
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe molecular mechanism of light-induced bond formation and breakage in the cyanobacteriochrome TePixJ†
Jeannette
Ruf
 a,
Flavia
Bindschedler
a and
David
Buhrke
*ab
a,
Flavia
Bindschedler
a and
David
Buhrke
*ab
aDepartment of Chemistry, University of Zurich, Zurich, Switzerland. E-mail: david.buhrke@hu-berlin.de
bInstitute of Biology, Humboldt University Berlin, Germany
First published on 8th February 2023
Abstract
Cyanobacteriochromes (CBCRs) are small and versatile photoreceptor proteins with high potential for biotechnological applications. Among them, the so-called DXCF-CBCRs exhibit an intricate secondary photochemistry: miliseconds after activation with light, a covalent linkage between a conserved cysteine residue and the light-absorbing tetrapyrrole chromophore is reversibly formed or broken. We employed time-resolved IR spectroscopy over ten orders of magnitude in time in conjunction with 2D-IR spectroscopy to investigate the molecular mechanism of this intriguing reaction in the DXCF-CBCR model system TePixJ from T. elongatus. The crosspeak pattern in the 2D-IR spectrum facilitated the assignment of the dominant signals to vibrational modes of the chromophore, which in turn enabled us to construct a mechanistic model for the photocycle reactions from the time-resolved IR spectra. Here, we assigned the time-resolved signals to several proton transfer steps and distinct geometric changes of the chromophore. We propose a model that describes how these events lead to the rearrangement of charges in the chromophore binding pocket, which serves as the trigger for the light-induced bond formation and breakage with the nearby cysteine.
I Introduction
Throughout evolution, many organisms evolved the ability to adapt to specific environmental light conditions by utilizing light-sensitive proteins, i.e. photoreceptors.1–3 One family of photoreceptors, cyanobacteriochromes (CBCRs), regulate a variety of light responses in cyanobacteria.4–6 CBCRs have a modular domain structure and often consist of multiple photosensory GAF (cGMP-phosphodiesterase/adenylate cyclase/FhlA) domains that are linked together in a chain and connected to a catalytic output module such as a kinase or cyclase domain. The output modules can be swapped in a modular fashion, thus CBCRs are emerging tools for the optogenetic control of slow processes such as gene expression in microorganisms.7–10 The photosensory GAF domains incorporate tetrapyrrole co-factors such as biliverdin, phycocyanobilin (PCB), or phycoviolobilin (PVB) with a broad variety of absorption properties ranging from the UV to the NIR.6 Among the different CBCR subfamilies, DXCF-CBCRs (named after a conserved Asp-Xaa-Cys-Phe motif) harbour two conserved cysteine residues that play a crucial role in their photochemical reaction cascade.11,12 DXCF-CBCRs incorporate the red-absorbing pigment PCB, which is covalently attached to one conserved Cys residue at the A-ring vinyl group. To reach the photoactive form, the protein autocatalytically reduces the PCB chromophore in a second step to the blue shifted pigment PVB.11,13,14 In a third step, the second conserved cysteine (from the DXCF motif) covalently attaches to the PVB, breaking the conjugated system a second time and resulting in a second blue-shift of the absorption.Here, we studied the photosensory GAF domain from the DXCF-CBCR TePixJ (abbreviated as TePixJ for simplictiy in the following), a protein that is responsible for positive phototaxis in the thermophilic cyanobacterium Thermosynechococcus elongatus.15 The crystal structure of this protein in the dark adapted blue-absorbing “Pb” parent state, including the attachment to the two cysteine residues 494 and 522 (numbering related ot the full-length protein) is shown in Fig. 1A.12 Upon photoexication, TePixJ is known to undergo a complex photoreaction that involves isomeristaion of the pyrrole ring D and the breaking of the thioether bond to C494 to reach the green absorbing “Pg” parent state (Fig. 1B and C).12,16,17 The characteristic absorption spectra of the two parent states are shown in panel D.
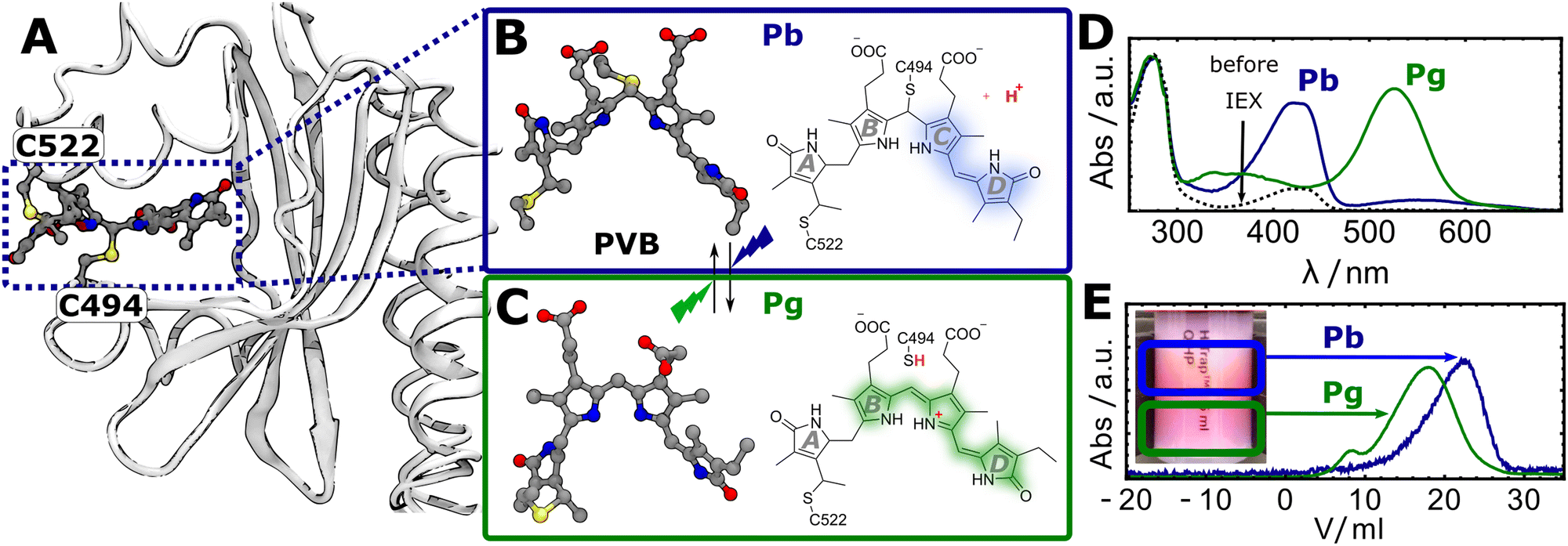 | ||
| Fig. 1 (A) Crystal structure of TePixJ in the Pb state (PDB: 6PRU), highlighting the attachment of the PVB chromophore to the two cysteine residues C494 and C522.12 (B and C) Lewis structures of PVB in the Pb and Pg states, respectively.12,16 (D) UV-Vis absorption spectra of TePixJ before and after purification with the IEX method. (E) IEX chromatogram showing the elution profiles of the Pb and Pg states. Inset: Photograph of the IEX column highlighting the two fractions. | ||
Over the last years, TePixJ was expressed and purified heterologously in E.Coli by different laboratories around the world, and some aspects of this proteins intriguing photochemistry are already understood to a certain extent. In these works, the efficiency of PCB incorporation and conversion to PVB differed dramatically between different labs. Results by Burgie et al. suggest that high incorporation and conversion efficiencies can be reached,12,18 but other labs obtained samples with lower incorporation albeit the used expression and purification protocols were quite similar.14,19,20 Another issue that occured in some studies are impurities caused by unconverted PCB that are typically detected as a sharp band with maximum at 570 nm. Further, a detailed description of the molecular mechanism and the driving force for the reversible light-induced thiol bridge formation and breaking still remains elusive. To investigate these processes, vibrational spectroscopies are the method of choice, because they combine a high temporal and spatial resolution.21–24 Although two time-resolved spectroscopic studies recently established the time-scales of different events in the photocycle of TePixJ, such as the formation and breaking of the covalent Cys-attachment,19,20 it was up to now not possible to assign and track the evolution of different vibrational marker modes to deduce a mechanistic model with atomic resolution.
