
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Entropic contributions to the stability of electrochemically adsorbed anion layers on Au(111): a microcalorimetric study†
Marco
Schönig
 * and
Rolf
Schuster
*
* and
Rolf
Schuster
*
Institute of Physical Chemistry, Karlsruhe Institute of Technology, Kaiserstraße 12, 76131, Karlsruhe, Germany. E-mail: marco.schoenig@kit.edu; rolf.schuster@kit.edu
First published on 25th November 2022
Abstract
We measure the entropy of formation of the interface upon anion adsorption (Cl−, Br− I− and SO42−) on Au(111) as an important indicator for the structure, order and composition of the interface. The entropy of formation of the interface exhibits a rather universal behaviour for all anions with a steep decrease upon initial adsorption followed by a shallow minimum at intermediate anion coverages and a strong increase close to the completion of the adsorbate adlayer. The strong variation of the entropy signals significant entropic contributions to the free enthalpy of the adsorption process and thus the stability of the adsorbed phase. At low anion coverages, close to the potential of zero charge, we attribute the entropy variations to the rearrangement of the interfacial water structure. At intermediate and high anion coverages, a comparison with the results of a lattice-gas model, considering pairwise repulsive interactions within the quasi-chemical approximation, shows that the entropy changes upon anion adsorption can be explained by the configurational entropy of the adsorbed phase. Thus, entropic contributions from both the solvent and the adsorbate are important for the stability of surface phases, particularly for disordered systems.
Introduction
Recently, the influence of an electrolyte solution on the conversion rates of electrocatalytic reactions has received increasing research attention, dedicated to improve the efficiency of these catalytic reactions.1 To understand how an electrolyte influences these reactions, knowledge about the structure and composition of the interfacial layer between the electrode and the electrolyte, e.g., on the rigidity of the interfacial water network2 or on the surface concentration and surface phases of adsorbed ions,3 is of crucial importance. In this respect, the entropy of formation of the interface, i.e., the entropy change during the polarization of an electrode, is particularly interesting, since it contains contributions from both the interfacial water structure and the ions in the double layer (DL).4–6 It was therefore proposed as a descriptor for the electrolytes’ influence on the electrode activity.7 The entropy of formation of the interface was first measured by Harrison et al. using Hg electrodes, where the measurement of the surface tension provided direct access to thermodynamic quantities using the electrocapillary equation.8 In contrast, for solid electrodes, the entropy of formation of the interface is harder to obtain. The currently most used methods are laser-induced temperature jump techniques4 or the analysis of the temperature dependence of the voltammogram based on a generalized adsorption isotherm.9 Despite both methods providing significant insight into electrochemical systems such as hydrogen adsorption on platinum,6 they cannot straightforwardly provide the entropy of formation of the interface. For example, the laser-induced temperature jump technique, in which the potential response to fast heating of the working electrode using a laser pulse is determined, provides only qualitative information on the entropy, because the absolute temperature variation is not known. Nonetheless, the potential at which the entropy exhibits an extremum can be obtained precisely from the zero-crossing of the temperature coefficient of the potential, at least in electrolytes in which the thermodiffusion potential is negligible. The analysis using a generalized adsorption isotherm on the other hand is limited to systems, where this isotherm is applicable.6An alternative approach to measure the entropy of formation of the interface is provided by measuring the heat exchanged at a single electrode upon polarization, which is realized by electrochemical microcalorimetry (ECM).10 The reversible part of the exchanged heat, also called Peltier heat, corresponds to the partial molar entropy change at the interface caused by the corresponding electrochemical process, that is to its reaction entropy. Using this method, we have recently shown that the partial molar entropy varies significantly during sulfate adsorption on Au(111), in the potential region where the sulfate coverage increases from 0 to around 0.2 monolayers (MLs). Anion adsorption on Au(111) provides an almost perfect model system for studying the formation of an interface since this surface system is nearly ideally polarizable over an at least 1 V wide potential interval, allowing the investigation of surface adlayers up to high coverages, without interferences from faradaic processes such as metal dissolution or others.11 In addition, the Au electrode constitutes a promising surface for electrocatalysis,12e.g., as a catalyst for CO-selective CO2 reduction,13 and ions have proven to play a critical role in its reactivity.14 Thus, we extended this study towards a comparative investigation of anion adsorption (SO42−, Cl−, Br− and I−) on Au(111). We will discuss the contributions of solvent and electrolyte to the entropy of formation of the interface and employ statistical mechanics models to describe the measured data.
