
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Direct measurement of single-molecule dynamics and reaction kinetics in confinement using time-resolved transmission electron microscopy†‡
Kayleigh L. Y.
Fung
a,
Stephen T.
Skowron
 a,
Ruth
Hayter
a,
Stephen E.
Mason
a,
Benjamin L.
Weare
a,
Nicholas A.
Besley§
a,
Quentin M.
Ramasse
bc,
Christopher S.
Allen
de and
Andrei N.
Khlobystov
*a
a,
Ruth
Hayter
a,
Stephen E.
Mason
a,
Benjamin L.
Weare
a,
Nicholas A.
Besley§
a,
Quentin M.
Ramasse
bc,
Christopher S.
Allen
de and
Andrei N.
Khlobystov
*a
aSchool of Chemistry, University of Nottingham, University Park, Nottingham NG7 2RD, UK. E-mail: Andrei.Khlobystov@nottingham.ac.uk
bSuperSTEM Laboratory, SciTech Daresbury Campus, Keckwick Lane, Daresbury WA4 4AD, UK
cSchool of Chemical and Process Engineering and School of Physics and Astronomy, University of Leeds, Leeds LS2 9JT, UK
dElectron Physical Sciences Imaging Centre, Diamond Light Source Ltd., Oxfordshire OX11 0DE, UK
eDepartment of Materials, University of Oxford, Parks Road, Oxford OX1 3PH, UK
First published on 17th February 2023
Abstract
We report experimental methodologies utilising transmission electron microscopy (TEM) as an imaging tool for reaction kinetics at the single molecule level, in direct space and with spatiotemporal continuity. Using reactions of perchlorocoronene (PCC) in nanotubes of different diameters and at different temperatures, we found a period of molecular movement to precede the intermolecular addition of PCC, with a stronger dependence of the reaction rate on the nanotube diameter, controlling the local environments around molecules, than on the reaction temperature (−175, 23 or 400 °C). Once initiated, polymerisation of PCC follows zero-order reaction kinetics with the observed reaction cross section σobs of 1.13 × 10−9 nm2 (11.3 ± 0.6 barn), determined directly from time-resolved TEM image series acquired with a rate of 100 frames per second. Polymerisation was shown to proceed from a single point, with molecules reacting sequentially, as in a domino effect, due to the strict conformational requirement of the Diels–Alder cycloaddition creating the bottleneck for the reaction. The reaction mechanism was corroborated by correlating structures of reaction intermediates observed in TEM images, with molecular weights measured by using mass spectrometry (MS) when the same reaction was triggered by UV irradiation. The approaches developed in this study bring the imaging of chemical reactions at the single-molecule level closer to traditional concepts of chemistry.
Introduction
The development of low-dimensional nanomaterials that are macroscopic in fewer than three dimensions have boosted new methodologies for studying chemical reactions at the nanoscale. For example, nanomaterials have proved to be useful as supports for molecules1,2 and templates for controlling the morphology of products of their reactions.3,4 Notably, carbon nanomaterials, such as graphene and single-walled carbon nanotubes (SWNTs), have been employed as atomically thin membranes and reaction vessels, respectively, with the latter providing a platform for synthesizing and performing nanoconfined reactions of a wide range of different reactants.5 In addition to providing morphological control, this type of encapsulation or confinement on the nanoscale can have a substantial impact on chemical reactions, affecting their rates and selectivities, activities, and stabilities of catalytic centres.6–8 Reaction rates in nanoreactors can be affected by local concentrations of reactants or by stabilisation of transition states, both enthalpically (by favourable product–host interactions) and entropically (by pre-organising reactants and restricting their degrees of freedom);9 or by non-classical behaviour due to different thermodynamics10 or kinetics11–13 in confinement. Studying chemical reactions in nanoreactors therefore presents opportunities to gain insight into fundamental phenomena in addition to providing useful routes to control of reactions.Reaction mechanisms are typically studied using macroscale reaction kinetics measurements where changes of molecular concentrations are followed by techniques such as nuclear magnetic resonance (NMR), absorption, scattering or luminescence spectroscopies, or X-ray diffraction (XRD). Surface analysis techniques such as X-ray photoelectron spectroscopy (XPS), scanning electron microscopy (SEM), scanning tunnelling microscopy (STM), and atomic force microscopy (AFM) allow a closer glimpse of reaction mechanisms confined to a surface, such as metals or graphite. Indeed, the scanning probe methods (STM and AFM) can achieve atomic resolution, with the ability to detect structures of reaction products and intermediates.14–16 Transmission electron microscopy (TEM) is another imaging technique capable of atomic resolution, coupled with a high temporal resolution relative to scanning probe methods.
In the context of studying reaction mechanisms, TEM is potentially an ideal imaging tool due to its atomic spatial resolution, ability to image the entirety of 3D molecular structures, fast time resolution (thousands of fps), as well as a rich range of associated in situ spectroscopies. Importantly, controlled interactions of the incident electrons with atoms in the sample can provide a stimulus for inducing reactions. Although this is traditionally considered unwanted ‘beam damage’ during structural characterisation, it is increasingly being used as an intentional stimulus for driving reactions that can be simultaneously followed with atomic or molecular resolution imaging. In addition to the formation of new and previously inaccessible materials and structures,17–22 the combined use of the electron beam as an imaging probe and a stimulus of chemical reactions (ChemTEM approach) is an exciting technique for probing fundamental physical and chemical phenomena.23–25 With carefully designed experiments, statistical information can be acquired directly from images to give kinetic information about the reactions.26–32 The application of TEM to studying reactions of molecules has great potential to become a new analytical tool for chemists. However, further development of methodologies is required in order to link the chemistry of single molecules observed by TEM to the traditional quantitative chemical kinetics that most chemists are familiar with.
Here, we have developed experimental methodologies that connect single-molecule imaging and analysis with core concepts of chemical kinetics. We selected beam-induced polymerisation reactions of perchlorocoronene (PCC) molecules confined in nanotubes as a test system for our methodology. Firstly, we have examined the impact of the electron beam on reactions of PCC@SWNT at room temperature, followed by cryogenic and elevated temperature imaging to decouple the role of thermal energy from the energy delivered to the molecules by the electron beam. Secondly, studying the polymerisation reaction over time at high temporal resolution allowed us to perform a kinetic analysis of the electron beam induced reaction, measuring the order and cross-section of the reaction. Finally, we have correlated the reaction of PCC at the single-molecule level with a bulk analysis technique (matrix-assisted laser desorption/ionisation time of flight mass spectrometry; MALDI-ToF MS), complementing the understanding gained from TEM and providing further corroboration of the proposed reaction mechanisms. Overall, the approaches developed in this study push the imaging of chemical reactions in direct space and real time by TEM closer to traditional concepts of chemistry.
