
Unveiling the high reactivity of experimental pseudodiradical azomethine ylides within molecular electron density theory†
Mar
Ríos-Gutiérrez
 *a,
Luis R.
Domingo
a and
Radomir
Jasiński
*b
*a,
Luis R.
Domingo
a and
Radomir
Jasiński
*b
aDepartment of Organic Chemistry, University of Valencia, Dr Moliner 50, Burjassot, 46100 Valencia, Spain. E-mail: rios@utopia.uv.es
bFaculty of Chemical Engineering and Technology, Department of Organic Chemistry and Technology, Cracow University of Technology, Warszawska 24, 31-155 Cracow, Poland. E-mail: radomir@chemia.pk.edu.pl
First published on 22nd November 2022
Abstract
The [3+2] cycloaddition (32CA) reactions of N-methyl azomethine ylide (AY) with styrene, benzaldehyde and methyl 2-formyl-benzoate (MFB) were studied within molecular electron density theory (MEDT), at the ωB97X-D/6-311G(d) computational level, in order to characterize the reactivity of an experimental pseudodiradical TAC for the first time. ELF topological analysis indicates that AY presents a pseudodiradical structure. Analysis of CDFT reactivity indices allows classifying AY as a supernucleophile; while styrene is classified as a moderate electrophile, benzaldehyde and MFB are classified as strong electrophiles. The 32CA reaction with MFB is the most favorable one with a relatively low activation Gibbs free energy of 6.9 kcal mol−1, being irreversible and completely endo stereo- and chemo-selective towards the carbonyl group, a behavior predicted by the analysis of the Parr functions. The bonding evolution theory (BET) study indicates that while the 32CA reaction of AY with styrene is characterized as a pdr-type 32CA reaction, the one involving benzaldehyde follows a pmr-type mechanism prompted by the presence of the carbonyl group. The present MEDT study describes in detail the tunable high reactivity of one of the few experimentally available pseudodiradical TACs, showing that the mechanism of 32CA reactions can be modified not only by changing the electronic structure of TACs through proper substitution but also by the nature of their opposing ethylene derivative.
1. Introduction
[3+2] cycloaddition (32CA) reactions are one of the most efficient synthetic methods for the construction of five-membered heterocyclic compounds, due to their ability to build organic cyclic motifs regio- and/or stereoselectively.1Understanding of the 32CA reactions is a challenge for organic chemists as a consequence of the variable electronic structures of the three-atom-components (TACs) participating in these cycloaddition reactions. Recent molecular electron density theory2 (MEDT) studies have allowed four different types of TACs to be characterized,3i.e. pseudodiradical, pseudo(mono)radical, carbenoid and zwitterionic TACs, participating in four types of different reactivity models towards ethylene 5, i.e. pdr-, pmr-, cb-, and zw-type reactions, respectively (see Chart 1),3 in such a manner that while pdr-type 32CA reactions take place very easily,4 zw-type 32CA reactions demand appropriate nucleophilic/electrophilic activation of the reagents (see Chart 1).5
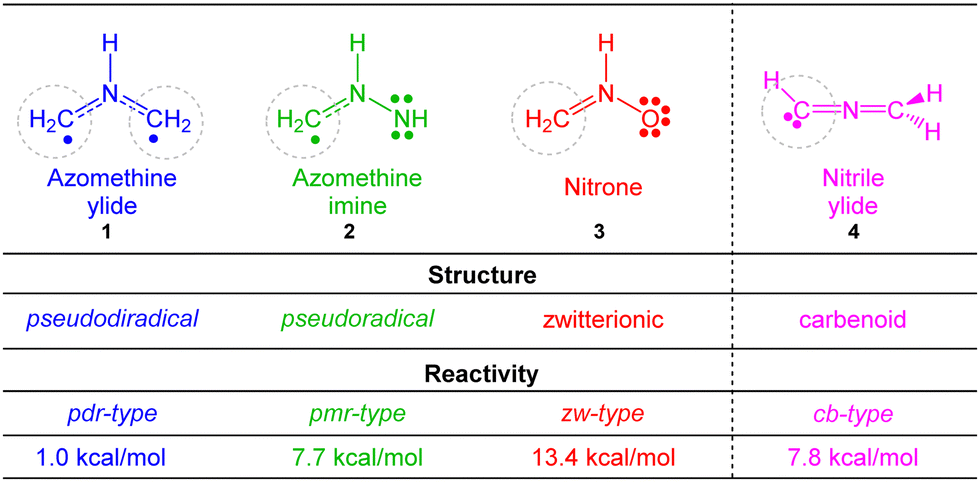 | ||
| Chart 1 Electronic structure of the simplest TACs and proposed reactivity types in 32CA reactions. MPWB1K/6-311G(d) gas phase activation energies of the non-polar 32CA reactions between the four simplest TACs 1–4 and ethylene 5, relative to the corresponding molecular complexes, are given in kcal mol−1. | ||
However, the simplest TACs such as those shown in Chart 1 do not participate in experimental 32CA reactions. In general, TACs are very reactive intermediates usually generated in situ. The adequate substitution that stabilizes TACs can change their electronic structure and, consequently, their reactivity. Therefore, it is desirable to know how substitution modifies their chemical properties.
Azomethine ylides (AYs) are a relevant class of TACs participating in 32CA reactions exhibiting the most universal protocol for the preparation of the pyrrolidine and the oxazolidine molecular segments (Scheme 1).6
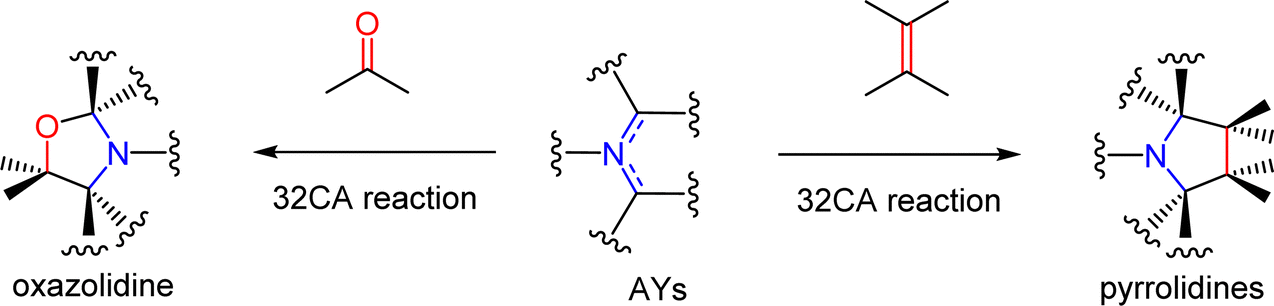 | ||
| Scheme 1 Synthesis of pyrrolidines and oxazolidines via 32CA reactions of AYs. | ||
The importance of these processes is greatly supported by their high selectivity and full atom economy. In this sense, a wide range of usable bioactive compounds can be prepared, such us (±)-aspidospermidine,7a spirotryprosatine B,7b rhynchofylline,7b and many others. They are also applied as starting materials for the stereoselective synthesis of biologically important naturally occurring alkaloids.8
The simplest AY 1, with a pseudodiradical structure,9 is one of the most reactive TACs (see Chart 1 and Scheme 2).3 However, the presence of one electron-withdrawing (EW) carbonyl C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O group in the isatin derivative AY 7, or two EW carbomethoxy CO2Me groups in one of the two methylene carbons of AY 13, stabilizes the corresponding pseudodiradical center, modifying their reactivity to that of a pseudo(mono)diradical TAC10 or even a zwitterionic TAC (see Scheme 2).11 On the other hand, the presence of two fluorine nuclei at the terminal methylene of fluorinated AY 10 changes its electronic structure to that of a carbenoid TAC participating in cb-type 32CA reactions toward electrophilic carbonyl compounds (see Scheme 2).12 Consequently, depending on the substitution present at AYs, four types of structures and reactivities are observed. However, the simplest AY 1 is not an experimental TAC, and very few experimental pdr-type 32CA reactions have been observed.