Results and discussion
The upper part of Fig. 1 shows typical cyclic voltammograms (CVs) obtained for a Au(111) surface in 0.1 M H2SO4 (yellow), 0.1 M KCl (green), 0.1 M KBr (red) and 0.1 M KI (purple). Note that in our calorimeter we measure the heat at the backside of a thin working electrode. We are thus restricted to the use of (111)-textured Au films on 50 μm thick sapphire sheets instead of Au(111) single crystals (details on the experiment and surface preparation are provided in the ESI†). The CVs recorded in solutions of different anions show very similar shapes and agree well with previously reported CVs for Au(111) electrodes,11 justifying the usage of the employed gold films as a model for the Au(111) surface. The horizontal shift between the CVs is attributed to the different enthalpies of the adsorption of different anions, which is caused by the hydration enthalpy of these ions in solution and increases in the order of SO42− < Cl− < Br− < I−.15 All CVs exhibit a sharp, tall peak in the anodic sweep, resulting from the lifting of the surface reconstruction of the Au(111) surface.16 It is well established that the onset potential of this peak coincides with the potential of the zero charge (pzc) of the Au(111) electrode in contact with the respective solution.17 The second, broader peak reflects the charge flowing due to the increasing coverage of the specifically adsorbed anions.11 Finally, at potentials close to the positive inflection point of the CV, a small third peak is visible for SO42− (0.875 V), Cl− (0.785 V) and Br− (0.375 V), which is ascribed to the formation of an ordered adlayer of the adsorbed anions. Around this peak, the anion coverage reaches saturation, with the surface concentration of the halides being close to the maximum packing density, given by their van der Waals radii. For sulfate, the packing density is only one third of its possible maximum value11 and it has been shown that water is incorporated in the sulfate adlayer.18 In I−-containing solutions, this peak is typically barely visible on a pure single crystal19 and in our experiments, due to the limited terrace size of our Au(111) films, this peak was not observed.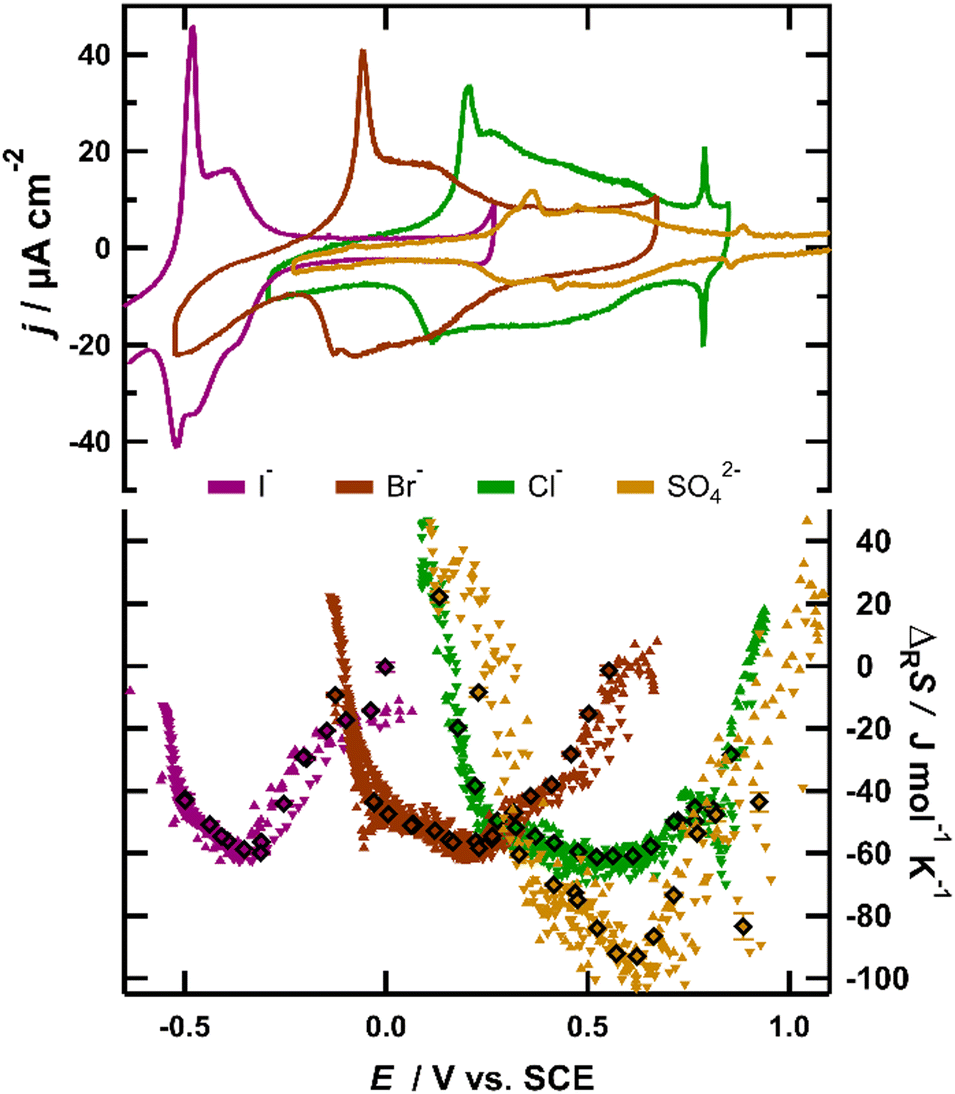 | ||
| Fig. 1 Comparison of I− (purple), Br− (red), Cl− (green) and SO42− (yellow) adsorption from 0.1 M KI, KBr, KCl and 0.1 M H2SO4 solutions. Left axis: cyclic voltammograms at a sweep rate of 50 mV s−1; right axis: reaction entropy obtained by series of 10 ms, 50 and 100 μA positive and negative current pulses (upward and downward triangles) and by potential pulses of varying amplitude and polarity (diamonds). | ||
The reaction entropy of the interface formation process derived from our ECM measurements is depicted in the lower part of Fig. 1. The reaction entropy was obtained by two different experimental procedures. The diamonds represent data, where the electrochemical reaction was driven by 10 ms potential pulses with different amplitudes of alternating polarity, all starting from a fixed rest potential. The data represented by the upward and downward triangles originate from a continuous series of 10 ms positive (upward) or negative (downward) current pulses of a constant amplitude of 50 or 100 μA. Using this method, we were able to sweep the potential and thus the anion coverage quasi continuously through the adsorption region (step size ca. 20 mV). The positive sweep typically started slightly before the sharp current peak in the respective CV of the involved anion, while the negative sweep started positive of the adsorption region. The example potential-, current- and heat transients for both procedures are given in the ESI† (for more details on the experimental methods, see the ESI† or ref. 20). In the present work, the reaction entropy is always referring to the anodic reaction, i.e. the anion adsorption process.