Results and discussion
Pre-reaction and polymerisation dynamics of confined molecules
Under 80 keV electron beam irradiation, PCC molecules encapsulated within SWNTs (Fig. 1(a)) are known to undergo a slow process of dissociation of C–Cl bonds via direct knock-on transfer of energy from the electron beam to the carbon atom (up to 15.8 eV). Previously, using experimental measurements and computational modelling we described the mechanism of beam-induced reactions of PCC, which occur on a timescale commensurate with typical imaging rates used in TEM.33 Our time-resolved imaging, using a continuous electron beam flux of 2.17 × 106 e− nm−2 s−1, revealed that the reaction does not begin instantaneously. In a typical experiment at 23 °C, we observed a latent period of tens of seconds where no intermolecular reactions occurred, preceding polymerisation of PCC under these conditions (Fig. 1(b)), due to the steric requirement of the Diels–Alder cycloaddition. In the SWNT, the PCC molecules pack tightly together in ‘stacks’, forming unfavourable orientations for the cycloaddition (Fig. 2(a)); there is a requirement for an angle of ca. 100° between two PCC molecules for the Diels–Alder cycloaddition to take place (Fig. 2(b)). Therefore, sufficient pre-reaction movement has to take place before any polymerisation can begin (Fig. 2(c) and (d)), resulting in the ‘latent fluence’ which contributes to the exceptional beam stability of PCC@SWNTs. | ||
| Fig. 1 (a) Left: Face-on and edge-on structural models of perchlorocoronene (PCC), illustrating its non-planar structure due to the steric bulk of chlorine atoms. Right: A model of PCC encapsulated within a single walled carbon nanotube (SWNT) and a micrograph of PCC@SWNT where the molecules appear as straight lines. The scale bar is 1 nm. (b) A series of micrographs illustrating the stack of PCC molecules changing orientations inside the nanotube. Yellow arrows are used to highlight the angle of the molecules with respect to the nanotube axis. Movement of the stack is required to create sufficient space for intermolecular Diels–Alder cycloadditions. The electron flux used was 2.17 × 106 e− nm−2 s−1. The scale bar is 1 nm. | ||
 | ||
| Fig. 2 (a) Tightly packed PCC molecules, separated by van der Waals distances, inside SWNTs (black parallel lines represent nanotube sidewalls) are in an unfavourable orientation for Diels–Alder cycloaddition. (b) Once sufficient pre-reaction movement has taken place to increase the intermolecular space, PCC molecules can adopt more favourable orientations for the cycloaddition (ca. 100° between two PCCs). (c) Reaction scheme of the Diels–Alder cycloaddition between a PCC molecule (diene) and the aryne species (dienophile) after loss of Cl2. (d) A schematic diagram of the Diels–Alder cycloaddition of PCC molecules within a SWNT. (e) A molecule of PCC within the stack changes orientation over a second of imaging. The red arrow on the right of the nanotube indicates a region with no distinct molecular structure where motion is rapid, and polymerisation had begun. The polymer is able to twist, freeing up space for additional movement of molecules. This can be seen as a Y-shaped feature, indicated by the red arrow on the left, due to movement of a PCC molecule within the stack during image capture. (f) A series of micrographs showing the planarisation of two PCC adducts, indicated by red arrows, appear as two parallel wiggly lines at 554.20 s. The lines have two kinks, suggesting two joints between three molecules, i.e. the adducts are both trimers. Over a period of imaging (approximately 2.49 s), the two lines became parallel (red arrow at 556.69 s). The adducts appeared to planarise sequentially; the left sides of both of the angular adducts became planar before the right sides (red arrow at 555.45 s). The electron flux used in both (e) and (f) was 2.17 × 106 e− nm−2 s−1. (g) STEM imaging of PCC@SWNTs reveals the S-shape of PCC (in HRTEM, PCC appears as a straight line). The point probe of STEM allows us to capture a more accurate image of the shape of PCC. (h) After 260.18 s and a total electron fluence of 7.56 × 108 e− nm−2, PCC had polymerised into a nanoribbon. The chlorine substituents terminating the nanoribbon can be seen in the STEM image. Scale bars are 1 nm. | ||
Once the reaction was initiated, there was a slow (over several minutes) “domino” effect where the polymerisation continued along the nanotube from the location where the first PCC dimer was formed (Fig. 2(e)). PCC molecules on the right side of the nanotube began to transform into elongated structures, corresponding to a nascent nanoribbon product, which can twist and allow space for the stack of molecules to translate and react. Y-Shaped molecules in the middle of the stack have been observed at this stage of the process (Fig. 2(e)), which is likely a result of a PCC molecule moving from one position to another during the image capture, similar to that observed for PCC on graphene.34 Given that the capture rate is 100 fps in our experiments, the observed delocalisation of molecules highlights the increased freedom of movement of PCC in the nanotube once polymerisation had been initiated.
The next step is rearrangement and planarisation of Diels–Alder adducts, driven by aromatisation stabilising the product.33 The fast image capture rate allowed investigation of the dynamics of angular PCC trimers. These precursors of nanoribbons (Fig. 2(f); red arrow at 554.20 s) underwent a series of rearrangements and planarisation over approximately 2.49 s while translating during image capture, resulting in blurring of some images. Additional blurring was observed in the parts of the trimers undergoing rearrangement, suggesting increased movement or vibration (the semi-planarised trimers are indicated by the red arrow at 555.45 s). This suggested that the rearrangements occurred sequentially along the trimers rather than simultaneously. Stacking of the two trimers together appear to facilitate the process, as sequential planarisation would enable maximum π–π stacking between the two trimers. After the rearrangements, the planar trimers appeared as two distinct parallel lines again (Fig. 2(f); red arrow at 556.69 s). The overall reaction of PCC transformation into the final nanoribbon product took place over 9 minutes and 29.09 seconds (or 569.09 s), which corresponds to a total fluence of 1.23 × 109 e− nm−2.
Further structural information can be gained from STEM (scanning TEM) imaging of PCC@SWNTs. In STEM, the electron beam (here a 60 keV beam energy) is rastered as a sub-Angstrom probe across the sample. 60 keV was used in order to eliminate beam damage to the nanotube structure since a locally stronger electron flux is necessary for higher signal to noise ratio in STEM mode. The molecules were not under continuous parallel illumination, reducing molecular vibration, and allowing us to capture the S-shape of the PCC molecule (Fig. 2(g)). After a flunce of 7.56 × 108 e− nm−2, the molecules had reacted to form a nanoribbon, where the chlorine atoms are terminating the zigzag edges (Fig. 2(h), recorded 260.18 s after the initial image in Fig. 2(g)). It is interesting to note that electron beam-induced reactions in STEM vary depending on scan rate, or instantaneous pixel charge, even when total fluence and flux are fixed.35 This suggests that the dynamics of PCC polymerisation in STEM may differ from those in TEM.
Effects of temperature on electron beam activated polymerisation in confinement
Measuring reaction rates at different temperatures is a classical way of probing reaction mechanisms. In the case of PCC polymerisation in confinement, thermal activation may also have an effect on the pre-reaction movement and reorientation of molecules (Fig. 1 and 2). We carried out a series of experiments to determine the effects of temperature on the pre-reaction dynamics and electron beam induced polymerisation of PCC, by measuring the latent fluence at −175 °C, 200 °C, and 400 °C using electron fluxes on the order of 105 e− nm−2 s−1. It is important to note that PCC molecules are thermally stable at all temperatures used in TEM experiments (PCC was inserted into SWNTs at 400 °C) and therefore no thermally induced reactions of PCC may occur without the influence of the electron beam.32In a recent study we established the range of the latent fluence for PCC polymerisation at room temperature to be between 5.4 × 106–6.3 × 107 e− nm−2 (Fig. 3(a)).32 An additional important factor to consider is the initial stacking of the molecules with respect to their neighbours and the SWNT sidewalls: for PCC molecules this is determined by the diameter of the nanotube in a similar fashion to examples of diameter-dependent stacking of coronene36,37 and C60 in nanotubes.38 Nanotubes with diameters wider than a PCC molecule contained stacks of molecules that were aligned perpendicular to the nanotube wall (Fig. 3(c)), maximising the van der Waals interactions between PCC molecules as well as between molecules and the nanotube. In contrast, nanotubes with narrower diameters contained stacks of molecules that were slanted at an angle (between 10–40°) with respect to the nanotube axis (Fig. 3(b)). Given the steric requirements of the Diels–Alder reaction, it is plausible that the latter case would exhibit lower latent fluences, requiring less movement to achieve the near-orthogonal orientation required for Diels–Alder cycloadditions. However, plotting the latent fluence vs. nanotube diameter (Fig. 3(a)) reveals no clear trend, with latent fluences falling in the range 5.4 × 106–6.3 × 107 e− nm−2.
