
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSulfur–arene interactions: the S⋯π and S–H⋯π interactions in the dimers of benzofuran⋯sulfur dioxide and benzofuran⋯hydrogen sulfide†
Yan
Jin
ab,
Wenqin
Li
 b,
Rizalina Tama
Saragi‡
b,
Marcos
Juanes‡
b,
Cristóbal
Pérez
b,
Alberto
Lesarri
*b and
Gang
Feng
*a
b,
Rizalina Tama
Saragi‡
b,
Marcos
Juanes‡
b,
Cristóbal
Pérez
b,
Alberto
Lesarri
*b and
Gang
Feng
*a
aSchool of Chemistry and Chemical Engineering, Chongqing University, Daxuecheng South Rd. 55, Chongqing 401331, China. E-mail: fengg@cqu.edu.cn
bDepartamento de Quimica Fisica y Quimica Inorganica, Facultad de Ciencias – I.U. CINQUIMA, Universidad de Valladolid, Paseo de Belen, 7, 47011 Valladolid, Spain. E-mail: alberto.lesarri@uva.es
First published on 18th April 2023
Abstract
Non-covalent interactions between sulfur centers and aromatic rings play important roles in biological chemistry. We examined here the sulfur–arene interactions between the fused aromatic heterocycle benzofuran and two prototype sulfur divalent triatomics (sulfur dioxide and hydrogen sulfide). The weakly-bound adducts were generated in a supersonic jet expansion and characterized with broadband (chirped-pulsed) time-domain microwave spectroscopy. The rotational spectrum confirmed the detection of a single isomer for both heterodimers, consistent with the computational predictions for the global minima. The benzofuran⋯sulfur dioxide dimer exhibits a stacked structure with sulfur closer to benzofuran, while in benzofuran⋯hydrogen sulfide the two S–H bonds are oriented towards the bicycle. These binding topologies are similar to the corresponding benzene adducts, but offer increased interaction energies. The stabilizing interactions are described as S⋯π or S–H⋯π, respectively, using a combination of density-functional theory calculations (dispersion corrected B3LYP and B2PLYP), natural bond orbital theory, energy decomposition and electronic density analysis methods. The two heterodimers present a larger dispersion component, but nearly balanced by electrostatic contributions.
Introduction
Sulfur-containing compounds are key components of living organisms, playing important biochemical and metabolical roles. As an example, the sulfur amino acids methionine and cysteine are incorporated into proteins, with the first one initiating the synthesis of virtually all eukaryotic proteins and the latter critically influencing protein folding by formation of disulfide bonds.1 For this reason, the analysis of sulfur-centered non-covalent interactions2 is of fundamental interest to understand the biological functions and reactivity of these compounds. Among the multiple non-covalent interactions observed in protein tertiary structures, the presence of sulfur–arene contacts was recognized early as a significant contributor,3,4 as later confirmed from crystal data mining from the Brookhaven Protein Data Bank (PDB) and the Cambridge Crystallographic Database (CCD).5 However, it may be difficult to extract accurate information from individual intermolecular interactions using large macromolecular structures with conventional (ca. 2.5 Å) crystallographic resolution. In these cases, both computational modelling and gas-phase experimental studies can provide complementary chemically specific structural information and a physical description of the molecular forces at play. Previous computational studies concerning the interaction between aromatic rings and sulfur compounds have focused on small prototype heterodimers like methanethiol⋯benzene6 or hydrogen sulfide⋯benzene,7,8 characterized by a S–H⋯π weak hydrogen bond. The use of sulfur non-covalent interactions in drug design and larger examples of arene complexation have also been reviewed elsewhere.9,10 However, the number of high-resolution gas-phase spectroscopic studies of aggregation between sulfur compounds and π-systems is small. Examples include the dimers of hydrogen sulfide with benzene,11,12 phenylacetylene,13 phenol,14,15p-cresol16 or indole,17 or the sulfur dioxide dimers with benzene,18–20 toluene21 or furan,22 loosely classified as van der Waals adducts. Here, we will explore the interactions between divalent sulfur and aromatic rings using broadband (chirped-pulse) microwave spectroscopy and jet expansions, extending previous rotationally resolved investigations on sulfur dioxide aggregation23 and thiol microsolvation24,25 and dimerization.26,27 We chose heterodimers of sulfur dioxide (SO2) and hydrogen sulfide (H2S) to simultaneously explore both the S⋯π and the hydrogen bonded S–H⋯π interactions. In the present work the fused heterocycle benzofuran was selected as aggregation partner because of the presence of three plausible binding sites at the two six- and five-membered rings and the oxygen heteroatom. The observation of sulfur clusters is expected to offer comparison with the equivalent oxygen partners, gauging the influence of the lower electronegativity, larger polarizability and reduced hydrogen bond capacity of sulfur. This information will contribute to the description of weak non-covalent interactions involving chalcogens, which has expanded considerably in the last decade.28,29Methods
Experiment
The dimers of benzofuran with sulfur dioxide (BF⋯SO2) and hydrogen sulfide (BF⋯H2S) were prepared by supersonic expansion of the two precursors, highly diluted with neon as carrier gas. For this purpose, benzofuran was heated to ca. 310 K inside a solenoid-driven pulsed-nozzle (Parker Series 9) and pressurized with a gas line containing each of the sulfur compounds at typical concentrations of 0.5%. The expansion from stagnation pressures of ca. 2-3 bar to the vacuum chamber ultimate pressure of ca. 10−6–10−7 mbar created a vertically moving molecular jet. The nozzle orifice was 1 mm. All chemical samples were obtained commercially.The microwave spectrum was recorded in the 2–8 GHz region with a direct-digital broadband (chirped-pulse) Fourier transform microwave spectrometer.30 This spectrometer uses a fast (20 GS/s) digital arbitrary waveform generator to create a series of short (4 μs) microwave chirped-pulses, which are amplified to 20 W. The exciting pulse propagates perpendicularly to the jet molecular expansion, inducing a broadband fast-passage31 electric-dipole excitation. The resulting transient emission or free-induced decay is amplified, collected in the time-domain with a digital oscilloscope for ca. 40 μs per pulse, and Fourier-transformed to yield the frequency spectrum. The resonance frequencies have typical linewidths (FWHM) below 100 kHz after apodization with a Kaiser–Bessel window. The spectra were averaged for about 1 M cycles. The final frequency measurements have estimated accuracies of 10 kHz or better.