From Fig. 1, it is clearly visible that the reaction entropy as a function of potential follows the same trend for all anions. The individual curves are displaced according to the shift of the characteristic peaks of the CVs of the individual anions. Around the sharp anodic peak in the CV, where the anion coverage is still small, the reaction entropy decreases steeply from initially positive values to negative values with increasing potential. Note that within a potential interval of roughly 0.2 V the reaction entropy decreases by about 70 J mol−1 K−1, corresponding to an entropy driven increase of the free enthalpy of the adsorption process of TΔΔRS ≈ 70 J mol−1 K−1 × 300 K = 21 kJ mol−1 ≙ 0.22 eV. Thus at low anion coverage the variation of the electrode potential or in other words the change of the free enthalpy of the cell reaction is essentially of entropic origin. After the adsorption of typically 10–20% of the maximum anion coverage, the steep decrease levels off and reaches a shallow minimum at intermediate anion coverages. This means that variations of the enthalpic contributions to the free enthalpy of the cell reaction now become relevant. Repulsive dipole–dipole interactions among the adsorbed anion/substrate dipoles may provide a straightforward explanation for the variation of the free enthalpy of the cell reaction with anion coverages in this potential range.21 Since with increasing anion coverage the repulsive energy increases, it has to be compensated by a higher driving force for the anion adsorption, i.e., a more positive electrode potential.15 At more positive potentials, where the anion coverage reaches its maximum value, the reaction entropy increases again, exhibiting another zero-crossing. Later, in this contribution, we show that this increase of the adsorption entropy may also be attributed to repulsive interactions between the anion/substrate dipoles.
The resemblance of the reaction entropy curves of the different anions is intriguing and strongly suggests that the variation of the reaction entropy with potential has a common physicochemical origin. For further discussion of the potential dependence of the reaction entropy, we formally separate it into contributions stemming from the immobilization of the ions on the surface during the adsorption process and those from changes of the arrangement of the surrounding solvent molecules. The entropic contributions of the electrons in the metal electrode are much smaller than the observed entropy changes (typically well below 1 J mol−1 K−1)22 and thus are neglected in the following discussion.
The entropy change upon immobilization of the ions is given by the difference of the partial molar entropy of the ions in solution without solvent contributions, which remains constant at a constant ion concentration in solution,23 and the partial molar entropy of the adsorbed ions, which is determined by their configurational entropy in the surface phase plus vibrational contributions. The configurational contribution will depend strongly on the anion coverage and thus on the potential.6,10 In contrast, since it was found that the vibrational frequency of the metal–halide bond only negligibly changes with potential,24 the entropic contributions from the vibrational degrees of freedom are expected to depend only weakly on the anion coverage or potential and will only add a constant entropy offset to the ionic adsorption entropy. Therefore, we start our discussion with the configurational entropy of the ions adsorbed on the surface. For this purpose, it is useful to depict the reaction entropy as a function of the anion coverage. In Fig. 2, the averaged reaction entropy in 0.1 M KCl from Fig. 1 is plotted as green circles as a function of the anion coverage θ (information on the averaging and the corresponding plots for the other ions can be found in the ESI† Fig. S2a–c). We calculated the anion coverage using the surface excess concentration as a function of potential obtained by Shi et al., employing the Gibbs-adsorption isotherm.19,25,26 As a maximum anion coverage (θ = 1), the packing density calculated by the van der Waals radius of the respective anion is used,11 leading to a maximal coverage of one Cl− per two Au surface sites.27Fig. 2 in general shows the same trend as Fig. 1, but appears spread at small coverages, due to the nonlinear potential dependence of the coverage.25 The minimum of the reaction entropy is found around a coverage of θ = 0.75.