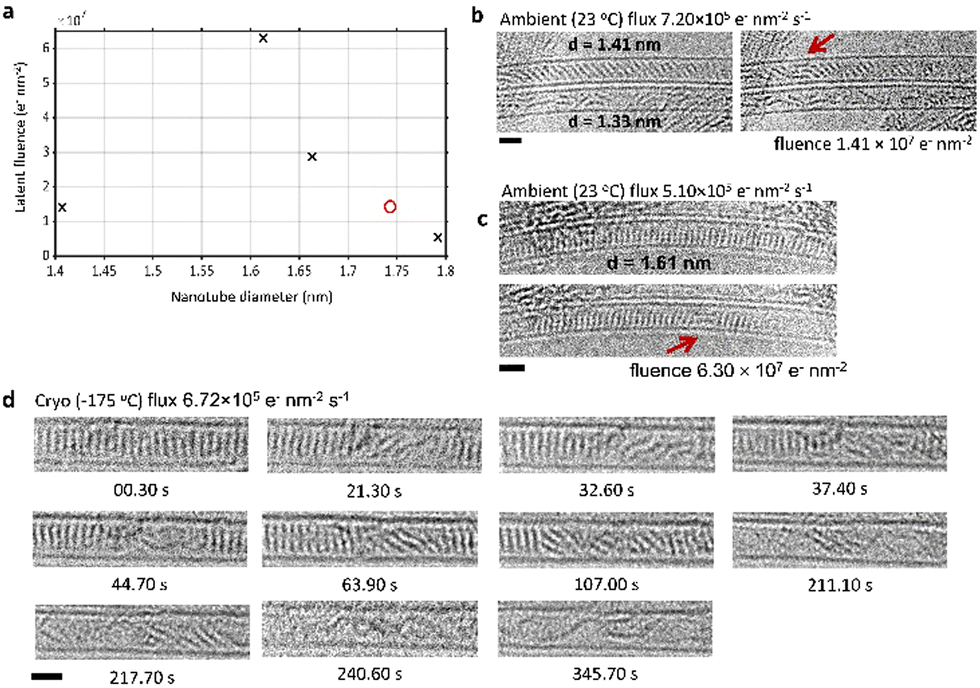 | ||
| Fig. 3 (a) A plot of the latent fluence against SWNT diameter for 23 °C (black X data points) and −175 °C (red O data point) does not have a clear trend. (b) and (c) Several image series of PCC@SWNT in nanotubes of different diameters (indicated on images) were acquired at 23 °C for comparison to heating experiments. For each series of images, the first image in the series is shown alongside the image of the first dimer (red arrows). The slant of the stack of molecules can be seen in (b). Electron fluxes, latent fluences, nanotube diameters, and time stamps are shown. (d) A time series images of PCC@SWNT acquired at −175 °C. An electron flux of 6.72 × 105 e− nm−2 s−1 was used over a period of 288.40 s which corresponded to a total fluence of 1.94 × 108 e− nm−2. First dimer formation was observed at 21.3 s or a fluence of 1.43 × 107 e− nm−2. Scale bars are 1 nm. | ||
Given the requirement of molecular motion prior to the start of the reaction, it could be expected that varying the temperature would have a substantial impact on the latent fluence, especially at lower temperatures where this motion could be ‘frozen out’. However, even at the cryogenic temperature of −175 °C, molecules were still observed to move and shuffle, sufficient for the electron beam induced polymerisation to take place (Fig. 3(d)). Time-resolved imaging of PCC polymerisation at −175 °C showed first dimer formation after 21.3 s, or a fluence of 1.43 × 107 e− nm−2, which is in the same fluence range as the experiments at 23 °C (Fig. 3(b) and (c)). We have calculated the interaction energy of two PCC molecules in a stack confined in the SWNT as 83.1 kJ mol−1, using density functional theory and the AIRBED method (ESI,‡ Section 1).39 At this energy, the separation of two PCC molecules cannot appreciably occur thermally at either 23 °C or cryogenic temperatures, implying that the observed pre-reaction dynamics are driven by the electron beam. The latent fluence therefore corresponds to the fluence required for the electron beam to induce sufficient molecular motion.
In contrast to 23 °C and below, the calculated interaction energy implies that thermal separation of PCC molecules can occur to an appreciable degree (with a characteristic timescale on the order of seconds) at temperatures elevated above approximately 90 °C, which would potentially lead to qualitatively different reaction progress as thermal contributions to the pre-reaction dynamics become significant. Measurements of the latent fluence at two temperatures above this (200 and 400 °C) indeed exhibit very different behaviour to lower temperatures (Fig. 4). A strong dependence of the latent fluence on nanotube diameter appears at high temperature: in all of our observed image series, in SWNTs with diameters below ∼1.55 nm the PCC stack begins reacting at a fluence lower than we are able to record (i.e. dimerisation has occurred prior to the first interpretable image); while in SWNTs with diameters above ∼1.55 nm the PCC stack does not react at all, even at fluences above the established room temperature range (>6.3 × 107 e− nm−2). Notably, there was no apparent difference between the two elevated temperatures 200 and 400 °C.
 | ||
| Fig. 4 (a) A log plot of the latent fluence against SWNT diameter for 200 °C (black square data points) and 400 °C (red asterisk data points), showing elevated temperature limits (indicated with vertical lines) compared to room temperature range established in Fig. 3 (blue shaded region on the plot). (b) and (c) time series TEM images of PCC@SWNT acquired at (b) 200 °C and (c) 400 °C. Electron fluxes, latent fluences, nanotube diameters, and time stamps are shown. Adducts of PCC are indicated with red arrows. All scale bars are 1 nm. | ||
The elimination of the latent fluence at elevated temperatures in narrow SWNTs is consistent with our calculations – the thermal energy becomes significant enough to drive the molecular separation and shuffling, such that there is no longer a latent fluence due to this step being driven the electron beam. At both 200 and 400 °C, the molecules will have sufficient energy to overcome the separation barrier on a much faster timescale than TEM imaging, making the results at these temperatures comparable. However, the enhanced electron beam stability of the PCC stacks in wider nanotubes at elevated temperatures is unexpected. This change in behaviour occurs above the diameter where PCC molecules can stack perfectly perpendicularly to the nanotube walls, which would be expected to maximise the molecular interaction energy and increase the barrier to molecular separation – indeed, the largest latent fluence we measured at room temperature lies in this range of diameters – but there is no clear reason why additional thermal energy would increase the latent fluence. There are a number of possible explanations for this. One, although we observe the molecular stacks persisting for large electron fluences, individual molecules may undergo reversible reactions that cannot be observed in the projected images. Two, at these high temperatures, the electron beam-induced reaction has a recombination/diffusion component that is important, where the initial C–Cl bond breaking induced by the e-beam can be reversed by thermal activation (similar to C–C bond recombination accelerated at high temperatures under electron irradiation40), thus introducing a dependence on the nanotube diameter. Three, the electron beam-induced reaction could follow a different mechanism at such high temperatures i.e. the reactions between PCC molecules could become electron beam and thermally-induced, leading to different reaction pathways. The combination of the electron beam, temperature, and nanotube diameter affecting relative molecular orientations is complex, and the interplay of these factors could be affecting diffusion or recombination dynamics of emitted atoms, and hence observed molecular stabilities and latent fluences. The nano-confined reactions studied here therefore show an extreme sensitivity to small changes in their exact local environment and their reaction vessel, in contrast to bulk, unconfined reactions. This is a result of every single molecule being in close contact with the nanotube host, and highlights the importance of scalable methods for the production of single-chirality nanotubes.
