
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSatellite ligand effects on magnetic exchange in dimers. A structural, magnetic and theoretical investigation of Cu2L2X4 (L = methylisothiazolinone and X = Cl−, Br−)†
Stefan
Coetzee
a,
Mark M.
Turnbull
 b,
Christopher P.
Landee
c,
Jeffrey C.
Monroe
b,
Mercè
Deumal
d,
Juan. J.
Novoa
e and
Melanie
Rademeyer
*a
b,
Christopher P.
Landee
c,
Jeffrey C.
Monroe
b,
Mercè
Deumal
d,
Juan. J.
Novoa
e and
Melanie
Rademeyer
*a
aDepartment of Chemistry, University of Pretoria, Pretoria, 0002, South Africa. E-mail: melanie.rademeyer@up.ac.za
bCarlson School of Chemistry and Biochemistry, Clark University, 950 Main St., Worcester, Massachusetts 01610, USA
cDepartment of Physics, Clark University, 950 Main St., Worcester, Massachusetts 01610, USA
dSerra-Húnter Fellow, Departament de Ciència de Materials i Química Física and IQTCUB, Facultat de Química, Universitat de Barcelona, Martí i Franquès 1, 08028, Barcelona, Spain
eDepartament de Ciència de Materials i Química Física and IQTCUB, Facultat de Química, Universitat de Barcelona, Martí i Franquès 1, 08028, Barcelona, Spain
First published on 28th February 2023
Abstract
Halide-bridged polymers have gained significant interest due to their diverse properties and potential applications. Stacked Cu2L2X4 dimers, where L is an organic ligand and X can be Cl− or Br−, are of interest because a chloride analogue where L = 2-pyridone, had previously been reported to exhibit bulk ferromagnetism, which augured great potentiality for this class of compounds. The synthesis, structural characterization, magnetic susceptibility measurements, and computational studies of two isostructural CuClMI (MI = methylisothiazolinone) and CuBrMI polymers of Cu(II), along with a related CuClPYR (PYR = 2-pyridone) is reported. CuClMI and CuBrMI were found to exhibit AFM bulk properties, due to FM/AFM alternating chains along the halide-bridged polymer axis, while FM bulk properties were confirmed for CuClPYR exhibiting a FM spin ladder. In combination with a benzamide analogue, CuClBA, three O-donor amides, CuClMI, CuClBA and CuClPYR were analyzed and revealed that the kinetic exchange is affected by the identity, but more importantly, the orientation of the satellite ligands. The torsional angle of the ligand with the dimer plane is shown to significantly affect the magnetic exchange in the dimer, and between dimers, explaining the reported FM bulk properties of CuClPYR. This finding is exceedingly important, as it suggests that a spin device can be constructed to flip between singlet/triplet states by manipulating the orientation of the satellite/terminal ligand.
Introduction
Halide-bridged polymers of Cu(II) are known to exhibit a myriad of structural permutations. Selected examples of common halide-bridged polymers are illustrated in Scheme 1. Of specific interest in the current study are double-halide-bridged polymers (Scheme 1(d)) consisting of stacked, planar dimers of the formula Cu2L2X4, where L is an organic ligand, and X = Cl− or Br−, as depicted in Fig. 1.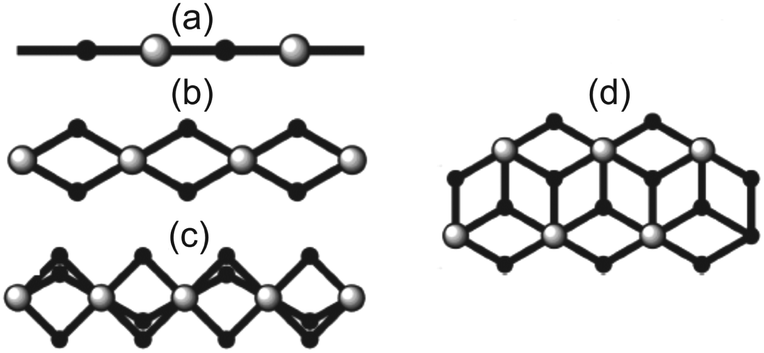 | ||
| Scheme 1 Examples of different types of halide-bridged polymers are (a) corner-sharing, (b) simple edge-sharing, (c) face-sharing, and (d) complex edge-sharing. Black and white spheres represent transition metals and halide ligands respectively. | ||
 | ||
| Fig. 1 Stacked dimers in Cu(II)-based halide bi-bridged compounds, whose magnetic interactions can potentially occur within the dimer (J1), or via interactions between dimers (J2–4). Colour code: Cu in blue, X in green, L in red, where X = Cl− or Br− and L = organic ligand. | ||
The current study will focus on the magnetic properties of stacked-dimer halide-bridged polymers that form low-dimensional magnetic materials as a result of the presence of the inorganic polymer in the structure, with the bridging halide ions resulting in various Cu–X⋯X–Cu and Cu–X⋯Cu magnetic exchange pathways between Cu(II) cations. Low-dimensional magnetic materials are of interest in the modeling and understanding of room-temperature superconductors1 and can assist in the fundamental understanding of magneto-structural relationships. In addition, the Cu(II) ion represents a simple magnetic ion, with S = 1/2 and quenching of the orbital angular momentum. This makes it an ideal system to study both experimentally and computationally. The magnetic exchange interactions in these stacked-dimer halide-bridged polymers have been the subject of several studies reported in the literature, with different magnetic properties reported for polymers containing different organic ligands.2–5 The parent compound, CuCl2, has been found to exhibit incommensurate long-range magnetic phase transitions at low temperatures into an antiferromagnetic (AFM) spin spiral.8 and have been shown to be sensitive to the nature of the halide, and geometric parameters of the inorganic bridge.9 Rodríguez et al.10 studied multiple parameters’ effects on the magnetic exchange in polymeric (Cu2X2(NH3)4)2+ dimers through the use of density functional theory (DFT). They demonstrated that the magnetic exchange was determined by the way the Cu(II) ions were linked, including the connectivity, bond angles, bond lengths, and variation in the bridging geometry, but of more interest in this context, that the type of ligand attached to the terminal positions of the polymers (red atoms in Fig. 1) have a large influence on the magnitude and sign of the magnetic exchange in these dimers. Cl− terminal ligands favored AFM exchange across the dimers, while N-donor ligands favored FM exchange. Importantly, their work focussed on the dimers in isolation, only considering the magnetic exchange in the dimer (blue arrow in Fig. 1). Coetzee et al. demonstrated that the magnetic exchange of these compounds is actually determined by the inter-dimer exchange (green arrow in Fig. 1) if the exchange across the dimer is FM.6 Inspired by the abovementioned contributions, the current authors examined a related set of CuXL compounds,6 where X = Cl− or Br− and L = O-donor amides, shown in Scheme 2. The Heisenberg Hamiltonian used throughout all studies is of the form:  where positive JAB corresponds to a FM interaction. When the amide is benzamide (BA), the exchange in the dimers (J1) was found to be strongly FM (red arrow for Cu⋯Cu in-plane magnetic exchange in Fig. 1) with JCuClBA1 = +34.70 cm−1 and JCuBrBA1 = +49.67 cm−1. Dimers had very small FM (∼0.3 cm−1) inter-dimer interactions (blue (J2) and orange (J4) arrows in Fig. 1) competing with stronger AFM inter-dimer interactions (green arrows in Fig. 1) of JCuClBA3 = −3.32 cm−1 and JCuBrBA3 = −7.15 cm−1 (see Table 1) resulting in an 1D magnetic topology, which consists of alternating chains of FM dimers (effective S = 1) antiferromagnetically connected. When the amide is 2-pyridone (PYR), J1 was experimentally fitted to be weakly FM (+9.4 cm−1), with the overall exchange between dimers AFM (−3.2 cm−1) for CuClPYR, resulting in an overall FM low-temperature response.7 The authors did comment that the model did not fit well, which may indicate that the magnetic structure of CuClPYR is more complex. When the amide is methylisothiazolinone (MI), the fitted magnetic exchange of CuClMI was reported to have a FM J1 of +5.03 cm−1, a FM J2 = +1.96 cm−1 and an AFM J3 of −1.36 cm−1 resulting in an overall FM response.2