O group in the isatin derivative AY 7, or two EW carbomethoxy CO2Me groups in one of the two methylene carbons of AY 13, stabilizes the corresponding pseudodiradical center, modifying their reactivity to that of a pseudo(mono)diradical TAC10 or even a zwitterionic TAC (see Scheme 2).11 On the other hand, the presence of two fluorine nuclei at the terminal methylene of fluorinated AY 10 changes its electronic structure to that of a carbenoid TAC participating in cb-type 32CA reactions toward electrophilic carbonyl compounds (see Scheme 2).12 Consequently, depending on the substitution present at AYs, four types of structures and reactivities are observed. However, the simplest AY 1 is not an experimental TAC, and very few experimental pdr-type 32CA reactions have been observed.
 | ||
| Scheme 2 Four different AYs with four different types of reactivities. | ||
Some authors have reported that the non-stabilized N-methyl AY 18, derived in situ from sarcosine 16 and paraformaldehyde 17, undergoes 32CA reactions with aromatic aldehydes to form the corresponding oxazolidines (see Scheme 3).13 Thus, in 2001, Nyerges et al. reported the 32CA reactions of AY 18 with aryl aldehydes 19 yielding the corresponding 1,3-oxazolidines 20 with high yields.14
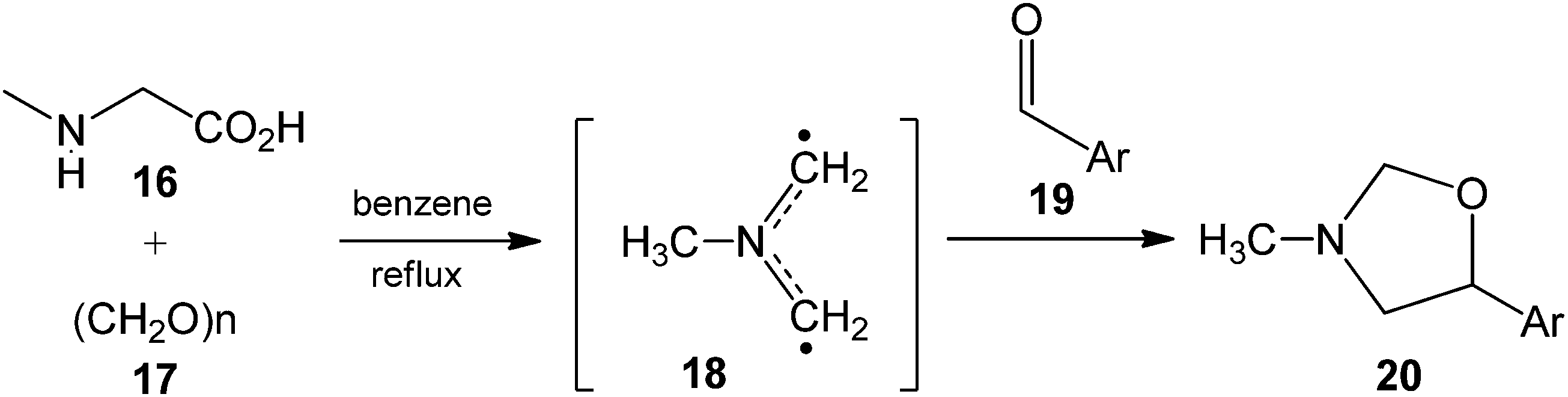 | ||
| Scheme 3 Generation of non-stabilized AY 18in situ, and its 32CA reactions with aromatic aldehydes 19 yielding 1,3-oxazolidines 20. | ||
More recently, in 2015, Moshkin et al. reported the 32CA reaction of AY 18 with methyl 2-formyl-benzoate (MFB) 21 yielding oxazolidine 22 with high yield and total chemoselectivity (see Scheme 4).15
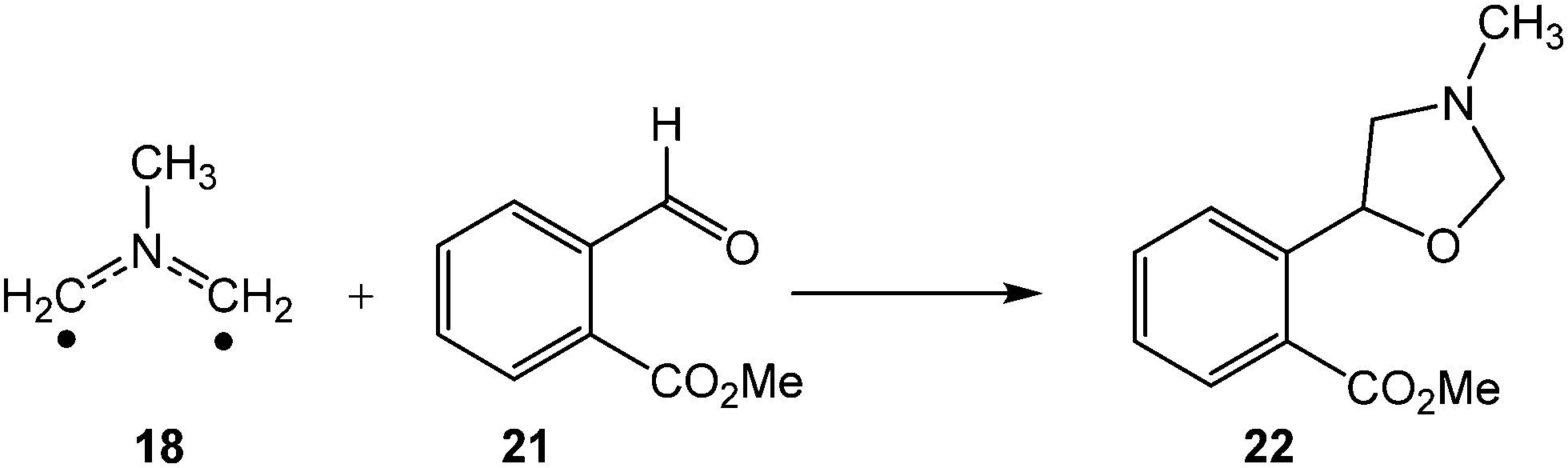 | ||
| Scheme 4 Chemoselective 32CA reaction of AY 18 with MFB 21. | ||
Several non-stabilized AYs such as N-phenyl and N-benzyl AYs 23 and 24 were also used in the synthesis of pyrrolidines and oxazolidines (see Chart 2).15
 | ||
| Chart 2 Experimental non-stabilized N-phenyl and N-benzyl AYs 23 and 24. | ||
A preliminary Electron Localization Function16 (ELF) topological analysis of the electronic structure of N-methyl AY 18 revealed its pseudodiradical structure. Due to the absence of any MEDT study of experimental pdr-type 32CA reactions of AYs, the 32CA reactions of AY 18 with styrene 25, benzaldehyde 26 and MFB 21 are herein theoretically studied for the first time in order to corroborate the high reactivity and selectivity of an experimental pseudodiradical AY participating in pdr-type 32CA reactions (see Scheme 5). The origin of the chemoselectivity in the 32CA reactions of MFB 21 is also analyzed.