II Materials and methods
Sample preparation
Competent E. coli BL21 cells were transformed simultaneously with a pET-15b vector (ampicilin resistance) containing the sequence for the TePixJ GAF domain (amino acids 437 to 588 of the full-length protein)12 with C-terminal 6xHis-tag, and a pCOLADuet-1 vector (kanamycin resistance) containing the sequence for a heme oxygenase and phycocyanobilin:ferredoxin oxidoreductase from synechocystis (GenScript Biotech, Piscataway Township NJ, USA). Cells were grown in TB (terrific broth) medium that contained ampicilin and kanamycin to an OD of 0.5 at 37 and expression of both vectors was induced at the same point in time by adding 1 mM IPTG. The temperature was lowered to 20 and protein expressed for 2 h. Cells were lysed by sonication, and a mixture of TePixJ apo- and holoprotein was obtained after standard Ni-affinity purification. To separate the apo- from the holoprotein and a residual fraction of PCB-bound protein, anion-exchange chromatography (IEX) with a NaCl gradient was performed under green light to convert all TePixJ to the Pb form which eluted at significantly higher salt concentrations then the apoprotein. All three fractions eluted between 10–15% IEX B buffer that contained 2 M NaCl. Finally samples were desalted into HEPES buffer (pH = 7.8) using a gel-filtration column and the solvent was exchanged to D2O by two cycles of lyophilization in the dark.Spectroscopic measurements
To cover the total timeframe from ten ps to hundreds of ms, two experimental setups were used. For the time span ranging from 10 ps to μs, two electronically synchronized laser systems running at 2.5 kHz and centered at 840 nm were applied. The mid-IR probe pulses (≈120 μm) were generated in a homebuilt optical parametric amplifier.25 To induce the photoisomerization of the chromophore in the Pb state, second harmonic generation in a BBO crystal was used. The resulting pump pulses were focused on the sample on ≈140 μm with an energy of 600 nJ and a pulse length of ≈120 fs. To initiate the Pg → Pb conversion, white light generation in a flowing water cell was applied, followed by a 550 ± 8 nm bandpass filter. This generated green pump pulses with a pulse energy of 125 nJ and were focused to 300 μm at the sample position. We estimate excitation densities of 19% and 1% for the Pb and Pg states, respectively.To cover the timerange from μs to ms, a vis-pump-IR-multiprobe with 100 kHz system as described earlier,26 combined with a stop-flow sample delivery system27 was used. To initiate the Pg → Pb conversion a green ns laser (532 nm, AO-S532, CNI, Changchun, China) and for the Pb → Pg direction a blue laser (447 nm, QB-447-25, CrystaLaser, Reno, NV, USA) were used. Both pump pulses were focused on the sample on ≈110 μm with an energy of 10 m J (green) and 12 m J (blue). Here, a 2 × 32 MCT array detector with a spectral resolution of ca. 4 cm−1 per pixel was used. For both techniques, the pump polarization was set to magic angle (54.7°) compared to the probe beam and multichannel referencing28 was used for noise suppression.
For the stationary 2D-IR experiments, the same 100 kHz system was used. The two IR pump pulses were generated with a pulse shaper (PhaseTech). We applied phase cycling in four states to suppress scattering and the number of laser shots needed to measure a full 2D-IR spectrum was minimized by undersampling in a rotating frame.26 The waiting time (t2) was set to 200 fs. In all spectroscopic experiments, the sample path length was 50 μm.
