Effect of total charge on the electronic structure of thiolate-protected X@Ag12 superatoms (X = Ag, Au)†
Received
29th October 2022
, Accepted 5th January 2023
First published on 6th January 2023
Abstract
Electronic structures of chemically synthesized silver-based clusters [XAg16(TBBT)12]3− (X = Ag or Au; TBBT = 4-tert-butylbenzenethiolate) having an icosahedral X@Ag12 superatomic core were studied by gas-phase photoelectron spectroscopy and density functional theory calculations. The electron binding energy of the highest occupied molecular orbital (HOMO) with a 1P superatomic nature was determined to be 0.23 and 0.29 eV for X = Ag or Au, respectively. Resonant tunnelling electron emission through the repulsive Coulomb barrier (RCB) was observed. From the kinetic energy of the tunnelling electrons, it was estimated that the lowest unoccupied molecular orbital (LUMO) was supported at 1.51 and 1.62 eV above the vacuum level by the RCB for X = Ag or Au, respectively. The HOMO of [XAg16(TBBT)12]3− (X = Ag or Au) was destabilized by 3.74 and 3.71 eV, respectively, compared with those of [XAg24(DMBT)18]− (DMBT = 2,4-dimethylbenzenethiolate) having the icosahedral X@Ag12 core due to the larger negative charge imparted by the ligand layers.
1. Introduction
Thiolate (SR)-protected gold clusters [Aux(SR)y]z have been studied extensively as ideal systems to elucidate how their structures and properties depend on the descriptors x, y, and z.1–8 Atomically-precise synthesis and single-crystal X-ray diffraction (SCXRD) revealed that most of the hitherto isolated [Aux(SR)y]z are structurally divided into the Au cores and the protecting layers with –SR–(AuSR)n– (n = 1–4) structures.9–16 Theoretical studies have established a simple picture of their electronic structures: the valence electrons with the number of x–y–z are accommodated in quantized orbitals (1S, 1P, 1D, 2S,…) distributed over the Au cores.17 According to this picture, the representative cluster [Au25(SR)18]− can be formally viewed as an icosahedral (Au13)5+ core with the closed electron configuration (1S)2(1P)6 fully protected by six units of [Au2(SR)3]−. From this viewpoint, [Aux(SR)y]z can be described as chemically modified superatoms.
Recently, thiolate-protected silver clusters [Agx(SR)y]z have attracted growing interest.18–25 As expected from the similar electron configuration of Au and Ag, [Agx(SR)y]z and [Aux(SR)y]z exhibit common structural features. For example, [XAg24(DMBT)18]− (1− and 2− for X = Ag and Au, respectively; DMBT = 2,4-dimethylbenzenethiolate) has an icosahedral (X@Ag12)5+ core capped by six units of [Ag2(DMBT)3]− (Table 1).22,23 Meanwhile, recent SCXRD studies have shown that [Agx(SR)y]z clusters take more diverse interfacial structures than [Aux(SR)y]z. For example, an icosahedral (Au@Ag12)5+ core was protected by four units of tridentate [Ag(TBBT)3]2− in [AuAg16(TBBT)12]3− (43−; TBBT = 4-tert-butylbenzenethiolate) (Table 1).24 A pure Ag analogue [Ag17(TBBT)12]3− (33−) has been theoretically proposed to consist of an icosahedral (Ag13)5+ core and four units of [Ag(TBBT)3]2− (Table 1) although the SCXRD data is not available.25 Clusters 1−/2− and 33−/43− provide ideal platforms for investigating how the ligand layers affect the electronic structure of (X@Ag12)5+ by modulating the potential for confining the valence electrons. More specifically, we can gain insight into how the electronic structure of 33− and 43− is affected by the formation of the repulsive Coulomb barrier (RCB) originating from the electrostatic repulsion between the detached electrons and remaining anions 32− and 42−.26–29