where positive JAB corresponds to a FM interaction. When the amide is benzamide (BA), the exchange in the dimers (J1) was found to be strongly FM (red arrow for Cu⋯Cu in-plane magnetic exchange in Fig. 1) with JCuClBA1 = +34.70 cm−1 and JCuBrBA1 = +49.67 cm−1. Dimers had very small FM (∼0.3 cm−1) inter-dimer interactions (blue (J2) and orange (J4) arrows in Fig. 1) competing with stronger AFM inter-dimer interactions (green arrows in Fig. 1) of JCuClBA3 = −3.32 cm−1 and JCuBrBA3 = −7.15 cm−1 (see Table 1) resulting in an 1D magnetic topology, which consists of alternating chains of FM dimers (effective S = 1) antiferromagnetically connected. When the amide is 2-pyridone (PYR), J1 was experimentally fitted to be weakly FM (+9.4 cm−1), with the overall exchange between dimers AFM (−3.2 cm−1) for CuClPYR, resulting in an overall FM low-temperature response.7 The authors did comment that the model did not fit well, which may indicate that the magnetic structure of CuClPYR is more complex. When the amide is methylisothiazolinone (MI), the fitted magnetic exchange of CuClMI was reported to have a FM J1 of +5.03 cm−1, a FM J2 = +1.96 cm−1 and an AFM J3 of −1.36 cm−1 resulting in an overall FM response.2
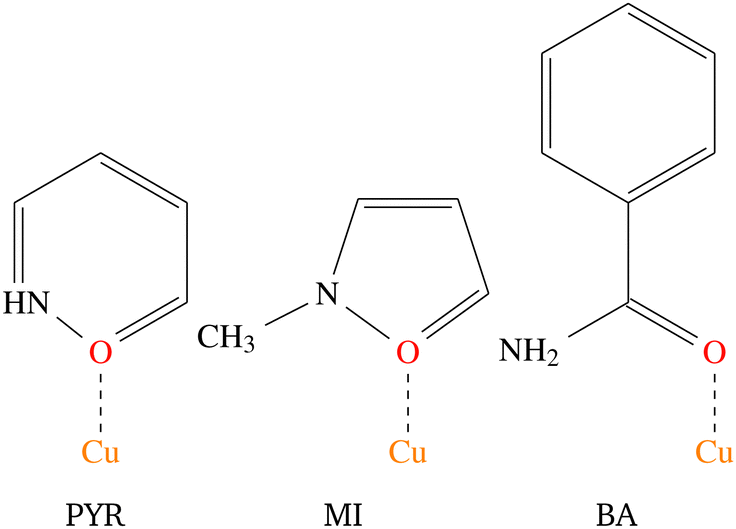 | ||
| Scheme 2 The structures of the three amides, 2-pyridone (PYR), methylisothiazolinone (MI) and benzamide (BA) ligands considered in this study, with the coordination modes indicated. | ||
Therefore, when the amide is MI and PYR, the bulk properties are FM, while BA produces AFM bulk properties at low temperatures. J1 is FM in all cases, while J2 and J3 vary significantly. This is interesting since all of the terminal ligands are similar and is even more intriguing when comparing the structures of the bridges, where differences in the Cu–Cl distances are all within 0.01 Å, Cu⋯Cl distances within 0.05 Å, Cl–Cu⋯Cl and Cu–Cl⋯Cu angles are within 1° respectively, while the interdimer Cu–Cl⋯Cu angles are within 2° of one another. The inorganic polymer backbone, is, therefore effectively the same for CuClBA, CuClMI, and CuClPYR. Yet, these compounds produce different magnetic properties.
The differentiating structural feature between these three compounds is the Clterm–Cu–O–Ccarbonyl–C torsional angle, which are 135°, 55° and 58° for CuClPYR, CuClMI, and CuClBA, respectively. CuClMI, as we will show, actually exhibits AFM properties, similar to CuClBA, which points to the Clterm–Cu–O–Ccarbonyl–C torsional angle as the parameter that has an effect on one or more of the exchange interactions that results in the different observed properties.
To understand how this occurs, a computational study using the First-Principles Bottom-Up (FPBU)11 method, and other calculations were conducted on CuClMI, CuClBA, CuClPYR, and the newly synthesized CuBrMI compound. The calculated Ji values are validated against experimental data, and a rigorous magnetic model is obtained for these compounds. It must be stressed that the effect of coordination angle on individual magnetic exchange interactions is explored as a target for spin manipulation.
Experimental section
Chemicals and reagents
All chemicals were used as purchased without further purification: CuBr2 (99%, Sigma Aldrich), 2-methyl-1,2-thiazol-3(2H)-one (methylisothiazolinone) (99%, Aldrich), ethanol (99.8%, Sigma-Aldrich).Data acquisition
The structure of CuClMI was published by Kato et al.2 and the structure files were obtained from the Cambridge Structural Database (CSD)12 with the CSD refcode: [OPABAJ],2 which was used as the input structure for the computational investigation. An experimental magnetic susceptibility data set for CuClMI was obtained from the original author, M. Kato.2Instrumental studies
Computational details
The First Principles Bottom-Up (FPBU)11 working strategy allows for a systematic theoretical analysis of the magnetic topology and the calculation of the bulk magnetic properties of a material. The only input into the analysis is the experimental single-crystal structure. The FPBU analysis was completed for CuClMI,2CuClPYR,7 and the new crystal structure of CuBrMI determined in this study.Potential pairs of radicals are identified from the crystal structure, up to a cut-off distance of ca.10 Å between Cu(II) ions. Once all the radical pairs have been identified, the radical⋯radical pair magnetic exchange interactions (JAB) for all the unique radical pairs are calculated. Cluster models used to evaluate magnetic coupling between Cu-moieties must account for the coordination and environment of both Cu(II) ions, i.e. for X and L ligands in CuXL compounds. Using the Heisenberg Hamiltonian  and broken-symmetry (BS) approximation,22 the magnetic exchange interaction JAB between any two radicals A and B is calculated at the level of unrestricted density functional theory (DFT/UB3LYP)23–26 through the difference in energy between the triplet (ET) and open-shell singlet state using the broken-symmetry approach (EBSS) employing a dimer cluster model (see eqn (1)).
and broken-symmetry (BS) approximation,22 the magnetic exchange interaction JAB between any two radicals A and B is calculated at the level of unrestricted density functional theory (DFT/UB3LYP)23–26 through the difference in energy between the triplet (ET) and open-shell singlet state using the broken-symmetry approach (EBSS) employing a dimer cluster model (see eqn (1)).
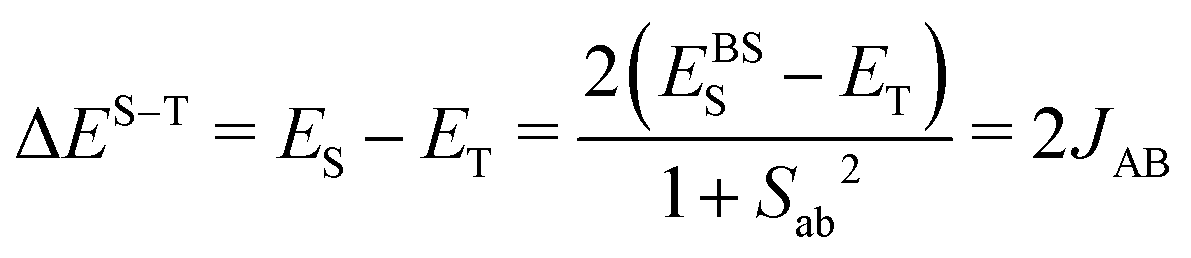 | (1) |
In eqn (1), Sab represents the overlap between molecular spin-carrying orbitals and is obtained from the DFT calculations, to produce spin-projected energy for the BS state. This approximation can be extended to cluster models containing more than two radicals, which generally improves the description of the chemical environment around the radicals and provide a more accurate estimation of JAB (see ESI,† Section S2 for a detailed discussion). For this work, a finite cluster comprised of two stacked Cu2L2X4 dimeric units (tetramer model) was used in addition to the dimer model, where several spin permutations were calculated to determine the individual JAB values. The dimer and tetramer finite cluster models were required to calculate the different JAB values for all compounds (hereafter, Ji). All energies were calculated using the Ahlrich TZVP basis set.27 The energy calculations for the pairs of radical dimers were carried out using Gaussian 09 Rev. D01,28 and for the tetrameric structures, ORCA 4.2.029 was used.