III Results
Anion exchange chromatography
In our first attempt to express TePixJ heterologously in E. Coli, the sample showed a strong absorption at 280 nm relative to the characteristic Pb-PVB band at 420 nm, suggesting that the sample contained a large excess of apo-protein compared to results from other groups (Fig. 1D, dashed black line).12,18 We tried several chromatographic methods to separate the desired holo- from the apoprotein and were successful with anion-exchange chromatography (IEX) with a NaCl gradient. Here, the apoprotein eluted at lower NaCl concentration then the holo-protein, indicating that the fractions bear increasingly negative charge. After this purification step, most of the apo-protein was successfully removed (Fig. 1D). Surprisingly, we could also separate the Pb and Pg states of an already purified sample by IEX when starting under conditions where both states were initially populated to approximately 50%. The two states separated visibly on the column into a red (Pg) and a yellow (Pb) fraction, and the elution was tracked by recording the absorption at 420 and 520 nm as a function of elution volume (Fig. 1E). This indicates that the Pb state is more negatively charged than Pg, which can be explained by the release of the thiol proton in Pg (highlighted in red in Fig. 1B and C).Time-resolved IR spectroscopy
Once pure samples were obtained, the photoreaction of TePixJ was monitored by time-resolved infrared (TRIR) spectroscopy over more than ten orders of magnitude in time by combining two conceptually different setups for two different time windows (see materials and methods for details). The merged data was analyzed with two complementary methods: lifetime analysis was used in conjunction with a global fit to a sequential reaction scheme (Fig. 2).29,30 The frequency-integrated amplitude from the lifetime analysis (dynamical content D31,32) is shown in panel B. Briefly, this plot allows to estimate the time points where spectral changes take place, which are reflected by the local maxima in the plot. A more detailed description of lifetime analysis and dynamical content can be found in ESI† S1. These time points were used as initial values for the time constants in the global fit to a unidirectional sequential model, which resulted in six so-called evolution-associated difference spectra (EADS, panel D), whose time-dependent concentration profiles c(EADS) are shown in panel C. If the presumption of a strictly unidirectional and sequential reaction scheme of discrete states is correct, the EADS represent the real spectra and concentration profiles of six species. If the presumption is not correct, the EADS still represent the most relevant spectral changes in the different temporal stages of the photoreaction in a compact way.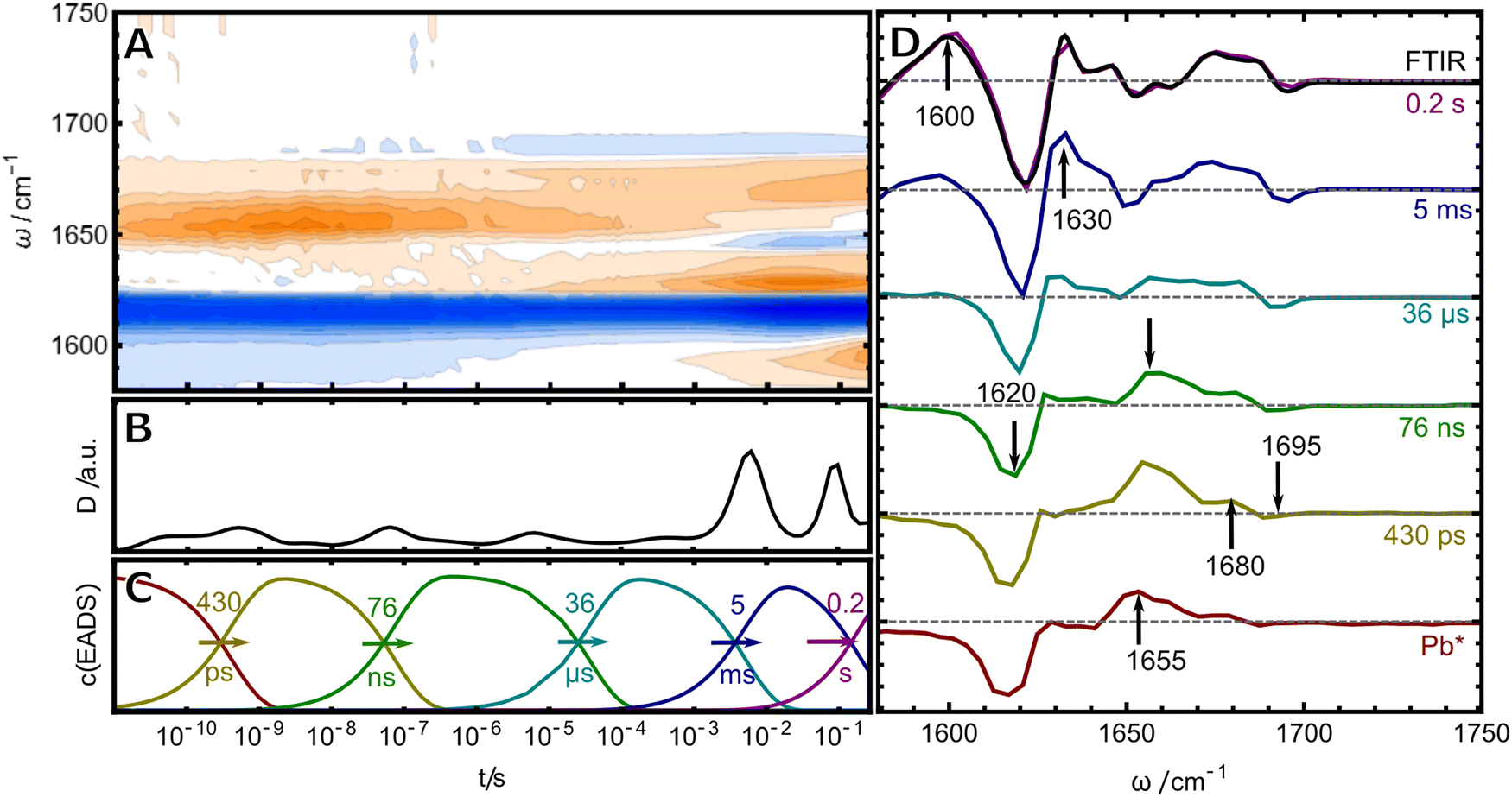 | ||
| Fig. 2 Photochemistry after excitation of Pb-TePixJ monitored by TRIR spectroscopy. (A) Data represented as a contour plot. (B) Dynamical content. (C) Concentration profiles of the (D) EADS. The last (violet) EADS corresponding to the 0.2 s process was overlaid with the steady-state FTIR difference spectrum (black line). | ||
Excitation of the dipyrrole chromophore
The first (red) EADS corresponds to the electronic excited state Pb* and is dominated by a bleach signal at 1610–1620 cm−1, accompanied by positive excited state absorption at 1655 cm−1.19 In Pb, only the dipyrrolic unit of rings C and D can be excited by blue light, and thus, the bleach signal must originate from this relatively small part of the PVB chromophore (highlighted in blue in Fig. 1B). For band assignment, hexamethylpyrromethene is a useful model compound because its structure is very similar to the dipyrrolic unit. This molecule exhibits only two vibrational modes in the investigated frequency window, that correspond to the symmetric and asymmetric combination of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C stretches inside ring D and of the C/D methine bridge.33 In resonance Raman (RR) experiments of TePixJ in the Pb state, a strong CC stretching signal is also observed at 1620 cm−1, validating the assignment of this signal to a CC stretch.18 Another very small bleach signal is found at 1690 cm−1, which we assign to the CO stretch of the ring D carbonyl. We identify only one time constant of 430 ps for the decay of Pb*, while the authors of ref. 19 measured this process with higher time resolution and retrieved a bi-phasic decay with two time constants of 11.9 and 868 ps. The decay of Pb* is accompanied by the rise of two positive features at 1655 and 1680 cm−1. We assign these signals to the same vibrational modes as the bleaches, that are shifted in frequency due to the Z/E isomerisation of the C/D methine bridge.
C stretches inside ring D and of the C/D methine bridge.33 In resonance Raman (RR) experiments of TePixJ in the Pb state, a strong CC stretching signal is also observed at 1620 cm−1, validating the assignment of this signal to a CC stretch.18 Another very small bleach signal is found at 1690 cm−1, which we assign to the CO stretch of the ring D carbonyl. We identify only one time constant of 430 ps for the decay of Pb*, while the authors of ref. 19 measured this process with higher time resolution and retrieved a bi-phasic decay with two time constants of 11.9 and 868 ps. The decay of Pb* is accompanied by the rise of two positive features at 1655 and 1680 cm−1. We assign these signals to the same vibrational modes as the bleaches, that are shifted in frequency due to the Z/E isomerisation of the C/D methine bridge.