Table 1 Structures of thiolate-protected X@Ag12 superatoms
| Cluster |
[XAg24(DMBT)18]− |
[XAg16(TBBT)12]3− |
| X |
Ag (1−)a |
Au (2−)b |
Ag (33−) |
Au (43−)c |
| DMBT = 2,4-dimethylbenzenethiolate. TBBT = 4-tert-butylbenzenethiolate. Color code: silver = Ag, yellow = S, orange = Ag or Au. Phenyl rings are depicted by black sticks. Hydrogen atoms were omitted for clarity. Ref. 22. Ref. 23. Ref. 24. Adiabatic electron affinity of the corresponding oxidized form. Ref. 32. This study. Repulsive Coulomb barrier height. |
| Ligand unit |
|
|
| Superatomic core |
|
| AEA (eV)d |
3.97e |
4.00e |
0.23f |
0.29f |
|
E
RCB (eV)g |
— |
— |
∼2.8f |
∼2.8f |
In the present study, we conducted gas-phase anion photoelectron spectroscopy (PES) with the help of density functional theory (DFT) calculations on 33− and 43−. The results showed that the 1P superatomic orbitals localized on the (X@Ag12)5+ cores of 33− and 43− were located at 0.23 ± 0.04 and 0.29 ± 0.02 eV below the vacuum level. In addition, we observed that 33− and 43− emitted the photoelectrons with a constant kinetic energy, regardless of the energies of photodetachment lasers. This novel phenomenon is ascribed to resonant electron tunnelling via unoccupied 1D orbital. This observation indicated that the 1D orbitals of 33− and 43− were located at 1.51 and 1.62 eV, respectively, above the vacuum level. In conclusion, the 1P and 1D orbitals of 33− and 43− were significantly upshifted compared to those of 1− and 2− by the increased total negative charge due to the ligand layers.
2. Experimental
2.1. Synthesis and characterization
Samples of 33− and 43− were synthesized according to the protocols reported by Bigioni et al.25 The chemical identity and purity of the samples were confirmed by electrospray ionization (ESI) mass spectrometry and UV-Vis absorption spectroscopy in N,N-dimethylformamide (DMF). Mass spectra were measured by using a JMS-T100LP (JEOL Ltd) mass spectrometer. The water used was Milli-Q grade (>18 MΩ). UV-Vis absorption spectra were recorded using a Jasco V-670 spectrophotometer.
[Ag17(TBBT)12]3−(Na+)3 (33−(Na+)3).
121 mg (0.714 mmol) of AgNO3 was dissolved in 7.2 mL of DMF. After stirring for 5 min, 86 μL (0.500 mmol) of TBBT-H was added to the solution. The solution was stirred for 15 min, and then 108 mg (2.85 mmol) of NaBH4 dissolved in 28.6 mL of DMF was added dropwise under vigorous stirring. The solution color rapidly changed to dark purple. After the solution was stirred for 3 h, 4.2 mL of water was added. The solution was stirred for an additional 2 min and then stored in a freezer at −20 °C for 16 h. Finally, the solution was filtered using a syringe filter. The DMF solution of the product was diluted to one-sixth with acetonitrile before the photoelectron spectroscopy measurements.
[AuAg16(TBBT)12]3−(Na+)3 (43−(Na+)3).
114 mg (0.672 mmol) of AgNO3 was dissolved in 4 mL of DMF to which was added 17.3 mg (0.042 mmol) of HAuCl4·4H2O dissolved in 3.2 mL of DMF. After stirring for 5 min, 86 μL (0.500 mmol) of TBBT-H was added to the solution. The solution was stirred for 15 min, and then 108 mg (2.85 mmol) of NaBH4 dissolved in 28.6 mL of DMF was added dropwise under vigorous stirring. The solution color rapidly changed to dark purple. After the solution was stirred for 3 h, 4.2 mL of water was added. The solution was stirred for an additional 2 min and then stored in a freezer at −20 °C for 16 h. Finally, the solution was filtered using a syringe filter. The DMF solution of the product was diluted to one-sixth with acetonitrile before the photoelectron spectroscopy measurements.