Based on the value of the significant Ji interactions, a magnetic topology is constructed, and magnetic models extracted accordingly. It must be stressed that a good magnetic model, in addition to include the most relevant Ji interactions, should have topological connectivity similar to the bulk of the material. Therefore, from the diagonalization of the appropriate magnetic model, the energy eigenvalues and the total spin number of each respective state are determined. No interactions are excluded a priori and multiple arrangements and connectivities are tested to obtain a model that best mimics the experimental data. A good model should display a convergence toward the experimental values as the number of radicals is increased, for example, from 2 to 8 to 16, in the effective Hamiltonian (see Section S3 for further discussion, ESI†).
Finally, the macroscopic magnetic properties can be calculated. The bulk magnetic susceptibility (χm) is calculated using the usual statistical mechanics expression.30 The value of g is 2.1 for CuClMI, as determined by EPR spectroscopy,2 and, based on this, g = 2.1 is assumed for CuBrMI and CuClPYR. Note that all calculated magnetic data were compared to the experimental data.
Decomposition of JAB
J AB exchange interactions consist of physical components and can be decomposed into contributions from direct exchange (J0), kinetic exchange (ΔJKE), core-electron polarisation (ΔJCP) and other exchange mechanisms (JOth) to yield eqn (2):| JAB = J0 + ΔJKE + ΔJCP + JOth | (2) |
These contributions to the JAB magnetic exchange interaction were discovered through the use of advanced wave-function based methods.31–34 The decomposition of JAB is intrinsic to wave-function based methods,35 and was also implemented in DFT based methods.35,36 A recent implementation of this J-decomposition scheme was included in ORCA 4.0,29 based on the work of Coulaud et al.35,36
J 1T, J1D and J3T were decomposed for CuClMI, CuBrMI, CuClPYR, CuBrBA, CuClBA, and compared to Cu2Cl62−.35 These decomposition calculations were performed using RO-KSDFT (B3LYP, using def-2-QZVPP as the basis for Cu(II), while for all other atoms, SVP was used) in ORCA 4.2.029 at a convergence criteria of 1 × 10−9 Ha (VeryTightSCF). Convergence of the self-consistent field (SCF) required the following settings: “Grid6 FinalGrid7”, “DIISMaxEq 25”, “directresetfreq 1”, and “DecompositionPath Strict” to obtain reliable singlet states. See ESI,† Section S4 for a full discussion.
Results and discussion
Crystallographic discussion of structures
To understand why FM interactions were reported for CuClMI,2 the new structure of CuBrMI was determined in this study (see Table S1.1 (ESI†) for the crystallographic parameters of the two structures and Table S1.2 (ESI†) for selected bond lengths and angles in Section S1, ESI†). The two compounds di-chloro-bis[chloro(2-methyl-1,2-thiazol-3(2H)-one)copper(II)], CuClMI, and di-bromo-bis[bromo(2-methyl-1,2-thiazol-3(2H)-one)copper(II)], CuBrMI, are isostructural (see Fig. 2(a) and (b) for dimeric units). Comparison of CuClMI and CuBrMI reveals that the Cu–X bond lengths and X–Cu–X, Cu–X–Cu, X⋯Cu–X, and Cu⋯X–Cu angles are all similar. With similar geometries in the dimers and polymers, similar magnetic properties are expected for the chloride and bromide analogues.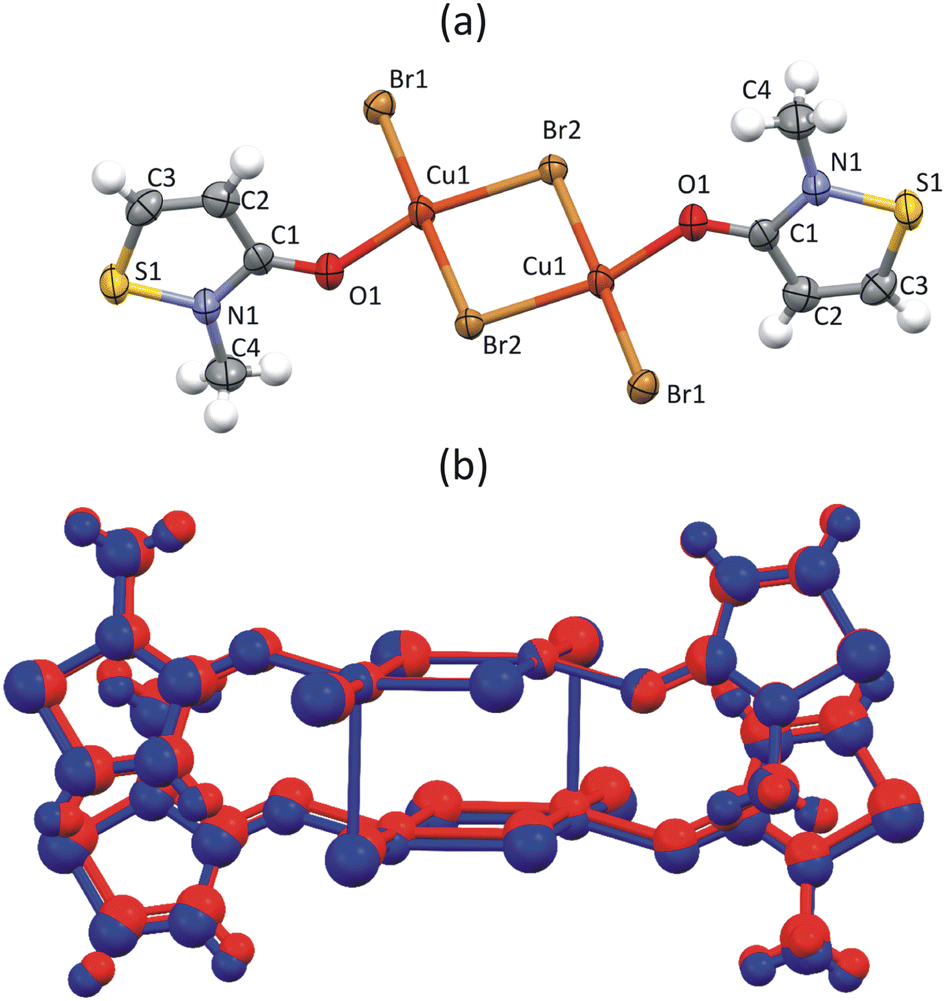 | ||
| Fig. 2 Dimeric unit of (a) CuBrMI. Atoms are drawn at a 50% probability, while hydrogen atoms are drawn as spheres of arbitrary radii. (b) Overlap of two isostructural stacked dimers of CuClMI (red) and CuBrMI2 (blue), plotted as a ball and stick representation. | ||
Assessing the nature of the magnetic interactions
A computational First-Principles Bottom-Up11 FPBU study was completed for CuClMI, CuBrMI and CuClPYR to obtain an improved fundamental understanding of the magnetic behaviour of these amide-containing polymers. For brevity, most of the analysis of CuClPYR is documented in Section S5 (ESI†), while there is some additional discussion of CuXMI results given in Section S3 (ESI†). In the present study, nine potential pairs of radicals (d1–d9) were identified from the crystal structures of CuClMI and CuBrMI, and ten pairs for CuClPYR, up to a cut-off distance of ∼10 Å between Cu(II) ions. Specifically, for CuXL, d1 is the radical pair comprised of the Cu(II) ions in the Cu2(L)2X4 (where L = MI, PYR or BA) dimeric unit, while the radical pair in d2 contains fragments of two neighbouring stacked dimers along the polymer axis (see radicals connected by red and blue lines in tetramer model in Fig. 3, respectively). d3 and d4 involve the Cu(II) ions from two neighbouring dimeric units diagonally across the dimeric unit (in green and orange in tetramer model, see Fig. 3). The interactions d1–d4 are potential magnetic couplings within the 1D polymers, whereas d5–d10 are potential interactions between the polymers. Since d5–d10 are computed to have a very small J5–10 exchange interaction, their effect on the overall magnetic properties is small, as discussed in ESI,† Section S3, and will not be discussed here. Finite tetramer cluster models for both compounds were constructed from the crystal structures in such a way that the chemical environments around these square planar clusters are chemically representative of the environment around the Cu(II) ions (i.e. halides and L ligands coordinated to the Cu(II) ions, as shown in Fig. 3). For CuClPYR, each PYR ligand forms a hydrogen-bonded dimer with another PYR molecule from a neighbouring chain. To consider whether this hydrogen bond has an effect on the magnetic properties, the tetrameric structure, CuClPYRHB was also considered (see Section S5, ESI†). | ||
| Fig. 3 Tetrameric finite cluster models used for CuClMI, CuBrMI and CuClPYR showing potential intra-dimer in-plane interactions J1 to J4. Coloured lines indicate the exchange interactions: J1 (red), J2 (blue), J3 (green), and J4 (orange). Atomic colors: Cu (blue), Cl (green), Br (brown), C (black), O (red), N (light blue), S (yellow) and H (white). | ||
The magnetic exchange interaction in a radical pair di will be indicated by Ji. A tetrameric cluster containing four Cu(II) ions was used to calculate J1–J4, as shown in Fig. 3, using several different spin configurations. Dimer clusters consisting of only two Cu(II) ions were used to calculate J5–J10. The calculated magnitudes of J1–J4 are larger for CuBrMI than for CuClMI (see Table 2), which can be explained by the higher magnetic exchange through bromide ligands compared to chloride ligands.6,9 For all compounds, the intra-dimer J1 interaction is FM, with J1 for CuClPYR significantly smaller than that of CuClMI.
