
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The E3 state of FeMoco: one hydride, two hydrides or dihydrogen?†
Yunjie
Pang
ab and
Ragnar
Bjornsson
 *bc
*bc
aCollege of Chemistry, Beijing Normal University, 100875, Beijing, China
bMax-Planck Institute for Chemical Energy Conversion, Stiftstrasse 34-36, 45470 Mülheim an der Ruhr, Germany
cUniv. Grenoble Alpes, CNRS, CEA, IRIG, Laboratoire de Chimie et Biologie des Métaux, 17 Rue des Martyrs, F-38054 Grenoble, Cedex, France. E-mail: ragnar.bjornsson@cea.fr
First published on 31st July 2023
Abstract
Hydrides are present in the reduced states of the iron-molybdenum cofactor (FeMoco) of Mo nitrogenase and are believed to play a key mechanistic role in the dinitrogen reduction reaction catalyzed by the enzyme. Two hydrides are present in the E4 state according to 1H ENDOR and there is likely a single hydride in the E2 redox state. The 2-hydride E4 state has been experimentally observed to bind N2 and it has been speculated that E3 may bind N2 as well. However, the E3 state has not been directly observed and very little is known about its molecular and electronic structure or reactivity. In recent computational studies, we have explored the energy surfaces of the E2 and E4 by QM/MM modelling, and found that the most stable hydride isomers contain bridging or partially bridging hydrides with an open protonated belt sulfide-bridge. In this work we systematically explore the energy surface of the E3 redox state, comparing single hydride and two-hydride isomers with varying coordination and bridging vs. terminal sulfhydryl groups. We also include a model featuring a triply protonated carbide. The results are only mildly dependent on the QM-region size. The three most stable E3 isomers at the r2SCAN level of theory have in common: an open belt sulfide-bridge (terminal sulfhydryl group on Fe6) and either 2 bridging hydrides (between Fe2 and Fe6), 1 bridging-1-terminal hydride (around Fe2 and Fe6) or a dihydrogen ligand bound at the Fe2 site. Analyzing the functional dependency of the results, we find that functionals previously found to predict accurate structures of spin-coupled Fe/Mo dimers and FeMoco (TPSSh, B97-D3, r2SCAN, and B3LYP*) are in generally good agreement about the stability of these 3 E3 isomers. However, B3LYP*, similar to its parent B3LYP method, predicts a triply protonated carbide isomer as the most stable isomer, an unlikely scenario in view of the lack of experimental evidence for carbide protonation occurring in reduced FeMoco states. Distinguishing further between the 3 hydride isomers is difficult and this flexible coordination nature of hydrides suggests that multiple hydride isomers could be present during experimental conditions. N2 binding was explored and resulted in geometries with 2 bridging hydrides and N2 bound to either Fe2 or Fe6 with a local low-spin state on the Fe. N2 binding is predicted to be mildly endothermic, similar to the E2 state, and it seems unlikely that the E3 state is capable of binding N2.
Introduction
The nitrogenase enzymes are responsible for the reduction of dinitrogen to ammonia in a complex 2-protein system that consists of the Fe protein and the MoFe protein (in the more active and better understood Mo nitrogenase).1–6 These proteins contain multiple iron–sulfur clusters that are responsible for electron-transfer that end up in the active-site iron-molybdenum cluster, FeMoco (a [MoFe7S9C] cluster), where dinitrogen binding and reduction occurs. Despite decades of detailed kinetic and spectroscopic studies, crystallography and theoretical studies, there is no consensus on a detailed reaction mechanism. Part of the complexity comes from the nature of multi-electron reduction of N2 substrate to the product NH3, which the enzyme accomplishes by an 8-electron mechanism:1–6| N2 + 8e− + 8H+ + 16MgATP → 2NH3 + H2 + 16MgADP | (1) |
Since the original seminal work by Thorneley and Lowe, the E3 state has been part of the kinetic schemes of nitrogenase such as the one shown in Fig. 1, yet very little is actually known about this state.1,7–9,24 The original Thorneley-Lowe model suggested N2 could bind to either E3 or E4 but that E4 was the state for which N2 reduction could occur from.1,24 Since then, regrettably, very little additional knowledge has emerged on the identity or reactivity of this state. Direct spectroscopic or kinetic determinations of E3 are mostly absent as mentioned in recent reviews on the spectroscopy and substrate reductions of nitrogenase.5,6 This is due to its lack of a Kramers EPR signal from an integer-spin cluster. E3 was once attributed to a spectroscopic signal,5,13 however, that is now known to be an EPR signal from E2. Kinetic studies on acetylene reduction in native MoFe protein and variants have suggested inhibition by N2 at the E3 redox level, however, the evidence for E3 involvement in these reactions is indirect.25,26
 | ||
| Fig. 1 The Thorneley-Lowe kinetic scheme relating E0, E1, E2, E3 and E4 redox states.24 N2 binding to E3 was originally suggested in the work by Thorneley and Lowe but the claim has not been substantiated. Also shown are recent suggested computational models for the E2 and E4 redox states.9,34–37 | ||
Computational studies of FeMoco offer the opportunity to fill in these types of gaps in the experimental literature. Unfortunately, FeMoco is arguably the most challenging enzyme cofactor known in biology featuring 8 open-shell metal ions spin-coupled to an S = 3/2 spin state. Considerable effort has been spent on detailed electronic structure investigations into the resting E0 state. There is some consensus on the overall redox state as well as individual oxidation states although the electronic structure is still deeply complicated and a detailed understanding and connection to the magnetic spectroscopy literature remains elusive. The determination of the interstitial atom of FeMoco (as carbide),15,27 the high-resolution X-ray crystal structure15 as well as new determinations of metal oxidation states of the cofactor27–30 have inspired multiple computational studies (or joint experimental-computational) into reduced states of FeMoco9,18,31–37 and attempts to understand the mechanism of nitrogen binding and reduction at FeMoco.32,34,38–50 Special efforts have been devoted to studies of the E4 state, known to contain 2 bridging hydrides.9,32–35
Considerably fewer studies have considered the structural possibilities in the E3 redox state, due no doubt to the lack of experimental data to compare to. In a systematic computational study by Cao and Ryde multiple structural possibilities were considered at the QM/MM level for various reduced states (E1–E4) of FeMoco, including E3.31 The most favorable E3 isomers found in that study consisted of both bridging and terminal hydrides as predicted by the TPSS functional or a triply carbide ion as predicted by B3LYP. A very recent study, also by Ryde and coworkers, suggests a model containing two bridging hydrides between Fe2 and Fe6 as the most favourable when using TPSS, r2SCAN, and TPSSh functionals, while B3LYP predicts a model containing a triply protonated carbide as the lowest-energy model.51 Additionally, Dance has studied the E3 redox state with large QM-cluster calculations and found the most favorable isomer to contain dihydrogen bound to the Fe6 site with a closed protonated belt sulfide-bridge and a model consisting of two terminal/bridging hydrides and an open belt sulfide-bridge.33
Overall, previous experimental and computational work strongly implies the existence of 1 hydride in the E2 state13,19–21,31,33,36,37 and direct ENDOR-observation has convincingly demonstrated the existence of 2 hydrides (and 2 sulfur-bound protons) in the E4 state.8,9,23 It seems likely to assume that the E3 redox state bears a structural similarity to either E2 or E4, however, it is not obvious whether the state should contain 1 or 2 hydrides. A single hydride E3 isomer would suggest a highly simplified electron-counting scheme of 2 electrons present in the hydride and 1 electron in the Fe part, while a 2-hydride isomer implies 4 electrons associated with the hydrides and an oxidized Fe part.
We have previously shown that the binding affinity of N2 at an Fe site in FeMoco depends strongly on the number of hydrides bound to the Fe (which appears to be connected to the ability of the Fe site to acquire a low spin-state).50 The question of whether the E3 state is capable of N2 binding or not may be related to the number and coordination of hydrides present in the structure.
In this study we systematically study the energy landscape of the E3 state by a previously established computational approach that combines QM/MM methodology to reliably treat the protein environment and previously benchmarked DFT methods together with large triple-zeta basis sets and inclusion of scalar relativistic effects and dispersion corrections. We compare 3 categories of E3 isomers: 1-hydride isomers, 2-hydride isomers with an open belt sulfide-bridge and 2-hydride isomers with a closed belt sulfide-bridge, and a model featuring a triply protonated carbide is also included. We test 35 broken-symmetry states for 3 different spin multiplicities for each isomer with a small QM-region and the r2SCAN density functional. We assess the reliability of the resulting energy ranking of isomers by comparing results using small and large QM-regions, and discuss the functional dependency of the results. Finally, we discuss the electronic structure of the most stable isomers as well as the possibility of N2 binding to E3.
Computational details
Our QM/MM model is partially based on the model we previously used to study E0, E1, E2, and E4 states of FeMoco.18,34,36,52–56 However, in this work, as in a recent study on N2 binding to these redox states,50 we utilize a model of the full solvated protein consisting of 320817 atoms in total. An active region of 993 atoms, centered on one of the FeMoco active sites, is used. There are 59 atoms (link-atoms included) in the small QM-A region that consists of FeMoco, sidechains of α-Cys275, α-His442, the singly protonated homocitrate ligand52,57,58 attached to Mo, three additional hydrogens compared to the E0 state, and two added link-atom hydrogens. The QM-region is visualized in Fig. S1 in the ESI.† The QM-B region (145 atoms in total; link-atoms included) contains eight more residues than the QM-A region: α-Val70, α-Arg96, α-Gln191, α-His195, α-Ser278, α-Glu380, α-Phe381, and α-Arg359. In the QM-C region, there are 191 atoms (link-atoms included) and 12 more residues (10 H2O molecules, α-Gly356, and α-Gly357) than the QM-B region (see Fig. S1 in Section 1 of the ESI†). The 10 water molecules included are present in the X-ray structure,15 as shown in Fig. S2 of the ESI.† The α-His195 residue is described as singly protonated on Nε (see Fig. S1 in the ESI†).As discussed in our recent study,50 the use of these QM-A, QM-B, and QM-C regions (same as the QM-I, QM-V, and QM-VI regions, respectively, in that article) converge quite well for reaction energies with even the small and cheap QM-A region being capable of fairly accurate results compared to the larger and more expensive QM-C region (see Table S2 of the ESI† in that article50).