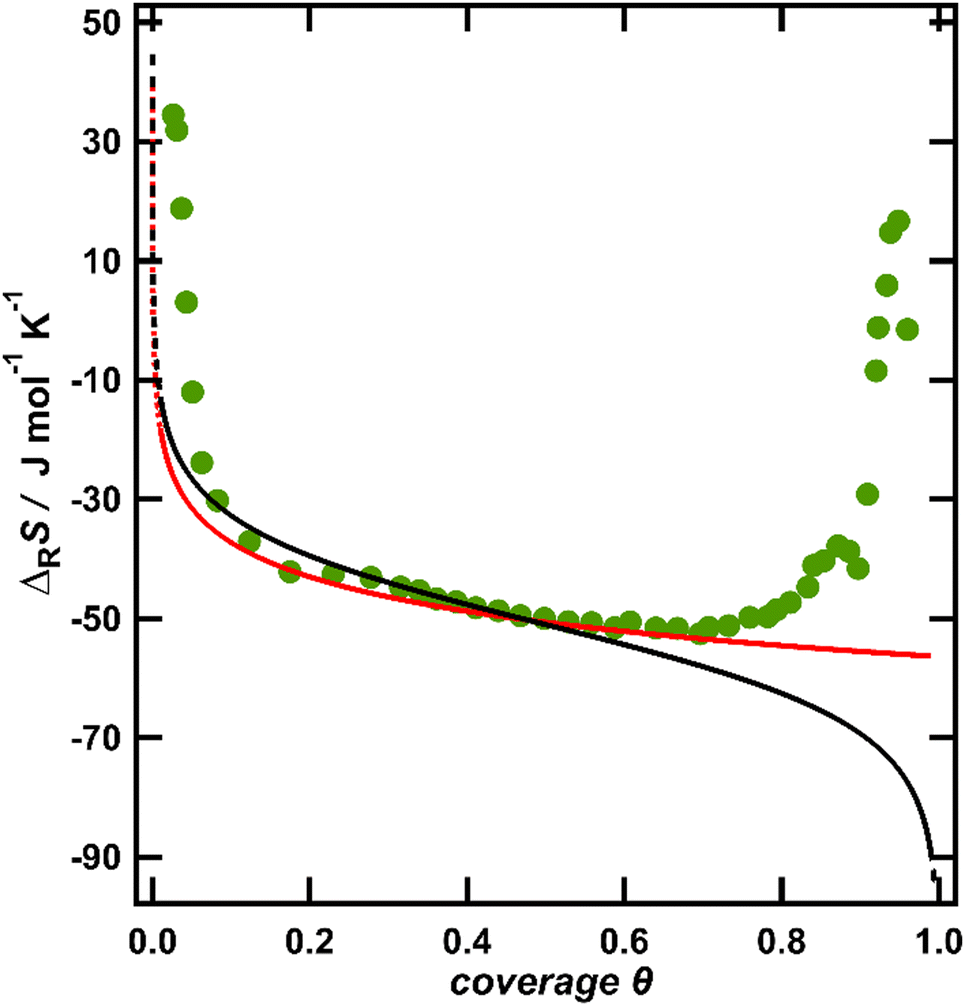 | ||
| Fig. 2 Averaged reaction entropy data of Au(111) in 0.1 M KCl from Fig. 1 as a function of the anion coverage of the chloride anions (green circles). The translational entropy of a 2D gas in a confined space calculated according to eqn (1) (red) and the configurational entropy of a Langmuir-like lattice-gas according to eqn (2) (black). Note that the model data are vertically shifted to coincide with the experimental data at θ = 0.5. | ||
The configurational entropy of the ion adlayer can be considered within two extreme model cases, either as a 2D gas of mobile adsorbed anions with two translational degrees of freedom or by adsorption of the anions on fixed sites of the surface lattice. In the case of a mobile anion adlayer, the partial molar translational entropy of the adsorbed anions can be calculated from an ideal 2D gas as described by eqn (1):6
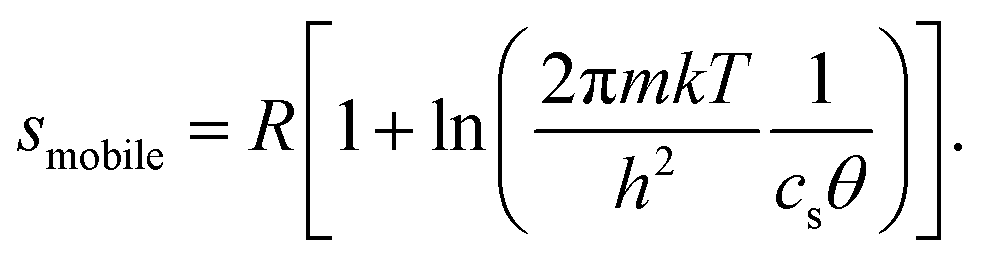 | (1) |
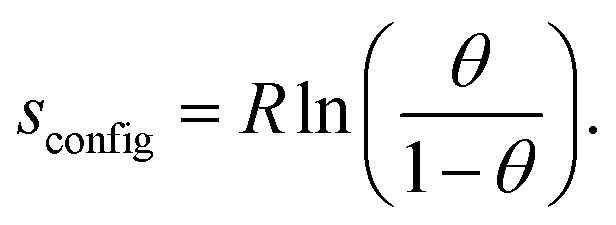 | (2) |
The partial molar entropies obtained from eqn (1) and (2) for the case of chloride adsorption are included in Fig. 2 as black and red lines. Since we only discuss the variation of the reaction entropy with coverage, the entropy axes are adjusted to the experimental reaction entropy at θ = 0.5 for better comparison.
At coverages θ < 0.6, the calculated partial molar entropy functions apparently agree well with the coverage dependence of the measured reaction entropy. However, the divergence of the calculated entropy curves at coverages below θ ≈ 0.1 (see dotted lines in Fig. 2) is somewhat hypothetical, due to the finite terrace size of the employed Au films. To obtain an estimate for the average terrace width, we studied the voltammetric response of our Au films in H2SO4 upon the formation of an ordered anion adlayer at 0.875 V (see Fig. 1 and the discussion above). The corresponding peak in the CV is very sensitive to the surface orientation and terrace size. By comparing the CV of our Au films in H2SO429 to the voltammetric profiles of a series of stepped Au[n(111)–(110)] single crystals in the same solution by Wandlowski et al.,30 we conclude that the electrochemical behaviour of our Au films corresponds to the response of a stepped Au(111) single crystal with an average size of ideally smooth terraces of about 20 atoms. An analogous conclusion was drawn for Au films on mica by Wandlowski et al. The 2D ideal gas model will certainly break down, as soon as the average area per ion, which is given by 1/csθ, approaches the terrace size. With a terrace diameter of about 20 atoms, this situation is reached for θ ≈ 0.005. Therefore, the above models for the configurational entropy are probably not applicable as explanation for the divergence of the experimental entropy at coverages below one percent of a ML. In addition, none of the two models can explain the behaviour of the experimental data at coverages above ca. θ = 0.75, where the experimental reaction entropy increases with increasing coverage. Such an increase may point to effective interactions between the anions, which have to be accounted for. Note that mean-field approximations, such as the Frumkin isotherm, approximate the entropy by that of an ideal gas and therefore exhibit the same partial molar entropy as the Langmuir isotherm.28 To account for the influence of adsorbate–adsorbate interactions on the configurational entropy of the ion adlayer, we employ the quasi-chemical approximation (QCA), where pair interactions between nearest neighbours are considered.
A representation of the chemical potential of the adsorbed anionic species in the framework of the QCA was given by Koper.31 From the chemical potential, the partial molar entropy is obtained by differentiation with respect to the temperature T (see the ESI†). In the quasi-chemical approximation, the pair interactions are considered via an interaction parameter w. Considering the dipole–dipole repulsion between dipoles composed of the ion and its image–charge in the metal to be the dominant interaction, as proposed, e.g., by Ocko and Wandlowski,21 this parameter becomes32
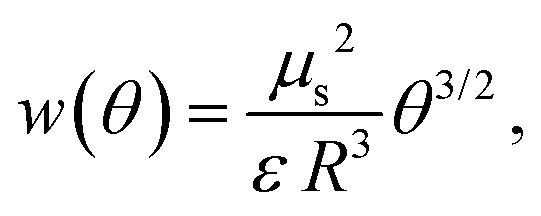 | (3) |
 phase on (111) surfaces,34 but these models are outside of the scope of the current publication.
phase on (111) surfaces,34 but these models are outside of the scope of the current publication.