2.2. Photoelectron spectroscopy (PES)
PES was conducted using a home-built apparatus consisting of an ESI source, a quadrupole linear ion trap (QLIT), a time-of-flight mass spectrometer (TOF-MS), and a magnetic-bottle type photoelectron spectrometer (MB-PES) (Fig. S1, ESI†). The samples of 33− and 43− dissolved in DMF/acetonitrile (1/5, v/v) solution were introduced into vacuum via the ESI source (a spray voltage −3.0 kV; flow rate 4.0 μL min−1) and desolvated by a heated capillary at ∼80 °C. The desolvated anions of 33− and 43− were trapped in the QLIT and were injected into the TOF-MS at a repetition rate of 10 or 50 Hz depending on the laser used for the PES. The mass-selected ion beams of 33− and 43− were guided by deflectors and a set of Einzel lenses to the MB-PES and irradiated with the second, third, or fourth harmonics of a nanosecond-pulsed Nd:YAG laser (532, 355, or 266 nm, respectively; Quanta-Ray GCR130, Spectra-Physics, US) operated at 10 Hz, or an ArF excimer laser (193 nm, PSX-100, MPB Communications Inc.) operated at 50 Hz. The detached electrons were detected by MB-PES. The electron kinetic energy (Ekin) was determined based on the TOF and converted to the electron binding energy (Ebin). The energy axis was calibrated against the known spectrum of I−. The value of Ebin was determined by the energy of the spectral onset where the tangential line at the inflection point intersected the baseline. The average value and standard deviation of Ebin were obtained by statistical analysis of multiple data sets.
2.3. Theoretical calculation
The geometric and electronic structures of 33− and 43− were studied by density functional theory (DFT) calculations at the PBE/def-SV(P) level. The scalar relativistic effect was included by using def-ecp as an effective core potential.30 The authentic ligands (TBBT) were used in the calculations. All calculations were carried out using the TURBOMOLE program.31 Structural optimization was conducted by using reported SCXRD structure24 as the initial structures. Vibrational frequency calculation did not show any negative frequencies, indicating that the optimized structures were located at the local minima.
3. Results and discussion
3.1. Structures of 33− and 43− by DFT calculations
ESI mass spectrometry (Fig. S2a, ESI†) confirmed that high-purity 33− and 43− could be synthesized. A comparison of the optical spectrum (Fig. S2b, ESI†) with that in ref. 24 indicated that our sample of 43− had the same geometric structure as that determined by SCXRD. To gain insight into the structure of our 33−, we conducted structural optimization of 43− and 33− by DFT calculations. The optimized structure of 43− (Fig. 1b and Table S2, ESI†) agreed well with the crystal structure of 43−, indicating the reliability of the calculation method. The optimized structure of 33− (Fig. 1a and Table S1, ESI†) was similar to that of 43− except that the central Au atom was replaced by Ag. In addition, the energy levels of the Kohn–Sham orbitals of 33− and 43− were very similar (Fig. 1c and d). The similarity in the UV-Vis absorption spectra of 43− and 33− (Fig. S2b, ESI†) indicated their structural similarity. We concluded that 33− consisted of an icosahedral (Ag13)5+ core and four units of [Ag(TBBT)3]2− (Table 1) as previously predicted theoretically.25 Fig. S3 (ESI†) indicated that the nearly degenerated highest occupied molecular orbital (HOMO) to HOMO−2 corresponded to the 1P orbitals, whereas the lowest unoccupied molecular orbital (LUMO) to LUMO+4 corresponded to the 1D orbitals.