The sign and magnitude of J1 in these compounds (see Table 2) can be attributed to the significant orthogonal overlap of the singly occupied (SOMO) orbitals, in agreement with the Goodenough–Kannamori rules,37,38 (see ESI,† Section S6) and agrees with previous findings for CuClMI2 (J = +28.6 cm−1) and other related compounds (e.g. +34.70 cm−1 for CuClBA and +49.67 cm−1 for CuBrBA).6 For CuClPYRJ1 = +11.67 cm−1, while the inclusion of the hydrogen-bonded pair almost doubles the value of J1, highlighting the importance of including the broader chemical environment of a magnetic molecule when calculating the value of J interactions. CuClPYR has a surprisingly small value for J1 compared to CuClMI and CuClBA, considering that the inorganic portions are very similar, and the organic ligands are all O-donor amides.
Due to the strength of J1, the d1 radical pair (which is the Cu2L2X4 dimeric unit) can be considered as the magnetic building block of these compounds which form S = 1 dimers at low temperature. These primary building blocks are linked by three interactions, namely J2, J3 and J4, and -depending on the strength of these secondary magnetic couplings- become either alternate links in a 1D chain magnetic topology or rungs in a 1D spin ladder topology. These interactions generally comprise short Cu⋯Cu, Cu–X⋯Cu, and Cu–X⋯X–Cu contacts that are potentially involved in the magnetic exchange between the Cu2L2X4 building blocks.
J 2 is FM for CuXL, and involves two Cu–Xcis⋯Cu contacts, where Xcis refers to the halide ligand cis to MI/PYR. One interaction comprises the Cu–Xcis⋯Cu pathway involving the bridging halide ligand and another a Cu–Xcis⋯Cu pathway involving the terminal halide ligand. This asymmetry in the exchange would not be fully accounted for if using a dimer model (see ESI,† Section S2) because it does not account for the change in electron density on the bridging halide ligand. However, this effect is included in the tetramer model, where the bridging and terminal halide ligands are appropriately represented (see tetramer structures in Fig. 3) and explains why the tetrameric models generally perform better at estimating magnetic exchange interactions.39
The J3 interaction of the MI and BA analogs are all AFM, while J3 is FM for CuClPYR. The absolute magnitude of J3 for CuClPYR is also significantly smaller than the other amides. Note that the geometries of the inorganic portions of CuClL are essentially the same, and therefore cannot explain why CuClPYR has a FM J3 interaction, and the other amides have an AFM J3 value. Thus, the organic ligand must be the cause of the observed differences in J3. Since all the L-ligands are chemically similar O-donor amides, it is unlikely that PYR itself is the cause, and it is more probable that the orientation of PYR is the cause for the lower value of J1, and for the FM J3 value.
Based on these results, an AFM ground state is expected for CuXMI. This finding is in contrast to the experimental bulk FM properties reported previously for CuClMI.2 A FM ground state is expected for CuClPYR, agreeing with previous experimental properties.7
Since an AFM ground state was predicted for CuClMI, a multitude of computational analyses were performed, including testing the effects of ligand exchange, magnetic field effects upon calculating the magnetic susceptibility, the effects of thermal contraction on J3, and testing whether a small difference in the inter-dimer exchange could have produced a FM response (see ESI,† Section S7, for a detailed discussion). The tests demonstrated that the computational model and method used were indeed robust, and concluded that strong FM interactions occur between Cu(II) ions in the dimer and weak AFM interactions are calculated between dimers. This prompted a re-examination of the experimental magnetic properties of CuClMI, presented below.
Experimental magnetic results
Experimental data recorded for CuClMI in the current study agrees well, not only with the data recorded for the isostructural compound CuBrMI shown in Fig. 4(a)–(c), but also with exhaustive computational analyses, as mentioned in the previous section. Therefore, the magnetic susceptibility data recorded in the current study were used exclusively for further analysis of the magnetic properties of CuClMI. | ||
| Fig. 4 Magnetic susceptibility plots for newly recorded data for CuClMI (Δ in red) and CuBrMI (O in blue), where (a) χm(T), (b) 1/χm(T), and (c) χmT(T). | ||
χ m(T) data were recorded on phase pure powder samples of CuClMI and CuBrMI, as documented in the Experimental section. The χm(T) data (Fig. 4(a)) show an increase in susceptibility to a maximum of 0.0815 emu Oe−1 mol−1 at 2.69 K and 0.026 emu Oe−1 mol−1 at 7.89 K for CuClMI and CuBrMI, respectively. This is indicative of weak AFM interactions. The experimental data recorded in this study for CuClMI show that the bulk magnetic properties of CuClMI are AFM.
The 1/χmT data, shown in Fig. 4(b), were fitted to a Curie Weiss model (T > 150 K) and indicate the presence of dominant FM interactions, as expected from the values of Ji magnetic couplings listed in Table 2. The large upward deviation from the Curie Weiss law around 100 K and 50 K for CuBrMI and CuClMI, respectively, is indicative of dominant FM interactions being present at this temperature. A Curie constant value of 0.477(5) emu K Oe−1 mol−1 was obtained from a Curie fit of the susceptibility data measured in this study for CuClMI and a value of 0.418(1) emu K Oe−1 mol−1 was found for CuBrMI (see Table 3).
| J 1T | J 2T | J 3T | J 4T | |
|---|---|---|---|---|
| CuClBA | 34.70 | 0.95 | −6.22 | 1.21 |
| CuClMI | 33.51 | 0.61 | −5.55 | 1.12 |
| CuClPYR | 11.67 | 1.11 | 0.36 | 0.10 |
| CuClPYRHB | 19.26 | 0.82 | 0.52 | 0.09 |
| CuBrMI | 49.16 | 0.03 | −6.09 | 0.09 |
| CuBrBA | 49.67 | 1.61 | −10.79 | 2.16 |
| C | θ (K) | R 2 | |
|---|---|---|---|
| a C is given in units of emu K Oe−1 mol−1. | |||
| CuClMI | 0.477(5) | 9(2) | 0.997 |
| CuBrMI | 0.418(1) | 20.7(8) | 0.999 |
The χmT product data recorded in the current study increase from 0.488 emu K Oe−1 mol−1 and 0.450 emu K Oe−1 mol−1 at room temperature to a maximum of 0.542 emu K Oe−1 mol−1 at 48 K and 0.494 emu K Oe−1 mol−1 at 86 K for CuClMI and CuBrMI, respectively (see Fig. 4(c)). This again indicates the presence of strong FM interactions, while the rapid decrease at lower temperatures indicates the presence of weaker AFM interactions.