Computations
Initial guess structures were generated with a simple molecular mechanics force field (MMFF).32 These structures were then optimized with quantum mechanical density functional theory (DFT) methods. In this work, the adduct structures were first obtained using the B3LYP hybrid method and D3 two-body empirical dispersion corrections33 (Becke-Johnson damping function34). The most stable structures were later reoptimized with the B2PLYP double-hybrid method.35 The Ahlrich's triple-ζ polarized basis set (def2-TZVP),36 which performs reasonably well in similar clusters,27 was used initially in combination with an ultrafine integration grid and normal convergence criteria. For comparison, the calculations were repeated using Dunning's correlation-consistent cc-pVTZ basis set37 and the jun-cc-pVTZ variation,38 removing the highest angular momentum diffuse functions. The vibrational frequencies were evaluated within the harmonic approximation. For the calculation of complexation energies the basis-sets superposition errors were accounted for within the Boys–Bernardi approximation.39 The natural-bond-orbital analysis40 used NBO version 3. All calculations were implemented in Gaussian 16.41 For the non-covalent interactions, the Johnson-Contreras analysis42 of the reduced electronic density function used NCIPlot.43 An energy decomposition analysis was conducted with symmetry-adapted perturbation theory44 (SAPT) at the SAPT 2 + 3(CCD)/aug-cc-pVDZ level, implemented in PSI4.45Results and discussion
Conformational landscape
The predictions for BF⋯SO2 confirmed the preference for stacked structures, similar to the heterodimers with benzene18 or furan.22 However, the larger number of non-equivalent binding sites on benzofuran results in four different isomers below 5 kJ mol−1 (B3LYP-D3(BJ)), shown in Fig. 1. For the B2PLYP method the last isomer converged to the global minimum. In these structures the sulfur atom of SO2 sits on top of the benzene unit, with the oxygen atoms pointing outwards the ring (rotatable 3D Fig. S1, ESI†). The SO2 moeity is not coplanar with the heterocyle, instead displaying a tilted orientation with sulfur closer to the ring and the two oxygens pointing outwards the ring. The predicted rotational and energetic parameters are presented in Table 1 and Tables S1, S2 (ESI†). In the case of the BF⋯H2S dimer the situation is different, as the predictions in Table 2 and Tables S3, S4 (ESI†) suggest a tendence to form S–H⋯π hydrogen bonds. In the most stable isomer of Fig. 2 the sulfur atom is located in between the two rings, with the two S–H bonds pointing down, respectively, to the six- and the five-membered ring (rotatable 3D Fig. S2, ESI†). A second isomer ca. 2 kJ mol−1 less stable has only one hydrogen atom oriented to the ring. | ||
| Fig. 1 The predicted most stable isomers of the dimer benzofuran⋯sulfur dioxide (B3LYP-D3(BJ)/def2-TZVP electronic energies in parentheses, see Table 1 and Tables S1, S2, ESI†). | ||
| Theoryd | Experimente | ||||
|---|---|---|---|---|---|
| Isomer I | Isomer II | Isomer III | 32S species | 34Species | |
| a Rotational constants (A, B, C), Watson's A-reduction centrifugal distortion constants (ΔJ, ΔJK, ΔK, δJ, δK) and electric dipole moments (μα, α = a, b, c). b Number of measured transitions (N) and standard deviation of the fit (σ). c Relative energies uncorrected (ΔE) and corrected with the zero-point energy (ΔE0), Gibbs energy (ΔG, 298K, 1 atm) and complexation energy (ΔEc, including BSSE corrections). d Calculations using B2PLYP-D3(BJ) and the def2-TZVP basis set, see also Tables S1 and S2 (ESI). e Standard errors in parentheses in units of the last digit. f Values fixed in the fit. | |||||
| A/MHza | 1070.1 | 1066.0 | 1244.1 | 1056.036(3) | 1054.90(1) |
| B/MHz | 830.5 | 830.1 | 666.0 | 811.9523(8) | 802.077(1) |
| C/MHz | 658.9 | 629.4 | 559.1 | 642.2063(9) | 635.9851(9) |
| ΔJ/kHz | 0.53 | 0.62 | 0.53 | 0.26(2) | [0.26] |
| ΔJK/kHz | −0.16 | 2.0 | −1.9 | 2.7(1) | [2.7] |
| ΔK/kHz | −0.19 | −2.3 | 2.9 | −2.7(3) | [−2.7] |
| δ J/kHz | 0.003 | −0.04 | 0.18 | [0.0]f | [0.0] |
| δ K/kHz | 0.54 | 3.7 | 0.90 | 1.1(1) | [1.1] |
| |μa|/D | 1.5 | 1.8 | 2.2 | + | + |
| |μb|/D | 0.8 | 0.7 | 0.2 | + | + |
| |μc|/D | 0.1 | 1.5 | 0.1 | − | − |
| N | 53 | 17 | |||
| σ/kHz | 8.0 | 8.2 | |||
| ΔE/kJ mol−1c | 0.0 | 1.9 | 0.2 | ||
| ΔE0/kJ mol−1 | 0.0 | 1.7 | 2.1 | ||
| ΔG/kJ mol−1 | 1.5 | 0.3 | 0.0 | ||
| ΔEC/kJ mol−1 | −21.30 | −19.25 | −19.41 | ||
| Theory | Experiment | |||||
|---|---|---|---|---|---|---|
| 32S species | 34S species | |||||
| Isomer I | Isomer II | v = 0+ | v = 0− | v = 0+ | v = 0− | |