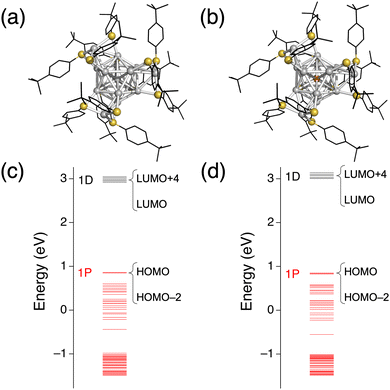 |
| | Fig. 1 DFT optimized structures of (a) 33− and (b) 43−. Energy diagrams of (c) 33− and (d) 43−. Phenyl rings and tertiary butyl groups are depicted as black sticks, and H atoms are omitted for simplicity. Color code: orange, Au; gray, Ag; yellow, S. | |
3.2. PES on 33− and 43−
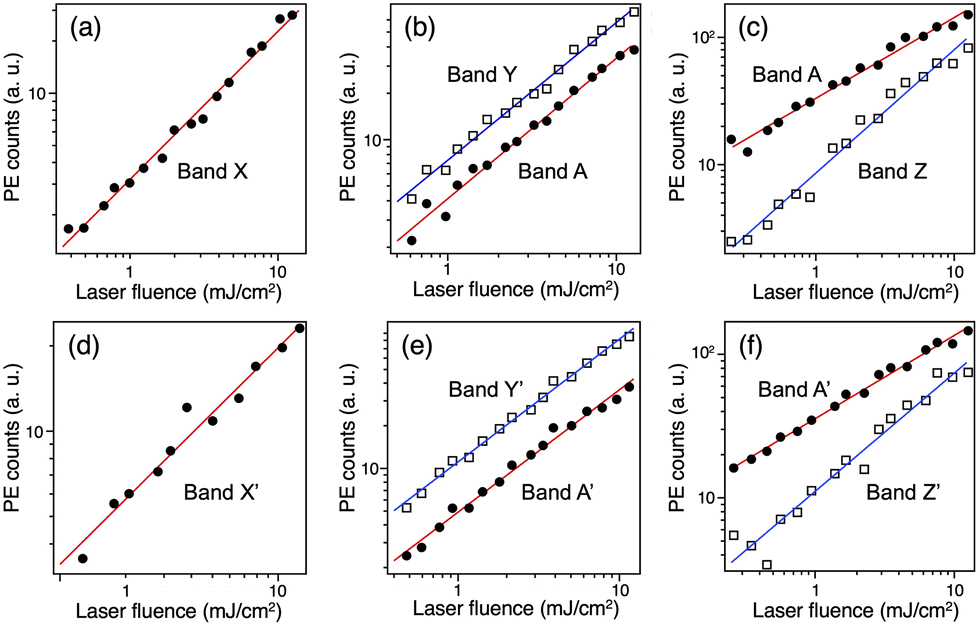
The PE spectra of 33− and 43− recorded at various wavelengths are presented as a function of Ebin in Fig. 2a and b, respectively. The PE spectra of 33− and 43− show similar profiles, indicating that the electronic structures of the (X@Ag12)5+ cores are not affected by whether X is Au or Ag as reported for 1− and 2−.32 This spectral similarity also supports the structural similarity between 33− and 43−. In the following, the broad bands in the PE spectra of 33− are labelled A and B, whereas the relatively sharp bands are labelled X, Y, and Z. The corresponding bands in the spectra of 43− are labelled A′, B′, X′, Y′, and Z′. To facilitate the assignment, the intensities of the selected bands were measured at 532, 355, and 266 nm and plotted in double logarithmic plots as a function of the laser fluence in Fig. 3. The slopes of the linear lines determined by the least squares fitting method are presented in Table 2. Band A (A′) at 355 nm and bands X (X′) and Y (Y′) are assigned to a one-photon process based on the slopes. At 266 nm, the slope for band A (A′) is significantly smaller than that for band Z (Z′). These results are explained by assuming the involvement of a two-photon electron detachment process in band Z (Z′) at 266 nm.