In this work we used the QM-A region to systematically explore all considered E3 models (Fig. 2) in three spin states (MS = 0, 1 and 2) and 35 BS solutions per spin state. Different orientations of protons on bridging or terminal sulfhydryl groups S2B and S5A were investigated using the lowest-energy state of each model with the QM-A region (see Section 7 in the ESI†). The QM-B region was then used to optimize the lowest-energy state for each model based on the QM-A data (the lowest-energy orientation for protons on sulfides). Finally, single-point calculations using the QM-C region were performed on top of QM-B region optimized geometries in order to evaluate the most accurate relative energies of E3 isomers. Broken-symmetry states of FeMoco were systematically investigated by considering all possible 35 BS solutions that can be obtained by flipping 3 of 7 open-shell Fe ions, following the original work by Noodleman and coworkers.59,60 We note that the open-shell Mo(III) ion (present in an unusual non-Hund configuration28) is part of the spin-coupling problem but the configuration arises naturally during the SCF procedure. The BS states were obtained by converging first the high spin state (MS = 34/2) of FeMoco and then flipping the spin-density of three selected Fe ions (using a procedure present in ORCA), and finally converging the SCF to the desired state. For example, BS147 (MS = 2) is found by flipping the Fe1, Fe4, and Fe7 to be down spin and converging to MS = 2. Additional information on BS states is included in Section 8 of the ESI.†
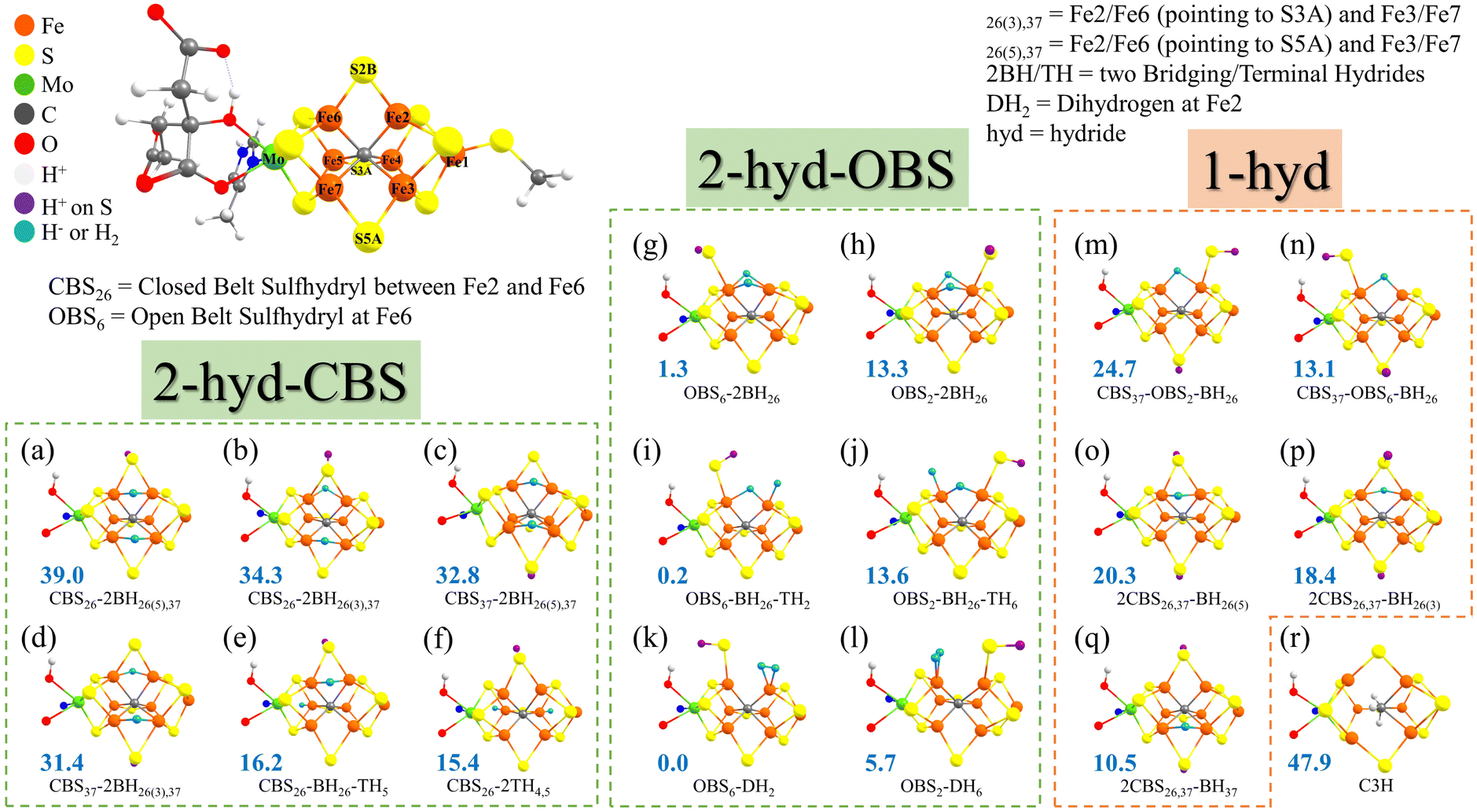 | ||
| Fig. 2 The eighteen structural models for the E3 state that were systematically explored in this work, divided into categories of 2-hyd-CBS (2 hydrides with a closed belt sulfide bridge), 2-hyd-OBS (2 hydrides with an open belt sulfide bridge), 1-hyd (single hydride) and finally the sole protonated carbide model (r). Also shown are relative QM/MM energies (lowest-energy BS state and spin state) in blue, calculated using the r2SCAN functional and the small QM-A region. All the models are isomers of the common E3 redox state surface. The lowest-energy spin state and BS state of a–r isomers are BS245 (MS = 1), BS137 (MS = 1), BS345 (MS = 1), BS345 (MS = 1), BS236 (MS = 2), BS345 (MS = 2), BS347 (MS = 1), BS2356 (MS = 0), BS235 (MS = 2), BS147 (MS = 1), BS147 (MS = 1), BS346 (MS = 2), BS147 (MS = 1), BS247 (MS = 2), BS14 (MS = 2), BS14 (MS = 2), BS346 (MS = 2), and BS125 (MS = 1), respectively. | ||
The QM/MM program ASH,61 developed in our group, was used. It has a flexible interface to the OpenMM molecular mechanics library62 and the ORCA quantum chemistry code.63 The CHARMM36 force field64 and standard electrostatic embedding QM/MM with link atoms as well as a charge-shifting scheme65 were utilized as in our previous papers.18,34,36,52–56 Geometry optimizations were performed using an interface to the geomeTRIC optimization library66 using HDLC internal coordinates.67
Four density functionals: r2SCAN,68 TPSSh,69,70 B97-D3,71–73 and B3LYP*74,75 were primarily used in this work to evaluate the functional dependency of the isomer energies (Results and Discussion section C). This choice is based on a recent benchmarking study on the geometries of spin-coupled iron–sulfur dimers and FeMoco compared to high-resolution X-ray crystallographic data.56 These 4 functionals emerged as the most reliable for Fe–Fe and Mo–Fe distances and as discussed, this was found to be primarily related to the covalency of the bridging M–S bonds between metal ions. The r2SCAN functional was the most reliable functional in that study56 (though only marginally) and it was subsequently used in our recent study on N2 binding to multiple redox states of FeMoco.50 In this work we used r2SCAN for all QM-A region geometry optimizations when evaluating models for E3 in all three spin states and 35 BS solutions (Results and Discussion section B). The five lowest-energy states were then selected for each model and were QM/MM optimized using the same QM-A region with the other functionals (Results and Discussion section C). To further analyze the functional dependency we have also included results using B3LYP76–78 and TPSS69 and this is discussed in Section 9 of the ESI.†
An all-electron scalar relativistic treatment (ZORA79) with recontracted segmented triple-zeta basis sets was used in this study, which has been shown to be close to the all-electron scalar relativistic basis set limit of structures of iron–sulfur compounds.56 The D4 dispersion correction80,81 was used in all calculations involving r2SCAN, TPSS and TPSSh while the B97-D3 and B3LYP* used the D3BJ dispersion correction (dispersion parameters for B3LYP were used). The Split-RI-J82 approximation for Coulomb integrals was used for all functionals and additionally the semi-numerical COSX approximation for Exchange integrals (for TPSSh and B3LYP*). A decontracted auxiliary basis set by Weigend et al.83,84 (named SARC/J in ORCA) was used with the RI approximation. The relativistically recontracted ZORA-def2-TZVP83,85 basis set was used on FeMoco and additional H-atoms (compared to the E0 state) on FeMoco as well as the N2 ligand in N2-bound states while Mo used the SARC-ZORA-TZVP86 basis set. The ZORA-def2-SVP basis set was used for other atoms in the QM region.
Relative energies were calculated from total electronic energies at 0 K. The effect of zero-point vibrational contributions on the relative energies of the lowest energy isomers was briefly investigated and the results are shown in Section 10 of the ESI;† the ZPVE effect is generally sufficiently small that it can be neglected, at least for E3 isomer energies. The effects of entropy were not considered in this work. For N2 binding energies one would expect a sizeable entropy penalty associated with the free energy of binding (∼10 kcal mol−1 based on a gas phase estimate or ∼ 4 kcal mol−1 if estimated from a diffusible position within the protein as suggested by Dance49) but in this work our aim is not to derive realistic estimates of the free energy of binding. N2 and H2 were optimized in vacuum when calculating binding energies and dissociation energies.
Hirshfeld87 and Mulliken88 population analysis methods were used to derive atomic charges and spin populations. A numerical partial Hessian approach89 was used to calculate harmonic N–N vibrational frequencies with N2 and the binding Fe as the partial Hessian. The calculated vibrational frequencies of N2 are shown as relative to the stretching vibration of free N2 in the vacuum using r2SCAN/ZORA-def2-TZVP (2432 cm−1).
Localized orbital analysis was performed using the Pipek–Mezey method,90 which was used to derive FeMoco electronic configurations for the lowest-energy isomers. Approximate ligand-field diagrams of Fe-bound N2 were obtained as described in a recent study, by the combination of diamagnetic substitution, localized orbital analysis and quasi-restricted orbital (QRO) transformation.50
Results and discussion
We start by introducing all models considered for the E3 state in section A. In section B we present the results of QM/MM-calculated energies using the r2SCAN density functional and a small QM-region (QM-A). In section C we explore the functional dependency of the results using 3 other density functionals (B97-D3, TPSSh and B3LYP*) while Section D compares the previous QM-A results to results calculated with the larger QM-C region. In section E we discuss the molecular and electronic structure with the help of localized orbital analysis of the 3 lowest-energy models. Finally, section F discusses N2 binding to the E3 redox state and comparison is made to N2 binding in the other En states.A. The E3 models investigated
The models for E3 discussed primarily in this work can be grouped into 3 categories as shown in Fig. 2: models with 2 hydrides with a closed belt sulfide-bridge (2-hyd-CBS, Fig. 2a–f), 2-hydride/H2 models with an open belt sulfide-bridge (2-hyd-OBS, Fig. 2g–l) and single-hydride models (1-hyd, Fig. 2m–q). We also discuss a model containing a triple protonated carbide (C3H, Fig. 2r) for comparison as previous studies have indicated some QM protocols favouring carbide protonation in reduced FeMoco states. All 18 models contain the same number of electrons and are on the same energy surface.Some of the 2-hydride models can be thought of as derived from E4 models discussed in the literature (shown in Fig. 1) but lacking an S-bound H-atom. For example, models shown in Fig. 2a–d in the 2-hyd-CBS category are derived from E4 models discussed by e.g. Ryde, Raugei and Hoffman;9,32,35 while models in Fig. 2g and h in the 2-hyd-OBS category are more related to E4 models from our own work.34 Ryde and coworkers also calculated models CBS26-2BH26(5),37 (a), OBS6-2BH26 (g) and OBS2-2BH26 (h) in a recent study.51 The 1-hyd models (m–q) are more similar to E2 models discussed in the literature36,37 but feature an extra S-bound proton (and electron). We note that models CBS37-OBS2-BH26 (m), 2CBS26,37-BH26(5) (o), and 2CBS26,37-BH26(3) (p) were included in a recent study.51
Additionally, we include models for E3 suggested in the QM/MM investigations by Ryde and coworkers:31,51CBS26-BH26-TH5 (e), CBS26-2TH4,5 (f), and C3H (r), shown in Fig. 2e, f and r. The CBS26-BH26-TH5 model contains a protonated closed belt sulfide-bridge between Fe2 and Fe6, a bridging hydride between Fe2 and Fe6, and a terminal hydride at Fe5. The CBS26-2TH4,5 also has a protonated closed sulfide-bridge but two terminal hydrides on both Fe4 and Fe5. Three hydrogens were added directly to the carbide in the C3H model (Fig. 2r).