 | ||
| Scheme 5 32CA reactions of the experimental non-stabilized pseudodiradical AY 18 studied herein. | ||
2. Computational methods
DFT calculations were performed using the hybrid ωB97X-D functional,18 which includes long-range exchange and semiclassical London-dispersion corrections, together with the standard 6-311G(d) basis set.19 A similar theory level is commonly used in the theoretical study of cycloaddition reactions. Optimizations were carried out by using the Berny analytical gradient optimization method.20 The stationary points on the potential energy surfaces (PES) were characterized through frequency analysis; transition states (TSs) were ensured to present one and only one imaginary frequency. The intrinsic reaction coordinate (IRC) paths21 were computed to find the unique connection between the TSs and minima using the second order González–Schlegel integration method.22 Solvent effects of benzene were considered by full optimization of the gas phase structures at the same computational level using the polarizable continuum model23 (PCM) in the framework of the self-consistent reaction field24 (SCRF). ωB97X-D/6-311G(d) enthalpies, entropies and Gibbs free energies in benzene were calculated with standard statistical thermodynamics at the boiling point temperature of benzene, 353.25 K, and 1 atm.19The global electron density transfer25 (GEDT) of the reactions was estimated using the equation 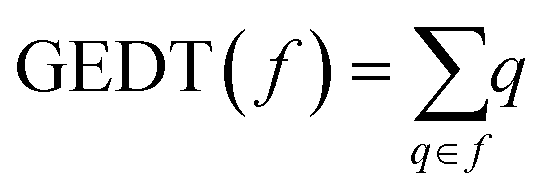 , were q are the charges, computed by natural population analysis26 (NPA), of all the atoms belonging to one of the two frameworks (f) at the TSs. Conceptual DFT27 (CDFT) reactivity indices were calculated at the B3LYP/6-31G(d) computational level because the electrophilicity and nucleophilicity scales were established at that level by using the equations given in ref. 27b. All computations were carried out with the Gaussian 16 suite of programs.28
, were q are the charges, computed by natural population analysis26 (NPA), of all the atoms belonging to one of the two frameworks (f) at the TSs. Conceptual DFT27 (CDFT) reactivity indices were calculated at the B3LYP/6-31G(d) computational level because the electrophilicity and nucleophilicity scales were established at that level by using the equations given in ref. 27b. All computations were carried out with the Gaussian 16 suite of programs.28
The topological analysis of the ELF17 was performed with the TopMod package29 using the ωB97X-D/6-311G(d) monodeterminantal wavefunctions with a cubical grid of step size of 0.1 bohr. GaussView program30 was used to visualize the molecular geometries of all the systems as well as the position of the ELF basin attractors.
3. Results and discussion
The following study has been divided into four sections: (i) first, the electronic structure of experimental AY 18 and aryl derivatives 21, 25, and 26 is characterized through ELF topological analysis; (ii) then, the CDFT reactivity indices of the reagents are analyzed in order to predict the preferred reaction paths; (iii) the PESs associated with the competitive reaction paths related to the 32CA reactions between AY 18 and aryl derivatives 21, 25, and 26 are studied; (iv) finally, a BET study is performed along the 32CA reactions of AY 18 with styrene 25 and benzaldehyde 26 in order to characterize the molecular mechanism of these reactions involving a pseudodiradical TAC.3.1. ELF topological analysis of the electronic structure of the reagents
The topological analysis of the ELF provides a straightforward connection between the electron density distribution and the chemical structure.31 Thus, in order to characterize the electronic structure of experimental AY 18, and predict its reactivity according to the structure–reactivity relationship of TACs participating in 32CA reactions,3 a topological analysis of the ELF was performed. The structures of styrene 25 and aryl aldehydes 21 and 26 were also studied in order to investigate whether there is any bonding change affecting their reactivity. ELF basins and the most relevant valence basins are given in Fig. 1. For comparison, ELF topology of the experimental non-stabilized AYs 23 and 24 is shown in Fig. S1 in the ESI.†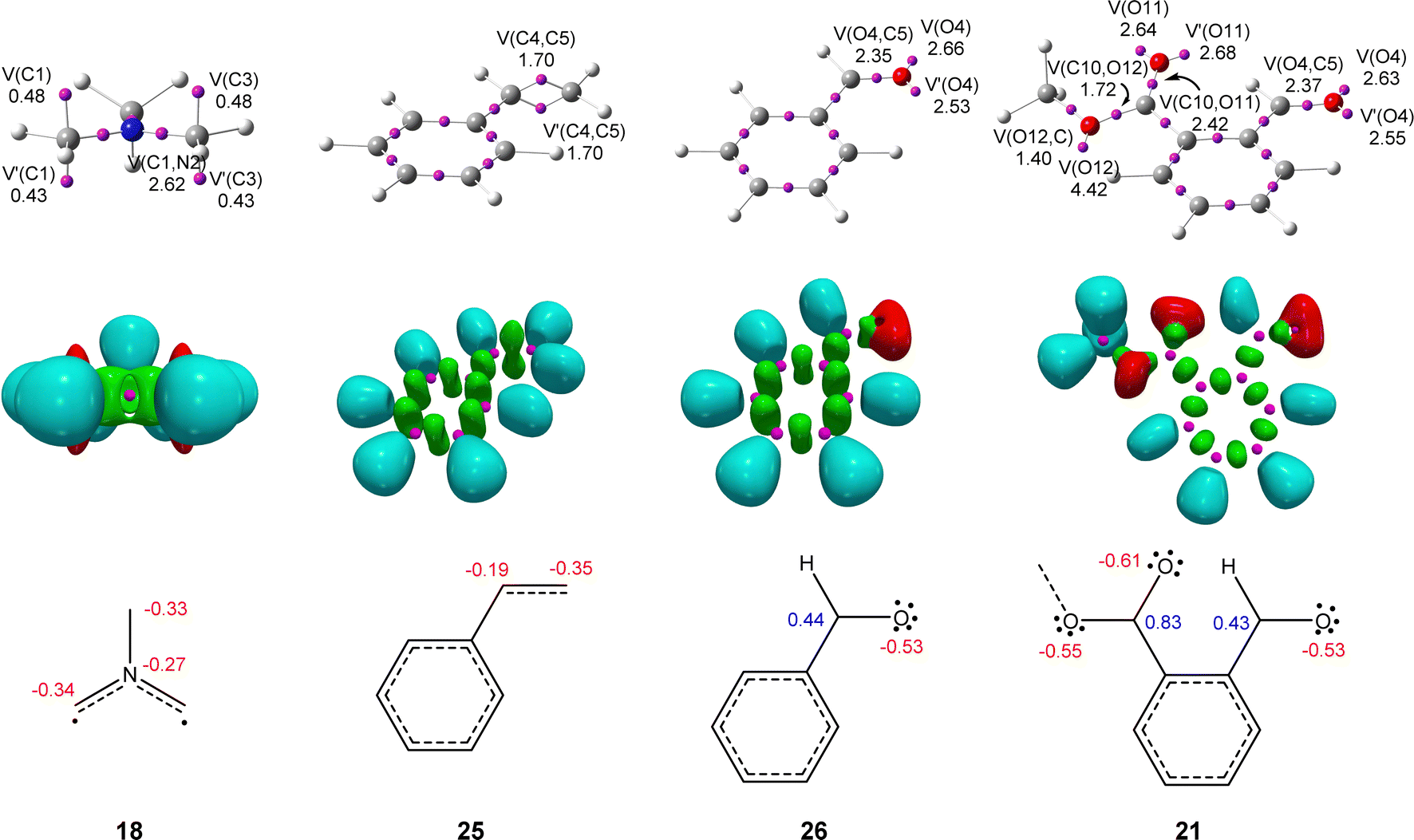 | ||
| Fig. 1 ωB97X-D/6-311G(d) ELF basin attractor positions together with the most relevant valence basin populations, ELF localization domains represented at an isosurface value of ELF = 0.75, and ELF-based Lewis-like structures together with natural atomic charges, for AY 18, styrene 25, and aryl aldehydes 21 and 26. Valence basin populations and natural atomic charges are given as the average number of electrons, e. Negative charges are colored in red and positive charges in blue. | ||