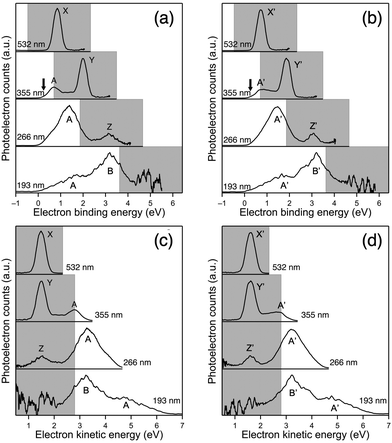 |
| | Fig. 2 PE spectra of (a) 33− and (b) 43− recorded at 532, 355, 266, and 193 nm plotted against the electron binding energy and PE spectra of (c) 33− and (d) 43− plotted against the electron kinetic energy. The gray shaded area is the cutoff region due to the RCB. | |
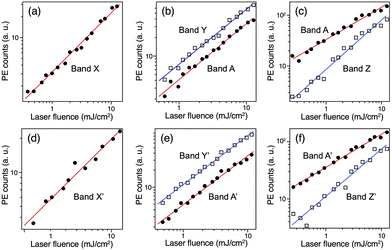 |
| | Fig. 3 Double-logarithmic plots of the laser fluence dependence of bands A, B and X–Z in the PE spectra of 33− at (a) 532, (b) 355, and (c) 266 nm, and 43− at (d) 532, (e) 355, and (f) 266 nm. | |
Table 2 Slopes of double-logarithmic plots of the laser fluence dependence
| Wavelength (nm) |
3
3−
|
4
3−
|
| A |
X |
Y |
Z |
A′ |
X′ |
Y′ |
Z′ |
| 532 |
|
0.85 ± 0.02 |
|
|
|
0.76 ± 0.05 |
|
|
| 355 |
0.91 ± 0.03 |
|
0.89 ± 0.02 |
|
0.86 ± 0.02 |
|
0.87 ± 0.02 |
|
| 266 |
0.64 ± 0.02 |
|
|
0.97 ± 0.04 |
0.58 ± 0.01 |
|
|
0.82 ± 0.05 |
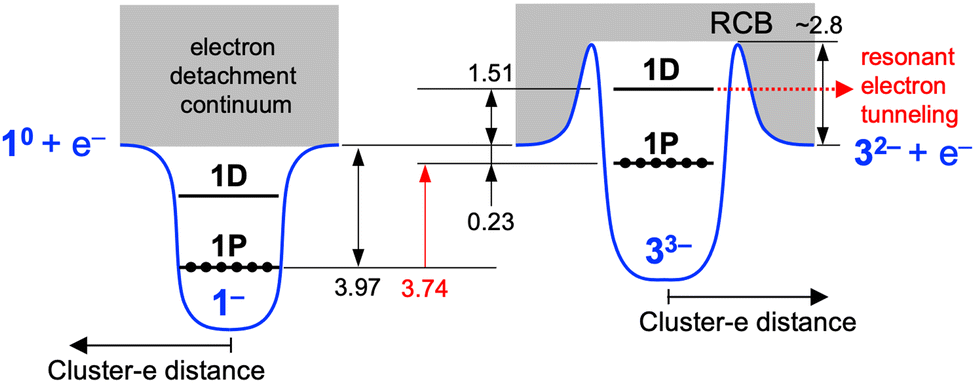
In the following, we first focused on the assignment of the PE spectra of 33−. Given that the height of the RCB (ERCB) of triply charged anions such as 1-hydroxy-3,6,8-pyrene-trisulfonate and monoprotonated copper phthalocyanine tetrasulfonate was 3.3 and 2.5 eV, respectively,28,33 bands X–Z are possibly located in the cutoff region due to the RCB and are not assigned to the direct electron detachment from the occupied orbitals. Therefore, band A with an onset of ∼0 eV was assigned to the electron detachment from the HOMO (1P orbitals based on the DFT calculation, Fig. 1c) of 33−. The Ebin value of 1P of 33− was determined to be 0.23 ± 0.04 eV from the spectral onset at 355 nm (indicated by an arrow in Fig. 2a): the onset was determined using the spectrum at 355 nm rather than those at 266 and 193 nm because the energy resolution was highest due to a lower electron kinetic energy.34 Then, the ERCB of 33− was estimated by comparing the profiles of band A at 355 and 266 nm: band A in the PE spectrum at 266 nm exhibited a peak at Ebin ∼ 1.33 eV, whereas that in the 355 nm spectrum showed a cutoff behavior at Ebin > ∼0.73 eV due to the suppression of the PE signals by the RCB. This comparison led us to conclude that the ERCB value of 33− was estimated to be ∼2.8 (= 3.49 − 0.73) eV. This ERCB value is comparable to those of other triply charged anions28,34 and confirms that bands X–Z are located in the cutoff region colored gray in Fig. 2a and b.