We note that Dance has previously suggested OBS6-BH26-TH2 as a model for the E3 state (named 3H.2x.26.2b in the original work).33 For comparison we also include OBS2-BH26-TH6 by exchanging the sulfhydryl and hydrides positions (compare Fig. 2i and j). Dance also reported on a model with a similar hydride geometry as OBS2-BH26-TH6 but with a CBS configuration.33 Such a geometry could not be stabilized by us but is discussed in the ESI† (see Fig. S4).
Models featuring a dihydrogen group in the E3 state have previously been speculated on91,92 and suggested by calculations.33,91 Inspired by OBS6-BH26-TH2 and OBS2-BH26-TH6 (Fig. 2i and j) we constructed the OBS6-DH2 and OBS2-DH6 models (shown in Fig. 2k and l). Dance previously suggested a model with a protonated closed sulfide-bridge (S2B) and H2 at Fe6 as a candidate for E3,33 discussed in the ESI† and shown in Fig. S5, but such a model could either not be stabilized or found to be very high in energy.
B. Energy ranking of E3 isomers using r2SCAN and the 59-atom QM-A region
The 18 E3 models (a–r in Fig. 2) were optimized by QM/MM calculations using the small QM-A region, and the r2SCAN functional. For each structural isomer we explored three spin states (MS = 0, 1, 2) and 35 possible broken-symmetry state alignments for each spin state, giving a total of 105 electronic states calculated for each structural isomer. A comparison of spin-state energies can be found in Fig. S18 for each isomer in section 8 of the ESI† and we additionally show the type of BS class that tends to be the most favorable in Table S10 (ESI†). While here we opted to systematically study the BS-state problem rigorously, it appears that for the hydride-based isomers in this work it may be sufficient for future studies to primarily focus on the BS7, BS8 and BS10 classes of broken-symmetry solutions (and to a lesser extent BS6 and BS9). Furthermore Section 12 of the ESI† shows the energies and spin populations of every BS-state calculated for each isomer.Fig. 2 shows the relative QM/MM energy of the lowest state calculated for each isomer. The C3H model (Fig. 2r) containing a triply protonated carbide, previously discussed by Ryde and coworkers,31 is predicted to be much higher than other isomers and would seemingly be ruled out at the r2SCAN level of theory (note that Section C discusses the functional sensitivity of this model). Comparing the 3 different categories of hydride-based E3 models, it is clear that 2-hyd-CBS models are predicted to be in general higher in energy than models from the other categories, featuring both the highest-energy structural isomers in the comparison (e.g. structures in Fig. 2a–d, >30 kcal mol−1) and the lowest-energy structures (shown in Fig. 2e and f) are 15–16 kcal mol−1 higher in energy than the lowest-energy isomer (Fig. 2k). The lowest-energy isomers in the 2-hyd-CBS category, featuring either terminal or bridging hydrides: CBS26-BH26-TH5 and CBS26-2TH4,5 were previously suggested by Ryde and coworkers.31 Interestingly, the E3 models (a–d) inspired by some previous E4 models9,32,35 are here predicted to be very high in energy (31–39 kcal mol−1).
The single-hydride 1-hyd isomers are on average lower in energy than the 2-hyd-CBS isomers but are still at least 10 kcal mol−1 above the lowest energy isomer (Fig. 2k) in the 2-hyd-OBS category. The most favorable 1-hyd isomer features a bridging hydride between Fe3 and Fe7 and with 2 protons on S2B and S5A.
The overall stability of the isomers in the 2-hyd-OBS category suggests an overall improved thermodynamic preference associated with an isomer having 2 hydrides instead of just 1, but only if those hydrides are allowed more flexible coordination, here by having an open belt sulfide-bridge (i.e. protonated S2B converting to a terminal sulfhydryl group on Fe2 or Fe6). This improved stability of hydride-based isomers upon allowing a protonated belt sulfide to become a terminal sulfhydryl group, is consistent with our previous work on the E2 and E4 states.34,36
Three 2-hyd-OBS models emerge overall as the lowest in energy: OBS6-DH2 (lowest), OBS6-BH26-TH2 (+0.2 kcal mol−1) and OBS6-2BH26 (+1.3 kcal mol−1). These 3 models have a terminal sulfhydryl group on Fe6 in common but differ in terms of the precise H-atom coordination. OBS6-DH2 furthermore differs where the H-atoms have merged to form an H2 ligand bound side-on to Fe2.
Symmetrically related versions of these 3 models are: OBS2-DH6, OBS2-BH26-TH6 and OBS2-2BH26; they are, however, not as stable, due to the position of the sulfhydryl group on Fe2 instead of Fe6. This causes a steric clash with the His195 residue as confirmed by comparison with simple cluster-continuum calculations (Fig. S3 in the ESI†). We note that this preference may also be affected by the protonation state of His195 as e.g. discussed in recent work50 but here we chose to model His195 as singly protonated on the Nε atom (as in the E0 state).
The results in this section differs from previous computational work on the E3 state by including more diverse isomers than were previously studied or by employing considerably different computational protocols. In previous QM/MM work by Ryde and coworkers,312-hyd-OBS type models for E3 were simply not included (as the hemilability of protonated sulfide bridges was not known at the time). In very recent work by Jiang and Ryde,51 the OBS6-2BH26 model was found to be the lowest-energy model (using TPSSh, r2SCAN, and TPSS) which is consistent with our results. Dance has also previously studied various E3 isomers,33 and an OBS6-BH26-TH2 model was found by him to be the most stable, which is also consistent with our results. The OBS6-2BH26 and OBS2-DH2 isomers were not explored in Dance's work; he did, however, find a CBS26-DH6 model to be very stable. Dance also reported that CBS26-BH26-TH6 is about 5 kcal mol−1 higher than OBS6-BH26-TH2. Attempts at reproducing calculations on CBS26-DH6 and CBS26-BH26-TH6 models (see Fig. S4 and S5 in the ESI†), however, were unsuccessful and we attribute these differences to the different computational protocols employed (QM-cluster vs. QM/MM and BLYP vs. r2SCAN).
The energy of evolving H2 from the E3-OBS6-DH2 model, i.e. the reaction E3-OBS6-DH2 → E1-S2B + H2 has an energy of −15.2 kcal mol−1 using the QM-A region (−9.9 kcal mol−1 using the QM-C region), meaning there is a reasonable driving force for E3 → E1 relaxation (this can be compared to a calculated E2 → E0 relaxation energy of −19.4 kcal mol−1 using a QM-C region reported in ref. 36). The enzyme would have to prevent this favorable H2 relaxation process by utilizing a high electron flux rate in order to reach the E4 state (or alternatively N2 binding) faster than the rate for H2 evolution. Barriers for H2 evolution were not calculated in this work but may be lower than that previously calculated for E2 → E0 (∼21 kcal mol−136). It is also possible that the initial E3 state formed, right after e−/H+ transfer (the details of which are unknown), is not one of the favorable 2-hyd-OBS isomers but perhaps a state closer in nature to an E2-like isomer, perhaps one of the 1-hyd isomers shown e.g. in Fig. 2n or q. The relevant rate for H2 evolution may then involve complex proton and/or hydride reorganization (with unknown barriers), perhaps going from 1-hyd → 2-hyd-OBS or some other pathway that leads to H2 evolution.
C. The functional dependency of the energy ranking
A critical part of an electronic structure study of FeMoco is the choice of methodology to describe the electronic structure. In this work we utilize a scalar relativistic (using the ZORA Hamiltonian) all-electron approach with polarized triple-zeta basis sets on all FeMoco atoms, that should result in very small basis-set incompleteness errors and no errors due to effective core potential approximations (that can be large for FeS systems as discussed in our previous benchmarking study56). This means that the primary error in the electronic structure comes from the use of broken-symmetry density functional theory and in particular the choice of functional. The choice of functional is critical for FeMoco chemistry as is well demonstrated for FeMoco E4 isomer energies in the work by Ryde and coworkers35 where different non-hybrid and hybrid functionals gave completely different results for E4 isomer energies, in particular regarding the stability of hydride vs. protonated carbide formation.In recent work from our group56 we benchmarked different functionals on their ability to describe the geometries of spin-coupled Fe–Fe and Mo–Fe dimers (mostly with bridging sulfides) as well as FeMoco itself. Four functionals emerged as the most accurate: r2SCAN, B97-D3, TPSSh, B3LYP*, having considerably lower errors than other non-hybrid functionals (that have a tendency to underestimate metal–metal distances) and other hybrid functionals (that have a tendency to overestimate metal–metal distances).56 The trends in metal–metal distances were found to correlate with covalency parameters of the Fe–S bridging bonds, suggesting that the geometric errors are reflecting directly errors made in the covalency of the metal–ligand bonds that are responsible for spin-coupling interactions in this complex spin-coupled cofactor. r2SCAN was found to have a slight edge in the study and being a non-hybrid functional (lower computational cost) we used it to generate the results in section B. We note that other FeMoco studies from our group previously used TPSSh18,28,30,34,36,52,55 while other researchers have utilized functionals such as TPSS, BP86, BLYP, PBE, B3LYP, B3LYP* and M06-2X.9,31–33,35,37–49,58,93–99
Here, we test whether density functionals that predict similar geometric structures of spin-coupled FeS systems (including E0 FeMoco), predict similar energies of FeMoco E3 isomers or not. We use the same QM/MM setup and same QM-region as in section B and every isomer was optimized with each functional to allow a fair comparison and to prevent bias. The five lowest-energy BS-states for each structural isomer (from the r2SCAN comparison in Section B) were used in this comparison (see Section 9 in the ESI† for more information).