| a Parameter definition as in Table 1. The theoretical predictions used the B2PLYP-D3(BJ) method and the def2-TZVP basis set, see also Tables S3 and S4 (ESI). | ||||||
| A/MHza | 1167.9 | 1197.7 | 1174.3380(9) | 1173.5247(9) | 1172.814(4) | 1172.033(4) |
| B/MHz | 1087.7 | 1112.7 | 1037.3505(7) | 1037.7417(6) | 1004.705(1) | 1005.059(1) |
| C/MHz | 788.6 | 812.4 | 763.8964(5) | 764.0018(4) | 745.390(1) | 745.495(2) |
| ΔJ/kHz | 0.64 | 2.5 | 1.17(2) | 1.10(4) | ||
| ΔJK/kHz | 4.1 | −6.9 | 2.47(8) | 3.6(2) | ||
| ΔK/kHz | −4.5 | 4.8 | −2.6(1) | −3.8(4) | ||
| δ J/kHz | −0.20 | −0.28 | 0.449(9) | [0.449] | ||
| δ K/kHz | 18.2 | −13.0 | 1.81(4) | [1.81] | ||
| |μa|/D | 1.4 | 0.1 | + | + | ||
| |μb|/D | 0.2 | 0.5 | − | − | ||
| |μc|/D | 0.9 | 0.2 | + | + | ||
| N | 30 | 30 | 15 | 19 | ||
| σ/kHz | 3.9 | 1.3 | ||||
| ΔE/kJ mol−1 | 0.0 | 1.3 | ||||
| ΔE0/kJ mol−1 | 0.0 | 1.5 | ||||
| ΔG/kJ mol−1 | 0.0 | 1.0 | ||||
| ΔEC/kJ mol−1 | −14.02 | −13.10 | ||||
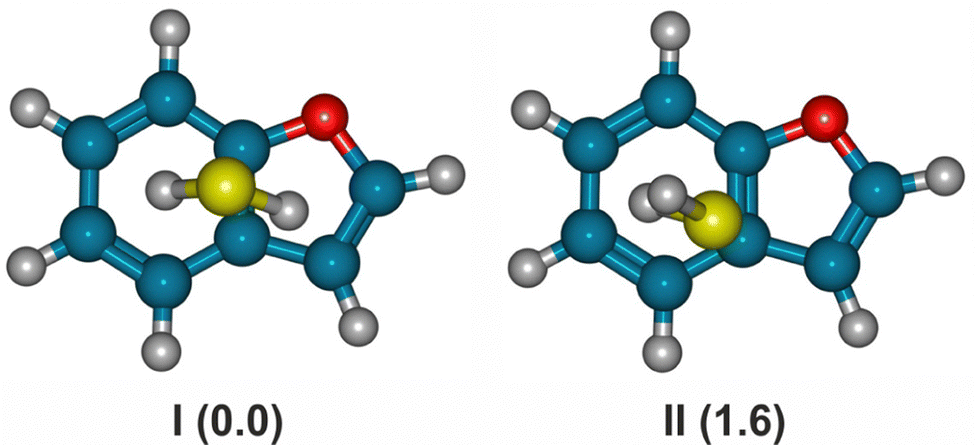 | ||
| Fig. 2 The predicted most stable isomers of the dimer benzofuran⋯hydrogen sulfide (B3LYP-D3(BJ)/def2-TZVP electronic energies in parentheses, see Table 2 and Tables S3 and S4, ESI†). | ||
Rotational spectra
The experimental microwave spectra of the two benzofuran heterodimers are shown in Fig. S3 and S4 (ESI†), with the most prominent transitions being easily attributable to the benzofuran monomer.46–48 The transitions of the two adducts, generally of 1/20–1/30 intensity with respect to the monomer, had appreciable signal-to-noise ratios (SNR < 150![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) and revealed a single isomer for both aggregates. The spectrum of BF⋯SO2, was made of R- (J + 1 ← J) and Q-branch (J ← J) μa- and μb-type transitions (J = 1–5, K−1 = 0–4). As shown in Fig. 3, the spectral intensity permitted the additional assignment of the 34S isotopologue in natural abundance (ca. 4%). In both cases, the Watson A-reduced Hamiltonian reproduced the experiment to experimental accuracy (Table 1). Conversely, the spectrum of BF⋯H2S showed evidence of a large-amplitude motion within the cluster, as the rotational transitions were split into two asymmetric hyperfine components, illustrated in Fig. 4. The exchange of two half-spin fermions would result in a 1:3 statistical weight for the 0+ and 0− torsional states, respectively, which is close to the observations. For this reason, the tunnelling splitting is ascribed to the exchange of the two hydrogen atoms by internal rotation of H2S around its symmetry axis. This effect has been observed in microsolvation clusters where water behaves as weak proton donor,49 as in benzene⋯water.50 In our case the two torsional components were fitted independently to semirigid models, floating the rotational constants of each state, but sharing the centrifugal distortion parameters. The observations included R-branch μa- and μc-transitions (J = 0–4, K−1 = 0–3) for the parent and the 34S isotopologue in natural abundance. The results of the fit are collected in Table 2. The observed transitions are listed in Tables S5–S8 (ESI†). No other dimer species were detected in the spectra.
:1) and revealed a single isomer for both aggregates. The spectrum of BF⋯SO2, was made of R- (J + 1 ← J) and Q-branch (J ← J) μa- and μb-type transitions (J = 1–5, K−1 = 0–4). As shown in Fig. 3, the spectral intensity permitted the additional assignment of the 34S isotopologue in natural abundance (ca. 4%). In both cases, the Watson A-reduced Hamiltonian reproduced the experiment to experimental accuracy (Table 1). Conversely, the spectrum of BF⋯H2S showed evidence of a large-amplitude motion within the cluster, as the rotational transitions were split into two asymmetric hyperfine components, illustrated in Fig. 4. The exchange of two half-spin fermions would result in a 1:3 statistical weight for the 0+ and 0− torsional states, respectively, which is close to the observations. For this reason, the tunnelling splitting is ascribed to the exchange of the two hydrogen atoms by internal rotation of H2S around its symmetry axis. This effect has been observed in microsolvation clusters where water behaves as weak proton donor,49 as in benzene⋯water.50 In our case the two torsional components were fitted independently to semirigid models, floating the rotational constants of each state, but sharing the centrifugal distortion parameters. The observations included R-branch μa- and μc-transitions (J = 0–4, K−1 = 0–3) for the parent and the 34S isotopologue in natural abundance. The results of the fit are collected in Table 2. The observed transitions are listed in Tables S5–S8 (ESI†). No other dimer species were detected in the spectra.