Bands X–Z are assigned as follows based on the previous observation of PE signals in the cutoff region for multiply charged anions of phthalocyanine tetrasulfonate, a dinuclear Pt(II) complex, and fluorescein dianion.35–37 These PEs had the same kinetic energy regardless of the photon energy and were assigned to resonant tunnelling from the electronically excited state through the RCB. To test such a possibility for bands X–Z, we plotted the PE spectra as a function of Ekin (Fig. 2c). The Ekin values for bands X–Z were nearly the same regardless of the photon energy: 1.49 ± 0.01, 1.50 ± 0.01, and 1.54 ± 0.01 eV, respectively. This result indicated that bands X–Z originated from the spontaneous emission of an electron, promoted to the common MO located at 1.51 eV above the vacuum level, via tunnelling through the RCB (Scheme 1). The MO responsible for bands X–Z is 1D superatomic orbital, given that the intense absorption band of 33− at 2.92 eV (Fig. S2b, ESI†) was assigned to the optical transition from the phenyl ring to the 1D orbital.25 Nearly the constant Ekin values for bands X–Z indicate that the relaxation of 1D orbital of 33− after photoexcitation was negligibly small. From the discussion above, we conclude that the 1D and 1P orbitals of 33− were located at 1.51 eV above and 0.23 eV below the vacuum level, respectively (Scheme 1). Therefore, the HOMO–LUMO gap energy (EHL) of 33− was estimated to be 1.74 (= 1.51 + 0.23) eV, which is comparable to the optical EHL (Fig. S2b, ESI†). The power dependence of band Z suggested that two-photon electron detachment from the doubly charged state 32− was involved in addition to the direct detachment at 266 nm:
| | | 33− + hν (266 nm) → 32− + e− | (1) |
| | | 32− + hν (266 nm) → 3− + e− | (2) |
Finally, the PE spectra of
43− were assigned similarly. The
Ebin of 1P of
43− was determined to be 0.29 ± 0.02 eV from the spectral onset at 355 nm (indicated by an arrow in
Fig. 2b). The
Ekin values for bands X′–Z′ were 1.61 ± 0.01, 1.63 ± 0.01, and 1.62 ± 0.01 eV, respectively (
Fig. 2d). Thus, the
EHL of
43− was estimated to be 1.91 (= 1.62 + 0.29) eV based on these PES results.
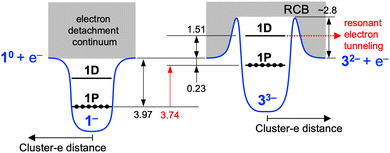 |
| | Scheme 1 Schematic illustration of energy levels of 1− and 33−. The numbers are the energies in eV. The energies are not scaled. | |
On the basis of the above results, the electronic structures of 33− and 43− are compared with those of 1− and 2−, respectively. The HOMO of 33− is located at 0.23 eV below the vacuum level, while that of 1− is located at 3.97 eV below the vacuum level (Scheme 1).32 In contrast, the HOMO of 43− is located at 0.29 eV below the vacuum level, while that of 2− is located at 4.00 eV below the vacuum level.32 This result indicates that the HOMO of (X@Ag12)5+ superatomic core was upshifted by 3.74 and 3.71 eV for X = Ag and Au, respectively, by increasing the total negative charge from −1 to −3.