The results in Fig. 3 reveal overall a highly similar trend for 3 of the functionals: r2SCAN, B97-D3 and TPSSh. The 3 functionals are in good agreement about the 2-hyd-OBS isomers being more favorable than 2-hyd-CBS as well as 1-hyd isomers. Furthermore, these 3 functionals agree on the 3 most stable isomers: OBS6-DH2, OBS6-BH26-TH2 and OBS6-2BH26.
 | ||
| Fig. 3 A comparison of the relative energies of E3 isomers with different density functionals using QM-A region. The x-axis indicates the structural isomer from Fig. 2. r2SCAN, B97-D3, TPSSh consistently predict that the 3 most stable isomers are OBS6-2BH26,OBS6-BH26-TH2 and OBS6-DH2 while B3LYP* favours the r isomer. Note that all functionals include either D3 or D4 dispersion correction (see Computational details). Information about spin states and BS solutions of each model are available in Tables S12–S29 of the ESI.† | ||
While the results for B3LYP* are somewhat similar, they differ in an important way: the carbide-protonated model C3H is found to be the lowest energy isomer (4.5 kcal mol−1 lower than OBS6-DH2), in sharp contrast to r2SCAN (where C3H is the least favorable model) and TPSSh and B97-D3 (unfavorable by 25.1 and 29.8 kcal mol−1). This mild preference for carbide-protonation with B3LYP* follows the well-established preference of carbide-protonation in the E4 state by the regular 20% B3LYP hybrid. As shown in the ESI,† B3LYP favors the carbide-protonation in E3 by ∼20 kcal mol−1. The B3LYP* result is hence not entirely unexpected (differing by 15% instead of 20% HF exchange). The B3LYP*/B3LYP results are also consistent with the recent study by Jiang and Ryde51 where B3LYP predicts C3H to be the lowest energy model. Their work, however, also show TPSSh predicting the C3H model to be almost isoenergetic with OBS6-2BH26 (0.9 kcal mol−1). Our TPSSh results in contrast show C3H to be unfavorable by 25.1 kcal mol−1. The reason for the difference is likely rooted in the larger basis sets (ZORA-def2-TZVP) together with a ZORA relativistic treatment used in our work, while a small def2-SV(P) basis set was used by Ryde and coworkers.51
In the ESI† we have included data with the non-hybrid functional TPSS for comparison (see Fig. S19 in section 9 of the ESI†). TPSS gives a slightly different picture from r2SCAN, B97-D3, and TPSSh. The functional is overall in agreement about isomers OBS6-2BH26 and OBS6-BH26-TH2 being low-energy but predicts isomer OBS6-DH2 as being considerably less stable. TPSS (like most other non-hybrid functionals) has a tendency to systematically underestimate Fe–Fe distances in spin-coupled Fe–S dimers and to give worse structures for FeMoco overall, apparently due to overestimating Fe–S bond covalency.
Overall, the results of this functional comparison show that r2SCAN, B97-D3 and TPSSh functionals predict very similar energetics for E3 isomers. These 3 functionals have previously been found to describe well the metal–metal distances of spin-coupled iron–sulfur dimers as well as E0 FeMoco itself. This is despite the fairly large variability in hydride isomers (1 vs. 2 hydrides or Fe-bound H2), terminal vs. bridging hydrides and open vs. closed terminal sulfide bridges. In contrast, the B3LYP* functional, which was previously56 found to behave similarly to r2SCAN, B97-D3 and TPSSh, favours the triply protonated carbide model (similar to its parent B3LYP functional).
There is no experimental evidence to suggest that the interstitial carbide ion of FeMoco can be protonated. In fact, a recent 13C ENDOR study on several reduced En and ligand-bound FeMoco states shows that the carbide hyperfine coupling remains unchanged, suggesting that the carbide coordination environment remains intact, consistent with a stabilizing role of the carbide. In view of this, the prediction of protonated carbide isomers by B3LYP* (and B3LYP) likely represent artifacts from these functionals. We suggest OBS6-2BH26 and OBS6-BH26-TH2 and OBS6-DH2 models as the most likely models for the E3 state.
D. The QM-region size dependency of the energy ranking
Another critical aspect of modelling metalloenzymes is the treatment of the protein environment. In the QM/MM model used, the whole solvated protein is included in the calculation, however, only a small part is described by quantum mechanics (DFT) and the rest of the system is described by an MM forcefield (CHARMM36). The QM and MM subsystems interact via electrostatic embedding and Lennard-Jones potentials (and bonded interactions at the interface). There are always potential errors made by the definition of the QM-region, in particular residues strongly interacting with the cofactor should ideally be present in the QM-region (to avoid imperfect quantum-classical interactions). By increasing the size of the QM-region one can reduce the magnitude of such effects.In Fig. 4 we compare the effect of using a small 59-atom QM-A region (used in Sections B and C) and using the larger 191-atom QM-C region (single-point calculations performed on the 145-atom QM-B optimized geometries). This comparison was only performed using the r2SCAN functional and only for isomers e to q (see Fig. 2) as multiple functionals consistently found isomers a–d and r to be much higher in energy than other isomers in energy. The results in Fig. 4 reassuringly reveal that the energy landscape does not change very much overall and the stability of 2-hyd-OBS isomers holds and both QM-regions predict the same 3 isomers (models OBS6-2BH26 (g), OBS6-BH26-TH2 (i) and OBS6-DH2 (k)) as the most favourable. However, the larger QM-C region slightly increases the energy gap between these 3 isomers, further stabilizing OBS6-DH2 (3.1 and 3.3 kcal mol−1 more stable than OBS6-2BH26 and OBS6-BH26-TH2 at the r2SCAN level).
 | ||
| Fig. 4 The comparison of relative QM/MM energies using r2SCAN for models (e–q) between using the QM-A and QM-C regions. The labels of the x-axis indicate the models defined in Fig. 2. QM-A region contains 59 atoms while QM-C contains 191 atoms. The spin states and BS solutions are the same as Fig. 2. | ||
Considering both the functional and the QM-region dependency we conclude that the OBS6-DH2, OBS6-BH26-TH2 and OBS6-2BH26 are consistently predicted to be the most stable E3 isomers. The dihydrogen isomer (OBS6-DH2) is predicted to be the most stable, with the energy gap differing depending on precisely which functional and QM-region are used. However, there is certainly uncertainty associated with these energies and we suggest any of these 3 isomers is likely to be encountered experimentally. In order to check whether these 3 isomers might be distinguished by other factors we performed additional calculations for these 3 isomers (see Section 10 in the ESI†) where we checked how much the D4 dispersion correction plays a role (a very small effect of 0.1–0.3 kcal mol−1) and whether inclusion of a zero-point vibrational energy (ZPVE) correction would affect the energy ordering. The ZPVE correction was also found to be relatively small in magnitude, ∼0.7 kcal mol−1 (see Section 10 in the ESI†).
E. The electronic structure of the 3 lowest energy isomers
Comparing the geometric features of the common [Fe2H2C] core of these isomers reveals interesting differences (see Fig. 5) when evaluated using the r2SCAN functional and QM-region C. The Fe2–Fe6 distance is considerably shorter in the OBS6-2BH26 isomers (2.41 Å) than in the OBS6-BH26-TH2 (2.68 Å) and OBS6-DH2 (2.63 Å), a geometric feature likely a result of the 2 bridging hydrides. There are also considerable changes in Fe–C bond lengths in these 3 isomers, suggesting that the carbide might be pushing electron density to different degrees depending on the hydride/H2 coordination. The Fe–H distances of the bridging hydrides are generally shorter for Fe2 than for Fe6 in OBS6-BH26-TH2 and OBS6-2BH26, that suggest that these hydrides are not equally shared between Fe ions. The Fe2–H distances in OBS6-DH2 are considerably longer (1.71 Å) than the Fe–hydride distances, and with a relatively short H–H distance of 0.84 Å, a dihydrogen ligand description seems appropriate. | ||
| Fig. 5 Top: Electronic configurations of OBS6-2BH26, OBS6-BH26-TH2 and OBS6-DH2 derived from localized orbital analysis of the QM/MM calculation using r2SCAN and the QM-C region. The spin states and BS solutions of these three isomers are the same as Fig. 4. The localized orbitals of these three models are shown in Fig. S12–S14 in the ESI.† The green and purple numbers indicate localized orbital populations of Mo, Fe and H-atoms. Bottom: Close-up of the geometries associated with the Fe–H interactions at Fe2 and Fe6. HSP(atom) indicates the Hirshfeld spin populations of Fe2 and Fe6. | ||
The electronic structure of these 3 isomers is also compared in Fig. 5 by electronic configuration diagrams derived from localized orbital analysis of the broken-symmetry determinants. The E3 state has three more e−/H+ pairs than the E0 state, having 22 d-electrons and 19 d-electrons in the Fe4 sub-cubane and MoFe3 sub-cubane, respectively. Analogous electron configuration diagrams for E0 are found in Fig. S11 in the ESI† for comparison. In OBS6-DH2, we find that two additional electrons are present in the form of the H2 ligand while an additional localized electron is present on Fe2 (suggesting a high-spin ferrous ion). OBS6-BH26-TH2 and OBS6-2BH26 models have interestingly very similar electronic structures which offers a possible explanation for the very similar energies for these isomers. This is despite the different hydride coordination and the different Fe2–Fe6 distance. Unlike the dihydrogen isomer, 4 electrons are associated with the H-atoms, with localized orbitals indicating considerable Fe-character. Interestingly, there is an unusual electronic configuration for Fe2: most of the Fe-character in the hydride orbitals is associated with Fe2 rather than Fe6, which together with the asymmetric Fe–H distances previously discussed, suggests that these hydrides are not completely bridging (see localized orbital visualizations in Fig. S14 of the ESI†). The use of 4 electrons to stabilize the Fe–H bonds at first glance suggests that the cofactor has been effectively oxidized but the strong covalency of the Fe–H bonds in these states makes it difficult to make a clear assignment of oxidation states.
The comparison of electronic configurations and geometries for different functionals (B3LYP*, TPSSh, B97-D3, r2SCAN, and TPSS) is shown in Fig. S20 to S25 in the ESI.† The five functionals give similar electronic configurations and geometries for OBS6-DH2. For OBS6-BH26-TH2, B3LYP*, TPSSh, B97-D3 give a similar description as r2SCAN, with an electron-deficient Fe4 subcubane, but TPSS in contrast predicts an electron-deficient MoFe3 sub-cubane. The electronic structure of the OBS6-2BH26 isomer is more complex; B3LYP* predicts similar electronic and geometric structures to r2SCAN, and two bridging hydrides share four electrons with Fe2, correlating with shorter Fe2-hydride distances than Fe6-hydride (see Fig. S21, ESI†), and one electron has moved from the Fe4 sub-cubane to the hydrides (compared to the E0 state, see Fig. S11, ESI†). In contrast, TPSSh predicts four electrons associated with Fe6–H bonds which correlates with shorter Fe6–H distances than Fe2–H distances, and an electron-deficient MoFe3-cubane. TPSS and B97-D3 behave intermediate to the other functionals, predicting four almost equally shared electrons between Fe2 and Fe6, resulting in similar Fe2–H and Fe6–H distances and unusually Fe2 and Fe6 share an electron.