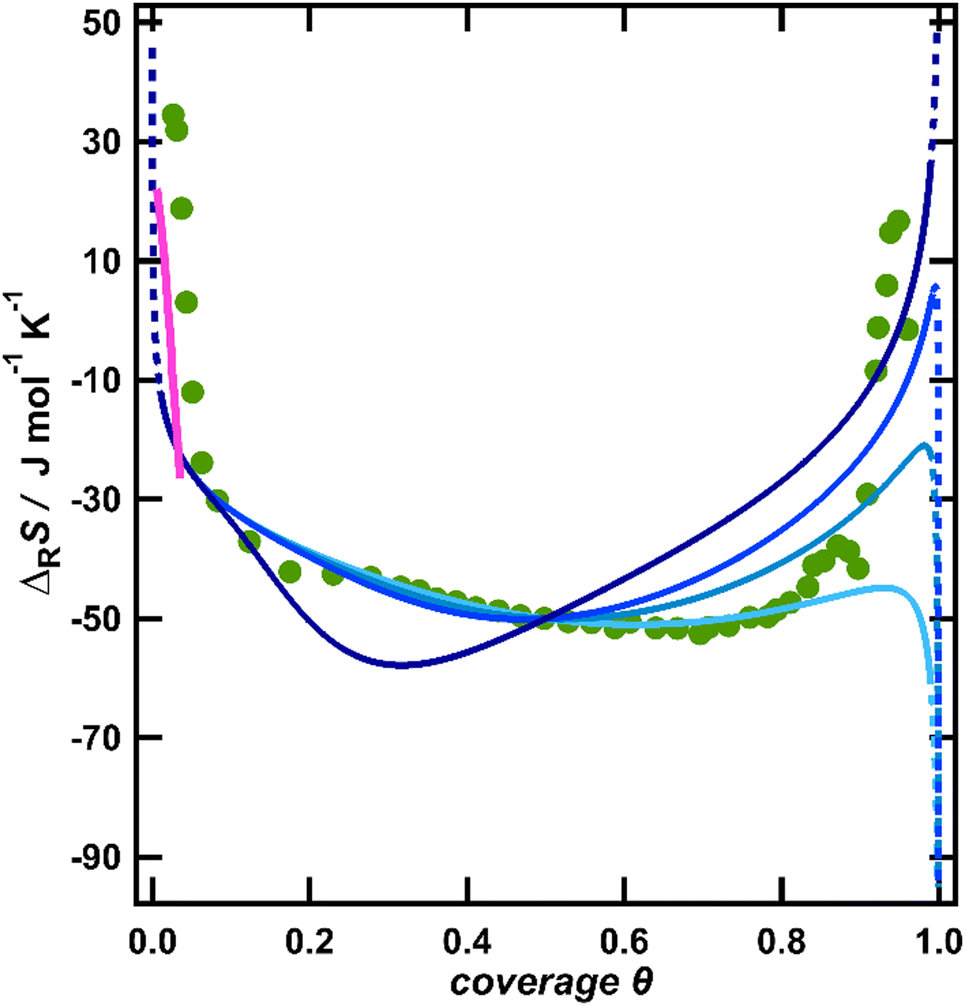 | ||
| Fig. 3 Comparison of the experimental reaction entropy with the configurational entropy of a lattice-gas described by the quasi-chemical approximation according to eqn (S4) (ESI†) with different repulsive interaction energies w at a full coverage: 10 kJ mol−1 (light blue), 15 kJ mol−1 (blue), 20 kJ mol−1 (dark blue) and 75 kJ mol−1 (indigo). In addition, the reaction entropy from the reorientation of water, derived from a model by Bockris and Habib is included as a pink line. | ||
To estimate if the magnitude of this pair interaction is reasonable, we explicitly calculated w(θ = 1) from eqn (3) using a surface dipole moment μs of −0.3 D,11 the van der Waals radius of chlorine (R = 1.75 Å)35 for the minimum distance R and the vacuum permittivity as ε, which results in an interaction energy of around 13.9 kJ mol−1. Thus, the interaction energies presented in Fig. 3 seem reasonable. Wang et al. pointed out that instead of dipole–dipole interactions also surface-mediated interactions might dominate in adsorbate layers.36 Such interactions would exhibit the same θ3/2 coverage dependence as dipole–dipole interactions and thus are also consistent with our data. It should be noted that by using sole coulombic interactions, which would exhibit a θ1/2 coverage dependence,32 we could not achieve a reasonable agreement between the model and experiment (see the ESI,† Fig. S2).
One major detail not included in these considerations is that halide adlayers on Au(111) are known to form incommensurate structures,15 which cannot be described within a simple lattice-gas model.31 To our knowledge, no analytical model for the calculation of the entropy of incommensurate adlayers is known. While this may decrease the validity of the introduced lattice-gas models, we expect that the adsorbate–adsorbate interactions determine the configurations also in the incommensurate adlayer. Therefore, we think that the occurrence of incommensurate structures does not undermine the main conclusion, which is that repulsive interactions lead to an entropic stabilization of the anion adlayer at high coverages.
The configurational entropy of a lattice-gas with repulsive interactions thus provides a reasonable explanation for the reaction entropy at coverages above a few percent of a ML, but still lacks an interpretation of the data at smaller coverages. So far, we have not considered the second contribution to the reaction entropy, originating from the rearrangement of the solvent molecules upon polarization of the electrode. In particular, near the pzc, at low anion coverages, the electric field in the interface will strongly change with progressing polarization and thus the water dipoles are expected to orient significantly along the field,37 thereby lowering their orientational and librational freedom.38 Indeed, for mercury electrodes, Harrison et al. found a maximum of the entropy of formation of the interface, which is located at potentials slightly negative of the pzc.8 This small shift was explained by the water molecules having their natural orientation at the pzc with the oxygen atom towards the surface.39 Only at slightly negative potentials, the natural orientation is overcome and the water molecules obtain the highest degree of looseness reflected in a maximum of the total entropy and thus in a zero-crossing of the reaction entropy.38 Note that the reaction entropy is the derivative of the total entropy with respect to the conversion of the adsorption reaction.8 Similarly, a maximum of the entropy at potentials slightly negative of the pzc was found by Climent et al. on Au(111) by laser-induced temperature jump experiments in sulfate- and perchlorate-containing solutions.4 They also ascribed their observations to the reorientation of the interfacial water upon the polarization of the DL. To estimate how water reorientation would manifest itself in the overall reaction entropy, we utilized a thermodynamic model proposed by Bockris and Habib as the explanation of the entropy data on Hg electrodes.38 They calculated the total entropy of water in the interface as a function of charge. To obtain the corresponding partial molar entropy, i.e., the contribution of water to the reaction entropy, we numerically differentiated the data of Bockris and Habib with respect to the charge. The calculated reaction entropy is included in Fig. 3 as the pink line. Albeit the absolute position of this line is somewhat arbitrary, due to the insufficient knowledge of the pzc, its slope coincides well with the data. Therefore, the increasing restriction of the orientational and librational freedom of the water molecules in the interface with increasing DL polarization, i.e., water reorientation may explain the strong variation of the reaction entropy close to the pzc, at low anion coverages.