4. Conclusions
In summary, we examined the electronic structure of the (X@Ag12)5+ superatomic core of [XAg16(TBBT)12]3− (X = Ag or Au) by the gas-phase anion PES and DFT calculation. The Ebin value of the 1P orbital was determined to be 0.23 ± 0.04 and 0.29 ± 0.02 eV for X = Ag or Au, respectively. The resonant tunnelling electron emission through the RCB was observed and enabled us to estimate that the 1D orbital was located at 1.51 and 1.62 eV above the vacuum level by the RCB for X = Ag or Au, respectively. The HOMO of [XAg16(TBBT)12]3− (X = Ag or Au) was destabilized by 3.73 and 3.71 eV, respectively, compared with those of [XAg24(DMBT)18]−. This remarkable upshift of the superatomic orbitals was due to the increase in the total negative charge imparted by the ligand layers.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This research was financially supported by JST, CREST (Grant No. JPMJCR20B2) and JSPS KAKENHI (Grant No. JP20H00370, JP21J20631).
Notes and references
- R. Jin, C. Zeng, M. Zhou and Y. Chen, Chem. Rev., 2016, 116, 10346–10413 CrossRef CAS PubMed.
- I. Chakraborty and T. Pradeep, Chem. Rev., 2017, 117, 8208–8271 CrossRef CAS PubMed.
- Q. Yao, X. Yuan, T. Chen, D. T. Leong and J. Xie, Adv. Mater., 2018, 30, 1–23 Search PubMed.
- S. Jin, S. Wang and M. Zhu, Chem. – Asian J., 2019, 14, 3222–3231 CrossRef CAS PubMed.
- H. Hirai, S. Ito, S. Takano, K. Koyasu and T. Tsukuda, Chem. Sci., 2020, 11, 12233–12248 RSC.
- N. Xiaab and Z. Wu, Chem. Sci., 2021, 12, 2368 RSC.
- T. Omoda, S. Takano and T. Tsukuda, Small, 2021, 17, 1–18 Search PubMed.
- T. Kawawaki, A. Ebina, Y. Hosokawa, S. Ozaki, D. Suzuki, S. Hossain and Y. Negishi, Small, 2021, 17, 1–35 Search PubMed.
- P. D. Jadzinsky, G. Calero, C. J. Ackerson, D. A. Bushnell and R. D. Kornberg, Science, 2007, 318, 430–433 CrossRef CAS PubMed.
- M. Zhu, C. M. Aikens, F. J. Hollander, G. C. Schatz and R. Jin, J. Am. Chem. Soc., 2008, 130, 5883–5885 CrossRef CAS PubMed.
- M. W. Heaven, A. Dass, P. S. White, K. M. Holt and R. W. Murray, J. Am. Chem. Soc., 2008, 130, 3754–3755 CrossRef CAS PubMed.
- H. Qian, W. T. Eckenhoff, Y. Zhu, T. Pintauer and R. Jin, J. Am. Chem. Soc., 2010, 132, 8280–8281 CrossRef CAS PubMed.
- A. Das, T. Li, K. Nobusada, C. Zeng, N. L. Rosi and R. Jin, J. Am. Chem. Soc., 2013, 135, 18264–18267 CrossRef CAS PubMed.
- A. Das, C. Liu, H. Y. Byun, K. Nobusada, S. Zhao, N. Rosi and R. Jin, Angew. Chem., Int. Ed., 2015, 54, 3140–3144 CrossRef CAS PubMed.
- S. Chen, S. Wang, J. Zhong, Y. Song, J. Zhang, H. Sheng, Y. Pei and M. Zhu, Angew. Chem., Int. Ed., 2015, 54, 3145–3149 CrossRef CAS PubMed.
- A. Das, T. Li, G. Li, K. Nobusada, C. Zeng, N. L. Rosi and R. Jin, Nanoscale, 2014, 6, 6458–6462 RSC.
- M. Walter, J. Akola, O. Lopez-Acevedo, P. D. Jadzinsky, G. Calero, C. J. Ackerson, R. L. Whetten, H. Grönbeck and H. Häkkinen, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 9157–9162 CrossRef CAS PubMed.