Kinetics of electron beam induced polymerisation in confinement
The polymerisation of PCC molecules into a chlorine-terminated graphene nanoribbon was imaged over time in order to determine the experimental reaction cross section (Fig. 5). Conceptually, our approach is similar to traditional measurements of rate laws of bulk reactions (typically by spectroscopy or diffraction methods), but with one important difference – ChemTEM33 is a technique that allows us to study reaction kinetics on the single molecule level with spatiotemporal continuity. Changes in the number of individual molecules, countable directly in TEM images, can be related to the reaction time and electron fluence. This approach has the potential to elucidate the reaction mechanism and determine the kinetic parameters of the reaction. | ||
| Fig. 5 (a) Electron beam-induced reaction scheme where two PCC molecules react to form a dimer, with an observed reaction rate constant of kobs. (b) The overall electron beam-induced reaction from a stack of PCC molecules inside a nanotube to a chlorine-terminated graphene nanoribbon. The electron flux used was 2.17 × 106 e− nm−2 s−1 over a period of 569.09 s which corresponded to a total fluence of 1.23 × 109 e− nm−2. The scale bar is 0.5 nm. (c) Balanced equation of the electron beam induced reaction, from PCC molecules to a nanoribbon and a by-product of Cl2. (d) The electron beam-induced PCC polymerisation has a linear decrease in number of PCC molecules per total area of nanotube (nPCC/A0) over time. Each data point in this plot corresponds to each time stamped image in (a) apart from t = 0 s when irradiation had not begun (and there is no image). | ||
Using TEM images acquired at 100 fps, we followed the progress of PCC polymerisation by measuring changes in the number of PCC molecules over time. The number of unreacted molecules (which appear as uniform 1.41 nm straight lines; Fig. 5(b)) were counted in each micrograph. We followed loss of the reactants rather than formation of the products since the products are non-linear and flexible, making it difficult to precisely discern their increase over time.
The initial concentration of PCC molecules in the nanotube is 2.06 mol L−1. However, our reaction stimulus is quite different from those studied in bulk kinetics measurements. The electron beam strikes an area of the sample per unit time and the driving force for the reaction occurs along an axis perpendicular to the nanotube. Because of this, we operate with areal number density of the molecules, described as the number of molecules nPCC per area of nanotube A0 in nm2 (nPCC/A0). The area of nanotube A0 was calculated as the nanotube diameter multiplied by the length of nanotube occupied by molecules at the beginning of imaging (t = 0 s). Changes in nPCC/A0 over time can thus be measured directly from TEM images. Since the electron beam is both the imaging probe and the stimulus of reactions, the reaction proceeds proportionally with the electron beam flux than time t. However, as the electron beam flux linking the two was kept constant throughout this experiment, here they are equivalent, and we use time for better intuition from the viewpoint of traditional macroscale chemical kinetics.
There was a linear decrease in nPCC/A0 over time following the latent period at the beginning of the reaction (Fig. 5(b)). This suggests that the observed order of PCC polymerisation in SWNTs is zero with respect to PCC molecules; in striking contrast, a thermally induced polymerisation in the same material exhibited second order behaviour,32 while kinetics of solid-state polymerisation reactions measured in the bulk tend to be second or third order.41 A particular subset of solid-state reactions, topochemical polymerisations, involve molecules in a crystal reacting without the need for any significant movement or reorientation. These reactions can be studied with spectroscopic and diffraction methods (in the latter case, X-rays may act as a probe as well as a stimulus of the reaction),42 and exhibit reaction orders closer to one,43 much lower than the solid-state reactions of disordered molecules. Polymerisation of PCC confined in SWNTs is comparable to these topochemical reaction conditions, but with the reaction allowed to propagate in one direction only, i.e. along the nanotube axis. This imposes restrictions upon the reaction of PCC molecules which may explain why the reaction order decreases to zero within the confines of SWNTs. It is interesting to note that zero-order reactions are known to occur when heterogeneous catalysts are saturated with reactants. This creates a bottleneck where the rate of reaction is limited by how many reactant molecules can react at the same time. For example, in a system of enzymes saturated with substrate, the catalytic rate of the enzyme determines the rate of conversion rather than the concentration of the substrate.
In our system of PCC@SWNT, the reactant molecules are packed as closely as they physically can be to each other (separated by the atomic van der Waals radii). Therefore, the concentration or areal number density of reactants in the reaction zone is saturated and remains constant. The observed zero-order kinetics with respect to PCC molecules can be directly correlated to the reaction mechanism observed in the images. This appeared as a domino effect of polymerisation propagating from a single point along the nanotube due to the strict conformational requirement of the Diels–Alder cycloaddition creating the bottleneck for the reaction. In contrast, the previously studied32 thermally induced polymerisation of PCC@SWNT (at temperatures above 500 °C) is observed to occur from multiple points in the molecular stack, as there is sufficient thermal energy to overcome this effect, consistent with the measured higher order of reaction.
Therefore, based on our experimental observations, we can propose the electron beam-induced reaction as two PCC molecules reacting to form a dimer, with an observed reaction rate constant of kobs (Fig. 5(a)).
The differential form of this zero-order reaction can be written as follows:
 | (1) |
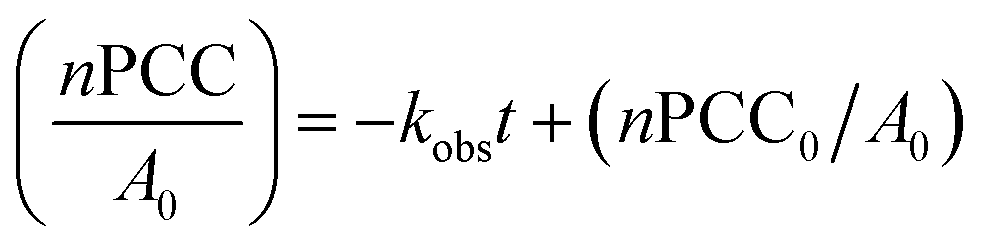 | (2) |
The gradient −kobs can be found using the linear plot of nPCC/A0 against time and linked to the observed reaction cross section σobs and flux of incident electrons j in TEM:
| kobs = j·σobs | (3) |
The linear plot of nPCC/A0 against t has a gradient of −0.002450 nm−2 s−1 (Fig. 5(d)), leading to σobs of 1.13 × 10−9 nm2 or 11.3 ± 0.6 barn for the reaction. Errors arise from measurements of the nanotube length which affects the values of nPCC/A0, as well as temporal resolution and fluctuation of the electron beam flux over the course of imaging. As shown in Fig. 5(a), kobs is a combination of the initial electron beam-induced bond cleavage and the subsequent intermolecular reaction (which will be dependent on the discussed steric effects), therefore, the observed reaction cross section σobs does not directly correspond to an elementary reaction step in PCC polymerisation. However, comparison of relative values of σobs measured in different experimental conditions (such as electron beam energy or temperature of the sample) can provide mechanistic insights, similar to how observed rates of enzyme catalysis can be compared to understand the roles of key amino acids in enzyme active sites. For example, heating (demonstrated in the previous section) may increase σobs as thermal energy is likely to increase the rate of PCC molecular movement and reorientation within the SWNT. If PCC polymerisation is dominated by direct kinetic energy transfer from incident electrons to atoms, lowering the temperature will have minimal effect on σobs.44 Thus, comparing values of σobs from different experimental conditions can help determine the driving forces behind the observed reaction mechanisms of PCC polymerisation.