The experimental magnetic data recorded for CuClMI in this study exhibited significant signal noise in the low-temperature region, which is evident in Fig. 4(c). Magnetic susceptibility data were recorded again for a second sample of CuClMI from the same batch with the same noise randomly appearing. An anomaly is observed in both of the CuClMI sample's χm data at ∼15 K, which is evident as a small “hump” in the χmT product data, shown in Fig. 4(c). (Also see Fig. S8.1 in ESI,† Section S8). It is unclear what caused this, therefore, some additional testing is required to understand its origin. This anomaly is especially interesting, since a potential magnetic phase transition was also observed for CuClBA at ∼45 K.6
The magnetic topology of CuXMI and CuClPYR
Taking into account all significant interactions, the magnetic topology of CuClMI and CuBrMI consists of FM J1 dimers that are connected by AFM J3 interactions, which then further couple through very weak competing FM and AFM interactions (J2 and J4) into a polymeric chain. Our calculations suggest the presence of an alternating magnetic chain with strong FM and weak AFM interactions, which can be considered to be well isolated from other chains, as there are no significant interactions between them (see Fig. 5(a) and Table 2). CuClPYR has FM dimers (rungs), with FM interactions along the polymer (rails) to form a FM spin ladder, with weak cross-rung interactions (see Fig. 5(b) and Table 2).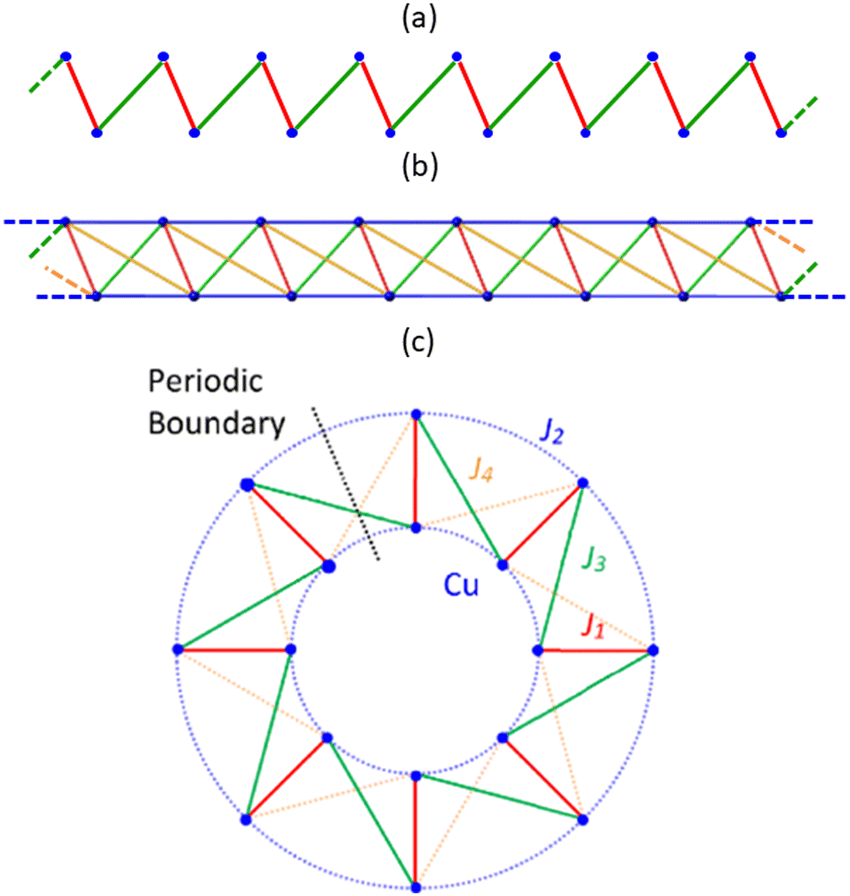 | ||
| Fig. 5 (a) The 2 × 8 alternating chain for CuClMI and CuBrMI. (b) The 2 × 8 spin ladder model for CuClPYR. (c) General 2 × 8 cyclic model valid for all three compounds by switching off the appropriate Ji interactions. The cyclic model is constructed by mathematically connecting the ends of the 2 × 8 models. Interactions that cross the black periodic boundary condition are connected to the other end of the 2 × 8 model to introduce pseudo-periodic boundary conditions. Note that each CuXL moiety has been replaced by the Cu atom in blue. J1 (red), J2 (blue), J3 (green), J4 (orange). | ||
After a comprehensive analysis of the performance of different potential open and cyclic magnetic models of CuXL with and without interactions between neighbouring polymeric chains (see ESI,† Section S3), it was concluded that the best magnetic model for calculating magnetic properties of CuXMI using statistical mechanics was a cyclic 16 Cu(II) cluster model using the Jtetrameri (see Fig. 5(c)), which introduces pseudo periodic boundary conditions, while the 16 Cu(II) 2 × 8 model in Fig. 5(b) was best to calculate the magnetic susceptibility of CuClPYR. Note that the structure of CuClPYR was reported with an R-factor of 23%, so the quality of the calculated data is not as good, which is likely why the 2 × 8 model performed a bit better. For CuBrMI, the calculated temperature-dependent magnetic susceptibility shown in Fig. 6 predicts a maximum susceptibility of 0.031 emu Oe−1 mol−1 at 6.5 K (Exp: 0.026 emu Oe−1 mol−1 at 7.5 K) and correctly predicts an AFM ground state (see Section S3 (ESI†), for results obtained using other models). For CuClMI, the computational results replicated the experimental magnetic data recorded in this study. The calculated magnetic susceptibility as a function of temperature data predict a maximum in the susceptibility of 0.070 emu Oe−1 mol−1 at 3 K (Exp: 0.082 emu Oe−1 mol−1 at 2.69 K). Therefore, it correctly predicts an AFM ground state for CuClMI as shown in Fig. 6. For CuClPYR, the tetramer models with (red), and without (blue) additional hydrogen bonds both predict a FM ground state, with the inclusion of additional hydrogen-bonded molecules improving the estimation of the bulk magnetic susceptibility slightly with respect to the experimental data (black).
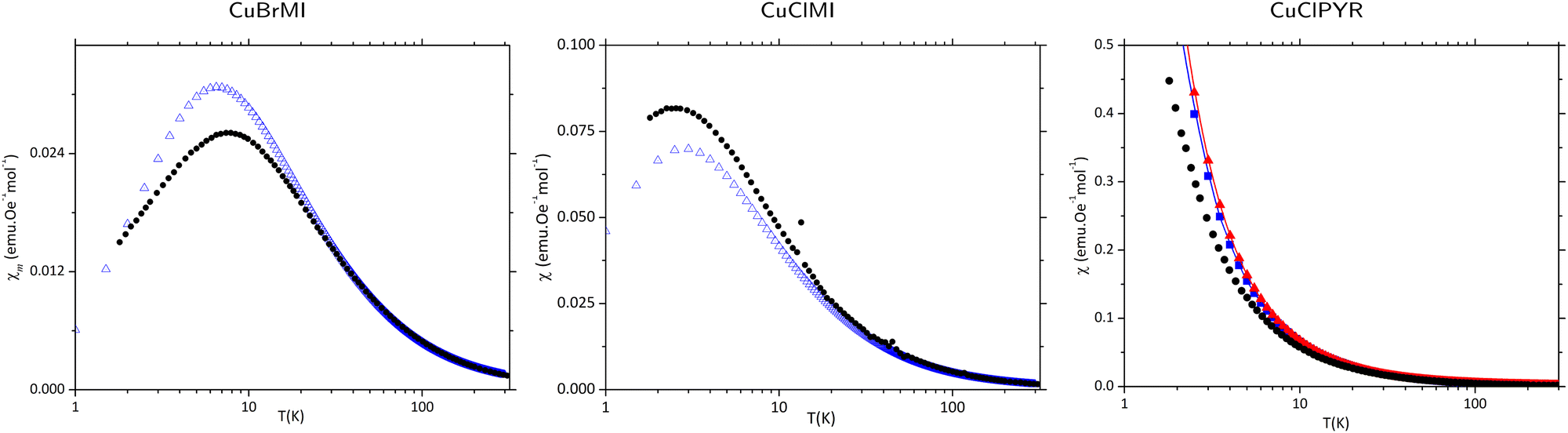 | ||
| Fig. 6 Calculated χm(T) data for CuBrMI and CuClMI. Experimental (•) data, and calculated data (blue triangles) using a 2 × 8 cyclic model with J1T. Calculated data of CuClPYR using the 2 × 8 model, for the hydrogen-bonded (solid blue triangles), and non-hydrogen-bonded (solid red triangles). Note that models are shown with interpolated data for clarity. | ||
The macroscopic energy state population of CuClMI and CuBrMI at the temperature where the χm is at a maximum (Tmax) reveals that many high spin states are responsible for the increase in the χmT product data at T > Tmax while at T < Tmax a singlet state becomes preferentially occupied, reducing χm at low temperature. This is discussed in more depth in Section S9 (ESI†). The high spin states at T > Tmax are due to FM coupling between Cu(II) ions in the dimers, with AFM interactions between the dimers resulting in the reduction of χm at low temperature. For CuClPYR, high spin states are more populated at low temperatures resulting in a FM ground state.