Water reorientation should however quickly saturate with the increasing electric field strength at the interface.40 Indeed, Ataka et al. found by the surface-enhanced infrared adsorption spectroscopy of Au in 0.5 M HClO4 that within 200 mV positive of the pzc the interfacial water arranged into an ice-like layer, where the water dipoles are ultimately inclined by at least 60° to the surface.41 This supports that variations of the reaction entropy caused by the reorientation of water will be limited to low anion coverages, as indicated by the pink line in Fig. 3. To further substantiate this argument, we estimated the hypothetical amount of water which had to reorganize from a maximally loosened state into a fully ordered ice-like water in order to account for the measured variation of the reaction entropy of over 50 J mol−1 K−1 between its zero-crossing and the minimum at θ = 0.75. For this, we assume that the change of the reaction entropy upon the transition from fully loosened water near the pzc to fully oriented ice-like water would correspond to the freezing entropy of water of −22 J mol−1 K−1.42 Note that this quantity refers to moles of water, whereas the reaction entropy is referenced towards the conversion of the cell reaction, i.e., moles of charge flowing in the outer cell circuit. By comparing the reaction entropy variation of 50 J mol−1 K−1 with the freezing entropy of water, we conclude that more than two layers of water would need to change their state from fully loosened to fully oriented ice-like in order to account for the measured variation or the reaction entropy. This seems to be an unreasonably high amount, since in situ X-ray diffraction and reflection studies by Wang et al. of Au(111) in F−, Cl− and Br− containing solutions with various concentrations show that already the second water layer is more disordered than the first layer.16 In addition, structure-breaking anions such as chloride and structure-making anions such as sulfate should have opposite effects on the hydrogen-bonding network.41 Therefore, if the contributions of the solvent to the reaction entropy variations were significant, we would expect differences of the shapes of the reaction entropy curves for different anions, in particular at higher anion coverages. From these considerations, we conclude that the potential-dependent water reorientation may significantly change the entropy of formation of the interface only at potentials close to the pzc, corresponding to anion coverages well below θ ≈ 0.1. Above this coverage, the configurational entropy of the anions and its coverage dependence may explain the observed entropy variations.
To corroborate the finding that essentially the anion contribution causes the variation of the reaction entropy at intermediate to high anion coverages, we conducted ECM measurements, where we substituted the electrolyte cation K+ with Li+ and Cs+. In Fig. 4, the reaction entropy obtained in 0.1 M LiCl, 0.1 M KCl and 0.1 M CsCl is shown as a function of potential. For better comparison, the potential is referenced to the first peak in the CV, signalling the lifting of the surface reconstruction, which was shown to be independent of the nature of the cation.16 The absolute variation of the reaction entropy with potential is within the same order of magnitude for all three solutions. This indicates that the coadsorption of cations is irrelevant, since due to the large differences in the ionic entropies in solution (for Cs+ and Li+, they differ by more than 120 J mol−1 K−1),43 huge differences of the adsorption entropies between solutions with different cations would be expected. Nevertheless, the minimum of the reaction entropy is shifted to lower potentials in the case of Cs+ and to higher potentials for Li+ in comparison with K+. The observed trend could straightforwardly be explained within the framework of our QCA lattice-gas model, if the cations influence the interactions between the anions. According to Fig. 3, a higher interaction parameter w(θ = 1) shifts the minimum of the reaction entropy curve to lower coverages. This implies that, due to the presence of cations, the repulsive interactions between the surface dipoles increase in the order of Li+ < K+ < Cs+, which is explicable by a stronger screening of the surface dipole in the presence of Li+ and a decreased screening in the