A comparison of the electron beam-induced behaviour of PCC@SWNTs and PCC in a three-dimensional crystal also provides insight into the reaction mechanisms of this molecule under the electron beam. PCC@SWNTs remained unchanged over a period of several minutes at an electron flux of 2.17 × 106 e− nm−2 s−1 while nanoscale crystals of PCC rapidly became amorphous after seconds of irradiation at a lower electron flux of 2.25 × 105 e− nm−2 s−1 (see ESI,‡ Section 2). The differences in behaviour under the electron beam between PCC@SWNTs and PCC molecule in crystal form is comparable to the stability of coronene (C24H12) in SWNTs and in crystal under an 80 keV electron beam.45 The host SWNT offers beam stability to the encapsulated PCC molecules because PCC@SWNTs are much thinner than PCC crystals, and are electrically as well as thermally conductive. Therefore, local charging and heating effects are minimised, and direct knock-on is known to dominate in conductive samples.44 In contrast, radiolysis, heating, and charging are the major mechanisms for reactions in insulating PCC crystals.44
The method presented here of studying the kinetics of electron beam induced reactions at the single-molecule level using our ChemTEM technique can be applied to any system where individual molecules can be imaged during their reactions over time. The TEM provides a unique laboratory environment within which reactions induced by the intense electron beam can be imaged simultaneously, offering an exciting opportunity to discover reaction mechanisms and to study kinetics at the molecular level.
Further corroboration of reaction mechanism: identification by-products and intermediates
The product of PCC polymerisation has been determined as a zigzag nanoribbon terminated with Cl-atoms in our previous study,33 and we have discussed intermediates in this study (Fig. 2). However, low molecular weight by-products are more difficult to detect using TEM. The time-resolved ChemTEM imaging at −175 °C (Fig. 3) revealed the formation of crystals outside of the nanotubes, growing progressively bigger as more PCC molecules reacted (Fig. 6). The observed static crystals had a lattice d-spacing of 2.30 Å or 4.03 Å, and 2.05–2.06 Å when rotating (see ESI,‡ Section 3), which are in good agreement with the (1 3 1), (0 0 4) and (0 0 2) crystal planes (2.20 Å, 2.07 Å, and 4.13 Å respectively) of orthorhombic solid Cl2.46–48 While our previous study postulated that Cl2 is a by-product of PCC polymerisation, our current ChemTEM approach applied under cryogenic conditions allowed freezing out of Cl2 (melting point −101.5 °C), and thus providing direct evidence for diatomic chlorine being formed as a result of C–Cl bond dissociation in PCC caused by the electron beam, significantly corroborating the proposed mechanism.33 | ||
| Fig. 6 (a) Time series TEM images of PCC@SWNTs under cryogenic conditions. An electron flux of 4.47 × 105 e− nm−2 s−1 was used over a period of 39.83 s which corresponded to a total fluence of 1.78 × 107 e− nm−2. A rectangular high-contrast crystal was formed on outside of nanotube. The blue rectangles indicate where the crystal is in each image. Line profiles were measured for each blue rectangle and are shown beside each image, with the d-spacings labelled. The scale bar is 0.5 nm. (b) A high contrast crystal formed during imaging under cryogenic conditions (Fig. 3(d)), shown in the inset. The scale bar is 0.5 nm. A line profile was measured from the green rectangle which shows a d-spacing of 0.23 nm, consistent with the (131) plane of orthorhombic Cl2 crystal. | ||
It is important to emphasise that carefully designed macroscale experiments can complement the single-molecule observations, enhancing the mechanistic understanding (as in the studies of solid-state polymerisation using X-rays).42 In the context of PCC polymerisation, matrix-assisted laser desorption/ionisation time of flight mass spectrometry (MALDI-ToF MS), using a UV laser (355 nm, 500 μJ, 66.7 Hz), can trigger reactions of PCC comparable to those in TEM.49
Under UV irradiation in the MALDI-ToF MS, PCC molecules exhibited a regular pattern of Cl2 loss; peaks at 642 (C24Cl10), 572 (C24Cl8), and 500 (C24Cl6) m/z with precipitously decreasing intensity (Fig. 7(a)). The major de-chlorination product, C24Cl10, correlates with the aryne intermediate after loss of Cl2 under the electron beam (Fig. 2(c) and 5(c)). The absence of loss of single Cl atom in mass spectra is significant because elimination of a single Cl atom would lead to an aryl radical which is less stable than an aryne.
 | ||
| Fig. 7 (a) The MALDI-ToF mass spectrum of PCC, taken using a UV laser (355 nm, 500 μJ, 66.7 Hz) at 20% laser power without the use of a matrix. The top spectrum is an expansion of the region showing the molecular ion peaks at 713 m/z as well as fragmentation of the parent ion via successive loss of Cl2 forming the arynes at 642, 572, and 500 m/z; the bottom spectrum shows the adducts due to PCC dimerisation under the UV laser, as well as fragmentation peaks due to loss of Cl2. (b) Proposed structures of the adducts formed in the MALDI-ToF. | ||
MALDI-ToF MS also revealed the formation of dimers via a Diels–Alder cycloaddition, giving rise to a series of molecules at higher molecular weight: 1356 m/z (C48Cl22, corresponding to the initial angular adduct observed as non-linear intermediates in TEM during the early stages of PCC polymerisation; Fig. 2(d) and (f)); C48Cl20 1285 m/z (a likely intermediate between the initial angular adduct and the aromatic adduct); and 1214 m/z (C48Cl18, the aromatic adduct observed forming through planarisation of the angular adduct in TEM; Fig. 2(f)). The planar, aromatic adduct has the highest intensity in the mass spectrum which suggests that C48Cl18 is much more stable compared to other adducts (Fig. 7(a)). Under the UV laser, C48Cl18 undergoes a similar pattern of Cl2 loss to PCC, indicating that reactive aryne groups may form on the edge of the adduct. These arynes can then participate in further reactions with PCC molecules, as we have seen previously in TEM (Fig. 5(b)).
This remarkable correlation and agreement of the reaction pathways observed in MALDI-ToF and TEM shows that these two different methods (one bulk and one local-probe) can complement one another in terms of understanding the reaction mechanism. Moreover, it appears that polymerisation of PCC takes place and follows a similar path under different sources of energy (i.e. a beam of photons in MALDI-ToF and electrons in TEM), thus suggesting that if a chemical process is thermodynamically possible, it can be induced by a variety of different stimuli. This point is reinforced by the fact that the formation of the same product can be achieved at a preparative bulk scale by heating.33 This example of dovetailing bulk analysis with a local-probe method illustrates a future potential for a deeper understanding of reaction mechanisms when ChemTEM is linked with other analytical tools.