Difference 2D-IR spectroscopy
A helpful technique to unwrap the complex pattern of positive and negative peaks in the difference spectra is difference 2D-IR spectroscopy. We recently showed that this advanced nonlinear method is sensitive to changes in coupling between adjacent CC and CO stretching modes in the tetrapyrrole co-factor of bacteriophytochrome Agp1.34 The characteristic crosspeak pattern reports on the changes in connectivity of certain functional groups. The “Pg-minus-Pb” difference 2D-IR spectrum is shown in Fig. 3 together with the linear “Pg-minus-Pb” FTIR-difference spectrum (the absolute 2D-IR spectra of Pb and Pg and details on the FTIR difference spectrum are given in Fig. S1 and S2, ESI†).
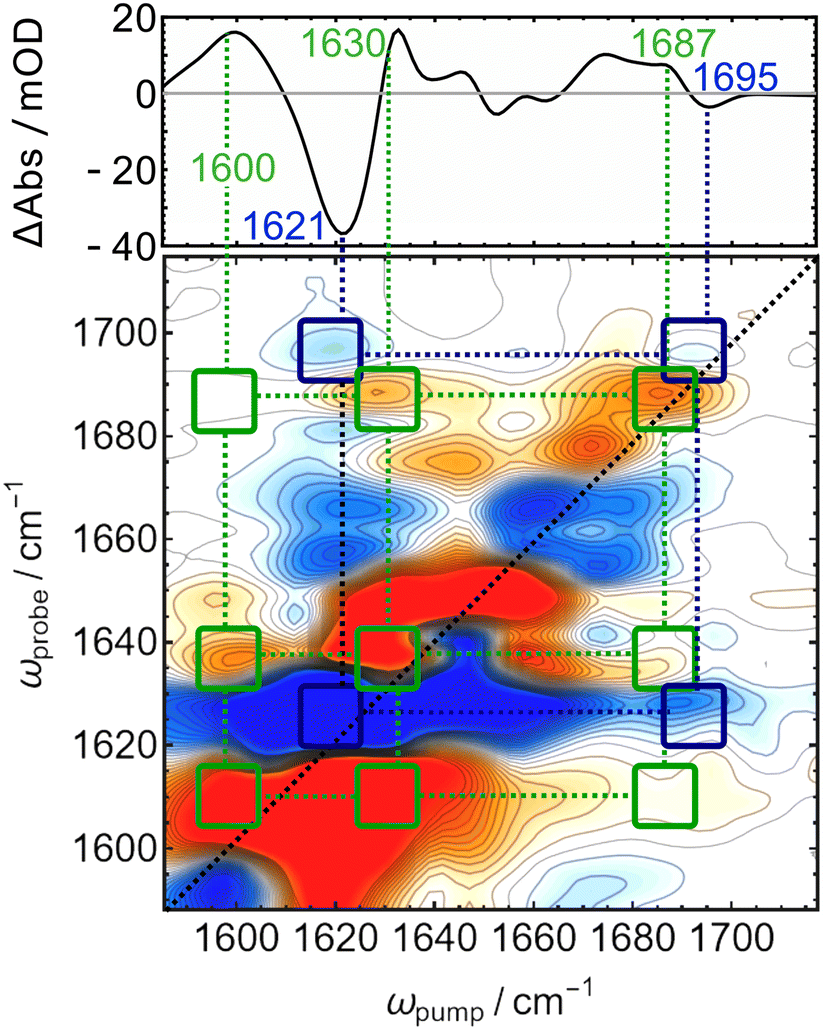 | ||
| Fig. 3 “Pg-minus-Pb” difference 2D-IR spectrum of TePixJ (lower panel) compared to the linear FTIR difference spectrum (top panel). | ||
All bands in the FTIR-difference spectrum are accompanied by counterparts on the main diagonal of the 2D-difference spectrum and correspond to the individual bleaches associated with Pb (blue, negative) and new absorption in Pg (red, positive). The assignement of the CC and CO bleach bands that originate from the relatively small dipyrrolic unit are expected to couple strongly to each other, which is confirmed by a pattern of negative (off-diagonal) crosspeaks connecting them (highlighted with dark blue squares and dotted lines). On the other hand, a pattern of positive crosspeaks is found that connects the diagonal bands at 1600, 1630, and 1687 cm−1 to each other, indicating that these are the frequencies of two different CC stretches and COD, which are coupling to each other in Pg. The overall downshift of the COD mode by ca. 10 cm−1 is consistent with crystal structures, where the carbonyl is coordinated by an asparagine and a histidine residue in Pb,12 but moves to a solvent exposed position in Pg,16 where a stronger coordination by water molecules and thus a lower frequency can be expected.35,36 The intensity of the positive crosspeaks indicates that the feature at 1630 couples more strongly to COD then the intensive diagonal band at 1600 cm−1, which indicates that the higher frequency mode corresponds to the C/D bridge, while latter likely originates from the B/C bridge that is only conjugated in Pg.
Secondary photochemistry and Cys-bond breaking
With this assignment in mind, we turn to investigate the secondary photochemistry of TePixJ after Pb excitation. The last EADS inf Fig. 2 is identical to the steady state “Pg-minus-Pb” FTIR-difference spectrum and indicates that all secondary photoreactions are completed within 0.2 s as the final Pg state is formed (black line overlapped with violet EADS). The EADS in between the first ground state photoproduct (second EADS) and the final reaction report on ground-state intermediates of the reaction cascade. The first two of those are formed with time constants of 76 ns and 36 μs and are dominated by the decay of the absorption at 1655 cm−1 and the concomitant rise of a positive band at 1630 cm−1, as well as an intensification of the difference signal in the COD region. On this time scale, the protein environment can adapt to the local structural changes caused by the photoisomerisation, therefore amide I contributions might play a role. Nevertheless, the overall spectral signature is dominated by signals at positions that we assigned beforehand to be characteristic of CC modes in the dipyrrolic unit. The predominant downshift of the large positive signal probably originates from a twisting motion around the C/D bridge which releases the strain of the primary photoproduct and is accompanied by a blue shift in the visible absorption.19 The entire process is not reflected by one sharp transition, but rather stretches from the ns to μs, which is consistent with the interpretation that these rearrangements are determinated by slow, complex and therefore non-exponential adaptations of the surrounding protein environment. The largest spectral changes occur in the late stages of the photoreaction, where two components with 5 ms and 0.2 s are detected. The 5 ms EADS is accompanied by intensification of signals that are associated with CC stretches in the region around 1610–1620 cm−1, which is indicative of rearrangements of the conjugated system. The last EADS is dominated by the rise of the band at 1600 cm−1 and happens on a time scale consistent with the large shift in the visible spectrum which was interpreted as the breaking of the Cys-bond (ref. 19 reported a time constant of 86.7 ms for this process in H2O).
