and A. F. Kip, Phys. Rev., 1955, 98, 337–348 CrossRef CAS.
- F. J. Dyson, Phys. Rev., 1955, 98, 349–359 CrossRef CAS.
- D. Pines and C. P. Slichter, Phys. Rev., 1955, 100, 1014–1020 CrossRef CAS.
- G. R. Eaton and S. S. Eaton, Nitroxides, Royal Society of Chemistry, Cambridge, 2021, pp. 551–579 Search PubMed.
- S. S. Eaton and G. R. Eaton, eMagRes, John Wiley & Sons, Ltd, Chichester, UK, 2016, vol. 5, pp. 1543–1556 Search PubMed.
- D. J. Lépine, Phys. Rev. B, 1970, 2, 2429–2439 CrossRef.
- D. V. Savchenko, E. N. Kalabukhova, A. Pöppl, E. N. Mokhov and B. D. Shanina, Phys. Status Solidi B, 2011, 248, 2950–2956 CrossRef CAS.
- D. V. Savchenko, J. Appl. Phys., 2015, 117, 045708 CrossRef.
- D. Savchenko, E. Kalabukhova, B. Shanina, S. Cichoň, J. Honolka, V. Kiselov and E. Mokhov, J. Appl. Phys., 2016, 119, 045701 CrossRef.
- A. Kulikov and G. Likhtenstein, Adv. Mol. Relax. Interact. Processes, 1977, 10, 47–79 CrossRef CAS.
- J. S. Hyde and K. V. Rao, J. Magn. Reson., 1969, 29, 509–516 Search PubMed.
- S. S. Eaton and G. R. Eaton, in Determination of Distances Based on T1 and Tm Effects, ed. L. J. Berliner, G. R. Eaton and S. S. Eaton, Springer US, Boston, MA, 2000, pp. 347–381 Search PubMed.
- G. Jeschke and Y. Polyhach, Phys. Chem. Chem. Phys., 2007, 9, 1895–1910 RSC.
- D. Bytchenkoff and S. Rodts, J. Magn. Reson., 2010, 208, 4–19 CrossRef PubMed.
- J. Granwehr and W. Köckenberger, Appl. Magn. Reson., 2008, 34, 355–378 CrossRef CAS.
- L. Venkataramanan, Y.-Q. Song and M. D. Hürlimann, IEEE Trans. Signal Process., 2002, 50, 1017–1026 CrossRef.
- Y. Q. Song, L. Venkataramanan, M. D. Hürlimann, M. Flaum, P. Frulla and C. Straley, J. Magn. Reson., 2002, 154, 261–268 CrossRef CAS PubMed.
- A. A. Istratov and O. F. Vyvenko, Rev. Sci. Instrum., 1999, 70, 1233–1257 CrossRef CAS.
- G. C. Borgia, R. J. S. Brown and P. Fantazzini, J. Magn. Reson., 1998, 132, 65–77 CrossRef CAS PubMed.
- J. Granwehr and P. J. Roberts, J. Chem. Theory Comput., 2012, 8, 3473–3482 CrossRef CAS PubMed.
- M. Prange and Y.-Q. Song, J. Magn. Reson., 2009, 196, 54–60 CrossRef CAS PubMed.
- S. Merz, J. Wang, P. Galvosas and J. Granwehr, Molecules, 2021, 26, 6690 CrossRef CAS PubMed.
- I. Cho, J. Choi, K. Kim, M. H. Ryou and Y. M. Lee, RSC Adv., 2015, 5, 95073–95078 RSC.
- H. B. Lin, W. Z. Huang, H. B. Rong, J. N. Hu, S. W. Mai, L. D. Xing, M. Q. Xu, X. P. Li and W. S. Li, J. Power Sources, 2015, 287, 276–282 CrossRef CAS.
- C. Gemperle, G. Aebli, A. Schweiger and R. Ernst, J. Magn. Reson., 1990, 88, 241–256 CAS.
- J. W. Eaton, D. Bateman, S. Hauberg and R. Wehbring, GNU Octave version 6.3.0 manual: a high-level interactive language for numerical computations, 2021.
- B. Ellis and J. F. Baugher, J. Polym. Sci., Part A-2, 1973, 11, 1461–1463 CrossRef CAS.
- P. Sheng, E. Sichel and J. Gittleman, Phys. Rev. Lett., 1978, 40, 1197–1200 CrossRef CAS.
- S. Misra and U. Orhun, Phys. Rev. B, 1990, 41, 2577–2580 CrossRef CAS PubMed.
- E. Bordignon, eMagRes, John Wiley & Sons, Ltd, Chichester, UK, 2017, pp. 235–254 Search PubMed.
- J. R. Klauder and P. W. Anderson, Phys. Rev., 1962, 125, 912–932 CrossRef CAS.
- Y. V. Toropov, S. A. Dzuba, Y. D. Tsvetkov, V. Monaco, F. Formaggio, M. Crisma, C. Toniolo and J. Raap, Appl. Magn. Reson., 1998, 15, 237–246 CrossRef CAS.
- S. Agnello, R. Boscaino, M. Cannas and F. M. Gelardi, Phys. Rev. B, 2001, 64, 174423 CrossRef.
- S. Merz, P. Jakes, R.-A. Taranenko, Svitlana Eichel and J. Granwehr, Phys. Chem. Chem. Phys., 2019, 21, 17018–17028 RSC.
- S. Stoll, eMagRes, 2017, 23–38 CAS.
- Q. Zhang, Z. Yu, P. Du and C. Su, Recent Pat. Nanotechnol., 2010, 4, 100–110 CrossRef CAS PubMed.
- B. Kasumaj, H. Dube, N. Zoelch, F. Diederich and G. Jeschke, J. Magn. Reson., 2012, 223, 187–197 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3cp00378g |
| This journal is © the Owner Societies 2023 |