presence of Cs+. This would be in-line with infrared-spectroscopy studies, showing that upon the substitution of K+ with Li+ or Na+ the vibration frequency of the O–H stretching bands associated with water molecules arranged around the anion is blue-shifted by up to 15 cm−1.44 This explanation implies a rather high concentration of cations in the proximity of the adsorbed anions, which at first glance contradicts the classical DL theory. However, recent results indicate that the DL is more compact on Au(111) and Pt(111) than on Hg electrodes,45 due to the attractive interaction between the electrolyte and the metal surface, and an accumulation of ions near the surface is expected.46
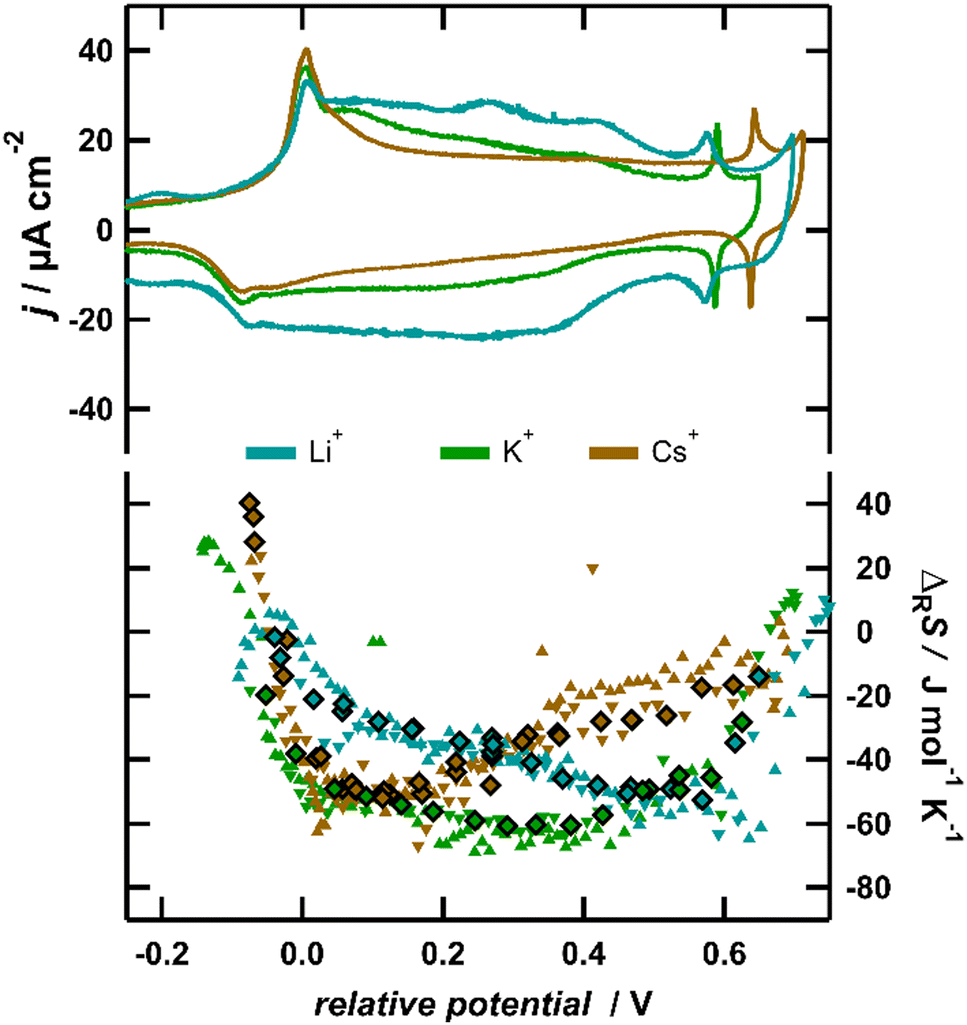 | ||
| Fig. 4 Left axis: cyclic voltammograms of Au(111) in 0.1 M LiCl (light blue), KCl (green) and CsCl (brown) at a sweep rate of 50 mV s−1; right axis: reaction entropy obtained by series of 10 ms, 50 and 100 μA positive and negative current pulses (upward and downward triangles) and by potential pulses of varying amplitude and polarity (diamonds). Note that the potential scale is referenced to the potential of the first current peak in the CV. | ||
Conclusions
Our experiments on the adsorption entropy of anions on Au(111) showed that for all investigated anions this quantity exhibits a universal, U-shaped variation with potential or anion coverage and that these entropy variations are of considerable magnitude. Particularly, at low anion coverages, the entropic contribution dominantly determines the free enthalpy of the adsorption reaction and thus the potential drop across the interface. This signals that entropy contributions should be explicitly considered in theoretical studies on the stability of electrochemical surface phases, especially for disordered surface systems. In our interpretation, for low anion coverages, we ascribe the entropy variations to the reorientation of solvent molecules near pzc. This process should be generally important, also for ordered phases, at a low total surface charge, i.e., at a low electric field in the interface. In contrast, at intermediate to high anion coverages, the configurational entropy of the surface phase of interacting anions could explain the entropy variations. Similar variations are expected for nearly every adsorbate system, where the adsorbate coverage varies continuously with potential, exhibiting a continuous variation of its structure without forming ordered phases. Thus, the entropy of formation of the interface, in our case that for anion adsorption, provides a rather general indicator of the structure, order and composition of the interface, which might motivate its use as a descriptor for electrocatalytic activity as mentioned above.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge funding by the Deutsche Forschungsgemeinschaft (SCHU 958/7-2). We further thank M. T. M. Koper for bringing ref. 33 and 34 to our attention.References
- (a) D. Strmcnik, K. Kodama, D. van der Vliet, J. Greeley, V. R. Stamenkovic and N. M. Marković, Nat. Chem., 2009, 1, 466 CrossRef CAS PubMed; (b) V. Colic, M. D. Pohl, D. Scieszka and A. S. Bandarenka, Catal. Today, 2016, 262, 24 CrossRef CAS; (c) B. Deng, M. Huang, X. Zhao, S. Mou and F. Dong, ACS Catal., 2022, 12, 331 CrossRef CAS; (d) J. Suntivich, E. E. Perry, H. A. Gasteiger and Y. Shao-Horn, Electrocatalysis, 2013, 4, 49 CrossRef CAS.
- T. Wang, Y. Zhang, B. Huang, B. Cai, R. R. Rao, L. Giordano, S.-G. Sun and Y. Shao-Horn, Nat. Catal., 2021, 4, 753 CrossRef CAS.
- D. S. Strmcnik, P. Rebec, M. Gaberscek, D. Tripkovic, V. Stamenkovic, C. Lucas and N. M. Marković, J. Phys. Chem. C, 2007, 111, 18672 CrossRef CAS.
- V. Climent, B. A. Coles and R. G. Compton, J. Phys. Chem. B, 2002, 106, 5258 CrossRef CAS.
- A. Ganassin, P. Sebastián, V. Climent, W. Schuhmann, A. S. Bandarenka and J. Feliu, Sci. Rep., 2017, 7, 1246 CrossRef PubMed.
- N. Garcia-Araez, V. Climent and J. Feliu, J. Phys. Chem. C, 2009, 113, 19913 CrossRef CAS.
- (a) X. Ding, D. Scieszka, S. Watzele, S. Xue, B. Garlyyev, R. W. Haid and A. S. Bandarenka, ChemElectroChem, 2022, 9, e202101088 CAS; (b) B. Huang, R. R. Rao, S. You, K. Hpone Myint, Y. Song, Y. Wang, W. Ding, L. Giordano, Y. Zhang, T. Wang, S. Muy, Y. Katayama, J. C. Grossman, A. P. Willard, K. Xu, Y. Jiang and Y. Shao-Horn, JACS Au, 2021, 1, 1674 CrossRef CAS PubMed.