Satellite ligand effects on J1 and J3
The lower value of FM J1 and the FM value of J3 for CuClPYR are not due to the structure of the inorganic scaffolding, as they are essentially the same. Further, if these differences were caused by the type of ligand alone, then CuClMI, CuClBA and CuClPYR should have a similar effect on the J1 and J3 values. Instead, we find that CuClMI and CuClBA behave similarly to one another, and happen to have similar coordination torsion angles (Cl–Cu–O–C) of ca. 50°, while CuClPYR behaves differently, and has a coordination torsion angle of ∼225°. Therefore, a study was carried out to assess the effect that this coordination torsion angle has on the magnetic exchange coupling between CuXL spin-carrying moieties.The effect of rotation of the MI, BA, and PYR ligands about the Cu–O bond on the two main magnetic exchange interactions, namely J1 and J3, was examined using a dimer cluster which enables 360° rotation of the ligand and a tetramer model, respectively. The magnetic exchange interactions at some angles were also decomposed to examine how the individual components are affected by the rotation of the organic ligand MI, using the magnetic exchange decomposition scheme40 as implemented in the software package ORCA.29 Note that J1D is generally 15% smaller than J1T, but the compositions of J1D and J1T appear to be comparable. Let us remind here that, according to this scheme (eqn (2)), JAB indicates the calculated magnetic exchange (namely, J1D or JiT with i = 1, 3) (see tetramer model in Fig. 3), J0 describes the direct magnetic exchange between two Cu(II) ions, while ΔJCP, the core-polarisation exchange, describes the extent of spin polarisation of the MO's. ΔJKE indicates the kinetic exchange, which describes the extent to which unpaired electrons are delocalised, and JOth describes the contribution to the magnetic exchange from other sources.
Dimer rotation
The effect of the rotation of the MI, BA, and PYR ligands about the Cl–Cu–O–C torsion angle on the J1D magnetic exchange interaction is shown in Fig. 7(a). At a torsion angle of 0° the amide plane is approximately co-planar with the Cu2Cl2 bridge plane, and at 90° the ligand is perpendicular to the inorganic bridge.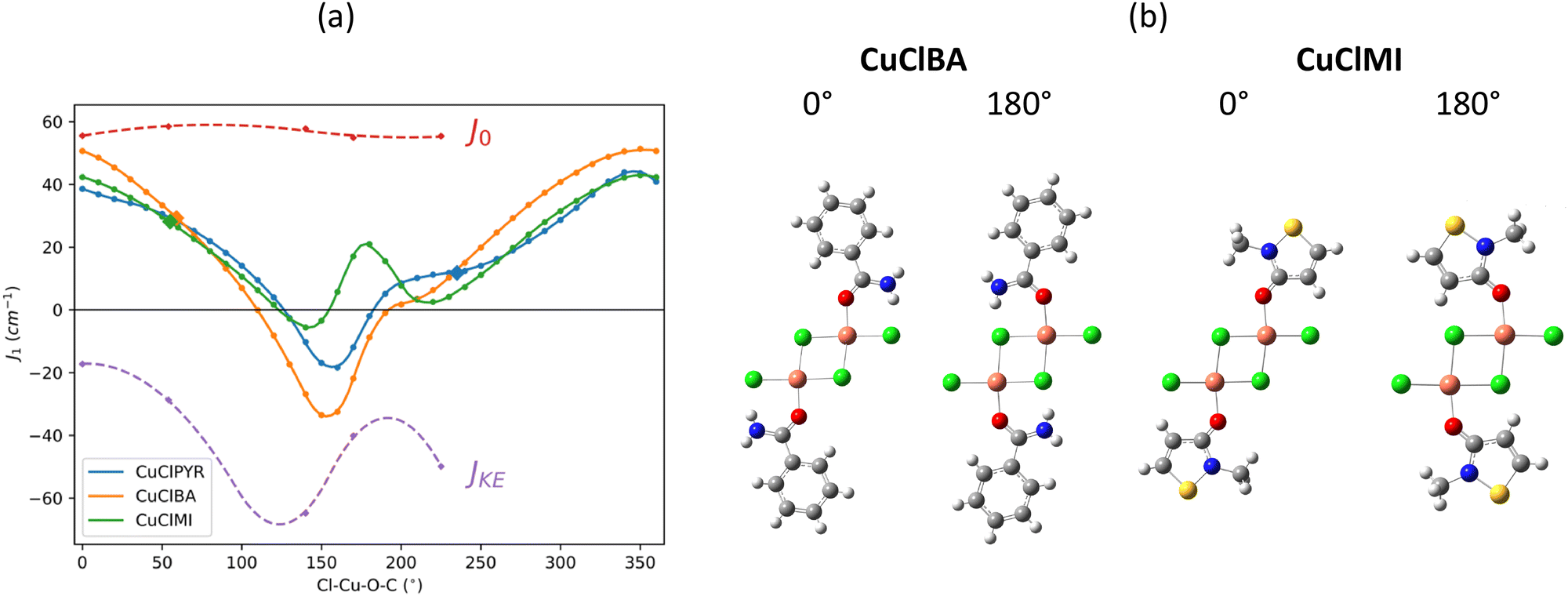 | ||
| Fig. 7 (a) Magnetic exchange decomposition data for J1D as a function of the Cl–Cu–O–C torsion angle with the values of J1D calculated at the experimental structure shown as large solid diamond symbols. J0 and JKE refer to the decomposed J1D for the various rotated CuClMI structures in Table 4. (b) Dimer structures of CuClBA and CuClMI at specific Cl–Cu–O–C angles. | ||
There is a major effect on J1D upon rotation. Importantly, all three amides behave similarly, and this explains why the J1 at the experimental positions (shown as larger solid symbols) is small for CuClPYR: the angle of the PYR in CuClPYR dampens the FM exchange across the dimer, but would result in an equal FM exchange to the other amides, if in a similar orientation (see blue line next to large solid orange and green symbols in Fig. 7(a)). To our knowledge, the effect of satellite orientation has not been reported before, with most of the focus being on either the bridge,9 or the identity10 of the terminal ligands.
While the three amides do behave similarly, there are some distinguishing features. CuClBA is distinct since it spans a larger range of values compared to the other two amides (−40 cm−1 < J +50 cm−1). J1D is at a maximum when the ligand is at 0° (see Fig. 7(b), co-planar with the inorganic bridge, while all of the curves have a minimum at ca. 180°, which is interrupted by either a local maximum/peak shoulder. To understand these features, J1D was decomposed and is illustrated as striped lines in ((Fig. 7(a))).
J CuClMI1D was decomposed at torsion angles of 0°, 54° (exp.), 140°, 180° and 225° (exp. value of CuClPYR at 135°–360° = −225°). From Table 4 it is clear that J0, ΔJCP, and JOth remain relatively constant across the rotation, while the kinetic exchange ΔJKE varies greatly, as shown in Fig. 7. ΔJKE matches JCuClMI1D well, meaning that the rotation only appreciably affects ΔJKE.
| Structure | J 1D | J 0 | ΔJKE | ΔJCP | J Oth |
|---|---|---|---|---|---|
a
 ![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 35 was decomposed using a Cu2Cl62− dimer (note values of original publication were divided by 2 to agree with Heisenberg Hamiltonian here used). 35 was decomposed using a Cu2Cl62− dimer (note values of original publication were divided by 2 to agree with Heisenberg Hamiltonian here used).
|
|||||
| CuClMI(0°) | 42.5 | 55.5 | −17.3 | 7.3 | −3.0 |
| CuClMI(140°) | −5.0 | 57.8 | −64.8 | 9.2 | −7.1 |
| CuClMI(180°) | 21.0 | 54.4 | −9.3 | 0.0 | 0.0 |
| CuClMI(225°) | 6.9 | 55.4 | −49.9 | 8.3 | −6.8 |
| CuClMI | 28.4 | 56.7 | −31.7 | 7.9 | −4.5 |
| CuBrMI | 42.3 | 70.4 | −29.8 | 7.9 | −6.2 |
| CuClPYR | 12.2 | 49.8 | −39.6 | 7.2 | −5.2 |
| CuBrBA 6 | 42.4 | 72.3 | −31.6 | 7.9 | −3.1 |
| CuClBA 6 | 29.7 | 55.6 | −29.5 | 7.6 | −2.0 |
|
Cu2Cl62−35a |
−10.4 | 53.5 | −70.3 | 6.5 | |
In the case of Cu2Cl62− (see Table 4),35 there is significantly more delocalisation of spin density onto two terminal chloride ligands, compared to the example shown here, which results in the massive ΔJKE overpowering J0 to produce an AFM dimer.