These geometric differences and electronic structure analysis clearly reveal a complex electronic structure which is not easily understood at present, requiring further studies. Also, despite similar energetics predicted by the different functionals (see Fig. 3), the electronic structure from different functionals is not always similar.
F. N2 binding to the E3 state
The original kinetic model by Thorneley and Lowe1,24 suggested N2 binding to the E3 state, although little additional data has since appeared to support or refute that hypothesis. The Fe2–Fe3–Fe6–Fe7 face has long been suggested as a site for N2 binding, primarily based on mutation studies.100,101 Additionally, crystallographic studies102–104 have shown displacement of the bridging sulfide (S2B) between Fe2 and Fe6 under certain conditions, which further implicates Fe2 or Fe6 as a likely site of ligand binding.105,106 A recent X-ray structure study104 appeared to show N2 replacing three possible sulfur sites (S2B, S3A and S5A) but this claim is controversial.107–109 Regardless of the precise interpretations of recent crystal structures, Fe2 and Fe6 are likely binding sites for N2.In previous work from our group34,50 we showed how calculations of N2 binding to Fe sites (specifically Fe2 and Fe6) at FeMoco is almost always connected with the formation of local low-spin states of Fe and furthermore hydrides appear to aid in the formation of such states, offering a possible explanation for why the E4 state has an improved affinity for N2.
As the results in this work suggest OBS6-DH2, OBS6-BH26-TH2 and OBS6-2BH26 as plausible models for the E3 state, and with all 3 models featuring either 2 hydrides or an Fe-bound H2 it is interesting to consider whether N2 binding might be favored to these E3 isomers or not. As OBS6-2BH26 has a more obvious accessible binding site, we focused primarily on this isomer.
We systematically investigated N2 binding to Fe2 of OBS6-2BH26 or Fe6 of OBS2-2BH26 in three spin states (MS = 0, 1, 2) and 35 BS solutions using the QM-A region, and the binding energies are shown in Fig. S15 in the ESI.† The QM-A region was then expanded to the QM-C region, and the binding energies of N2 to Fe2 or Fe6 were found to be +1.5 and +1.9 kcal mol−1 (relative to OBS6-DH2), respectively, shown in Fig. 6, which is similar to the r2SCAN results reported by Ryde and coworkers.51
 | ||
| Fig. 6 N2 binding energies using the QM-C region and r2SCAN. Also shown are relative N–N frequencies for N2 (relative to free N2: 2432 cm−1), Hirshfeld spin populations and quasi-restricted orbital (QRO)-based ligand field diagrams of binding site Fe. The N2@Fe2 and N2@Fe6 have a spin state of MS = 0 and BS states of BS147 and BS234, respectively. The spin states and BS solutions of (a) and (b) are BS347 (MS = 1) and BS2356 (MS = 0), respectively. | ||
We note that Dance previously found the OBS6-2BH26-N2@Fe2 isomer by binding N2 to CBS26-BH26-TH6 (unstable in the present study; see Fig. S4 in the ESI†), and reported that there is a barrier of 9 kcal mol−1 to making the terminal hydride become bridging.38,42
The direct binding of N2viaOBS2-2BH26 → OBS2-2BH26-N2@Fe6 is much stronger (−13.9 kcal mol−1) but the OBS2-2BH26 isomer is higher in energy due to clashes of the terminal sulfhydryl group with the His195 residue. The addition of a translational entropy penalty would make this binding even more unfavourable and hence unlikely to occur.
Dance suggested that an H2 ligand can enhance N2 binding to Fe.43,44 Therefore, we additionally explored N2 binding to OBS6-DH2, and N2 binding to OBS6-BH26-TH2 was also attempted for completeness. N2 binding scenarios where N2 binds directly to Fe2 in the OBS6-BH26-TH2 and OBS6-DH2 isomers were studied in either a cis or trans orientation relative to the Fe2–carbide bond. These calculations were performed using the QM-A region and we investigated only the low spin state (MS = 0) and we restricted the broken-symmetry state comparison to the BS6 class of solutions (BS156, BS157 and BS167) and the BS10 class of solutions (BS125, BS127, BS135, BS136, BS146 and BS147) as these were the lowest-energy BS states for the N2-bound isomers of OBS6-2BH26-N2@Fe2 and OBS2-2BH26-N2@Fe6 (see Fig. S15 and Table S10, ESI†). The results in Fig. 7b reveal that N2 binding to Fe2 in OBS6-BH26-TH2 in a cis orientation relative to the Fe2–C bond results in a stable N2-bound minimum but with an unfavourable binding energy (22.6 kcal mol−1). Starting an optimization with N2 instead trans to the Fe2–C bond (Fig. 7c), however, leads to the spontaneous formation of the OBS6-2BH26-N2@Fe2 isomer (see Fig. 7d and Fig. 6). Comparing the OBS6-DH2 isomer instead we find that N2 binding cis to the Fe2–C bond (Fig. 7f) is even more unfavourable (25.6 kcal mol−1) and binding trans (Fig. 7g) is still fairly unfavourable compared to the OBS6-2BH26 and OBS2-2BH26 structures. These results suggest that it is rather bridging hydrides between Fe2 and Fe6 that enhance N2 binding than a H2 ligand.
 | ||
| Fig. 7 The binding energies (B. E.) of N2 binding to OBS6-BH26-TH2 and OBS6-DH2 (using the QM-A region and r2SCAN), which are relative to the lowest-energy isomer OBS6-DH2 (Fig. 2k). The BS states of (b), (d), (f), and (g) are BS156, BS147, BS156, and BS157, respectively, and all these states are in low spin (MS = 0). Note the energy shown in (d) differs from Fig. 6, due to different QM regions. | ||
Comparing N2 binding energies across the FeMoco redox series: E0, E1, E2, and E4 states (using the models discussed in our recent work50), the binding energy of N2 to the E3 state is found to be marginally more favorable than E2 and not as favorable as binding to the E4 state (see Fig. 8), which is similar to trends reported by Ryde and Jiang.51 The ligand-field diagrams of Fe2 and Fe6 with bound N2 in Fig. 6 (obtained from diamagnetic substitution and QROs) show that both Fe ions are low-spin Fe3+ in an approximate octahedral ligand-field and two dxz and dyz orbitals overlap with π* orbitals of N2 (see Fig. S15, ESI†), which is very similar to that found for the E4 state in our previous study.50 We previously suggested that a low-spin electronic structure on the Fe site is generally encountered in almost all N2-bound FeMoco structures, and that such spin-paired electronic structure is likely assisted by the strong sigma-donor hydrides, especially when more favorable hydride ligation is made possible by an open belt sulfide bridge.50 While the E3 models OBS2-2BH26 and OBS6-2BH26 models have the same di-hydride geometric and electronic structure features, the binding energy is predicted to be less than the E4 state, probably related to an overall less-reduced Fe environment in E3 that decreases backbonding to the N2 ligand.
 | ||
| Fig. 8 The trend of N2 binding energies in going from models of E0 to E4 states according to calculations from previous work50 and this work (using r2SCAN). Also shown are approximate ligand-field diagrams, derived from a combination of localized-orbital analysis and QRO MO energies from a diamagnetically substituted model (see ref. 50). The results for the E3 state were calculated using the QM-C region that is the same as the QM-VI region in ref. 50. The N2 binding energy of the E3 state is relative to the lowest-energy model OBS6-DH2 (Fig. 2k). The spin states and BS solutions of these five models from E0 to E4 states are BS147 (MS = 1/2), BS346 (MS = 2), BS235 (MS = 1/2), BS147 (MS = 0), and BS234 (MS = 1/2), respectively. | ||
Conclusions
In this study we have discussed the energy surface of the E3 redox state of FeMoco and attempted to shed light on the possible forms that this complicated cofactor can take upon addition of 3 electrons and 3 protons (relative to the E0 state). This fills an important gap in the literature in our opinion as so little is known experimentally about this state, being EPR-silent by traditional EPR spectroscopy and hard to isolate. In fact, no spectroscopic data is available on this state and only indirect kinetic data is available.1,24 Furthermore, only a few computational studies31,33,51 have been devoted to the state.Our results suggest that the most likely structure of the E3-state cofactor involves structures with a protonated S2B sulfide bridge in an open form, i.e. a terminal sulfhydryl group on Fe6, which allows a few different Fe–H structures to form (all of them involving 2 hydrides or at least 2 Fe–H interactions). One of these models includes 2 bridging hydrides between Fe2 and Fe6 (Fig. 2g), a structure similar to an E4 model previously discussed by us and others,9,32,34 while another model (Fig. 2i) features 1 bridging, 1 terminal hydride instead (similar to the lowest energy E4 structure in a recent study by us50). The third structure is a bit different, instead of featuring individual Fe–hydrides, it takes a form best described as a side-on Fe–(H2) ligand at the Fe2 site. Energetically, these 3 structures are essentially equivalent when calculated using our QM/MM model with a small QM-region and using the r2SCAN density functional, being ∼1 kcal mol−1 of each other, with the Fe–H2 structure marginally favoured. Upon increasing the QM-region we find a slight shift in relative energies for these 3 isomers, favouring the Fe–H2 structure, OBS6-DH2 (being ∼3 kcal mol−1 lower in energy than the other two).
The electronic structures of these 3 states, analyzed via localized orbital analysis, show considerable differences as shown in Fig. 5. Structure OBS6-DH2 (k) in particular, with an Fe–H2 interaction, features 2 localized electrons associated with the H2, while structures OBS6-2BH26 (g) and OBS6-BH26-TH2 (i) are more similar and feature 4 electrons associated with the bridging/terminal hydride atoms. Such a distinct difference in electronic structure might be expected to give rise to considerable functional dependency.
The functional dependency on isomer energies is predicted to be fairly mild when evaluated using 3 different functionals that we have previously found to describe the molecular and electronic structures of spin-coupled iron–sulfur clusters well: r2SCAN, B97-D3, TPSSh. The results of these three functionals show that OBS6-2BH26 (g), OBS6-BH26-TH2 (i), and OBS6-DH2 (k) are the 3 most favourable models. However, the B3LYP* functional (previously found to behave similarly to the other 3 functionals for geometries)56 interestingly favours a triply protonated carbide.
The functional dependency of FeMoco isomers is actually known to be extremely sensitive in general, especially with respect to Hartree–Fock exchange, as discussed by Ryde and coworkers for energies110 and Harvey and coworkers for electron density changes.111 Functionals such as B3LYP and other hybrids with at least 20% HF exchange are e.g. known to predict interstitial carbide protonation in both E3 and E4 redox states31,110 and it seems that this also applies to the 15% B3LYP* functional.