The Cys-attachment reaction
As a next step, we studied the photochemistry in the reverse direction to access the mechanism of Cys-bond formation. Here, five EADS were identified in the relevant time window between 10 ps and 0.1 s (Fig. 4). The first (red) EADS corresponds to Pg* and shows two broad bleach bands at 1600 and 1680 cm−1 and a broad excited state absorption at 1655 cm−1. The bleach at 1680 cm−1 can be readily assigned to the CO stretch of ring D, while the signal at 1600 cm−1 was assigned beforehand to the B/C bridge CC stretching mode of the excited unit of the PVB chromophore. The second EADS shows the decay of these rather broad signals into three much weaker bleach bands and positive counterparts. The strong decrease of the difference signal in the second EADS might be interpreted in terms of a low photochemical quantum yield for the isomerisation. However, since the last EADS displays strong difference signals, we think this interpretation is misleading and that the weak signals rather originate from a strong overlap of positive and negative bands. The first two bleach bands in the 50 ps EADS are observed at 1600 and 1630 cm−1 with the second one having the larger intensity. This finding crossvalidates the assignment from the 2D-IR experiment, that there are indeed two distinct IR-active CC stretches in the Pg chromophore: one at 1600 cm−1 which can be readily seen in the first EADS and a second one at 1630 cm−1 that is obscured by the strong excited state absorption and thus only becomes evident in the later ground state intermediates. The next (green) EADS is formed within 2.8 μs and is characterized by the almost complete lack of positive bands and an additional bleach at 1730 cm−1. The positive bands re-appear in another EADS with 2 ms at the same time the 1730 cm−1 bleach disappears. The last EADS is identical with the “Pb-minus-Pg” FTIR difference spectrum (black line overlaid with purple EADS), indicating that the last step of the photoreaction takes place on a time scale of 12 ms, consistent with transient vis and CD results (ref. 19 and 20 reported time constants of 4.6 ms and for this process in H2O, respectively).
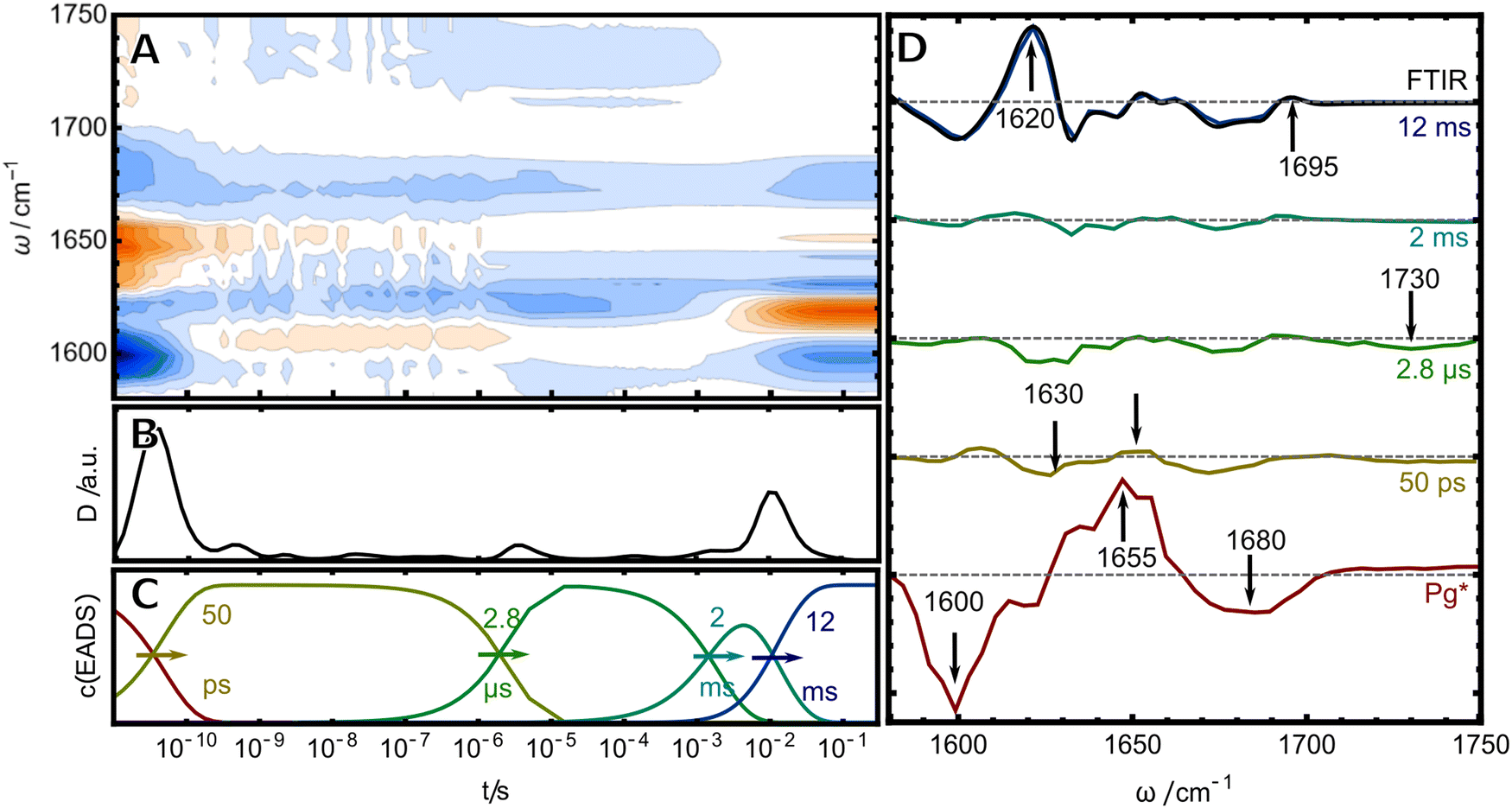 | ||
| Fig. 4 Photochemistry after excitation of Pg-TePixJ monitored by TRIR spectroscopy. (A) Data represented as a contour plot. (B) Dynamical content. (C) Concentration profiles of the (D) EADS. The last (blue) EADS corresponding to the 12 ms process was overlaid with the steady-state FTIR difference spectrum (black line). | ||
IV Discussion
Two distinct proton transfer steps
Over the last decade, several X-ray and NMR structures of TePixJ have been deposited in the protein data bank.12,16,17,37 The most recent Lewis structures of PVB in both parent states derived from these results (Fig. 1B and C)12 are suitable to explain the stationary blue/green photochemistry of TePixJ. However, it is evident that these structures can be only correct if the thiol proton leaves the chromophore in Pb. Up to now, it is not clear whether this proton is accepted by a residue inside the protein or released to the bulk solvent. Our IEX result shows that TePixJ in the Pb state is more negatively charged than in Pg. Therefore, we propose that the thiol proton is released to the bulk, which is not surprising since the crystal structures show that C494 is in a solvent accessible position (Fig. 1A).On top of this overall deprotonation between the two parent states, kinetic isotope effects in transient visible experiments19 indicated additional transient de- and reprotonation steps taking place on the microsecond time scale after triggering of the Pg photoreaction. Time- and