The magnitude of J1D is larger in CuBrBA and CuBrMI, compared to the chloride analogues. Magnetic exchange decomposition reveals that this is due to significantly larger direct exchange J0, while ΔJKE is largely unaffected, and can be explained by the larger orbital overlap of μ-Br− with Cu(II), facilitating better direct exchange.
ΔJCuXBAKE and ΔJCuXMIKE are similar, irrespective of the X halide bridge. A terminal chloride ligand, on the other hand, results in a 2-fold increase of  . From all of the evidence in this section, therefore, the terminal ligands exclusively modify the kinetic exchange, via their orientation and identity, while the identity of the bridging ligand affects the direct exchange exclusively.
. From all of the evidence in this section, therefore, the terminal ligands exclusively modify the kinetic exchange, via their orientation and identity, while the identity of the bridging ligand affects the direct exchange exclusively.
Tetramer rotation
While J1 is modulated by the orientation of the ligands, it is J3 that determines whether a structure has a FM or AFM ground state. The values of J3D were shown to be unreliable previously.6 To obtain a more reliable decomposition of J3, tetrameric clusters of CuClMI, CuClBA, CuClPYR were constructed. In addition, to illustrate that J3 too is modulated by the orientation of the ligand, the MI ligand in CuClMI was rotated CuClMI(Rot) to match the torsional angle of the PYR ligand in CuClPYR, the results of which are given in Table 5.| J i | J i | J 0 | ΔJKE | ΔJCP | J Oth |
|---|---|---|---|---|---|
| The decomposition of J2T and J4T was not done due to their small magnitudes. | |||||
| J CuClMI1T | 32.9 | 58.5 | −28.7 | 8.1 | −4.9 |
| J CuClMI(Rot)1T | 6.9 | 55.4 | −49.9 | 8.3 | −6.8 |
| J CuClPYR1T | 11.5 | 49.0 | −39.7 | 7.2 | −5.1 |

|
18.9 | 53.5 | −36.7 | 7.8 | −5.7 |
| J CuClMI3T | −2.5 | 1.2 | −3.0 | −0.3 | −0.4 |
| J CuClMI(Rot)3T | 0.5 | 1.0 | −0.1 | −0.3 | −0.1 |
| J CuClPYR3T | 2.0 | 3.1 | −0.6 | −0.2 | −0.3 |
| J CuClBA3T | −6.0 | 3.0 | −7.3 | −0.7 | −0.9 |
| J CuClMI(Rot)2T | 0.57 | ||||
| J CuClMI(Rot)4T | 0.1 | ||||
The magnitude of JCuClMI(Rot)1T is reduced by 80% upon rotation of MI, and matches the lower value of JCuClPYR1T at the same angle. This is due to a large increase in kinetic exchange across the dimer, dampening the FM direct exchange. The inclusion of hydrogen-bonded molecules to the O-donor atom also has a significant effect on J1T, and has an effect on both the kinetic and direct exchange components. This shows that the orientation of the ring, and weak interactions with non-magnetic molecules near the magnetic orbitals have a massive effect on the magnitude of J1. Importantly, there is a reversal in the sign of JCuClMI(Rot)3T to a weakly FM interaction. This matches the magnitude and sign of JCuClPYR3T, and shows that it is not the unique chemistry of PYR that causes the emergence of FM bulk properties, but it is simply a function of the angle at which the ligand is oriented, with respect to the magnetic orbitals. Decomposing JCuClMI3T and JCuClMI(Rot)3T shows that the change from AFM to FM is again due to ΔJKE, which demonstrates that the orientation of the non-magnetic orbitals in the case of CuClMI(Rot) and CuClPYR allows a larger amount of spin density to delocalise from the magnetic orbitals. The real space representation of ΔJKE can be visualised by the spin density differences between the Broken-Symmetry Restricted Open-Shell Ms = 0 (BS,RO) solution and the Broken-Symmetry solution with the Unrestricted, but Frozen non-magnetic Core orbital solution, (BS,UFC), shown in Fig. 8. This means that when the MI ligand is at its experimental torsion angle of 54°, (ΔJKE = −28.7 cm−1) the magnetic orbitals are confined to the O-, C-, and N-atoms, compared to when the angle is at 225°, (ΔJKE = −49.9 cm−1) where the spin is allowed to relax further into MI, propagating onto the O-, C- N-, CCarb.- and S-atoms. CuClPYR beaves similarly to CuClMI(Rot), with spin density propagating deep into the PYR ligand. Finally, let us mention that the local peaks at ca. 180° are due to opposite spin propagating into the portions of the ligand that is co-linear with the “opposite” Cu-μCl axis (shown as a blue volume in Fig. 8), reducing ΔJKE.
 | ||
| Fig. 8 Spin density difference of (BS,UFC) – (BS,RO) at various Cl–Cu–O–C torsion angles, which is a visual representation of ΔJKE. | ||
The manipulation of the terminal ligand can, therefore, lead to an alteration of the magnetic exchange in these materials, both in and between dimers. This means that isomorphs that have different ligand orientations can produce vastly different magnetic responses, and may even explain the anomaly observed for CuClMI at 15 K. In addition, ligand orientation disorder in structures, or doping can induce complex magnetic behavior. Finally, if the orientation of the rings can be manipulated in situ, a spin device can, in principle be constructed to flip between singlet/triplet states by just altering the orientation of the terminal ligand, for instance, by means of an electrical field as in the case of a prototypical biphenyl-based molecular system.41
Conclusions
The CuBrMI complex was structurally characterised and was found to be isostructural to the complex CuClMI reported in the literature.2 The structures of the inorganic scaffolding of CuClMI and CuBrMI are essentially identical with slight differences due to different Cu–X bond lengths. Within the FPBU11 strategy, the values of J1 agree well with previous calculations for CuClMI2 and are similar to those calculated for the benzamide analogue.6 The inclusion of hydrogen-bonded molecules in CuClPYR was shown to significantly affect the magnetic exchange in the dimer.The computed macroscopic magnetic susceptibility χm(T) curves of CuXMI agree excellently with the experimentally observed AFM behaviour of these compounds determined in this study, while the FM bulk properties of CuClPYR is, too, replicated well, especially if hydrogen-bonded molecules are added. The magnetic topology of CuXMI is described by alternating FM/AFM chains with limited FM NNN interactions, while CuClPYR is best described as a spin ladder with some cross-rung interactions.
The rotation of three amide-ligand dimers, extracted from CuClPYR, CuClBA, and CuClMI, shows that the magnetic interaction in, and between dimers can be modulated by the orientation of the ligands. Importantly, we showed that the FM properties of CuClPYR can be explained by the ligand's orientation, and would act similarly to other amides, if oriented similarly. Furthermore, it was shown through magnetic exchange decomposition that the identity, and orientation of the terminal/satellite ligands selectively affected the kinetic exchange in, and between dimers, while the bridging ligands only affect the direct exchange. This is due to the delocalization of spin density onto the terminal ligands to various degrees, which is dependent on the orientation of the ligand as well. When this parameter is targeted, the magnetic exchange of dimer pairs with terminal ligands can be tuned from AFM to FM by altering the coordination torsion angle.