 | ||
| Fig. 3 A section of the microwave spectrum of the dimer benzofuran⋯sulfur dioxide with transitions from the isotopologues with 32S (blue) and 34S (red). See full spectrum in Fig. S3 (ESI†). | ||
 | ||
| Fig. 4 A section of the microwave spectrum of the dimer benzofuran⋯hydrogen sulfide, showing the tunnelling splittings for the isotopologues with 32S (red/blue) and 34S (black/orange). See full spectrum in Fig. S4 (ESI†). | ||
The observed selection rules, rotational constants (Tables 1 and 2) and sulfur substitution coordinates51 (Tables S9 and S10, ESI†) were consistent with the observation of the global minimum species for the two dimers (predicted coordinates in Tables S11–S14, ESI†). Relative differences between the observed and calculated rotational constants are shown in Tables S1–S4 (ESI†). Discrimination of the different isomers is quite unambiguous. However, for the SO2 heterodimer the rotational constants of isomers I and II are relatively close. In this case, the absence of μc spectrum confirmed the observation of isomer I. Fig. S5 (ESI†) illustrates the increase of μc in isomer II of BF⋯SO2. A comparison of the centrifugal distortion constants is not significant in this case because not all parameters could be determined experimentally and because of the limitations of the harmonic approximation. The experiment also permitted the determination of a partial effective structure52 for both dimers, presented in Table 3. Assuming that the two moieties of the dimer47,53 remain unperturbed, six structural parameters are required to locate the triatomics over the benzofuran ring.22 However, since only two isotopic species are available, we fitted only the distance between the two centers of mass (rCM = r(XS–XBZ)) and the elevation (∠ (XS–XBZ–X)) and dihedral (τ (XS–XBZ–X–C5)) of each triatomic with respect to benzofuran. Other parameters relating to the orientation of the triatomics were kept fixed to the B2PLYP structure. The resulting structures reproduce the observed rotational constants within 0.5 MHz for BF⋯SO2 and 0.4 MHz for BF⋯H2S (Tables S15 and S16, ESI†). The effective structures are compared with theory in Table 3 and Tables S11, S13 (ESI†). The fitted center of mass distance in BF⋯SO2 (3.307(2) Å) is slightly shorter that those observed in the SO2 dimers with benzene (3.485(1) Å18) or furan (3.432(1) Å22), but they share a general stacked pattern characteristic of the S⋯π interaction, with the sulfur end of the triatomic closer to the ring. For the BF⋯H2S dimer the center of mass distance (3.660(3) Å) is also shorter than in benzene⋯H2S (3.771 Å11), where the S–H⋯π interaction produces a symmetric top. The shorter distances are indicative of stronger interaction energies in the dimers with benzofuran, as suggested by the computational binding energies below.
|
|
||||
|---|---|---|---|---|
| BF⋯SO2 | BF⋯H2S | |||
| r 0 | r e | r 0 | r e | |
| a XS, XBZ and X denote, respectively, the center of mass of the sulfur triatomic, the center of mass of the benzofuran monomer and the middle point between atoms C5 and C6 of benzofuran. b Monomer structures in ref. 47 and 53. c Equilibrium structures according to B2PLYP-D3(BJ)/cc-pVTZ. d Standard errors in parentheses in units of the last digit. e Derived parameters in square brackets. | ||||
| r CM = r(XS–XBZ)/Å a | 3.307(2)d | 3.237 | 3.660(3) | 3.567 |
| ∠ (XS–XBZ–X)/deg | 90.7(8) | 95.1 | 87.7(2) | 88.9 |
| τ (XS–XBZ–X–C5)/deg | −78.7(2) | −80.8 | −85.7(4) | −88.2 |
| r(H/O–XS)/Å | [1.286]e | 1.304 | [1.303] | 1.304 |
| ∠ (H/O–XS–XBZ)/deg | [108.7] | 108.6 | [45.9] | 45.8 |
| τ (H/O–XS–XBZ–X)/deg | [80.3] | 79.6 | [−0.6] | −0.6 |
| r(S–H/O)/Å | [1.431] | 1.458 | [1.336] | 1.341 |
| ∠ (S–H/O–XS)/deg | [14.0] | 14.3 | [1.7] | 1.7 |
| τ (S–H/O–XS–XBZ)/deg | [−66.9] | −66.4 | [−164.4] | −164.5 |

The presence of a single isomer for the two dimers contrasts with the prediction of low energy isomers below 2 kJ mol−1. This fact may be attributed to the presence of conformational relaxation pathways in the jet expansion,54 associated to small interconversion barriers. Considering the weak intermolecular forces (discussed below), and that the conversion between the stable isomers involves only small structural adjustments of the triatomics, the potential barriers are expected a priori to be small. We confirmed this fact with the calculation of interconversion barriers between the two most stable isomers using DFT (B3LYP-D3(BJ)/cc-pVTZ). The predicted direct/(reverse) barriers of 2.6/(0.5) and 2.4/(0.9) kJ mol−1 for BF⋯SO2 and BF⋯H2S, respectively, are much smaller than the empirical conversion thresholds of 5–7 kJ mol−1 for collisional relaxation,54 offering quantitative support for this hypothesis.
The non-covalent interactions in the observed dimers were analysed using several computational methods. The natural-bond-orbital (NBO) calculations in Tables S17 and S18 (ESI†) use atomic orbital overlap and density matrices to form a basis of orthonormal bond orbitals ordered by a maximum occupancy criterion. The charge transfer interactions are associated with occupancy changes from the filled to the unfilled orbitals. The most important donor–acceptor orbital associations in the BF⋯SO2 dimer correspond to π → σ*(S) interactions, involving the donor bonding orbital BD(C3a–C4) of benzofuran and three antibonding orbitals of SO2 (perturbative interaction energies E(2) = 1.0–2.7 kJ mol−1). This interaction is complemented by π(S) → σ*(C3a) and n(S) → σ*(C3a) back donation from SO2 (E(2) = 1.0 kJ mol−1), so they can be properly categorized as S⋯π bonding. For BF⋯H2S the main interactions are π → σ*(SH), involving three π donor orbitals in the ring and non-bonding orbitals of H2S (E(2) = 0.6–1.7 kJ mol−1). Weaker π⋯π* (in BF⋯SO2) and n⋯π* interactions originated by sulfur additionally contribute to dimer formation. As expected, the total interaction energies are much larger (ca. × 1.5) for the sulfur dioxide dimer (B2PLYP-D3(BJ): −20.5 to −22.2 kJ mol−1; B3LYP-D3(BJ): −25.4 to −25.7 kJ mol−1, see Table 1 and Tables S1, S2, ESI†) than for hydrogen sulfide (B2PLYP-D3(BJ): −14.0 to −14.8 kJ mol−1; B3LYP-D3(BJ): −16.0 to −16.3 kJ mol−1, see Table 2 and Tables S3, S4, ESI†). A similar trend is observed for a calculation of complexation energies for the benzene dimers with SO2 (B2PLYP-D3(BJ)/def2-TZVP: −17.9 kJ mol−1) and H2S (−11.6 kJ mol−1). Experimental complexation energies are available for the dimer of benzene⋯SO2,20 permitting an estimation of uncertainties for the computational values. The dissociation energy of −18.4(12) kJ mol−1 obtained from photoionization techniques suggests that the B2PLYP values are slightly underestimated (ca. 3%) respect to the experiment. To the best of our knowledge, no experimental dissociation energies for H2S dimers with benzene or furan have been determined.