Conclusions
We have developed methods allowing the assessment of intermolecular reactions at the single-molecule level, in direct space and with spatiotemporal continuity. The observation of each individual molecule taking part in the reaction enables us to directly investigate population heterogeneities, in sharp contrast to more traditional bulk techniques where this information is inaccessible due to ensemble-averaging.31 The reactions studied within carbon nanocontainers here exhibited a significant dependency on the precise nature of the local environment and molecular packing, with sub-nanometre differences in SWNT diameter resulting in more than two orders of magnitude difference in the required electron fluence to the initiation of the reaction. By linking the kinetic behaviour and rates of the reactions to the local environment experienced by each reactant molecule, we discovered the molecular dynamics preceding the reaction (the extent of which appears to be dictated by steric demands of the cycloaddition reaction rather than reaction temperature), determined the observed reaction cross section, and revealed a zero-order reaction kinetics with a “domino effect” propagating from a single point. All this was achieved through time-resolved TEM imaging of the intermolecular reactions.Furthermore, we have demonstrated that observed electron beam induced reactions can be replicated under UV irradiation in a MALDI-ToF MS, which allowed correlation of the local-probe data from TEM with this macroscale-probe analysis and corroborated the proposed reaction mechanism by identifying the chemical identity of key reaction intermediates, agreeing with the structures proposed on the basis of TEM images.
Intermolecular reactions of PCC have high activation barriers, making this type of molecule ideal for ChemTEM kinetics measurements at the single-molecule level. However, the same approach could be generally applicable to a wide range of diverse reaction types, provided the molecules can be tracked over time under TEM conditions. As higher temporal resolution and accurate control of incident electron flux on the specimens are required to advance these methodologies, the continued development of electron microscopy instrumentation, especially direct electron detection cameras, are expected to take the single-molecule reaction kinetics studies to the next level.
Methods
Reagents were purchased from Sigma Aldrich and used as received.High resolution transmission electron microscopy (HR-TEM) was carried out at the electron Physical Science Imaging Centre (ePSIC) at Diamond Light Source. The microscope JEOL ARM300F is Cs corrected (2-fold astigmatism = 1.688 nm, coma = 50.201 nm, 3-fold astigmatism = 54.478 nm, spherical aberration = 3.285 nm, star = 11.395 nm, 4-fold astigmatism = 2.224 nm) and was operated at 80 kV. The largest condenser aperture was used and no objective aperture was used. Micrographs were acquired using the Gatan OneView camera. In situ sample heating with single tilt DENS Solutions Wildfire heating holder, temperature range: ambient to 1200 °C. In situ sample cooling with liquid nitrogen single tilt Fischione cooling holder, operating temperature −175 °C. TEM samples were prepared via a drop casting technique, where samples were dispersed in HPLC-grade isopropanol, followed by deposition of several drops of the suspension onto copper grid mounted with “lacey” carbon films (Agar Scientific UK), and dried under ambient conditions. All micrographs were processed using Gatan Digital Micrograph and ImageJ Fiji software50–52 with the ImageJ plugins TurboReg and StackReg.53 Additional high-angle annular-dark field (HAADF) imaging was carried out at room temperature on a Nion UltraSTEM100 MC ‘Hermes’ microscope, operated at 60 kV. The beam convergence semi-angle was 32 mrad, resulting in a 0.1 nm probe with 30 pA of current; the HAADF detector angular range was 90–190 mrad.
Matrix assisted laser desorption/ionisation time-of-flight mass spectrometry (MALDI-ToF MS) analyses were performed on a Bruker ultraFlexIII instrument (Bruker Daltonik, Bremen, Germany). Samples were deposited on a stainless steel target plate (type MTP384; Bruker Daltonik, Bremen, Germany) and due to the nature of the samples, no matrix was required. A pulsed solid-state UV laser (355 nm, 500 μJ, 66.7 Hz) was used to ionise the sample. The instrument was operated in reflection mode. Data was acquired through the instrument's flexControl software (v3,B185; Bruker Daltonik, Bremen, Germany), and processed using Bruker's flexAnalysis software (v3,B96; Bruker Daltonik, Bremen, Germany).
Nuclear magnetic resonance (NMR) spectroscopy was carried out in deuterated chloroform using a Bruker AV(III)500 spectrometer by Analytical Chemistry at the University of Nottingham. All chemical shifts are quoted in ppm relative to neat trimethylsilate (TMS).
PCC was synthesised as described in literature.54,5513C NMR δ (500 MHz, CDCl3, ppm); 133.2 (Aryl C–Cl), 126.8 (Aryl C), 121.4 (Aryl C). See ESI,‡ Section 4 for the NMR spectrum. Positive-ion MALDI-ToF MS m/z; 713 (C24Cl12), 642 (C24Cl10), 571 (C24Cl8), 500 (C24Cl6).
Single-walled carbon nanotubes (50 mg) were refluxed in nitric acid (3 M, 50 mL) for 2 h. The nanotubes were then filtered over PTFE membrane filter (0.2 μm pore size) and washed with sodium hydroxide (1 M, 50 mL) as well as deionised water (50 mL) and dried under vacuum to give opened SWNTs (49.5 mg). Perchlorocoronene was added to the opened nanotubes in a 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mass ratio, sealed in a glass ampoule under vacuum (×10−4 mbar) and heated at 400 °C for 72 h. The resulting black powder was washed by sonicating in toluene and filtering over PTFE membrane filter (0.2 μm pore size).
:1 mass ratio, sealed in a glass ampoule under vacuum (×10−4 mbar) and heated at 400 °C for 72 h. The resulting black powder was washed by sonicating in toluene and filtering over PTFE membrane filter (0.2 μm pore size).
Author contributions
K. L. Y. F., S. T. S. and A. N. K. developed the original concept and experimental design. K. L. Y. F. prepared materials, performed the TEM experiments and analysed the data. R. H. performed the MALDI-ToF experiment and analysed the data. S. E. M. and N. A. B. carried out DFT calculations using the AIRBED method. Q. M. R. performed the STEM experiments and analysed the data. C. S. A. enabled HRTEM experiments and assisted data collection. K. L. Y. F., S. T. S., B. L. W., and A. N. K. interpreted the data and wrote the manuscript. All authors have given approval to the final version of the manuscript.Conflicts of interest
The authors declare no competing interests.Acknowledgements
We thank Diamond Light Source for access and support in the use of the electron Physical Science Imaging Centre (Instrument E02 and proposal numbers MG22887, MG23260, and MG25251) that contributed to the results presented here. Q. M. R. carried out the work at SuperSTEM, the National Research Facility for Advanced Electron Microscopy, supported by the Engineering and Physical Sciences Research Council (EPSRC). We also thank M. Cooper and S. Aslam from Analytical Services at the School of Chemistry, University of Nottingham, for MALDI-ToF and NMR characterisation of perchlorocoronene. A. N. K. acknowledges support from the Engineering and Physical Science Research Council (EPSRC; grant EP/R024790/1 and MASI Programme Grant EP/V000055/1).References
- T. W. Chamberlain, A. Camenisch, N. R. Champness, G. A. D. Briggs, S. C. Benjamin and A. Ardavan, et al., Toward Controlled Spacing in One-Dimensional Molecular Chains: Alkyl-Chain-Functionalized Fullerenes in Carbon Nanotubes, J. Am. Chem. Soc., 2007, 129(27), 8609–8614 CrossRef CAS PubMed.