- K. Zheng, X. Yuan, N. Goswami, Q. Zhang and J. Xie, RSC Adv., 2014, 4, 60581–60596 RSC.
- C. P. Joshi, M. S. Bootharaju and O. M. Bakr, J. Phys. Chem. Lett., 2015, 6, 3023–3035 CrossRef CAS PubMed.
- J. Yan, B.-K. Teo and N. Zheng, Acc. Chem. Res., 2018, 51, 3084–3093 CrossRef CAS PubMed.
- X. Kang and M. Zhu, Chem. Soc. Rev., 2019, 48, 2422–2457 RSC.
- C. P. Joshi, M. S. Bootharaju, M. J. Alhilaly and O. M. Bakr, J. Am. Chem. Soc., 2015, 137, 11578–11581 CrossRef CAS PubMed.
- M. S. Bootharaju, C. P. Joshi, M. R. Parida, O. F. Mohammed and O. M. Bakr, Angew. Chem., Int. Ed., 2016, 55, 922–926 CrossRef CAS PubMed.
- B. E. Conn, A. Atnagulov, B. Yoon, R. N. Barnett, U. Landman and T. P. Bigioni, Sci. Adv., 2016, 2, e1601609 CrossRef PubMed.
- S. Wickramasinghe, A. Atnagulov, B. Yoon, R. N. Barnett, W. P. Griffith, U. Landman and T. P. Bigioni, J. Am. Chem. Soc., 2015, 137, 11550–11553 CrossRef CAS PubMed.
- X.-B. Wang, C.-F. Ding and L.-S. Wang, Phys. Rev. Lett., 1998, 81, 3351–3354 CrossRef CAS.
- X.-B. Wang and L.-S. Wang, Phys. Rev. Lett., 1999, 83, 3402–3405 CrossRef CAS.
- X.-B. Wang and L.-S. Wang, Nature, 1999, 400, 245–248 CrossRef CAS.
- J. M. Weber, I. N. Ioffe, K. M. Berndt, D. Löffler, J. Friedrich, O. T. Ehrler, A. S. Danell, J. H. Parks and M. M. Kappes, J. Am. Chem. Soc., 2004, 126, 8585–8589 CrossRef CAS PubMed.
- D. Andrae, U. Haeussermann, M. Dolg, H. Stoll and H. Preuss, Theor. Chim. Acta, 1990, 77, 123–141 CrossRef CAS.
- F. Furche, R. Ahlrichs, C. Hättig, W. Klopper, M. Sierka and F. Weigend, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2014, 4, 91–100 CAS.
- S. Ito, Y. Tasaka, K. Nakamura, Y. Fujiwara, K. Hirata, K. Koyasu and T. Tsukuda, J. Phys. Chem. Lett., 2022, 13, 5049–5055 CrossRef PubMed.
- J. Yang, X.-P. Xing, X.-B. Wang, L.-S. Wang, A. P. Sergeeva and A. I. Boldyrev, J. Chem. Phys., 2008, 128, 091102 CrossRef PubMed.
- J. Fan and L.-S. Wang, J. Phys. Chem., 1994, 98, 11814–11817 CrossRef CAS.
- X.-B. Wang, K. Ferris and L.-S. Wang, J. Phys. Chem. A, 2000, 104, 25 CrossRef CAS.
- M. O. Winghart, J.-P. Yang, M. Vonderach, A. N. Unterreiner, D.-L. Huang, L.-S. Wang, S. Kruppa, C. Riehn and M. M. Kappes, J. Chem. Phys., 2016, 144, 054305 CrossRef PubMed.
- D. A. Horke, A. S. Chatterley and J. R. R. Verlet, Phys. Rev. Lett., 2012, 108, 1–5 CrossRef PubMed.
|
| This journal is © the Owner Societies 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  and
Tatsuya
Tsukuda
and
Tatsuya
Tsukuda