A spatial distribution of the non-bonding interactions in the dimer was obtained from a Johnson–Contreras42 electronic density analysis, based on a density (ρ) over gradient power (∇ρ) ratio. The reduced density gradient s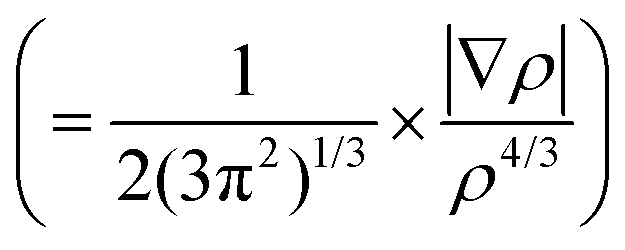 isosurfaces in Fig. 5 permit distinguishing attractive or repulsive regions by the sign of the second Hessian eigenvalue of the electronic density (sign(λ2ρ)), mapping the surfaces associated to attractive (coloured in blue) or weakly attractive (coloured in green) non-covalent interactions. Fig. 5 shows large delocalized regions between the two rings for both BF⋯SO2 and BF⋯H2S, compatible with the interactions noticed in the NBO analysis. For BF⋯SO2 the main interaction region is located above the C3a–C4 and C7a–C3a bonds, associated to the S⋯π interactions, while both the C3a–C7a bonds and the ring centers present more intense interactions in BF⋯H2S, corresponding to the SH⋯π interactions.
isosurfaces in Fig. 5 permit distinguishing attractive or repulsive regions by the sign of the second Hessian eigenvalue of the electronic density (sign(λ2ρ)), mapping the surfaces associated to attractive (coloured in blue) or weakly attractive (coloured in green) non-covalent interactions. Fig. 5 shows large delocalized regions between the two rings for both BF⋯SO2 and BF⋯H2S, compatible with the interactions noticed in the NBO analysis. For BF⋯SO2 the main interaction region is located above the C3a–C4 and C7a–C3a bonds, associated to the S⋯π interactions, while both the C3a–C7a bonds and the ring centers present more intense interactions in BF⋯H2S, corresponding to the SH⋯π interactions.
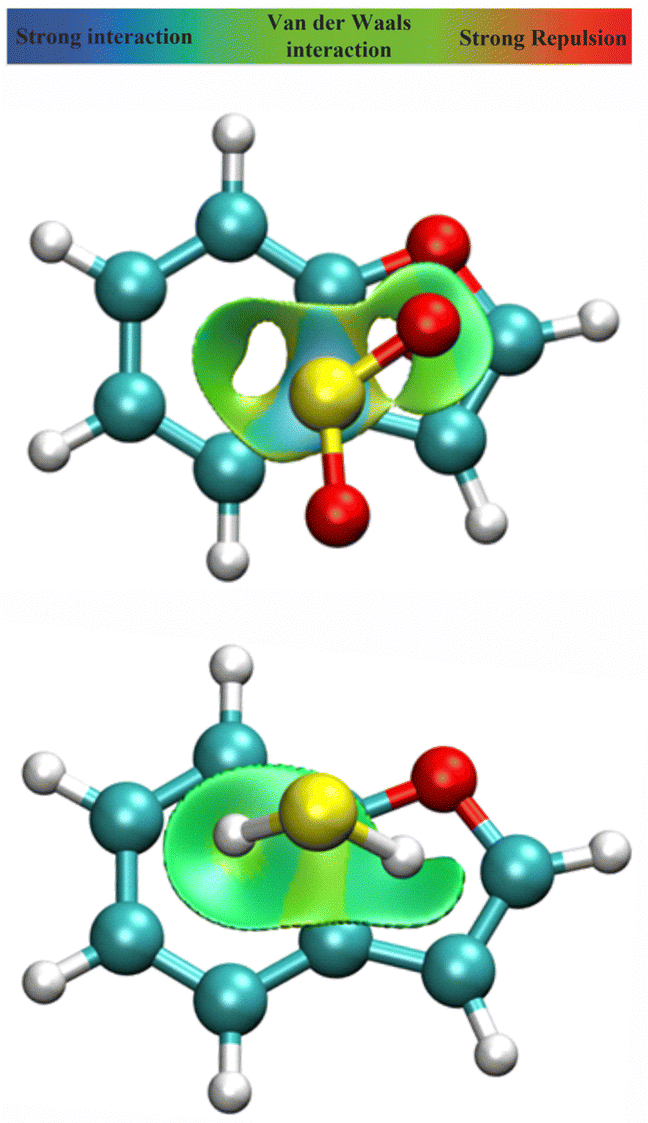 | ||
| Fig. 5 NCI plot39,40 for the dimers of benzofuran with sulfur dioxide (upper panel) and hydrogen sulfide (lower panel), producing a spatial description of attractive interactions in the dimer (isosurface s = 0.5 a.u.). | ||
The results of the SAPT energy decomposition analysis are presented in Table 4, giving insight into the components of the interaction energy. The two dimers similarly present a larger dispersion component (41% and 55%, respectively, of the total attractive energy) but nearly balanced by electrostatic contributions (34% and 35%, respectively). This energetic distribution is probably associated to the prevailing role in these complexes of the π ring electronic density and the large and polarizable molecular orbital distribution in the sulfur triatomics. In this sense both the S⋯π and S–H⋯π interactions occupy an intermediate position between the dominant dispersive character observed in π-stacking26,27,55 and dimers involving a primary hydrogen bond, particularly alcohols,56–58 but also some thiols like the prototypic hydrogen sulfide dimer.59 On the other hand, the dimer of benzyl mercaptan,60 which combines S–H⋯S and S–H⋯π interactions, presents an energy distribution close to the benzofuran dimers.