- S. Sinha and J. H. Warner, Recent Progress in Using Graphene as an Ultrathin Transparent Support for Transmission Electron Microscopy, Small Struct., 2021, 2, 2000049 CrossRef CAS.
- Y. Oaki and K. Sato, Crystal-controlled polymerization: recent advances in morphology design and control of organic polymer materials, J. Mater. Chem. A, 2018, 6(46), 23197–23219 RSC.
- D. A. Britz, A. N. Khlobystov, K. Porfyrakis, A. Ardavan and G. A. D. Briggs, Chemical reactions inside single-walled carbon nano test-tubes, Chem. Commun., 2005, 37–39 RSC.
- S. A. Miners, G. A. Rance and A. N. Khlobystov, Chemical reactions confined within carbon nanotubes, Chem. Soc. Rev., 2016, 45(17), 4727–4746 RSC.
- T. S. Koblenz, J. Wassenaar and J. N. H. Reek, Reactivity within a confined self-assembled nanospace, Chem. Soc. Rev., 2008, 37(2), 247–262 RSC.
- M. S. Koo, H. Kim, K. E. Lee and W. Choi, Photoconversion of Cyanide to Dinitrogen Using the Durable Electrode of a TaON Overlayer-Deposited WO3 Film and Visible Light, ACS EST Eng., 2021, 1(2), 228–238 CrossRef CAS.
- M. Nazari, A. Davoodabadi, D. Huang, T. Luo and H. Ghasemi, Transport Phenomena in Nano/Molecular Confinements, ACS Nano, 2020, 14(12), 16348–16391 CrossRef CAS PubMed.
- A. B. Grommet, M. Feller and R. Klajn, Chemical reactivity under nanoconfinement, Nat. Nanotechnol., 2020, 15(4), 256–271 CrossRef CAS PubMed.
- D. Bullara and Y. De Decker, Chemical Equilibrium on Low Dimensional Supports: Connecting the Microscopic Mechanism to the Macroscopic Observations, J. Stat. Phys., 2015, 161(1), 210–226 CrossRef CAS.
- M. Polak and L. Rubinovich, Nanochemical Equilibrium Involving a Small Number of Molecules: A Prediction of a Distinct Confinement Effect, Nano Lett., 2008, 8(10), 3543–3547 CrossRef CAS PubMed.
- Y. Khodorkovsky, L. Rubinovich and M. Polak, Stochastic Kinetics and Equilibrium of Nanoconfined Reactions, J. Phys. Chem. C, 2019, 123(40), 24949–24956 CrossRef CAS.
- Y. E. Ryabov, A. Puzenko and Y. Feldman, Nonmonotonic relaxation kinetics of confined systems, Phys. Rev. B: Condens. Matter Mater. Phys., 2004, 69(1), 014204 CrossRef.
- A. M. Bragança, A. Minoia, R. Steeno, J. Seibel, B. E. Hirsch and L. Verstraete, et al., Detection and Stabilization of a Previously Unknown Two-Dimensional (Pseudo)polymorph using Lateral Nanoconfinement, J. Am. Chem. Soc., 2021, 143(29), 11080–11087 CrossRef PubMed.
- F. Klappenberger, Y. Q. Zhang, J. Björk, S. Klyatskaya, M. Ruben and J. V. Barth, On-Surface Synthesis of Carbon-Based Scaffolds and Nanomaterials Using Terminal Alkynes, Acc. Chem. Res., 2015, 48(7), 2140–2150 CrossRef CAS PubMed.
- Z. Majzik, N. Pavliček, M. Vilas-Varela, D. Pérez, N. Moll and E. Guitián, et al., Studying an antiaromatic polycyclic hydrocarbon adsorbed on different surfaces, Nat. Commun., 2018, 9(1), 1198 CrossRef PubMed.
- S. T. Skowron, T. W. Chamberlain, J. Biskupek, U. Kaiser, E. Besley and A. N. Khlobystov, Chemical Reactions of Molecules Promoted and Simultaneously Imaged by the Electron Beam in Transmission Electron Microscopy, Acc. Chem. Res., 2017, 50(8), 1797–1807 CrossRef CAS PubMed.
- E. Nakamura, Movies of Molecular Motions and Reactions: The Single-Molecule, Real-Time Transmission Electron Microscope Imaging Technique, Angew. Chem., Int. Ed., 2013, 52(1), 236–252 CrossRef CAS PubMed.
- E. Nakamura, Atomic-Resolution Transmission Electron Microscopic Movies for Study of Organic Molecules, Assemblies, and Reactions: The First 10 Years of Development, Acc. Chem. Res., 2017, 50(6), 1281–1292 CrossRef CAS PubMed.
- T. Susi, J. C. Meyer and J. Kotakoski, Manipulating low-dimensional materials down to the level of single atoms with electron irradiation, Ultramicroscopy, 2017, 180, 163–172 CrossRef CAS PubMed.
- O. Dyck, M. Ziatdinov, D. B. Lingerfelt, R. R. Unocic, B. M. Hudak and A. R. Lupini, et al., Atom-by-atom fabrication with electron beams, Nat. Rev. Mater., 2019, 4(7), 497–507 CrossRef CAS.
- X. Zhao, J. Dan, J. Chen, Z. Ding, W. Zhou and K. P. Loh, et al., Atom-by-Atom Fabrication of Monolayer Molybdenum Membranes, Adv. Mater., 2018, 30(23), 1707281 CrossRef PubMed.
- S. Jeon, T. Heo, S. Y. Hwang, J. Ciston, K. C. Bustillo and B. W. Reed, et al., Reversible disorder-order transitions in atomic crystal nucleation, Science, 2021, 371(6528), 498–503 CrossRef CAS PubMed.
- K. Cao, S. T. Skowron, J. Biskupek, C. T. Stoppiello, C. Leist and E. Besley, et al., Imaging an unsupported metal–metal bond in dirhenium molecules at the atomic scale, Sci. Adv., 2020, 6(3), eaay5849 CrossRef CAS PubMed.
- K. Cao, S. T. Skowron, C. T. Stoppiello, J. Biskupek, A. N. Khlobystov and U. Kaiser, Direct Imaging of Atomic Permeation Through a Vacancy Defect in the Carbon Lattice, Angew. Chem., Int. Ed., 2020, 59(51), 22922–22927 CrossRef CAS PubMed.
- M. S. Isaacson, J. Langmore, N. W. Parker, D. Kopf and M. Utlaut, The study of the adsorption and diffusion of heavy atoms on light element substrates by means of the atomic resolution STEM, Ultramicroscopy, 1976, 1(3–4), 359–376 CrossRef CAS PubMed.
- J. C. Meyer, F. Eder, S. Kurasch, V. Skakalova, J. Kotakoski and H. J. Park, et al., Accurate Measurement of Electron Beam Induced Displacement Cross Sections for Single-Layer Graphene, Phys. Rev. Lett., 2012, 108(19), 196102 CrossRef PubMed.
- J. Kotakoski, C. Mangler and J. C. Meyer, Imaging atomic-level random walk of a point defect in graphene, Nat. Commun., 2014, 5(1), 3991 CrossRef CAS PubMed.
- S. T. Skowron, V. O. Koroteev, M. Baldoni, S. Lopatin, A. Zurutuza and A. Chuvilin, et al., Reaction kinetics of bond rotations in graphene, Carbon, 2016, 105, 176–182 CrossRef CAS.