However, there is no experimental evidence for carbide protonation occurring in any FeMoco state. In fact, according to recent 13C ENDOR studies of multiple trapped FeMoco states with labelled interstitial carbide,112 the carbide hyperfine coupling is practically unchanged. A likely interpretation is that the carbide appears more likely to have a general stabilizing role112 during catalysis. Together with the observation that most hybrid functionals also predict worse FeMoco structures (overestimating Fe–Fe, Fe–S and Fe–C distances) our interpretation is that only non-hybrid functionals and functionals with 0–10% HF exchange describe FeMoco qualitatively correctly.
Uncertainty about the functional dependency nonetheless remains that needs to be evaluated on a case-by-case basis. In recent previous work we compared the energies of E2-hyd and E2-nonhyd isomers that were found to be highly sensitive to the use of r2SCAN vs TPSSh.50 That sensitivity could be explained as the two isomers differing in the nature of the localization of added electrons (Fe-reduction vs. forming H−). Jiang and Ryde have also reported different trends for the E2 state comparing TPSS, r2SCAN and TPSSh.37 The nature of the electronic structure of reduced FeMoco isomers hence continues to present challenges to density functional treatments, both in terms of accuracy and interpretation of the complex electronic structure. The electronic structure interpretation of the E3 isomers discussed in Section E is far from straightforward and as shown in the ESI,† different functionals also give slightly different electronic structures (different BS solutions, different Fe–H covalency and different degrees of localization/delocalization). A systematic assessment of density functionals for the description of Fe–hydride bonds in open-shell Fe compounds is likely to be useful to shed further light on these matters. Overall, our understanding of the electronic structure of hydride-based FeMoco structures remains very much incomplete and continues to surprise us. The flexibility of hydride conformers found in this study is possibly part of a general phenomenon of dynamical flexibility of Fe-bound hydrides as e.g. discussed in recent temperature-dependent studies of an [Fe2H2] dimer studied by Holland and coworkers.113
N2 binding was explored to OBS6-2BH26 isomers and resulted in end-on N2-bound isomers similar to those studied in recent work.34,50 Conversely, N2 binding to OBS6-DH2 and OBS6-BH26-TH2 isomers was found to be unfavourable. Analysis of the electronic structure of the most favourable N2-bound state reveals that a low-spin Fe(III) state is found at the N2-binding Fe ion. However, the binding energy of N2 relative to the most stable E3 isomer (OBS6-DH2), is found to be slightly uphill, suggesting that the E3 redox isomer may not be reduced enough to favourable bind N2. Considering the overall uncertainty from factors such as QM model size, the lack of thermal contributions, as well as the functional dependency, the calculations suggest isomers OBS6-2BH26, OBS6-BH26-TH2 and OBS6-DH2 as the most favourable and we do not attempt to distinguish between them at this stage. Any of these 3 states may represent species formed under experimental conditions, however, E3 has never been isolated, so very little is actually known experimentally. Both E2 and E4 are known to evolve H2 if the electron-flux rate is slower than the rate for the H2 formation, and E3 is likely to be similar in this respect. This means that the E3 state would be meta-stable with respect to H2 formation. The stable OBS6-DH2 isomer found in this study, containing an H2 ligand, could actually represent a state on the path towards evolving H2 and falling back to E1. An isolable E3 state may then instead be better represented by either OBS6-2BH26 or OBS6-BH26-TH2, depending on the barriers present. Future work will have to consider the calculation of reaction pathways and barriers relating all E3 species from the initial formation of the state until the H2 formation.
Author contributions
The manuscript was written through contributions of all authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
R. B. acknowledges support from the Max Planck society. Y. P. acknowledges a scholarship from the China Scholarship Council and a grant from the National Natural Science Foundation of China (no. 22073010). Open Access funding provided by the Max Planck Society.References
- B. K. Burgess and D. J. Lowe, Mechanism of Molybdenum Nitrogenase, Chem. Rev., 1996, 96, 2983–3012 CrossRef CAS PubMed.
- O. Einsle and D. C. Rees, Structural Enzymology of Nitrogenase Enzymes, Chem. Rev., 2020, 120, 4969–5004 CrossRef CAS PubMed.
- A. J. Jasniewski, C. C. Lee, M. W. Ribbe and Y. Hu, Reactivity, Mechanism, and Assembly of the Alternative Nitrogenases, Chem. Rev., 2020, 120, 5107–5157 CrossRef CAS PubMed.
- H. L. Rutledge and F. A. Tezcan, Electron Transfer in Nitrogenase, Chem. Rev., 2020, 120, 5158–5193 CrossRef CAS PubMed.
- L. C. Seefeldt, Z. Y. Yang, D. A. Lukoyanov, D. F. Harris, D. R. Dean, S. Raugei and B. M. Hoffman, Reduction of Substrates by Nitrogenases, Chem. Rev., 2020, 120, 5082–5106 CrossRef CAS PubMed.
- C. Van Stappen, L. Decamps, G. E. Cutsail, R. Bjornsson, J. T. Henthorn, J. A. Birrell and S. DeBeer, The Spectroscopy of Nitrogenases, Chem. Rev., 2020, 120, 5005–5081 CrossRef CAS PubMed.
- Z. Y. Yang, N. Khadka, D. Lukoyanov, B. M. Hoffman, D. R. Dean and L. C. Seefeldt, On reversible H2 loss upon N2 binding to FeMo-cofactor of nitrogenase, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 16327–16332 CrossRef CAS PubMed.
- D. Lukoyanov, N. Khadka, Z. Y. Yang, D. R. Dean, L. C. Seefeldt and B. M. Hoffman, Reductive Elimination of H2 Activates Nitrogenase to Reduce the N identical withN Triple Bond: Characterization of the E4(4H) Janus Intermediate in Wild-Type Enzyme, J. Am. Chem. Soc., 2016, 138, 10674–10683 CrossRef CAS PubMed.
- V. Hoeke, L. Tociu, D. A. Case, L. C. Seefeldt, S. Raugei and B. M. Hoffman, High-Resolution ENDOR Spectroscopy Combined with Quantum Chemical Calculations Reveals the Structure of Nitrogenase Janus Intermediate E4(4H), J. Am. Chem. Soc., 2019, 141, 11984–11996 CrossRef CAS PubMed.
- E. Münck, H. Rhodes, W. H. Orme-Johnson, L. C. Davis, W. J. Brill and V. K. Shah, Nitrogenase. VIII. Mössbauer and EPR spectroscopy. The MoFe protein component from Azotobacter vinelandii OP, Biochim. Biophys. Acta, Protein Struct., 1975, 400, 32–53 CrossRef PubMed.
- J. Rawlings, V. K. Shah, J. R. Chisnell, W. J. Brill, R. Zimmermann, E. Münck and W. H. Orme-Johnson, Novel metal cluster in the iron-molybdenum cofactor of nitrogenase. Spectroscopic evidence, J. Biol. Chem., 1978, 253, 1001–1004 CrossRef CAS PubMed.
- T. V. Morgan, L. E. Mortenson, J. W. McDonald and G. D. Watt, Comparison of redox and EPR properties of the molybdenum iron proteins of Clostridium pasteurianum and Azotobacter vinelandii nitrogenases, J. Inorg. Biochem., 1988, 33, 111–120 CrossRef CAS PubMed.
- K. Fisher, W. E. Newton and D. J. Lowe, Electron Paramagnetic Resonance Analysis of Different Azotobacter vinelandii Nitrogenase MoFe-Protein Conformations Generated during Enzyme Turnover:
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Evidence for S = 3/2 Spin States from Reduced MoFe-Protein Intermediates, Biochemistry, 2001, 40, 3333–3339 CrossRef CAS PubMed.
Evidence for S = 3/2 Spin States from Reduced MoFe-Protein Intermediates, Biochemistry, 2001, 40, 3333–3339 CrossRef CAS PubMed. - O. Einsle, F. A. Tezcan, S. L. A. Andrade, B. Schmid, M. Yoshida, J. B. Howard and D. C. Rees, Nitrogenase MoFe-Protein at 1.16 Å Resolution: A Central Ligand in the FeMo-Cofactor, Science, 2002, 297, 1696–1700 CrossRef CAS PubMed.
- T. Spatzal, M. Aksoyoglu, L. Zhang, S. L. Andrade, E. Schleicher, S. Weber, D. C. Rees and O. Einsle, Evidence for interstitial carbon in nitrogenase FeMo cofactor, Science, 2011, 334, 940 CrossRef CAS PubMed.
- S. J. Yoo, H. C. Angove, V. Papaefthymiou, B. K. Burgess and E. Münck, Mössbauer Study of the MoFe Protein of Nitrogenase from Azotobacter vinelandii Using Selective 57Fe Enrichment of the M-Centers, J. Am. Chem. Soc., 2000, 122, 4926–4936 CrossRef CAS.
- C. Van Stappen, R. Davydov, Z. Y. Yang, R. Fan, Y. Guo, E. Bill, L. C. Seefeldt, B. M. Hoffman and S. DeBeer, Spectroscopic Description of the E1 State of Mo Nitrogenase Based on Mo and Fe X-ray Absorption and Mössbauer Studies, Inorg. Chem., 2019, 58, 12365–12376 CrossRef CAS PubMed.
- C. Van Stappen, A. T. Thorhallsson, L. Decamps, R. Bjornsson and S. DeBeer, Resolving the structure of the E1 state of Mo nitrogenase through Mo and Fe K-edge EXAFS and QM/MM calculations, Chem. Sci., 2019, 10, 9807–9821 RSC.
- D. Lukoyanov, B. M. Barney, D. R. Dean, L. C. Seefeldt and B. M. Hoffman, Connecting nitrogenase intermediates with the kinetic scheme for N2 reduction by a relaxation protocol and identification of the N2 binding state, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 1451–1455 CrossRef CAS PubMed.
- D. Lukoyanov, Z. Y. Yang, S. Duval, K. Danyal, D. R. Dean, L. C. Seefeldt and B. M. Hoffman, A confirmation of the quench-cryoannealing relaxation protocol for identifying reduction states of freeze-trapped nitrogenase intermediates, Inorg. Chem., 2014, 53, 3688–3693 CrossRef CAS PubMed.
- D. A. Lukoyanov, N. Khadka, Z. Y. Yang, D. R. Dean, L. C. Seefeldt and B. M. Hoffman, Hydride Conformers of the Nitrogenase FeMo-cofactor Two-Electron Reduced State E2(2H), Assigned Using Cryogenic Intra Electron Paramagnetic Resonance Cavity Photolysis, Inorg. Chem., 2018, 57, 6847–6852 CrossRef CAS PubMed.
- D. F. Harris, A. Badalyan and L. C. Seefeldt, Mechanistic Insights into Nitrogenase FeMo-Cofactor Catalysis through a Steady-State Kinetic Model, Biochemistry, 2022, 61, 2131–2137 CrossRef CAS PubMed.
- R. Y. Igarashi, M. Laryukhin, P. C. Dos Santos, H.-I. Lee, D. R. Dean, L. C. Seefeldt and B. M. Hoffman, Trapping H- Bound to the Nitrogenase FeMo-Cofactor Active Site during H2 Evolution: Characterization by ENDOR Spectroscopy, J. Am. Chem. Soc., 2005, 127, 6231–6241 CrossRef CAS PubMed.