Topological analysis of the ELF of experimental AY 18 shows the presence of two V(C) and V′(C) monosynaptic basins, integrating a total of 0.91 e, at each of the two methylene carbon atoms, and a V(C,N) disynaptic basin integrating 2.62 e. At styrene 25, two V(C,C) and V′(C,C) disynaptic basins, integrating a total population of 3.40 e, are observed in the C4–C5 ethylene region, while one V(C5,CAr) disynaptic basin, integrating 2.17 e, is observed in the C5–CAr bonding region. Finally, the carbonyl C–O regions of the aldehyde and ester groups in benzaldehyde 26 and MFB 21 are characterized by the presence of a V(O,C) disynaptic basin integrating ca. 2.4 e, and two V(O) and V′(O) disynaptic basins with a total population of ca. 5.2 e.
The ELF topology of AY 18 can be related to two pseudoradical carbon centers9 at the methylene groups and two C–N partial double bonds (see Fig. 1).4,9 At styrene 25, the ELF topological features indicate the delocalization of electron density within the aromatic phenyl ring and an underpopulated ethylene CC double bond; the population of the V(C6,CAr) disynaptic basin indicates that there is a small electron density delocalization between the ethylene and the aromatic ring. Finally, the ELF topological analyses of benzaldehyde 26 and MFB 21 show a strong polarization of the carbonyl C–O bonding region towards the oxygen, being rather described as populated C–O single bonds with a higher amount of non-bonding electron density at the oxygen nuclei.
The presence of two pseudoradical carbon centers in experimental AY 18 allows this species to be classified as a pseudodiradical TAC participating in pdr-type 32CA reactions,3 just as the simplest AY 1 (see Chart 1 and Scheme 2).4 On the other hand, there are no relevant bonding changes between benzaldehyde 26 and MFB 21; consequently, any reactivity difference between them will rather be attributed to their different electrophilic behaviors. It is also worth noting that, unlike benzaldehyde 26 and MFB 21, which present carbonyl C–O single bonds, the C–C ethylene region in styrene 25 has a double bond character which might require an extra energy cost to break. Note the C(O)4–C5 single bonds in the final cycloadducts 22, 27, and 28 (see Scheme 5).
Next, the charge distribution of these species was analyzed by NPA.26 NPA of AY 18 indicates that, except for hydrogen nuclei, the rest are negatively charged by ca. 0.3 e (see Fig. 1). This finding clearly proves that the electronic structure of TACs is not the result of a set of octet 1,2- and sextet 1,3-dipolar resonance structures, as Huisgen proposed.32 In styrene 25, all carbon atoms are also negatively charged; interestingly, the most electrophilic C4 carbon (see later) is the most negatively charged, −0.35 e. In benzaldehyde 26 and MFB 21, this effect is enhanced; while the more electrophilic aldehyde oxygen atoms are negatively charged by −0.53 e, the carbonyl carbons, which are positively charged by ca. 0.4 and 0.8 e, are either less or scarcely electrophilically activated (see later). This behavior contradicts the traditional conception that carbonyl carbons are electrophilic centers, and confirms Domingo's observation made in 2012 that charges do not control the electrophilic/nucleophilic behavior of molecules.33
3.2. Analysis of the conceptual DFT reactivity descriptors
CDFT27 is a branch of DFT that provides traditional reactivity concepts defined from experimental observation, such as electrophilicity and nucleophilicity, with a quantitative expression. In this sense, analysis of the CDFT indices has proved to be a useful tool to study polar cycloaddition reactions.34 Thus, in order to characterize, both qualitatively and quantitatively, the behavior of experimental AY 18 in the 32CA reactions with styrene 25, benzaldehyde 26, and MFB 21, the reactivity indicators defined within CDFT were analyzed. Table 1 shows the most relevant ones.| Structure | μ | η | ω | N |
|---|---|---|---|---|
| Simplest AY 1 | −1.77 | 4.37 | 0.35 | 5.15 |
| AY 18 | −1.76 | 4.32 | 0.35 | 5.19 |
| AY 24 | −2.15 | 3.81 | 0.61 | 5.06 |
| Ethylene 5 | −3.37 | 7.82 | 0.72 | 1.83 |
| AY 23 | −2.41 | 3.48 | 0.83 | 4.97 |
| Styrene 25 | −3.41 | 5.28 | 1.10 | 3.05 |
| Benzaldehyde 26 | −4.28 | 5.30 | 1.72 | 2.17 |
| MFB 21 | −4.33 | 4.76 | 1.96 | 2.39 |
An initial analysis of the results given in Table 1 indicates that the global reactivity indices of AY 18 and the simplest AY 1 are almost the same, consequently, a similar reactivity is expected based only on the electronic properties.