pH-resolved studies on a related blue/orange CBCR also came to the conclusion that formation of the C–S bond is accompanied by protonation changes of the conjugated system, and a model with a transiently deprotonated nitrogen at ring B was proposed.38 In our corresponding TRIR experiment, we observe a spectral signature that is dominated by bleach bands with very weak counterparts persisting up to 2 ms, which are consistent with a deprotonated chromophore and a neutral conjugated system with smaller dipole. Furthermore we observe an additional negative transient band at 1730 cm−1 which entirely disappears with 2 ms, concomitant with the appearance of positive features at 1620 and 1690 cm−1. This indicates that the fully-protonated state is re-established within 2 ms, and the negative signal at 1730 cm−1 is directly related to this process. In the spectral region around 1730 cm−1, there are only few possible functional groups that can absorb and cause such a signal in TePixJ, namely protonated carboxylic acids or the ring A carbonyl. An obvious mechanism would be that the chromophore deprotonates transiently, and a nearby carboxylate serves as proton acceptor, like in many retinal proteins.3,24 However, such a mechanism would cause a positive signal in this frequency window caused by the CO stretching of the formed carboxylic acid residue, and we observe a negative feature instead. The only other functional group that absorbs in the concerning frequency window is the ring A carbonyl of PCB.35 We therefore propose that the rearrangement of charges in the chromophore binding pocket caused by the deprotonation transiently reduces the transition dipole moment of either the ring A carbonyl or a protonated carboxylic acid, leading to the bleach at 1730 cm−1. In a second step, the reverse reaction occurs and the reprotonation of the conjugated system can serve as a trigger for the C–S bond formation, because the protonated conjugated system bears a larger partial positive charge at the methine bridge C atom, which is required for the nucleophilic attack of the thiol. The proposed mechanism of C–S bond formation is shown in the left half cycle of Fig. 5.
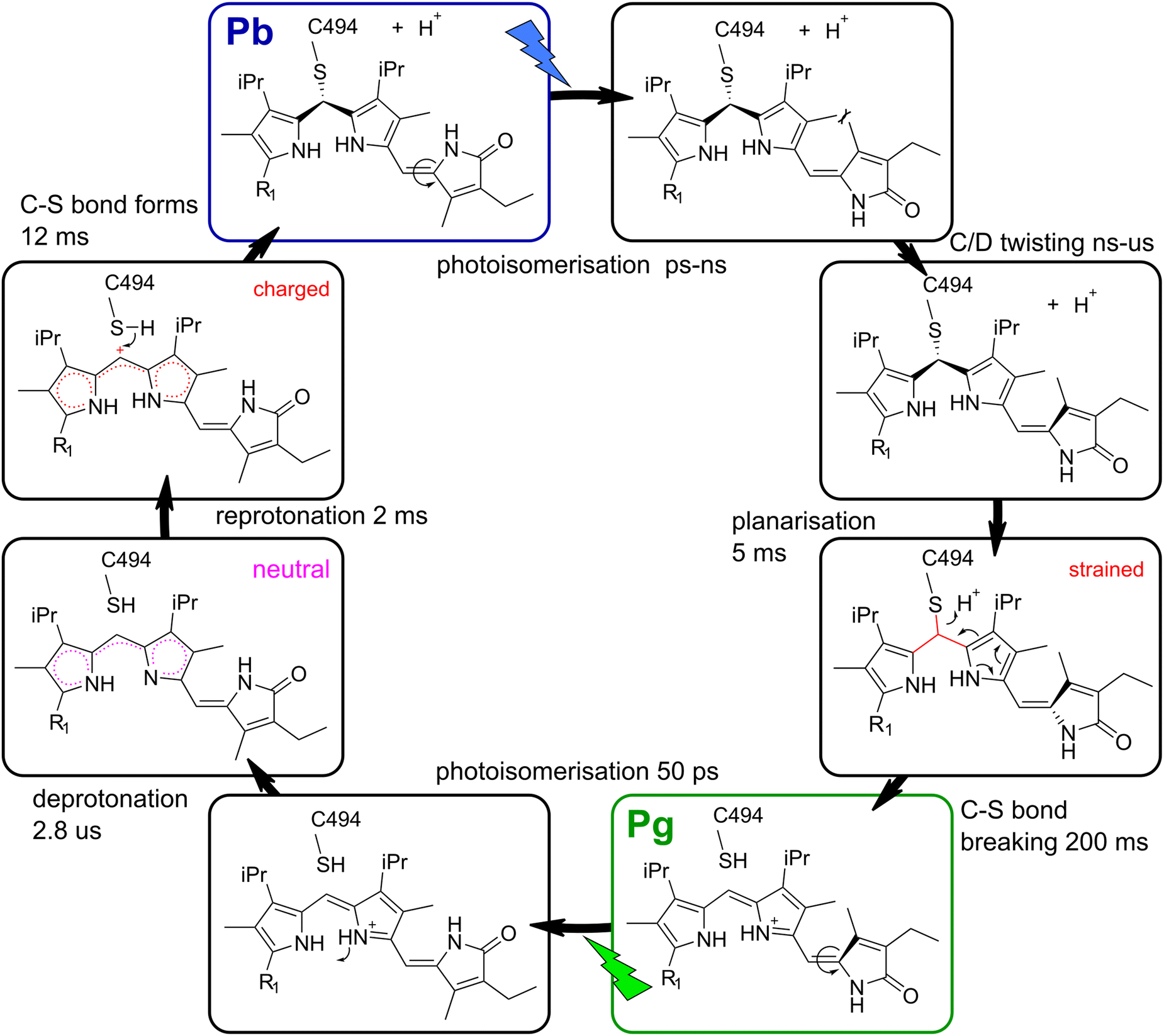 | ||
| Fig. 5 Proposed model for the TePixJ photocycle with Lewis structures of the intermediate stages. Important aspects of the intermediate states that drive the secondary photochemistry are highlighted in magenta and red. R1 indicates the position of ring A, compare Fig. 1B and C. | ||
Cys-bond breaking after planarisation
In the other half photocycle, no transient proton transfer was proposed by other groups, and we also found no signatures that indicate such a process in our transient IR data. However, in order to establish the Pg final geometry from the relaxed Pg photoproduct, the C–S bond needs to break and the B/C methine bridge planarises. The fact that the two CC bands at 1600 and 1630 cm−1 (Fig. 2) display different rise kinetics on the ms time scale allows to gain insights into this mechanism. The 5 ms EADS is already highly similar to the final photoproduct with only one prominent difference: the positive band at 1600 cm−1 is still almost completely missing. Since the results in the visible region clearly show that the characteristic Pg absorption is only established in the very last step and no green absorption is found after 5 ms,19 only the last process can be associated with the breaking of the C–S bond and extension of the conjugation length. This is in line with our assignment that the vibrational mode at 1600 cm−1 is associated with the B/C methine bridge, while the mode at 1630 cm−1 originates mainly from the C/D dipyrrolic unit.