Conclusions
Sulfur dioxide and hydrogen sulfide form weak dimers with benzofuran, as observed in a jet-cooled expansion by broadband rotational spectroscopy. In both aggregates a single most stable species is observed experimentally, despite DFT quantum mechanical predictions anticipating alternative low energy isomers below 2 kJ mol−1. Because the conversion between stable isomers was shown to proceed through small potential barriers (<5–7 kJ mol−1), the collisional relaxation mechanisms can explain the presence of a single isomer.54 The carriers of the spectrum are reproduced to experimental uncertainty with semirigid rotor Hamiltonian models. Unlike similar SO2 dimers of higher symmetry, like those with benzene or furan,18,22 no tunnelling motions associated to internal large-amplitude motions were observed in benzofuran⋯sulfur dioxide. Conversely, a tunnelling motion was detected for the H2S heterodimer, associated to the internal rotation in the triatomic. The experimental moments of inertia provided ground-state effective structures, consistent with the predicted global minima for the two dimers. In particular, SO2 adopts a stacked position with the sulfur atom closer to the ring, while H2S is depicted as hydrogen bonded to benzofuran. DFT D3-dispersion-corrected calculations at B3LYP and B2PLYP levels satisfactorily reproduced the observed molecular structures. The relative deviations for the rotational constants in the SO2 dimer are kept under 2.6% for both B3LYP and B2PLYP (Tables S1–S4, ESI†), despite the larger computational cost of the later method. Small structural variations are also observed for the three used basis sets. However, the stabilization energies are predicted differently, with B3LYP giving complexation energies ca. 14–18% larger than B2PLYP for BF⋯SO2. The structural predictions are worse for the H2S dimer, but kept under 4.8% for all six calculations methods. In the H2S dimer the B3LYP complexation energies are ca. 8–13% larger than for B2PLYP. Computational predictions using natural bond orbitals explain the dimer interactions as primarily originated from S⋯π and S–H⋯π interactions. This description, despite an increase in the binding energy, shows large similarities with the prototypic benzene dimers.11,12,18 New experiments are in progress to explore the intermolecular interactions between larger sulfur compounds and aromatic systems.Author contributions
Conceptualization, G. F. and A. L.; methodology, G. F. and A. L.; software, Y. J., W. L., R. T. S., M. J. and C. P.; validation, Y. J., W. L., R. T. S., M. J. and C. P.; formal analysis, Y. J., W. L., R. T. S., M. J., C. P., A. L., G. F.; investigation, Y. J., W. L., R. T. S., M. J.; resources, A. L. and G. F.; data curation, A. L. and G. F.; writing—original draft preparation, A. L. and G. F.; writing—review and editing, A. L., and G. F.; visualization, Y. J., W. L.; supervision, A. L. and G. F.; project administration, A. L. and G. F.; funding acquisition, A. L. and G. F.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Funding from the National Natural Science Foundation of China (No. 22273009), the Spanish Ministerio de Ciencia e Innovación (MCIN-AEI) and “ERDF A way of making Europe” (grant PID2021-125015NB-I00) and ERDF - Junta de Castilla y León (grants INFRARED IR2021-UVa13 and IR2020-1-UVa02) is gratefully acknowledged. J.Y. and W.L. thank the China Scholarship Council (CSC) for scholarships. M.J. thanks the Ministerio de Universidades and the Universidad de Valladolid for a “Margarita Salas” postdoctoral contract (CONREC-2021-265).Notes and references
- D. Voet and J. G. Voet, Biochemistry, Wiley and Sons, 4th edn, 2010 Search PubMed.
- A. Chand, D. K. Sahoo, A. Rana, S. Jena and H. S. Biswal, The prodigious hydrogen bonds with sulfur and selenium in molecular assemblies, structural biology, and functional materials, Acc. Chem. Res., 2020, 53, 1580–1592 CrossRef CAS PubMed.
- R. S. Morgan, C. E. Tatsch, R. H. Gushard, J. M. Mcadon and P. K. Warme, Chains of alternating sulfur and π-bonded atoms in eight small proteins, Int. J. Pept. Protein Res., 2009, 11, 209–217 Search PubMed.
- K. S. C. Reid, P. F. Lindley and J. M. Thornton, Sulphur-aromatic interactions in proteins, FEBS Lett., 1985, 190, 209–213 CrossRef CAS.
- R. J. Zauhar, C. L. Colbert, R. S. Morgan and W. J. Welsh, Evidence for a strong sulfur-aromatic interaction derived from crystallographic data, Biopolymers, 2000, 53, 233–248 CrossRef CAS PubMed.
- G. Duan, V. H. Smith and D. F. Weaver, Characterization of aromatic-thiol π-type hydrogen bonding and phenylalanine-cysteine side chain interactions through ab initio calculations and protein database analyses, Mol. Phys., 2001, 99, 1689–1699 CrossRef CAS.
- T. P. Tauer, M. E. Derrick and C. D. Sherrill, Estimates of the ab initio limit for sulfur–π interactions: the H2S–benzene dimer, J. Phys. Chem. A, 2005, 109, 191–196 CrossRef CAS PubMed.
- A. L. Ringer, A. Senenko and C. D. Sherrill, Models of S/π interactions in protein structures: Comparison of the H2S-benzene complex with PDB data, Protein Sci., 2007, 16, 2216–2223 CrossRef CAS PubMed.
- B. R. Beno, K. S. Yeung, M. D. Bartberger, L. D. Pennington and N. A. Meanwell, A survey of the role of noncovalent sulfur interactions in drug design, J. Med. Chem., 2015, 58, 4383–4438 CrossRef CAS PubMed.
- E. A. Meyer, R. K. Castellano and F. Diederich, Interactions with aromatic rings in chemical and biological recognition, 2003, vol. 42.
- E. Arunan, T. Emilsson, H. S. Gutowsky, G. T. Fraser, G. de Oliveira and C. E. Dykstra, Rotational spectrum of the weakly bonded C6H6–H2S dimer and comparisons to C6H6–H2O dimer, J. Chem. Phys., 2002, 117, 9766–9776 CrossRef CAS.
- D. Wang, P. Chopra, S. Wategaonkar and A. Fujii, Electronic and infrared spectroscopy of benzene-(H2S) n (n = 1 and 2): the prototype of the SH-π interaction, J. Phys. Chem. A, 2019, 123, 7255–7260 CrossRef CAS PubMed.
- M. Goswami and E. Arunan, Microwave spectrum and structure of C6H5CCH⋯H2S complex, J. Mol. Spectrosc., 2011, 268, 147–156 CrossRef CAS.