The electronic chemical potential35μ of AY 18, −1.76 eV, is above those of styrene 25, −3.41 eV, benzaldehyde 26, −4.28 eV, and MFB 21, −4.33 eV, suggesting that along a polar reaction, the GEDT25 will take place from AY 18 to the aryl derivatives via forward electron density flux (FEDF) reactions.36 Note, however, that the μ of styrene 25 and ethylene 5 are very similar, the latter being one of the least reactive species.34
The chemical hardness37η of the species participating in these selected 32CA reactions is quite similar, ranging between 4.32 (AY 18) and 5.30 (26) eV. While the AYs are the softest species, 3.5–4.4 eV, with closer values to that of MFB 21, 4.76 eV, styrene 25 and benzaldehyde 26 are the hardest species, 5.3 eV, after ethylene 5, 7.82 eV. This suggests that the reaction between AY 18 and MFB 21 should be easier than with styrene 25 and benzaldehyde 26.
The electrophilicity38ω and nucleophilicity39N indices of experimental AY 18 are 0.35 and 5.19 eV, respectively, being classified as a very poor electrophile and as a strong nucleophile, within the corresponding scales.27b The very high nucleophilic value of AY 18, higher than 4.0 eV, ranks it as a supernucleophile.34 As can be observed, the addition of a methyl group at the N2 nitrogen of the simplest AY 1 has no appreciable effect on the electronic properties of the experimental AY 18 other than increasing its nucleophilic character by a negligible amount. Finally, the non-stabilized phenyl and benzyl AYs 23 and 24 have electrophilicity ω indices of 0.83 and 0.62 eV, respectively, and nucleophilicity N indices of 4.97 and 5.06 eV. The presence of the aromatic ring in these AYs slightly increases their electrophilicity ω indices, but they are still classified as marginal electrophiles. On the other hand, the supernucleophilic character of these AYs indicates they will have a reactivity similar to that of AY 18 towards carbonyl compounds.
On the other hand, ethylene 5 is one of the most unreactive species,34 having electrophilicity ω and nucleophilicity N values of 0.72 and 1.83 eV, respectively. Inclusion of a phenyl ring in ethylene 5 increases both the electrophilicity and nucleophilicity of styrene 25, but has a much stronger effect on the latter, reaching ω and N values of 1.10 and 3.05 eV, respectively. These values allow classifying styrene 25 as a moderate electrophile and on the borderline of strong nucleophiles.
When the ethylene fragment in styrene 25 is substituted by an EW aldehyde –CHO group, the electrophilicity ω of benzaldehyde 26 somewhat increases to 1.72 eV and the nucleophilicity N significantly decreases to 2.17 eV, being classified as a strong electrophile and as a moderate nucleophile.
Inclusion of a methyl carboxylate group in the meta position of benzaldehyde 26 rises both the electrophilicity ω and the nucleophilicity N indices of MFB 21 to 1.96 and 2.39 eV, but it does not change its classification as a strong electrophile and moderate nucleophile.
A great number of theoretical studies of organic reactions have shown that the analysis of the Parr functions40 is one of the most accurate tools to predict and understand the local reactivity in reactions involving non-symmetric reagents. Thus, in order to predict the most favorable interaction along the experimental 32CA reactions of supernucleophilic AY 18 with electrophilic benzaldehyde 26 and MFB 21, the corresponding Parr functions were analyzed (see Fig. 2).
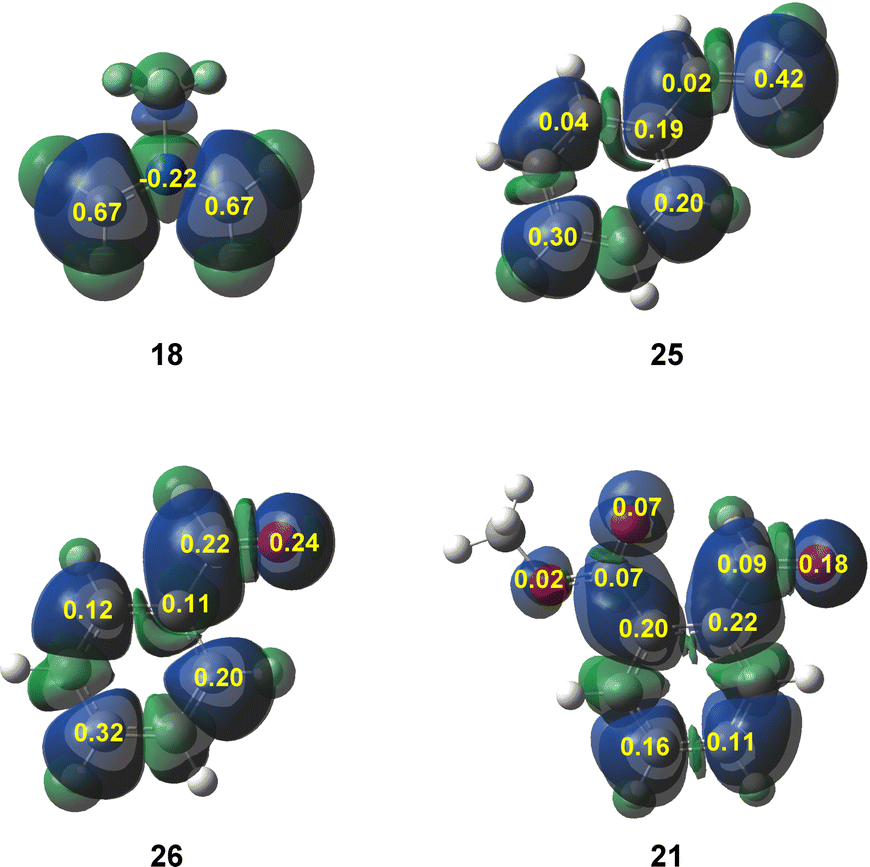 | ||
| Fig. 2 3D representations of the Mulliken atomic spin densities of the radical cation of AY 18 and the radical anions of 21, 25 and 26, and the corresponding nucleophilic and electrophilic Parr functions. | ||
The nucleophilic Parr functions at experimental AY 18, which are exactly the same as those at the simplest AY 1,3 indicate that the most nucleophilic centers are the two methylene carbons with a high Pk− value of 0.67. In the series of aryl derivatives, the analysis of the electrophilic activation is more complex. In the three molecules, the aromatic ring is more electrophilically activated than the unsaturated C5C[O]4 framework. Moreover, in the three cases, the terminal C[O]4 centers are more electrophilicity activated than the C5 ones. In the case of MFB 21, the carbonyl group is more electrophilically activated than the metoxycarboxyl one, accounting for the experimentally observed chemoselectivity.