The identity of the terminal ligand influences the amount of kinetic exchange, with terminal chloride ligands providing extensive delocalisation of magnetic orbitals which Rodríguez et al.10 examined previously, along with terminal ammonia and terminal amine ligands. The terminal halide ligands produced AFM exchange across the dimers, while ammonia and amine ligands produced FM exchange. Their work focussed on magnetic exchange in the dimer, while the identity of the terminal ligands between dimers is also presented here. This work also adds the amide ligand to the system, which indeed results in FM exchange in the dimer, and it is not very sensitive to structural differences beyond the carbonyl atom. Critically, the orientation of the terminal/satellite ligands is shown to affect the magnetic exchange in and between dimers. To the best of our knowledge, the effect of rotation of nonmagnetic ligands on magnetic exchange has not been shown, and the mechanism of this dependence was isolated to the kinetic exchange. This new information introduces an additional design tool to tune the magnetic exchange. The switch of magnetic exchange from FM to AFM is observed for the CuClMI system at a C–O–Cu–Cl angle of 225°, although it is very weakly AFM at that angle. It is possible that there exists a ligand system where the magnetic exchange in the dimer is switchable to produce a triplet or singlet state if the kinetic exchange of the ligand is stronger. This new discovery can lead to better control of magnetic exchange in molecules, or even allow for the manipulation of spin in quantum devices.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank Dr F. Malan for assistance with crystallographic aspects. MR acknowledges financial support from the University of Pretoria and Sasol. SC acknowledges a PhD bursary from the National Research Foundation (Grant no: 81614), and funding of a research visit to the University of Barcelona by the University of Pretoria Postgraduate Study Abroad Program. MMT and CPL are grateful for funds from PCI Synthesis, Inc. (now SEQENS) toward the purchase of the D8 Focus diffractometer, from the National Science Foundation (IMR-0314773) for purchase of the MPMS SQUID magnetometer, and from the Kresge Foundation for both instruments. MD and JJN thank the financial support from the Spanish Ministerio de Economía y Competitividad (Project No. PID2020-117803GB-I00 and CTQ2017-87773-P/AEI/FEDER), Spanish Structures of Excellence María de Maeztu program through grants MDM-2017-0767/CEX2021-001202-M, and Generalitat de Catalunya (Project No. 2017 SGR 348 and 2021 SGR 354). The authors thank the Institute of Theoretical and Computational Chemistry of the Universitat de Barcelona (IQTCUB) and the Centre for High-Performance Computing of South Africa (CHPC) for the use of their computational resources.References
- G. Givaja, P. Amo-Ochoa, C. J. Gómez-García and F. Zamora, Chem. Soc. Rev., 2012, 41, 115–147 RSC.
- M. Kato, K. Hida, T. Fujihara and A. Nagasawa, Eur. J. Inorg. Chem., 2010, 495–502 Search PubMed.
- C. Liu, D. R. Talham, J.-H. Park, E. Cizmar and M. W. Meisel, J. Chem. Phys., 2004, 120, 1140–1141 CrossRef CAS PubMed.
- S. N. Herringer, A. J. Longendyke, M. M. Turnbull, C. P. Landee, J. L. Wikaira, G. B. Jameson and S. G. Telfer, Dalton Trans., 2010, 39, 2785–2797 RSC.
- C. P. Landee and M. M. Turnbull, Eur. J. Inorg. Chem., 2013, 2266–2285 CrossRef CAS.
- S. Coetzee, M. M. Turnbull, C. P. Landee, J. J. Novoa, M. Deumal, S. Vela and M. Rademeyer, Polyhedron, 2020, 185, 114603 CrossRef CAS.
- K. C. Shortsleeves, M. M. Turnbull, C. B. Seith, E. N. Tripodakis, F. Xiao, C. P. Landee, L. N. Dawe, D. Garrett, G. D. de Delgado and B. M. Foxman, Polyhedron, 2013, 64, 110–121 CrossRef CAS.
- M. G. Banks, R. K. Kremer, C. Hoch, A. Simon, B. Ouladdiaf, J.-M. Broto, H. Rakoto, C. Lee and M.-H. Whangbo, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 80, 024404 CrossRef.
- O. Castell, J. Miralles and R. Caballol, Chem. Phys., 1994, 179, 377–384 CrossRef CAS.
- A. Rodrìguez-Fortea, P. Alemany, S. Alvarez and E. Ruiz, Inorg. Chem., 2002, 41, 3769–3778 CrossRef PubMed.
- J. J. Novoa, M. Deumal and J. Jornet-Somoza, Chem. Soc. Rev., 2011, 40, 3182 RSC.
- F. H. Allen and C. R. Groom, Angew. Chem., Int. Ed., 2014, 53, 662–671 CrossRef PubMed.
- Bruker, SAINT+, Bruker AXS Inc., Madison, Wisconsin, USA, 2012 Search PubMed.
- Bruker, SADABS, Bruker AXS Inc., Madison, Wisconsin, USA, 2001 Search PubMed.
- Bruker, APEXII, Bruker AXS Inc., Madison, Wisconsin, USA, 2012 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- L. J. Farrugia, J. Appl. Crystallogr., 2012, 45, 849–854 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- C. F. Macrae, P. R. Edgington, P. McCabe, E. Pidcock, G. P. Shields, R. Taylor, M. Towler and J. van de Streek, J. Appl. Crystallogr., 2006, 39, 453–457 CrossRef CAS.
- D. C. Palmer, Z. Kristallogr.-Crys. Mater., 2015, 230, 559–572 CrossRef CAS.
- V. Vreshch, J. Appl. Crystallogr., 2010, 44, 219–220 CrossRef.
- L. Noodleman, J. Chem. Phys., 1981, 74, 5737–5743 CrossRef CAS.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS PubMed.
- S. H. Vosko, L. Wilk and M. Nusair, Can. J. Phys., 1980, 58, 1200–1211 CrossRef CAS.
- P. J. Stephens, F. J. Devlin, C. F. Chabalowski and M. J. Frisch, J. Phys. Chem., 1994, 98, 11623–11627 CrossRef CAS.
- A. Schafer, H. Horn and R. Ahlrichs, J. Chem. Phys., 1992, 97, 2571–2577 CrossRef.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian∼09 Revision D.01, 2009 Search PubMed.
- F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2018, 8, e1327 Search PubMed.
- O. Kahn, Molecular Magnetism, VCH, 1993 Search PubMed.
- T. Terencio, R. Bastardis, N. Suaud, D. Maynau, J. Bonvoisin, J. P. Malrieu, C. J. Calzado and N. Guihery, Phys. Chem. Chem. Phys., 2011, 13, 12314–12320 RSC.
- C. J. Calzado, C. Angeli, D. Taratiel, R. Caballol and J.-P. Malrieu, J. Chem. Phys., 2009, 131, 044327 CrossRef PubMed.
- C. J. Calzado, J. Cabrero, J. P. Malrieu and R. Caballol, J. Chem. Phys., 2002, 116, 2728–2747 CrossRef CAS.
- C. J. Calzado, J. Cabrero, J. P. Malrieu and R. Caballol, J. Chem. Phys., 2002, 116, 3985–4000 CrossRef CAS.
- E. Coulaud, J.-P. Malrieu, N. Guihery and N. Ferre, J. Chem. Theory Comput., 2013, 9, 3429–3436 CrossRef CAS PubMed.
- E. Coulaud, N. Guihery, J.-P. Malrieu, D. Hagebaum-Reignier, D. Siri and N. Ferre, J. Chem. Phys., 2012, 137, 114106 CrossRef PubMed.
- J. B. Goodenough, Phys. Rev., 1955, 100, 564–573 CrossRef CAS.
- J. Kanamori, J. Phys. Chem. Solids, 1959, 10, 87–98 CrossRef CAS.
- S. Vela, A. Sopena, J. Ribas-Arino, J. J. Novoa and M. Deumal, Chem. – Eur. J., 2014, 20, 7083–7090 CrossRef CAS PubMed.
- G. David, F. Wennmohs, F. Neese and N. Ferr, Inorg. Chem., 2018, 57, 12769–12776 CrossRef CAS PubMed.
- K. Jutglar Lozano, R. Santiago, J. Ribas-Arino and S. T. Bromley, Phys. Chem. Chem. Phys., 2021, 23, 3844–3855 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Crystallographic data and powder X-ray diffraction patterns; dimer and tetramer cluster models; discussion of magnetic models and their calculated magnetic susceptibility; decomposition of JAB; analysis of CuClPYR; spin densities and SOMO's for dimer magnetic models; calculation of magnetic properties of CuClMI; comparison of magnetic data for CuClMI; energy spectra and thermal population. CCDC 2191590. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2cp05629a |
| This journal is © the Owner Societies 2023 |