- R. N. F. Thorneley and D. J. Lowe, in Molybdenum Enzymes, ed. T. G. Spiro, Wiley, New York, 1985, pp. 221–284 Search PubMed.
- D. J. Lowe, K. Fisher and R. N. F. Thorneley, Klebsiella pneumoniae nitrogenase. Mechanism of acetylene reduction and its inhibition by carbon monoxide, Biochem. J., 1990, 272, 621–625 CrossRef CAS PubMed.
- K. Fisher, M. J. Dilworth and W. E. Newton, Differential Effects on N2 Binding and Reduction, HD Formation, and Azide Reduction with α-195His- and α-191Gln-Substituted MoFe Proteins of Azotobacter vinelandii Nitrogenase, Biochemistry, 2000, 39, 15570–15577 CrossRef CAS PubMed.
- K. M. Lancaster, M. Roemelt, P. Ettenhuber, Y. Hu, M. W. Ribbe, F. Neese, U. Bergmann and S. DeBeer, X-ray Emission Spectroscopy Evidences a Central Carbon in the Nitrogenase Iron-Molybdenum Cofactor, Science, 2011, 334, 974–977 CrossRef CAS PubMed.
- R. Bjornsson, F. A. Lima, T. Spatzal, T. Weyhermüller, P. Glatzel, E. Bill, O. Einsle, F. Neese and S. DeBeer, Identification of a spin-coupled Mo(iii) in the nitrogenase iron–molybdenum cofactor, Chem. Sci., 2014, 5, 3096–3103 RSC.
- T. Spatzal, J. Schlesier, E. M. Burger, D. Sippel, L. Zhang, S. L. Andrade, D. C. Rees and O. Einsle, Nitrogenase FeMoco investigated by spatially resolved anomalous dispersion refinement, Nat. Commun., 2016, 7, 10902 CrossRef CAS PubMed.
- R. Bjornsson, F. Neese and S. DeBeer, Revisiting the Mössbauer Isomer Shifts of the FeMoco Cluster of Nitrogenase and the Cofactor Charge, Inorg. Chem., 2017, 56, 1470–1477 CrossRef CAS PubMed.
- L. Cao, O. Caldararu and U. Ryde, Protonation and Reduction of the FeMo Cluster in Nitrogenase Studied by Quantum Mechanics/Molecular Mechanics(QM/MM) Calculations, J. Chem. Theory Comput., 2018, 14, 6653–6678 CrossRef CAS PubMed.
- S. Raugei, L. C. Seefeldt and B. M. Hoffman, Critical computational analysis illuminates the reductive-elimination mechanism that activates nitrogenase for N2 reduction, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, E10521–E10530 CrossRef CAS PubMed.
- I. Dance, Survey of the Geometric and Electronic Structures of the Key Hydrogenated Forms of FeMo-co, the Active Site of the Enzyme Nitrogenase: Principles of the Mechanistically Significant Coordination Chemistry, Inorganics, 2019, 7, 8 CrossRef CAS.
- A. T. Thorhallsson, B. Benediktsson and R. Bjornsson, A model for dinitrogen binding in the E4 state of nitrogenase, Chem. Sci., 2019, 10, 11110–11124 RSC.
- L. Cao and U. Ryde, What Is the Structure of the E4 Intermediate in Nitrogenase?, J. Chem. Theory Comput., 2020, 16, 1936–1952 CrossRef CAS PubMed.
- A. T. Thorhallsson and R. Bjornsson, The E2 state of FeMoco: Hydride Formation versus Fe Reduction and a Mechanism for H2 Evolution, Chem. – Eur. J., 2021, 27, 16788–16800 CrossRef CAS PubMed.
- H. Jiang, O. K. G. Svensson and U. Ryde, QM/MM Study of Partial Dissociation of S2B for the E(2) Intermediate of Nitrogenase, Inorg. Chem., 2022, 61, 18067–18076 CrossRef CAS PubMed.
- I. Dance, How feasible is the reversible S-dissociation mechanism for the activation of FeMo-co, the catalytic site of nitrogenase?, Dalton Trans., 2019, 48, 1251–1262 RSC.
- P. E. M. Siegbahn, The mechanism for nitrogenase including all steps, Phys. Chem. Chem. Phys., 2019, 21, 15747–15759 RSC.
- L. Cao and U. Ryde, N2H2 binding to the nitrogenase FeMo cluster studied by QM/MM methods, J. Biol. Inorg. Chem., 2020, 25, 521–540 CrossRef CAS PubMed.
- L. Cao and U. Ryde, Putative reaction mechanism of nitrogenase after dissociation of a sulfide ligand, J. Catal., 2020, 391, 247–259 CrossRef CAS.
- I. Dance, Computational Investigations of the Chemical Mechanism of the Enzyme Nitrogenase, ChemBioChem, 2020, 21, 1671–1709 CrossRef CAS PubMed.
- I. Dance, Structures and reaction dynamics of N2 and H2 binding at FeMo-co, the active site of nitrogenase, Dalton Trans., 2021, 50, 18212–18237 RSC.
- I. Dance, Calculating the chemical mechanism of nitrogenase: new working hypotheses, Dalton Trans., 2022, 51, 12717–12728 RSC.
- I. Dance, Understanding the tethered unhooking and rehooking of S2B in the reaction domain of FeMo-co, the active site of nitrogenase, Dalton Trans., 2022, 51, 15538–15554 RSC.
- H. Jiang and U. Ryde, Thermodynamically Favourable States in the Reaction of Nitrogenase without Dissociation of any Sulfide Ligand, Chemistry, 2022, e202103933 CAS.
- H. Jiang, O. K. G. Svensson, L. Cao and U. Ryde, Proton Transfer Pathways in Nitrogenase with and without Dissociated S2B, Angew. Chem., Int. Ed., 2022, 61, e202208544 CrossRef CAS PubMed.
- W. J. Wei and P. E. M. Siegbahn, A Mechanism for Nitrogenase Including Loss of a Sulfide, Chemistry, 2022, 28, e202103745 CAS.
- I. Dance, The binding of reducible N(2) in the reaction domain of nitrogenase, Dalton Trans., 2023, 52, 2013–2026 RSC.
- Y. Pang and R. Bjornsson, Understanding the Electronic Structure Basis for N2 Binding to FeMoco: A Systematic Quantum Mechanics/Molecular Mechanics Investigation, Inorg. Chem., 2023, 62, 5357–5375 CrossRef CAS PubMed.
- H. Jiang and U. Ryde, N(2) binding to the E(0)-E(4) states of nitrogenase, Dalton Trans., 2023, 52, 9104–9120 RSC.
- B. Benediktsson and R. Bjornsson, QM/MM Study of the Nitrogenase MoFe Protein Resting State: Broken-Symmetry States, Protonation States, and QM Region Convergence in the FeMoco Active Site, Inorg. Chem., 2017, 56, 13417–13429 CrossRef CAS PubMed.
- B. Benediktsson, A. T. Thorhallsson and R. Bjornsson, QM/MM calculations reveal a bridging hydroxo group in a vanadium nitrogenase crystal structure, Chem. Commun., 2018, 54, 7310–7313 RSC.
- B. Benediktsson and R. Bjornsson, Quantum Mechanics/Molecular Mechanics Study of Resting-State Vanadium Nitrogenase: Molecular and Electronic Structure of the Iron-Vanadium Cofactor, Inorg. Chem., 2020, 59, 11514–11527 CrossRef CAS PubMed.
- N. Spiller, R. Bjornsson, S. DeBeer and F. Neese, Carbon Monoxide Binding to the Iron-Molybdenum Cofactor of Nitrogenase: a Detailed Quantum Mechanics/Molecular Mechanics Investigation, Inorg. Chem., 2021, 60, 18031–18047 CrossRef CAS PubMed.
- B. Benediktsson and R. Bjornsson, Analysis of the Geometric and Electronic Structure of Spin-Coupled Iron-Sulfur Dimers with Broken-Symmetry DFT: Implications for FeMoco, J. Chem. Theory Comput., 2022, 18, 1437–1457 CrossRef CAS PubMed.
- T. V. Harris and R. K. Szilagyi, Comparative Assessment of the Composition and Charge State of Nitrogenase FeMo-Cofactor, Inorg. Chem., 2011, 50, 4811–4824 CrossRef CAS PubMed.
- L. Cao, O. Caldararu and U. Ryde, Protonation States of Homocitrate and Nearby Residues in Nitrogenase Studied by Computational Methods and Quantum Refinement, J. Phys. Chem. B, 2017, 121, 8242–8262 CrossRef CAS PubMed.
- T. Lovell, J. Li, T. Liu, D. A. Case and L. Noodleman, FeMo Cofactor of Nitrogenase: A Density Functional Study of States MN, MOX, MR, and MI, J. Am. Chem. Soc., 2001, 123, 12392–12410 CrossRef CAS PubMed.
- T. Lovell, R. A. Torres, W.-G. Han, T. Liu, D. A. Case and L. Noodleman, Metal Substitution in the Active Site of Nitrogenase MFe7S9 (M = Mo4+, V3+, Fe3+), Inorg. Chem., 2002, 41, 5744–5753 CrossRef CAS PubMed.
- ASH - a multiscale modelling program, Version 0.9; Ragnar Bjornsson, 2022.
- P. Eastman, J. Swails, J. D. Chodera, R. T. McGibbon, Y. Zhao, K. A. Beauchamp, L. P. Wang, A. C. Simmonett, M. P. Harrigan, C. D. Stern, R. P. Wiewiora, B. R. Brooks and V. S. Pande, OpenMM 7: Rapid development of high performance algorithms for molecular dynamics, PLoS Comput. Biol., 2017, 13, e1005659 CrossRef PubMed.
- F. Neese, F. Wennmohs, U. Becker and C. Riplinger, The ORCA quantum chemistry program package, J. Chem. Phys., 2020, 152, 224108 CrossRef CAS PubMed.
- R. B. Best, X. Zhu, J. Shim, P. E. Lopes, J. Mittal, M. Feig and A. D. Mackerell, Optimization of the additive CHARMM all-atom protein force field targeting improved sampling of the backbone phi, psi and side-chain chi(1) and chi(2) dihedral angles, J. Chem. Theory Comput., 2012, 8, 3257–3273 CrossRef CAS PubMed.
- P. Sherwood, A. H. de Vries, M. F. Guest, G. Schreckenbach, C. R. A. Catlow, S. A. French, A. A. Sokol, S. T. Bromley, W. Thiel, A. J. Turner, S. Billeter, F. Terstegen, S. Thiel, J. Kendrick, S. C. Rogers, J. Casci, M. Watson, F. King, E. Karlsen, M. Sjøvoll, A. Fahmi, A. Schäfer and C. Lennartz, QUASI: A general purpose implementation of the QM/MM approach and its application to problems in catalysis, J. Mol. Struct. THEOCHEM, 2003, 632, 1–28 CrossRef CAS.