3.3. Analysis of the reaction paths associated with the 32CA reactions of AY 18 with styrene 25, benzaldehyde 26, and MFB 21
Due to the symmetry of AY 18, only two endo and exo stereoisomeric reaction paths are feasible along the 32CA reactions involving styrene 25 and benzaldehyde 26 (see Scheme 6); in the 32CA reaction with MFB 21, there are two additional chemoselective reaction paths involving either the aldehyde group, paths III, or the carboxylate group, paths IV (see Scheme 7). Analysis of the stationary points found along these reaction paths indicates that the 32CA reactions of AY 18 take place through one-step mechanisms involving a single TS. Relative energies in the gas phase and in benzene are given in Schemes 6 and 7, while absolute ones are given in Table S1 in the ESI.† | ||
| Scheme 6 Stereoisomeric reaction paths associated with the 32CA reactions of AY 18 with styrene 25 and benzaldehyde 26. Relative energies in the gas phase and in benzene (in parentheses) are given in kcal mol−1. | ||
 | ||
| Scheme 7 Chemo- and stereoisomeric reaction paths associated with the 32CA reaction of AY 18 with MFB 21. Relative energies in the gas phase and in benzene (in parentheses) are given in kcal mol−1. | ||
Most organic reactions start with the formation of molecular complexes (MCs) in which the two reagents are held together by weak intermolecular interactions. Many different MCs can be found for a single reaction depending on the relative approach of the reagents; however, due to the weak nature of the intermolecular forces between the two frameworks, the energy difference between them is usually in the range of that between conformational isomers and results in them being in thermodynamic equilibrium. Therefore, only the most stable MC among those found along the corresponding reaction paths was reported for each of the three 32CA reactions.
The formation of MC-Z is very favorable, releasing between 8.7 and 11.7 kcal mol−1. The GEDT25 at these MCs is 0.03 e at MC-I, and 0.10 e at MC-II and MC-III; consequently, only the two latter, with a GEDT > 0.10 e, may be considered as electron density transfer complexes (EDTCs).41 All TSs are energetically found below the separated reagents, the relative energies of TS-Z ranging between −0.1 (TS-IV-x) and −11.4 (TS-III-n) kcal mol−1; however, when the formation of MC-Z is considered, the activation energies of the preferred stereoisomeric pathways become positive by 3.8 (TS-I-x), 1.3 (TS-II-n), 0.3 (TS-III-n) and 8.3 (TS-IV-n) kcal mol−1. The three reactions are strongly exothermic, by between 74.8 (CA-I-x) and 37.6 (CA-IV-n) kcal mol−1.
Solvent effects of benzene stabilize all stationary points in a very small and similar amount, by between 1.0 and 4.1 kcal mol−1; the more electrophilic the aryl derivative, the stronger the stabilization. In the three reactions, the separated reagents are slightly more stabilized than MCs, TSs, and CAs by an average of 0.8 kcal mol−1 more, while the energies of TSs vary by only 0.3 kcal mol−1 in comparison to those of MCs. Consequently, the changes in both activation and reaction energies with the inclusion of benzene will be barely noticeable. On the other hand, except TS-III-x, the exo TSs are stabilized by between 0.1 and 0.8 kcal mol−1 more than the endo counterparts. Therefore, although benzene stabilizes the exo TSs slightly more, it does not produce any change neither in the stereo- nor in the chemoselectivity of the reactions.
Relative thermodynamic data are gathered in Table 2, while absolute values are given in Table S2 in the ESI.† Inclusion of thermal corrections to the electronic energies in benzene increases the relative enthalpies by between 1.0 and 2.3 kcal mol−1 for MCs and TSs, and from 3.5 to 5.1 kcal mol−1 for CAs. The effect on the activation enthalpies is negligible, which decreases by a maximum of 0.6 kcal mol−1. When the unfavorable negative entropies associated with these bimolecular reactions, which range from −32.5 to −54.6 cal mol−1 K−1, are added, the resulting relative Gibbs free energies considerably increase by 11.5–19.3 kcal mol−1. Now, the relative Gibbs free energies of MCs and TSs turn positive and the activation Gibbs free energies of the 32CA reactions of AY 18 with the series of aryl derivatives 21, 25 and 26 are 11.5 (TS-I-x), 8.6 (TS-II-n), 6.9 (TS-III-n) and 15.6 (TS-IV-n) kcal mol−1, the formation of CAs being strongly exergonic by between 11.7 (CA-IV-n) and 52.1 kcal mol−1 (CA-I-x). The Gibbs free energy profiles of the three reactions are represented in Fig. 3.