- S. Okada, S. Kowashi, L. Schweighauser, K. Yamanouchi, K. Harano and E. Nakamura, Direct Microscopic Analysis of Individual C60 Dimerization Events: Kinetics and Mechanisms, J. Am. Chem. Soc., 2017, 139(50), 18281–18287 CrossRef CAS PubMed.
- J. W. Jordan, K. L. Y. Fung, S. T. Skowron, C. S. Allen, J. Biskupek and G. N. Newton, et al., Single-molecule imaging and kinetic analysis of intermolecular polyoxometalate reactions, Chem. Sci., 2021, 12, 7377–7387 RSC.
- K. L. Y. Fung, S. T. Skowron, C. S. Allen and A. N. Khlobystov, Counting molecules in nano test tubes: a method for determining the activation parameters of thermally driven reactions through direct imaging, Chem. Commun., 2021, 57, 10628–10631 RSC.
- T. W. Chamberlain, J. Biskupek, S. T. Skowron, A. V. Markevich, S. Kurasch and O. Reimer, et al., Stop-Frame Filming and Discovery of Reactions at the Single-Molecule Level by Transmission Electron Microscopy, ACS Nano, 2017, 11, 2509–2520 CrossRef CAS PubMed.
- A. Markevich, S. Kurasch, O. Lehtinen, O. Reimer, X. Feng and K. Müllen, et al., Electron beam controlled covalent attachment of small organic molecules to graphene, Nanoscale, 2016, 8, 2711–2719 RSC.
- L. Jones, A. Varambhia, R. Beanland, D. Kepaptsoglou, I. Griffiths and A. Ishizuka, et al., Managing dose-, damage-and data-rates in multi-frame spectrum-imaging, Microscopy, 2018, 67(S1), 98–113 CrossRef PubMed.
- T. Okazaki, Y. Iizumi, S. Okubo, H. Kataura, Z. Liu and K. Suenaga, et al., Coaxially stacked coronene columns inside single-walled carbon nanotubes, Angew. Chem., Int. Ed., 2011, 50(21), 4853–4857 CrossRef CAS PubMed.
- Y. Sakane, K. Mouri and K. Shintani, Morphology of a columnar stack of coronene molecules encapsulated in a single-walled carbon nanotube, AIP Adv., 2015, 5, 117113 CrossRef.
- A. N. Khlobystov, D. A. Britz, A. Ardavan, G. Andrew and D. Briggs, Observation of Ordered Phases of Fullerenes in Carbon Nanotubes, Phys. Rev. Lett., 2004, 92(24), 245507 CrossRef PubMed.
- S. E. Mason, P. H. Beton and N. A. Besley, AIRBED: A Simplified Density Functional Theory Model for Physisorption on Surfaces, J. Chem. Theory Comput., 2019, 15(10), 5628–5634 CrossRef CAS PubMed.
- F. Banhart, T. Füller, P. Redlich and P. M. Ajayan, The formation, annealing and self-compression of carbon onions under electron irradiation, Chem. Phys. Lett., 1997, 269(3), 349–355 CrossRef CAS.
- S. N. Vouyiouka, E. K. Karakatsani and C. D. Papaspyrides, Solid state polymerization, Prog. Polym. Sci., 2005, 30(1), 10–37 CrossRef CAS.
- K. Tashiro, T. Kamae, M. Kobayashi, A. Matsumoto, K. Yokoi and S. Aoki, Structural Change in the Topochemical Solid-State Polymerization Process of Diethyl cis,cis-Muconate Crystal. 1. Investigation of Polymerization Process by Means of X-ray Diffraction, Infrared/Raman Spectra, and DSC, Macromolecules, 1999, 32(8), 2449–2454 CrossRef CAS.
- A. Matsumoto, T. Tanaka, T. Tsubouchi, K. Tashiro, S. Saragai and S. Nakamoto, Crystal Engineering for Topochemical Polymerization of Muconic Esters Using Halogen–Halogen and CH/π Interactions as Weak Intermolecular Interactions, J. Am. Chem. Soc., 2002, 124, 8891–8902 CrossRef CAS PubMed.
- R. F. Egerton, Mechanisms of radiation damage in beam-sensitive specimens, for TEM accelerating voltages between 10 and 300 kV, Microsc. Res. Tech., 2012, 75(11), 1550–1556 CrossRef CAS PubMed.
- T. W. Chamberlain, J. Biskupek, S. T. Skowron, P. A. Bayliss, E. Bichoutskaia and U. Kaiser, et al., Isotope substitution extends the lifetime of organic molecules in transmission electron microscopy, Small, 2015, 11(5), 622–629 CrossRef CAS PubMed.
- R. W. G. Wyckoff, Crystal structures, Interscience Publishers, New York, 2nd edn, 1963, vol. 1. [cited 2021 Apr 1]. Available from: https://onlinelibrary.wiley.com/doi/abs/10.1107/S0365110X65000361 Search PubMed.
- R. L. Collin, The crystal structure of solid chlorine, Acta Crystallogr., 1952, 5, 431–432 CrossRef CAS.
- J. Donohue and S. H. Goodman, Interatomic distances in solid chlorine, Acta Crystallogr., 1965, 18(3), 568–569 CrossRef CAS.
- R. K. E. Gover, T. W. Chamberlain, P. J. Sarre and A. N. Khlobystov, Piecing Together Large Polycyclic Aromatic Hydrocarbons and Fullerenes: A Combined ChemTEM Imaging and MALDI-ToF Mass Spectrometry Approach, Front. Chem., 2021, 9, 700562 CrossRef CAS PubMed.
- C. A. Schneider, W. S. Rasband and K. W. Eliceiri, NIH Image to ImageJ: 25 years of image analysis, Nat. Methods, 2012, 9(7), 671–675 CrossRef CAS PubMed.
- J. Schindelin, I. Arganda-Carreras, E. Frise, V. Kaynig, M. Longair and T. Pietzsch, et al., Fiji: an open-source platform for biological-image analysis, Nat. Methods, 2012, 9(7), 676–682 CrossRef CAS PubMed.
- J. Schindelin, C. T. Rueden, M. C. Hiner and K. W. Eliceiri, The ImageJ ecosystem: An open platform for biomedical image analysis, Mol. Reprod. Dev., 2015, 82(7–8), 518–529 CrossRef CAS PubMed.
- P. Thevenaz, U. E. Ruttimann and M. Unser, A pyramid approach to subpixel registration based on intensity, IEEE Trans. Image Process., 1998, 7(1), 27–41 CrossRef CAS PubMed.
- M. Ballester, C. Molinet and J. Castaner, Preparation of Highly Strained Aromatic Chlorocarbons. I. A Powerful Nuclear Chlorinating Agent. Relevant Reactivity Phenomena Traceable to Molecular Strain, J. Am. Chem. Soc., 1960, 82(16), 4254–4258 CrossRef CAS.
- M. Koshino, H. Kurata and S. Isoda, Study of structures at the boundary and defects in organic thin films of perchlorocoronene by high-resolution and analytical transmission electron microscopy, Ultramicroscopy, 2010, 110, 1465–1474 CrossRef CAS PubMed.
Footnotes |
| † This paper is dedicated to the memory of Professor Nick Besley. |
| ‡ Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2cp05183d |
| § Deceased author. |
| This journal is © the Owner Societies 2023 |