Interestingly, the final reaction is preceded by a distinct 5 ms event that establishes all marker bands of the C/D dypyrrolic unit in their final positions and relative intensities, which again might act as a trigger for the last step. It is consistent with our data that this process is the planarisation of the B/C methine bridge, which should be sensed strongly by the CC signals of the directly connected C/D bridge. Altogether, we propose that the planarisation of the B/C bridge within 5 ms acts as a trigger for the C–S bond breakage by introducing a conformational strain.
V Conclusions
By applying a combination of IEX chromatography, time-resolved and 2D-IR spectroscopy, we were able to investigate crucial proton transfer steps and the molecular triggers of C–S bond breaking and formation in the DXCF-CBCR TePixJ. There are three main findings in this study: (1) the thiol proton of C494 is released to the bulk solvent and not accepted by some residue inside the protein, increasing the net negative charge of TePixJ in the Pb state. (2) After excitation of Pg, a proton is transiently released from the conjugated system. The reprotonation reaction happens within 2 ms, establishing a positive partial charge at the B/C methine carbon and thereby triggering the C–S bond formation. (3) Breaking of the C–S bond in the reverse direction is preceded by the planarisation of the C/B unit, thereby establishing a mechanical strain. Taken together, these results provide a mechanistic understanding of the unique secondary photochemistry in DXCF-CBCRs and the reaction pathways that enable the fascinating color tuning in these proteins.Data availability statement
The data that support the findings are deposited at underthe DOI: https://doi.org/10.5281/zenodo.7602144.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was supported by the Swiss National Science Foundation (SNF) through Grant No. 200020B 188694/1. DB acknowledges a Liebig-Scholarship by the funds of the German chemical industry (Fonds der chemischen Industrie, FCI). We would like to thank Peter Hamm for mentorship and carful reading of the manuscript, Fritz Siebert, and Peter Hildebrandt for helpful discussions, and Jan Helbing, Bogdan Dereka and Wilhelm Redl for technical support in conducting the spectroscopic experiments.References
- A. Moglich, X. Yang, R. A. Ayers and K. Moffat, Structure and function of plant photoreceptors, Annu. Rev. Plant Biol., 2010, 61, 21–47 CrossRef CAS PubMed.
- M. A. van der Horst, J. Key and K. J. Hellingwerf, Photosensing in chemotrophic, non-phototrophic bacteria: let there be light sensing too, Trends Microbiol., 2007, 15(12), 554–562 CrossRef CAS PubMed.
- O. P. Ernst, D. T. Lodowski, M. Elstner, P. Hegemann, L. S. Brown and H. Kandori, Microbial and animal rhodopsins: Structures, functions, and molecular mechanisms, Chem. Rev., 2014, 114(1), 126–163 CrossRef CAS PubMed.
- M. Ikeuchi and T. Ishizuka, Cyanobacteriochromes: A new superfamily of tetrapyrrole-binding photoreceptors in cyanobacteria, Photochem. Photobiol. Sci., 2008, 7(10), 1159–1167 CrossRef CAS PubMed.
- N. C. Rockwell and J. C. Lagarias, A brief history of phytochromes, ChemPhysChem, 2010, 11(6), 1172–1180 CrossRef CAS PubMed.
- K. Fushimi and R. Narikawa, Cyanobacteriochromes: photoreceptors covering the entire UV-to-visible spectrum, Curr. Opin. Struct. Biol., 2019, 57, 39–46, DOI:10.1016/j.sbi.2019.01.018.
- M. Blain-Hartung, N. C. Rockwell, M. V. Moreno, S. S. Martin, F. Gan and D. A. Bryant, et al., Cyanobacteriochrome-based photoswitchable adenylyl cyclases (cPACs) for broad spectrum light regulation of cAMP levels in cells, J. Biol. Chem., 2018, 293(22), 8473–8483 CrossRef CAS PubMed.
- N. T. Ong and J. J. Tabor, A Miniaturized Escherichia coli Green Light Sensor with High Dynamic Range, ChemBioChem, 2018, 19(12), 1255–1258 CrossRef CAS PubMed.
- P. P. Hu, R. Guo, M. Zhou and K. H. Zhao, The Red-/Green- Switching GAF3 of Cyanobacteriochrome Slr1393 from Synechocystis sp. PCC6803 Regulates the Activity of an Adenylyl Cyclase, ChemBioChem, 2018, 19(17), 1887–1895 CrossRef CAS PubMed.
- J. Jang, S. McDonald, M. Uppalapati and A. Woolley, Green, orange, red, and far-red optogenetic 8 tools derived from cyanobacteriochromes, bioRxiv , 2019 DOI:10.1101/769422.
- N. C. Rockwell, S. S. Martin and J. C. Lagarias, Mechanistic insight into the photosensory versatility of DXCF cyanobacteriochromes, Biochemistry, 2012, 51(17), 3576–3585 CrossRef CAS PubMed.
- E. Burgie, J. A. Clinger, M. D. Miller, A. S. Brewster, P. Aller and A. Butryn, et al., Photoreversible interconversion of a phytochrome photosensory module in the crystalline state, Proc. Natl. Acad. Sci. U. S. A., 2020, 117(1), 300–307 CrossRef CAS PubMed.
- T. Ishizuka, R. Narikawa, T. Kohchi, M. Katayama and M. Ikeuchi, Cyanobacteriochrome TePixJ of Thermosynechococcus elongatus harbors phycoviolobilin as a chromophore, Plant Cell Physiol., 2007, 48(9), 1385–1390 CrossRef CAS PubMed.
- T. Ishizuka, A. Kamiya, H. Suzuki, R. Narikawa, T. Noguchi and T. Kohchi, et al., The cyanobacteriochrome, TePixJ, isomerizes its own chromophore by converting phycocyanobilin to phycoviolobilin, Biochemistry, 2011, 50(6), 953–961 CrossRef CAS PubMed.
- T. Ishizuka, T. Shimada, K. Okajima, S. Yoshihara, Y. Ochiai and M. Katayama, et al., Characterization of cyanobacteriochrome TePixJ from a thermophilic cyanobacterium Thermosynechococcus elongatus strain BP-1, Plant Cell Physiol., 2006, 47(9), 1251–1261 CrossRef CAS PubMed.