| Structure | ΔH | ΔS | ΔG |
|---|---|---|---|
| MC-I | −5.6 | −32.5 | 5.9 |
| TS-I-n | −1.9 | −40.5 | 12.4 |
| TS-I-x | −2.7 | −40.2 | 11.5 |
| CA-I-n | −68.8 | −47.1 | −52.1 |
| CA-I-x | −66.8 | −45.9 | −50.6 |
| MC-II | −7.8 | −39.1 | 6.0 |
| TS-II-n | −7.4 | −45.4 | 8.6 |
| TS-II-x | −6.0 | −42.8 | 9.1 |
| CA-II-n | −48.0 | −49.9 | −30.3 |
| CA-II-x | −48.2 | −49.3 | −30.8 |
| MC-III | −9.4 | −42.9 | 5.7 |
| TS-III-n | −9.6 | −46.8 | 6.9 |
| TS-III-x | −8.3 | −45.2 | 7.6 |
| TS-IV-n | −1.8 | −49.2 | 15.6 |
| TS-IV-x | 0.5 | −46.8 | 17.1 |
| CA-III-n | −52.4 | −53.2 | −33.6 |
| CA-III-x | −52.4 | −52.0 | −34.0 |
| CA-IV-n | −31.0 | −54.6 | −11.7 |
| CA-IV-x | −34.6 | −52.9 | −15.9 |
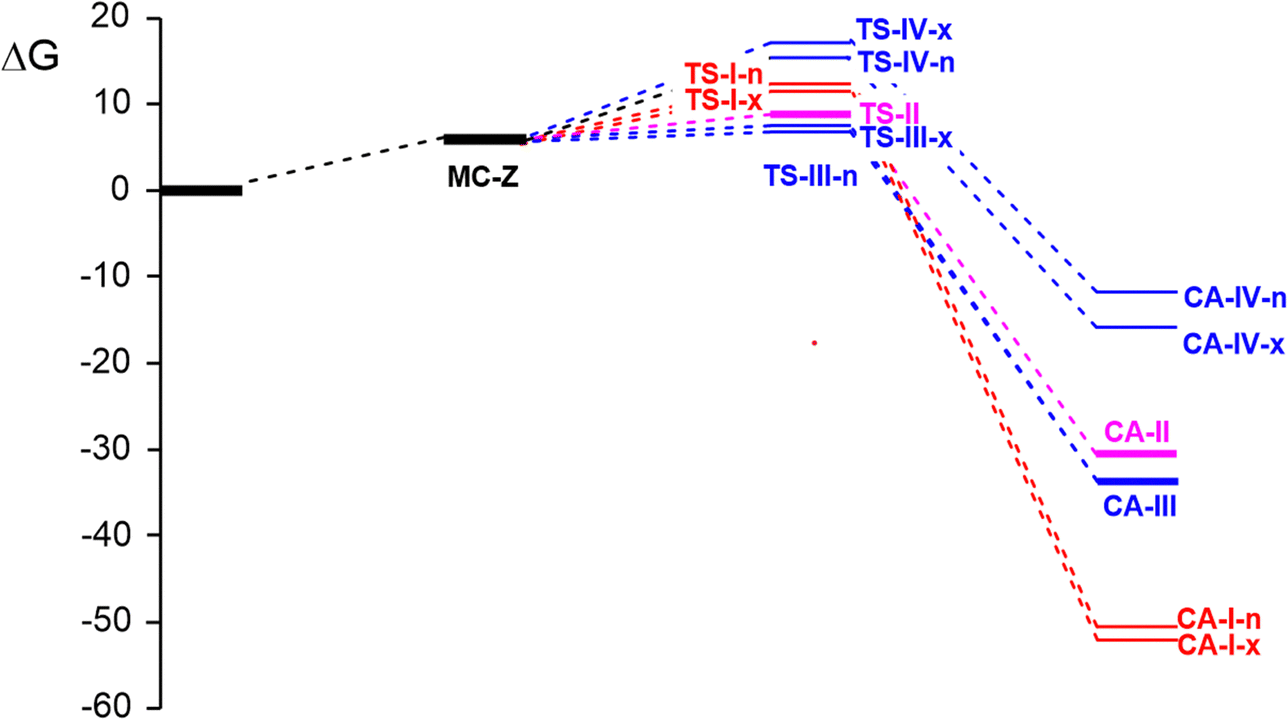 | ||
| Fig. 3 Gibbs free energy profiles, ΔG in kcal mol−1, of the stereo- and chemoisomeric reaction paths associated with the three 32CA reactions between AY 18 and stryrene 25 (in red), benzaldehyde 26 (in pink) and MFB 21 (in blue). | ||
Some concluding remarks can be extracted from these energy data: (i) despite the classification of MC-II and MC-III as EDTCs,41 their positive relative Gibbs free energies make them not experimentally isolable; (ii) while the 32CA reaction involving styrene 25 is exo stereoselective with TS-I-n being 0.9 kcal mol−1 higher in energy than TS-I-x, those involving benzaldehyde 26 and MFB 21 are endo selective with the corresponding exo TSs being 0.5 and 0.7 kcal mol−1 above the endo ones. These low energy differences between endo/exo TSs suggest the formation of a mixture of both stereoisomeric cycloadducts; (iii) the 32CA reactions of MFB 21 is strongly chemoselective towards the aldehyde group, with a huge difference of 8.7 kcal mol−1 between TS-III-n and TS-IV-n; (iv) the activation energies of these 32CA reactions decrease with the increase of the electrophilic character of the aryl derivatives; (v) the most favorable 32CA reaction is that involving the strongly electrophilic MFB 21, in agreement with the experimental outcomes. Note that the experimental reaction time for that reaction is at least 3h shorter than that involving benzaldehyde 26;15 (vi) the extremely low activation energies are in agreement with the pseudodiradical structure of AY 18 participating in pdr-type 32CA reactions, and with its supernucleophilic character;34 and finally, (vii) the strong exergonic character of the three 32CA reactions makes formation of the corresponding cycloadducts CA-I-x, CA-II-n and CA-III-n irreversible: consequently, these are the sole products under kinetic control.
Considering kinetic control of the reactions, the Eyring–Polanyi equation42 was used to estimate the composition of the reaction mixtures.

where ΔΔG‡ is the difference between the relative activation Gibbs free energies of the two TSs of interest, e.g. endo/exo TSs to ascertain stereoselectivity or TS-III/TS-IV for chemoselectivity. The expected percentages for each of the possible products of the 32CA reactions of AY 18 with styrene 25, benzaldehyde 26 and MFB 21 are gathered in Table 3. While the 32CA reaction involving styrene 25 is highly exo stereoselective with a 20.2(endo)
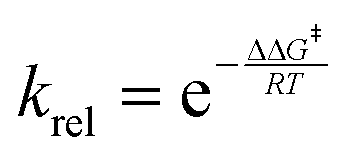
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :79.8(exo) ratio, those involving benzaldehyde 26 and MFB 21 are highly endo stereoselective with an inverse ratio of ca. 70(endo):30(exo). In addition, the 32CA reaction of MFB 21 is 100% chemoselective on the aldehyde group, in agreement with the experimental outcomes.15 These results indicate that in benzene solutions at 80 °C, the three 32CA reactions should yield a mixture of both endo/exo CAs. Unfortunately, experimental studies have not reported any endo:exo ratio to compare with, but the complete chemoselectivity in the latter reaction is in full agreement with the experimental outcomes.15
:79.8(exo) ratio, those involving benzaldehyde 26 and MFB 21 are highly endo stereoselective with an inverse ratio of ca. 70(endo):30(exo). In addition, the 32CA reaction of MFB 21 is 100% chemoselective on the aldehyde group, in agreement with the experimental outcomes.15 These results indicate that in benzene solutions at 80 °C, the three 32CA reactions should yield a mixture of both endo/exo CAs. Unfortunately, experimental studies have not reported any endo:exo ratio to compare with, but the complete chemoselectivity in the latter reaction is in full agreement with the experimental outcomes.15
| Z | CA-Z-n | CA-Z-x |
|---|---|---|
| I | 20.2 | 79.8 |
| II | 68.4 | 31.6 |
| III | 72.7 | 27.3 |
| IV | 0.0 | 0.0 |
Next, the geometries of the TSs involved in the studied competitive reaction paths were analyzed. Distances of the C–C(O)4 and C–C5 forming bonds are given in Table S3 in the ESI,† while the geometries of the most favorable TSs for each 32CA reaction are represented in Fig. 4. Analysis of these geometrical parameters in the gas phase and in benzene shows that (i) the shorter C–C4(5) distances in benzene are found in the range 2.42 Å (TS-I-x)–2.07 Å (TS-IV-n); (ii) the differences between the distances between the two pairs of interacting centers at the most favorable TSs in benzene are variable, with no particular trend: 0.2 (TS-I-x), 0.4 (TS-II-n), 0.3 (TS-III-n) and 0.7 (TS-IV-n) Å; (iii) while benzene makes no significant changes in TS-I-x, distances in TS-II-n and TS-IV-n slightly vary by ca. 0.03–0.09 Å and distances in the most favorable TS-III-n significantly decrease by 0.16–0.22 Å. Interestingly, only in the most favourable TS-III-n do both distances decrease. The asymmetry in benzene progressively increases by only 0.02 (TS-I-x) < 0.04 (TS-II-n) < 0.06 (TS-III-n) < 0.11 (TS-IV-n) Å; (iv) the geometrical asymmetry between endo/exo pairs of TSs is similar, with small average variations of only 0.08 Å. Only in the reaction of styrene 25, is the exo TS more asymmetric than the endo one; and (v) while the C–C distance involving the C4 carbon of styrene 25 is shorter at TS-I-x/n, that involving the carbonyl C5 carbon is shorter at the other TSs.