- L.-P. Wang and C. Song, Geometry optimization made simple with translation and rotation coordinates, J. Chem. Phys., 2016, 144, 214108 CrossRef PubMed.
- S. R. Billeter, A. J. Turner and W. Thiel, Linear scaling geometry optimisation and transition state search in hybrid delocalised internal coordinates, Phys. Chem. Chem. Phys., 2000, 2, 2177–2186 RSC.
- J. W. Furness, A. D. Kaplan, J. Ning, J. P. Perdew and J. Sun, Accurate and Numerically Efficient r(2)SCAN Meta-Generalized Gradient Approximation, J. Phys. Chem. Lett., 2020, 11, 8208–8215 CrossRef CAS PubMed.
- J. Tao, J. P. Perdew, V. N. Staroverov and G. E. Scuseria, Climbing the Density Functional Ladder: Nonempirical Meta--Generalized Gradient Approximation Designed for Molecules and Solids, Phys. Rev. Lett., 2003, 91, 146401 CrossRef PubMed.
- Y. Zhao and D. G. Truhlar, A new local density functional for main-group thermochemistry, transition metal bonding, thermochemical kinetics, and noncovalent interactions, J. Chem. Phys., 2006, 125, 194101 CrossRef PubMed.
- S. Grimme, Semiempirical GGA-type density functional constructed with a long-range dispersion correction, J. Comput. Chem., 2006, 27, 1787–1799 CrossRef CAS PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- S. Grimme, S. Ehrlich and L. Goerigk, Effect of the damping function in dispersion corrected density functional theory, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed.
- M. Reiher, O. Salomon and B. Artur Hess, Reparameterization of hybrid functionals based on energy differences of states of different multiplicity, Theor. Chem. Acc., 2001, 107, 48–55 Search PubMed.
- O. Salomon, M. Reiher and B. A. Hess, Assertion and validation of the performance of the B3LYP functional for the first transition metal row and the G2 test set, J. Chem. Phys., 2002, 117, 4729–4737 CrossRef CAS.
- A. D. Becke, Density-functional exchange-energy approximation with correct asymptotic behavior, Phys. Rev. A: At., Mol., Opt. Phys., 1988, 38, 3098–3100 CrossRef CAS PubMed.
- C. Lee, W. Yang and R. G. Parr, Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS PubMed.
- A. D. Becke, Density-functional thermochemistry. III. The role of exact exchange, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- C. van Wüllen, Molecular density functional calculations in the regular relativistic approximation: Method, application to coinage metal diatomics, hydrides, fluorides and chlorides, and comparison with first-order relativistic calculations, J. Chem. Phys., 1998, 109, 392–399 CrossRef.
- E. Caldeweyher, C. Bannwarth and S. Grimme, Extension of the D3 dispersion coefficient model, J. Chem. Phys., 2017, 147, 034112 CrossRef PubMed.
- S. Ehlert, U. Huniar, J. Ning, J. W. Furness, J. Sun, A. D. Kaplan, J. P. Perdew and J. G. Brandenburg, r(2)SCAN-D4: Dispersion corrected meta-generalized gradient approximation for general chemical applications, J. Chem. Phys., 2021, 154, 061101 CrossRef CAS PubMed.
- F. Neese, An improvement of the resolution of the identity approximation for the formation of the Coulomb matrix, J. Comput. Chem., 2003, 24, 1740–1747 CrossRef CAS PubMed.
- F. Weigend and R. Ahlrichs, Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- D. A. Pantazis and F. Neese, All-electron scalar relativistic basis sets for the 6p elements, Theor. Chem. Acc., 2012, 131 Search PubMed.
- D. A. Pantazis, X.-Y. Chen, C. R. Landis and F. Neese, All-Electron Scalar Relativistic Basis Sets for Third-Row Transition Metal Atoms, J. Chem. Theory Comput., 2008, 4, 908–919 CrossRef CAS PubMed.
- J. D. Rolfes, F. Neese and D. A. Pantazis, All-electron scalar relativistic basis sets for the elements Rb-Xe, J. Comput. Chem., 2020, 41, 1842–1849 CrossRef CAS PubMed.
- F. L. Hirshfeld, Bonded-atom fragments for describing molecular charge densities, Theor. Chem. Acc., 1977, 44, 129–138 Search PubMed.
- R. S. Mulliken, Electronic Population Analysis on LCAO–MO Molecular Wave Functions. I, J. Chem. Phys., 1955, 23, 1833–1840 CrossRef CAS.
- H. Li and J. H. Jensen, Partial Hessian vibrational analysis: the localization of the molecular vibrational energy and entropy, Theor. Chem. Acc., 2002, 107, 211–219 Search PubMed.
- J. Pipek and P. G. Mezey, A fast intrinsic localization procedure applicable for ab initio and semiempirical linear combination of atomic orbital wave functions, J. Chem. Phys., 1989, 90, 4916–4926 CrossRef CAS.
- L. C. Seefeldt, B. M. Hoffman and D. R. Dean, Electron transfer in nitrogenase catalysis, Curr. Opin. Chem. Biol., 2012, 16, 19–25 CrossRef CAS PubMed.
- B. M. Hoffman, D. Lukoyanov, D. R. Dean and L. C. Seefeldt, Nitrogenase: A Draft Mechanism, Acc. Chem. Res., 2013, 46, 587–595 CrossRef CAS PubMed.
- I. Dance, Misconception of reductive elimination of H2, in the context of the mechanism of nitrogenase, Dalton Trans., 2015, 44, 9027–9037 RSC.
- P. E. Siegbahn, Model Calculations Suggest that the Central Carbon in the FeMo-Cofactor of Nitrogenase Becomes Protonated in the Process of Nitrogen Fixation, J. Am. Chem. Soc., 2016, 138, 10485–10495 CrossRef CAS PubMed.
- I. Dance, New insights into the reaction capabilities of His(195) adjacent to the active site of nitrogenase, J. Inorg. Biochem., 2017, 169, 32–43 CrossRef CAS PubMed.
- L. Cao and U. Ryde, Influence of the protein and DFT method on the broken-symmetry and spin states in nitrogenase, Int. J. Quantum Chem., 2018, 118, e25627 CrossRef.
- I. Dance, What is the role of the isolated small water pool near FeMo-co, the active site of nitrogenase?, FEBS J., 2018, 285, 2972–2986 CrossRef CAS PubMed.
- P. E. M. Siegbahn, Is there computational support for an unprotonated carbon in the E(4) state of nitrogenase?, J. Comput. Chem., 2018, 39, 743–747 CrossRef CAS PubMed.
- W. J. Wei and P. E. M. Siegbahn, The active E4 structure of nitrogenase studied with different DFT functionals, J. Comput. Chem., 2021, 42, 81–85 CrossRef CAS PubMed.
- B. M. Barney, R. Y. Igarashi, P. C. Dos Santos, D. R. Dean and L. C. Seefeldt, Substrate interaction at an iron-sulfur face of the FeMo-cofactor during nitrogenase catalysis, J. Biol. Chem., 2004, 279, 53621–53624 CrossRef CAS PubMed.
- R. Sarma, B. M. Barney, S. Keable, D. R. Dean, L. C. Seefeldt and J. W. Peters, Insights into substrate binding at FeMo-cofactor in nitrogenase from the structure of an alpha-70(Ile) MoFe protein variant, J. Inorg. Biochem., 2010, 104, 385–389 CrossRef CAS PubMed.
- T. Spatzal, K. A. Perez, O. Einsle, J. B. Howard and D. C. Rees, Ligand binding to the FeMo-cofactor: Structures of CO-bound and reactivated nitrogenase, Science, 2014, 345, 1620 CrossRef CAS PubMed.
- D. Sippel, M. Rohde, J. Netzer, C. Trncik, J. Gies, K. Grunau, I. Djurdjevic, L. Decamps, S. L. A. Andrade and O. Einsle, A bound reaction intermediate sheds light on the mechanism of nitrogenase, Science, 2018, 359, 1484 CrossRef CAS PubMed.
- W. Kang, C. Lee Chi, J. Jasniewski Andrew, W. Ribbe Markus and Y. Hu, Structural evidence for a dynamic metallocofactor during N2 reduction by Mo-nitrogenase, Science, 2020, 368, 1381–1385 CrossRef CAS PubMed.
- K. L. Skubi and P. L. Holland, So Close, yet Sulfur Away: Opening the Nitrogenase Cofactor Structure Creates a Binding Site, Biochemistry, 2018, 57, 3540–3541 CrossRef CAS PubMed.
- T. M. Buscagan and D. C. Rees, Rethinking the Nitrogenase Mechanism: Activating the Active Site, Joule, 2019, 3, 2662–2678 CrossRef CAS PubMed.
- J. W. Peters, O. Einsle, D. R. Dean, S. DeBeer, B. M. Hoffman, P. L. Holland and L. C. Seefeldt, Comment on Structural evidence for a dynamic metallocofactor during N2 reduction by Mo-nitrogenase, Science, 2021, 371, eabe5481 CrossRef CAS PubMed.
- W. Kang, C. C. Lee, A. J. Jasniewski, M. W. Ribbe and Y. Hu, Response to Comment on Structural evidence for a dynamic metallocofactor during N2 reduction by Mo-nitrogenase, Science, 2021, 371, eabe5856 CrossRef CAS PubMed.
- J. Bergmann, E. Oksanen and U. Ryde, Critical evaluation of a crystal structure of nitrogenase with bound N2 ligands, J. Biol. Inorg. Chem., 2021, 26, 341–353 CrossRef CAS PubMed.
- L. Cao and U. Ryde, Extremely large differences in DFT energies for nitrogenase models, Phys. Chem. Chem. Phys., 2019, 21, 2480–2488 RSC.
- C. Martin-Fernandez and J. N. Harvey, On the Use of Normalized Metrics for Density Sensitivity Analysis in DFT, J. Phys. Chem. A, 2021, 125, 4639–4652 CrossRef CAS PubMed.
- D. A. Lukoyanov, Z. Y. Yang, A. Perez-Gonzalez, S. Raugei, D. R. Dean, L. C. Seefeldt and B. M. Hoffman, 13)C ENDOR Characterization of the Central Carbon within the Nitrogenase Catalytic Cofactor Indicates That the CFe6 Core Is a Stabilizing “Heart of Steel”, J. Am. Chem. Soc., 2022, 144, 18315–18328 CrossRef CAS PubMed.
- S. F. McWilliams, B. Q. Mercado, K. C. MacLeod, M. S. Fataftah, M. Tarrago, X. Wang, E. Bill, S. Ye and P. L. Holland, Dynamic effects on ligand field from rapid hydride motion in an iron(ii) dimer with an S = 3 ground state, Chem. Sci., 2023, 14, 2303–2312 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Additional information on QM-regions, electronic structure analysis, orbitals, geometries, broken-symmetry states, relative energies. Coordinates of QM-regions included as a ZIP-archive of XYZ-files. See DOI: https://doi.org/10.1039/d3cp01106b |
| This journal is © the Owner Societies 2023 |