- R. Narikawa, T. Ishizuka, N. Muraki, T. Shiba, G. Kurisu and M. Ikeuchi, Structures of cyanobacteriochromes from phototaxis regulators AnPixJ and TePixJ reveal general and specific photoconversion mechanism, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 918–923 CrossRef CAS PubMed.
- E. S. Burgie, J. M. Walker, G. N. Phillips and R. D. Vierstra, A photo-labile thioether linkage to phycoviolobilin provides the foundation for the blue/green photocycles in DXCFcyanobacteriochromes, Structure, 2013, 21(1), 88–97 CrossRef CAS PubMed.
- A. T. Ulijasz, G. Cornilescu, D. V. Stetten, C. Cornilescu, F. Velázquez Escobar and J. Zhang, et al., Cyanochromes Are Blue/Green Light Photoreversible Photoreceptors Defined by a Stable Double Cysteine Linkage to a Phycoviolobilin-type Chromophore, J. Biol. Chem., 2009, 284(43), 29757–29772 CrossRef CAS PubMed.
- S. J. O. Hardman, D. J. Heyes, I. V. Sazanovich and N. S. Scrutton, Photocycle of Cyanobacteriochrome TePixJ, Biochemistry, 2020, 59(32), 2909–2915 CrossRef CAS PubMed.
- J. A. Clinger, E. Chen, D. S. Kliger and G. N. Phillips, Pump-Probe Circular Dichroism Spectroscopy of Cyanobacteriochrome TePixJ Yields: Insights into Its Photoconversion, J. Phys. Chem. B, 2021, 125, 210, DOI:10.1021/acs.jpcb.0c04822.
- T. Kottke, V. A. Lórenz-Fonfría and J. Heberle, The grateful infrared: Sequential protein structural changes resolved by infrared difference spectroscopy, J. Phys. Chem. A, 2017, 121(2), 335–350 CrossRef CAS PubMed.
- A. A. Gil, S. P. Laptenok, J. B. French, J. N. Iuliano, A. Lukacs and C. R. Hall, et al., Femtosecond to Millisecond Dynamics of Light Induced Allostery in the Avena sativa LOV Domain, J. Phys. Chem. B, 2017, 121(5), 1010–1019 CrossRef CAS PubMed.
- D. Buhrke and P. Hildebrandt, Probing Structure and Reaction Dynamics of Proteins Using Time-Resolved Resonance Raman Spectroscopy, Chem. Rev., 2019, 120(7), 3577–3630 CrossRef PubMed.
- V. A. Lorenz-Fonfria, Infrared Difference Spectroscopy of Proteins: From Bands to Bonds, Chem. Rev., 2020, 120(7), 3466–3576 CrossRef CAS PubMed.
- P. Hamm, R. A. Kaindl and J. Stenger, Noise suppression in femtosecond mid-infrared light sources, Opt. Lett., 2000, 25(24), 1798 CrossRef CAS PubMed.
- P. Hamm, Transient 2D IR Spectroscopy from Micro- to Milliseconds, J. Chem. Phys., 2021, 154(104201), 1–6, DOI:10.1063/5.0045294.
- D. Buhrke, J. Ruf, P. Heckmeier and P. Hamm, A stop-flow sample delivery system for transient spectroscopy, Rev. Sci. Instrum., 2021, 92(12), 123001, DOI:10.1063/5.0068227.
- Y. Feng, I. Vinogradov and N. H. Ge, General noise suppression scheme with reference detection in heterodyne nonlinear spectroscopy, Opt. Express, 2017, 25(21), 26262 CrossRef CAS PubMed.
- I. H. M. Van Stokkum, D. S. Larsen and R. Van Grondelle, Global and target analysis of time-resolved spectra, Biochim. Biophys. Acta, Bioenerg., 2004, 1657(2–3), 82–104 CrossRef CAS PubMed.
- C. Slavov, H. Hartmann and J. Wachtveitl, Implementation and evaluation of data analysis strategies for timeresolved optical spectroscopy, Anal. Chem., 2015, 87(4), 2328–2336 CrossRef CAS PubMed.
- G. Stock and P. Hamm, A non-equilibrium approach to allosteric communication, Philos. Trans. R. Soc., B, 2018, 373, 20170187 CrossRef PubMed.
- D. Buhrke, K. T. Oppelt, P. J. Heckmeier, R. Fernández-Terán and P. Hamm, Nanosecond protein dynamics in a red/green cyanobacteriochrome revealed by transient IR spectroscopy, J. Chem. Phys., 2020, 153(24), 245101, DOI:10.1063/5.0033107.
- M. A. Mroginski, K. Németh, I. Magdó, M. Müller, U. Robben and C. Della Védova, et al., Calculation of the Vibrational Spectra of Linear Tetrapyrroles. 2. Resonance Raman Spectra of Hexamethylpyrromethene Monomers †, J. Phys. Chem. B, 2000, 104(46), 10885–10899 CrossRef CAS.
- D. Buhrke, N. Michael and P. Hamm, Vibrational couplings between protein and cofactor in bacterial phytochrome Agp1 revealed by 2D-IR spectroscopy Pr Agp1 Pfr Agp2, Proc. Natl. Acad. Sci. U. S. A., 2022, 119(31), e2206400119 CrossRef CAS PubMed.
- H. Foerstendorf, C. Benda, W. Gärtner, M. Storf, H. Scheer and F. Siebert, FTIR studies of phytochrome photoreactions reveal the CO bands of the chromophore: Consequences for its protonation states, conformation, and protein interaction, Biochemistry, 2001, 40(49), 14952–14959 CrossRef CAS PubMed.
- J. Ruf, P. Hamm and D. Buhrke, Needles in a Haystack: Hbonding in an Optogenetic Protein observed with Isotope Labeling and 2D-IR Spectroscopy, Phys. Chem. Chem. Phys., 2021, 23, 10267–10273 RSC.
- C. C. Cornilescu, G. Cornilescu, E. S. Burgie, J. L. Markley, A. T. Ulijasz and R. D. Vierstra, Dynamic structural changes underpin photoconversion of a blue/green cyanobacteriochrome between its dark and photoactivated states, J. Biol. Chem., 2014, 289(5), 3055–3065 CrossRef CAS PubMed.
- T. Sato, T. Kikukawa, R. Miyoshi, K. Kajimoto, C. Yonekawa and T. Fujisawa, et al., Protochromic absorption changes in the two-cysteine photocycle of a blue/orange cyanobacteriochrome, J. Biol. Chem., 2019, 294(49), 18909–18922, DOI:10.1074/jbc.RA119.010384.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2cp05856a |
| This journal is © the Owner Societies 2023 |