- S. Ghosh, S. Bhattacharyya and S. Wategaonkar, Dissociation energies of sulfur-centered hydrogen-bonded complexes, J. Phys. Chem. A, 2015, 119, 10863–10870 CrossRef CAS PubMed.
- S. Bhattacharyya and S. Wategaonkar, ZEKE photoelectron spectroscopy of p-fluorophenol⋯H2S/H2O complexes and dissociation energy measurement using the Birge-Sponer extrapolation method, J. Phys. Chem. A, 2014, 118, 9386–9396 CrossRef CAS PubMed.
- H. S. Biswal, P. R. Shirhatti and S. Wategaonkar, O–H···O versus O–H···S hydrogen bonding I: experimental and computational studies on the p-Cresol·H2O and p-Cresol·H2S complexes, J. Phys. Chem. A, 2009, 113, 5633–5643 CrossRef CAS PubMed.
- H. S. Biswal and S. Wategaonkar, Sulfur, not too far behind O, N, and C: SH···π hydrogen bond, J. Phys. Chem. A, 2009, 113, 12774–12782 CrossRef CAS PubMed.
- A. Taleb-Bendiab, K. W. Hillig and R. L. Kuczkowski, Microwave spectrum of benzene SO2: Barrier to internal rotation, structure, and dipole moment, J. Chem. Phys., 1992, 97, 2996–3006 CrossRef CAS.
- R. J. Wilson and R. L. Kuczkowski, The microwave spectrum and structure of fluorobenzene sulphur dioxide, J. Mol. Struct., 1996, 376, 1–10 CrossRef CAS.
- J. R. Grover, E. A. Walters, J. K. Newman, M. G. White, E. A. Walters and J. K. Newman, Measurement of the dissociation energies of gas-phase neutral dimers by a photoionization technique: values for trans-2-butene/sulfur dioxide, (trans-2-butene)2, and benzene/sulfur dioxide, J. Am. Chem. Soc., 1985, 107, 7329–7339 CrossRef CAS.
- A. Taleb-Bendiab, K. W. Hillig and R. L. Kuczkowski, Microwave spectrum of toluene SO2: Structure, barrier to internal rotation, and dipole moment, J. Chem. Phys., 1993, 98, 3627–3636 CrossRef CAS.
- J. J. Oh, L.-W. Xu, A. Taleb-Bendiab, K. W. Hillig and R. L. Kuczkowski, The microwave spectrum and structure of the furan · sulfur dioxide complex, J. Mol. Spectrosc., 1992, 153, 497–510 CrossRef CAS.
- Y. Jin, R. T. Saragi, M. Juanes, G. Feng and A. Lesarri, Interaction topologies of the S⋯O chalcogen bond: The conformational equilibrium of the cyclohexanol⋯SO2 cluster, Phys. Chem. Chem. Phys., 2021, 23, 10799–10806 RSC.
- M. Juanes, A. Lesarri, R. Pinacho, E. Charro, J. E. Rubio, L. Enríquez and M. Jaraíz, Sulfur hydrogen bonding in isolated monohydrates: furfuryl mercaptan versus furfuryl alcohol, Chem. – Eur. J., 2018, 24, 6564–6571 CrossRef CAS PubMed.
- M. Juanes, R. T. Saragi, R. Pinacho, J. E. Rubio and A. Lesarri, Sulfur hydrogen bonding and internal dynamics in the monohydrates of thenyl mercaptan and thenyl alcohol, Phys. Chem. Chem. Phys., 2020, 22, 12412–12421 RSC.
- R. T. Saragi, M. Juanes, C. Pérez, P. Pinacho, D. S. Tikhonov, W. Caminati, M. Schnell and A. Lesarri, Switching hydrogen bonding to π-stacking: the thiophenol dimer and trimer, J. Phys. Chem. Lett., 2021, 12, 1367–1373 CrossRef CAS PubMed.
- R. T. Saragi, C. Calabrese, M. Juanes, R. Pinacho, J. E. Rubio, C. Pérez and A. Lesarri, π-Stacking isomerism in polycyclic aromatic hydrocarbons: the 2-naphthalenethiol dimer, J. Phys. Chem. Lett., 2023, 14, 207–213 CrossRef CAS PubMed.
- I. Alkorta, J. Elguero and A. Frontera, Not only hydrogen bonds: other noncovalent interactions, Crystals, 2020, 10, 180 CrossRef CAS.
- M. Juanes, R. T. Saragi, W. Caminati and A. Lesarri, The hydrogen bond and beyond: perspectives for rotational investigations of non-covalent interactions, Chem. – Eur. J., 2019, 25, 11402–11411 CrossRef CAS PubMed.
- C. Pérez, S. Lobsiger, N. A. Seifert, D. P. Zaleski, B. Temelso, G. C. Shields, Z. Kisiel and B. H. Pate, Broadband Fourier transform rotational spectroscopy for structure determination: The water heptamer, Chem. Phys. Lett., 2013, 571, 1–15 CrossRef.
- J. C. McGurk, T. G. Schmalz and W. H. Flygare, Fast passage in rotational spectroscopy: Theory and experiment, J. Chem. Phys., 1974, 60, 4181–4188 CrossRef CAS.
- T. A. Halgren, MMFF VI. MMFF94s option for energy minimization studies, J. Comput. Chem., 1999, 20, 720–729 CrossRef CAS PubMed.
- S. Grimme, S. Ehrlich and L. Goerigk, Effect of the damping function in dispersion corrected density functional theory, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed.
- A. D. Becke and E. R. Johnson, A density-functional model of the dispersion interaction, J. Chem. Phys., 2005, 123, 154101 CrossRef PubMed.
- S. Grimme and F. Neese, Double-hybrid density functional theory for excited electronic states of molecules, J. Chem. Phys., 2007, 127, 1–18 CrossRef PubMed.
- F. Weigend and R. Ahlrichs, Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy, Phys. Chem. Chem. Phys., 2005, 7, 3297 RSC.
- R. A. Kendall, T. H. Dunning and R. J. Harrison, Electron affinities of the first-row atoms revisited. Systematic basis sets and wave functions, J. Chem. Phys., 1992, 96, 6796–6806 CrossRef CAS.
- E. Papajak, J. Zheng, X. Xu, H. R. Leverentz and D. G. Truhlar, Perspectives on basis sets beautiful: Seasonal plantings of diffuse basis functions, J. Chem. Theory Comput., 2011, 7, 3027–3034 CrossRef CAS PubMed.