- J. A. Harrison, J. E. B. Randles and D. J. Schiffrin, J. Electroanal. Chem., 1973, 48, 359 CrossRef CAS.
- R. Schuster, Curr. Opin. Electrochem., 2017, 1, 88 CrossRef CAS.
- J. Lindner, F. Weick, F. Endres and R. Schuster, J. Phys. Chem. C, 2020, 124, 693 CrossRef CAS.
- J. Lipkowski, Z. Shi, A. Chen, B. Pettinger and C. Bilger, Electrochim. Acta, 1998, 43, 2875 CrossRef CAS.
- P. Rodriguez and M. T. M. Koper, Phys. Chem. Chem. Phys., 2014, 16, 13583 RSC.
- Y. Hori, H. Wakebe, T. Tsukamoto and O. Koga, Electrochim. Acta, 1994, 39, 1833 CrossRef CAS.
- M. C. O. Monteiro, F. Dattila, B. Hagedoorn, R. García-Muelas, N. López and M. T. M. Koper, Nat. Catal., 2021, 4, 654 CrossRef CAS.
- O. M. Magnussen, Chem. Rev., 2002, 102, 679 CrossRef CAS PubMed.
- J. Wang, B. M. Ocko, A. J. Davenport and H. S. Isaacs, Phys. Rev. B: Condens. Matter Mater. Phys., 1992, 46, 10321 CrossRef CAS PubMed.
- Z. Shi, S. Wu and J. Lipkowski, Electrochim. Acta, 1995, 40, 9 CrossRef CAS.
- (a) T. Kondo, J. Morita, K. Hanaoka, S. Takakusagi, K. Tamura, M. Takahasi, J. Mizuki and K. Uosaki, J. Phys. Chem. C, 2007, 111, 13197 CrossRef CAS; (b) K. Ataka and M. Osawa, Langmuir, 1998, 14, 951 CrossRef CAS.
- A. Chen, Z. Shi, D. Bizzotto, J. Lipkowski, B. Pettinger and C. Bilger, J. Electroanal. Chem., 1999, 467, 342 CrossRef CAS.
- S. Frittmann, V. Halka and R. Schuster, Angew. Chem., 2016, 55, 4688 CrossRef CAS PubMed.
- B. M. Ocko and T. Wandlowski, MRS Online Proc. Libr., 1996, 451, 55 CrossRef.
- A. L. Rockwood, Thermochim. Acta, 2009, 490, 82 CrossRef CAS.
- B. E. Conway, J. Solution Chem., 1978, 7, 721 CrossRef CAS.
- P. Gao and M. J. Weaver, J. Phys. Chem., 1986, 90, 4057 CrossRef CAS.
- Z. Shi and J. Lipkowski, J. Electroanal. Chem., 1996, 403, 225 CrossRef.
- (a) Z. Shi, J. Lipkowski, S. Mirwald and B. Pettinger, Faraday Trans., 1996, 92, 3737 RSC; (b) Z. Shi, J. Lipkowski, M. Gamboa, P. Zelenay and A. Wieckowski, J. Electroanal. Chem., 1994, 366, 317 CrossRef CAS.
- O. M. Magnussen, B. M. Ocko, R. R. Adzic and J. X. Wang, Phys. Rev. B: Condens. Matter Mater. Phys., 1995, 51, 5510 CrossRef CAS PubMed.
- G. H. Findenegg and T. Hellweg, Statistische Thermodynamik, Springer; Berlin, Heidelberg, 2015 Search PubMed.
- M. Schönig, S. Frittmann and R. Schuster, Chem. Phys. Chem., 2022, 23, e202200227 CrossRef PubMed.
- T. Wandlowski, K. Ataka, S. Pronkin and D. Diesing, Electrochim. Acta, 2004, 49, 1233 CrossRef CAS.
- M. T. M. Koper, J. Electroanal. Chem., 1998, 450, 189 CrossRef CAS.
- B. E. Conway and H. Angerstein-Kozlowska, J. Electroanal. Chem., 1980, 113, 63 CrossRef CAS.
- R. J. Baxter, J. Phys. A: Math. Gen., 1980, 13, L61–L70 CrossRef.
- M. T. M. Koper and J. J. Lukkien, J. Electroanal. Chem., 2000, 485, 161 CrossRef CAS.
- A. Bondi, J. Phys. Chem., 1964, 68, 441 CrossRef CAS.
- X. Wang, R. Chen, Y. Wang, T. He and F.-C. Liu, J. Phys. Chem. B, 1998, 102, 7568 CrossRef CAS.
- W. R. Fawcett, S. Levine, R. M. deNobriga and A. C. McDonald, J. Electroanal. Chem., 1980, 111, 163 CrossRef CAS.
- J. Bockris and M. A. Habib, J. Electroanal. Chem., 1975, 65, 473 CrossRef CAS.
- R. Guidelli and W. Schmickler, Electrochim. Acta, 2000, 45, 2317 CrossRef CAS.
- D. C. Grahame, J. Chem. Phys., 1955, 23, 1725 CrossRef CAS.
- K. Ataka, T. Yotsuyanagi and M. Osawa, J. Phys. Chem., 1996, 100, 10664 CrossRef CAS.
- W. F. Giauque and J. W. Stout, J. Am. Chem. Soc., 1936, 58, 1144 CrossRef CAS.
- Y. Marcus, Ion solvation, Wiley; Chichester, 1985 Search PubMed.
- M. Futamata, Surf. Sci., 1999, 427–428, 179 CrossRef CAS.
- K. Ojha, N. Arulmozhi, D. Aranzales and M. T. M. Koper, Angew. Chem., 2020, 59, 711 CrossRef CAS PubMed.
- K. Doblhoff-Dier and M. T. M. Koper, J. Phys. Chem. C, 2021, 125, 16664 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2cp04680f |
| This journal is © the Owner Societies 2023 |