- S. F. Boys and F. Bernardi, The calculation of small molecular interactions by the differences of separate total energies. Some procedures with reduced errors, Mol. Phys., 1970, 19, 553–566 CrossRef CAS.
- A. E. Reed, R. B. Weinstock and F. Weinhold, Natural population analysis, J. Chem. Phys., 1985, 83, 735–746 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Revision A.03, 2016 Search PubMed.
- E. R. Johnson, S. Keinan, P. Mori-Sánchez, J. Contreras-García, A. J. Cohen and W. Yang, Revealing noncovalent interactions, J. Am. Chem. Soc., 2010, 132, 6498–6506 CrossRef CAS PubMed.
- J. Contreras-García, E. R. Johnson, S. Keinan, R. Chaudret, J. P. Piquemal, D. N. Beratan and W. Yang, NCIPLOT: A program for plotting noncovalent interaction regions, J. Chem. Theory Comput., 2011, 7, 625–632 CrossRef PubMed.
- B. Jeziorski, R. Moszynski and K. Szalewicz, Perturbation Theory Approach to Intermolecular Potential Energy Surfaces of van der Waals Complexes, Chem. Rev., 1994, 94, 1887–1930 CrossRef CAS.
- R. M. Parrish, L. A. Burns, D. G. A. Smith, A. C. Simmonett, A. E. DePrince, E. G. Hohenstein, U. Bozkaya, A. Y. Sokolov, R. Di Remigio, R. M. Richard, J. F. Gonthier, A. M. James, H. R. McAlexander, A. Kumar, M. Saitow, X. Wang, B. P. Pritchard, P. Verma, H. F. Schaefer, K. Patkowski, R. A. King, E. F. Valeev, F. A. Evangelista, J. M. Turney, T. D. Crawford and C. D. Sherrill, Psi4 1.1: An Open-Source Electronic Structure Program Emphasizing Automation, Advanced Libraries, and Interoperability, J. Chem. Theory Comput., 2017, 13, 3185–3197 CrossRef CAS PubMed.
- B. M. Giuliano and W. Caminati, Rotational spectrum of 2,3-benzofuran, Collect. Czech. Chem. Commun., 2003, 68, 1572–1578 CrossRef CAS.
- A. Maris, B. Michela Giuliano, S. Melandri, P. Ottaviani, W. Caminati, L. B. Favero and B. Velino, Structure, dipole moment and large amplitude motions of 1-benzofuran, Phys. Chem. Chem. Phys., 2005, 7, 3317 RSC.
- F. J. Lovas, Microwave Spectra of Molecules of Astrophysical Interest. XXII. Sulfur Dioxide (SO2), J. Phys. Chem. Ref. Data, 1985, 14, 395–488 CrossRef CAS.
- L. Evangelisti and W. Caminati, Internal dynamics in complexes of water with organic molecules. Details of the internal motions in tert-butylalcohol-water, Phys. Chem. Chem. Phys., 2010, 12, 14433–14441 RSC.
- S. Suzuki, P. G. Green, R. E. Bumgarner, S. Dasgupta, W. A. Goddard and G. A. Blake, Benzene forms hydrogen bonds with water, Science, 1992, 257, 942–945 CrossRef CAS PubMed.
- J. Kraitchman, Determination of Molecular Structure from Microwave Spectroscopic Data, Am. J. Phys., 1953, 21, 17–24 CrossRef CAS.
- H. D. Rudolph and J. Demaison, in Equlibrium Molecular Structures, ed. A. Demaison, J. Boggs, J. Csaszar, CRC Press, Boca Raton, Fl, 2011, pp. 125–158 Search PubMed.
- M. Nakata and K. Kuchitsu, Estimation of the equilibrium structures of polyatomic molecules using isotopic differences in vibrationally averaged structures, J. Mol. Struct., 1994, 320, 179–192 CrossRef CAS.
- P. D. Godfrey, F. M. Rodgers and R. D. Brown, Theory versus experiment in jet spectroscopy: Glycolic acid, J. Am. Chem. Soc., 1997, 119, 2232–2239 CrossRef CAS.
- M. Saeki, H. Akagi and M. Fujii, Theoretical study on the structure and the frequency of isomers of the naphthalene dimer, J. Chem. Theory Comput., 2006, 2, 1176–1183 CrossRef CAS PubMed.
- R. Medel, A. Camiruaga, R. T. Saragi, P. Pinacho, C. Pérez, M. Schnell, A. Lesarri, M. A. Suhm and J. A. Fernández, Rovibronic signatures of molecular aggregation in the gas phase: subtle homochirality trends in the dimer, trimer and tetramer of benzyl alcohol, Phys. Chem. Chem. Phys., 2021, 23, 23610–23624 RSC.
- A. Camiruaga, R. T. Saragi, F. Torres-Hernández, M. Juanes, I. Usabiaga, A. Lesarri and J. A. Fernández, The evolution towards cyclic structures in the aggregation of aromatic alcohols: the dimer, trimer and tetramer of 2-phenylethanol, Phys. Chem. Chem. Phys., 2022, 24, 24800–24809 RSC.
- M. Juanes, R. T. Saragi, C. Pérez, L. Enríquez, M. Jaraíz and A. Lesarri, Torsional chirality and molecular recognition: the homo and heterochiral dimers of thenyl and furfuryl alcohol, Phys. Chem. Chem. Phys., 2022, 24, 8999–9006 RSC.
- A. Das, P. K. Mandal, F. J. Lovas, C. Medcraft, N. R. Walker and E. Arunan, The H2S dimer is hydrogen-bonded: direct confirmation from microwave spectroscopy, Angew. Chem., Int. Ed., 2018, 57, 15199–15203 CrossRef CAS PubMed.
- R. T. Saragi, M. Juanes, R. Pinacho, J. E. Rubio, J. A. Fernández and A. Lesarri, Molecular recognition, transient chirality and sulfur hydrogen bonding in the benzyl mercaptan dimer, Symmetry, 2021, 13, 2022 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Additional Figures and Tables with experimental results and computations. See DOI: https://doi.org/10.1039/d3cp01146a |
| ‡ Present address: Institut für Ionenphysik und Angewandte Physik, Universität Innsbruck, Technikerstr. 25/4.OG, 6020 Innsbruck, Austria. |
| This journal is © the Owner Societies 2023 |