 | ||
| Fig. 4 ωB97X-D/6-311G(d) geometries of the more favorable stereoisomeric TSs involved in the competitive reaction paths associated with the 32CA reactions of AY 18 with styrene 25, benzaldehyde 26, and MFB 21. Distances are given in angstroms, Å. The values in benzene are given in parentheses. | ||
These data allow drawing some conclusions: (i) except for the unfavorable endo/exoTS-IV, all other TSs can be considered early TSs, in agreement with the very low activation energies;43 (ii) given that formation of C–C and C–O single bonds begin in the distance ranges 2.0–1.9 and 1.8–1.7 Å,3 respectively, these distances at the TSs suggest that bond formation has not started yet at any of them; (iii) the higher asymmetry of TS-IV-n might be due to a more advanced character of this less favorable TS; (iv) solvent effects of benzene have no impact on TS geometries, in agreement with the low energy differences in gas phase and in benzene (see above); and (v) the bond formation/mechanism in endo/exo pairs is expected to be similar.
Finally, the polar character of these 32CA reactions was assessed by computing the GEDT25 at all TSs (see Table 4). As can be observed, benzene has no significant impact on the gas phase GEDT values. The GEDT values computed using the AY framework at the most favorable TSs in benzene are 0.19 (TS-I-x), 0.28 (TS-II-n), 0.25 (TS-III-n) and 0.40 (TS-IV-n) e. These high values, which are a consequence of the supernucleophilic character of AY 18, confirm the classification of these polar reactions as FEDF.36 However, note that styrene 25 has a very similar chemical potential as unreactive ethylene 5, and therefore, the high computed GEDT should not be attributed to a favourable polar process but to the release of the high instability of supernucleophilic AY 18. Note that the reaction of styrene 25 is exo stereoselective, in agreement with the common stereoselectivity of non-polar reactions, while the reactions of benzaldehyde 26 and MFB 21 show the characterisitc endo stereoselectivity of polar reactions.5
| Structure | Gas phase | Benzene |
|---|---|---|
| TS-I-n | 0.20 | 0.21 |
| TS-I-x | 0.18 | 0.19 |
| TS-II-n | 0.29 | 0.28 |
| TS-II-x | 0.29 | 0.27 |
| TS-III-n | 0.17 | 0.25 |
| TS-III-x | 0.26 | 0.22 |
| TS-IV-n | 0.36 | 0.40 |
| TS-IV-x | 0.35 | 0.35 |
According to the polar mechanism of cycloaddition reactions,34 the higher the GEDT at the TS, the more stable the TS. However, the most stable TS-III-n presents a low GEDT value. Taking into account that GEDT values at the TSs depend on the electrophilic and nucleophilic character of the reagents and their position on the reaction path,44 this apparently inconsistent result is a consequence of the earlier character of the most favorable TS-III-n.
3.4. BET study of the molecular mechanisms of the 32CA reactions between AY 18 and the aryl derivatives 25 and 26
In order to characterize the mechanism of the 32CA reactions of AY 18 with aryl derivatives, a bonding evolution theory45 (BET) study of the bonding changes along the more favorable stereoisomeric reaction paths involving styrene 25 and benzaldehyde 26 was performed. Complete BET data are given in Tables S4 and S5 in the ESI,† while herein only the most relevant conclusions are commented on. ELF attractor positions of the structures participating in the bond formation processes are displayed in Fig. 5. | ||
| Fig. 5 ELF attractor positions of the structures involved in the C–C and C–O bond formation along the more favorable stereoisomeric reaction paths associated with the 32CA reactions of AY 18 with styrene 25 (top) and benzaldehyde 26 (bottom). The prime symbol marks the last structure of the corresponding BET phase. | ||
The BET study along the 32CA reaction involving styrene 25 reveals that: (i) there are no significant changes until passing the TS. The first change is the appearance of the N2 nitrogen non-bonding electron density at S2 as a consequence of the depopulation of the C1–N2 bonding region (see Table S4 in the ESI†); (ii) then, the C4 and C5 pseudoradical centers are formed at the styrene framework, in this order, from the depopulation of the C4–C5 bonding region. At S5, the four carbon pseudoradical centers required for the formation of the two new C–C single bonds are present;25 (iii) formation of the C1–C4 single bond, first, and the C3–C5 one, later, take place by the C-to-C coupling25 of the corresponding pseudoradical centers at C–C distances of 2.07 and 2.06 Å, and initial populations of 1.27 and 1.43 e, respectively; (iv) while the AY C1 pseudoradical center participates more than the C4 one in the formation of the C1–C4 single bond with a relation 65.4:31.5%, both C3 and C5 pseudoradical centers have an even participation in the formation of the C3–C5 single bond, 48.3:51.0%; (v) bond formation is asynchronous with a synchronicity index (see Section 2) of S = 0.5 and formation of the second C3–C5 single bond begins when the first C1–C4 single bond has reached ca. 90% of its final population; (vi) this single bond formation is in agreement with the analysis of the Parr functions, giving C4 as the most electrophilic center of styrene 25 (see Fig. 2); (vii) the presence of the two C1 and C3 pseudoradical centers of AY 18 until the formation of the first C1–C4 single bond indicates that the 32CA reaction between pseudodiradical AY 18 and styrene 25 follows a pdr-type mechanism.3,4
On the other hand, the BET study along the 32CA reaction involving benzaldehyde 26 shows that: (i) the first significant change is the unexpected disappearance of the C1 pseudoradical center facing the O4 carbonyl oxygen, with a population of 0.24 e at the end of Phase I (see Table S5 in the ESI†). The TS presents a similar structure with the presence of the C3 pseudoradical center only; (ii) then, the C5 pseudoradical center is created, and the formation of the C3–C5 single bond takes place, at a C–C distance of 2.05 Å and an initial population of 1.26 e, by donation of the non-bonding electron density of the AY C3 pseudoradical center to the benzaldehyde C5 one. Note that both C3 and C5 centers contribute 81.0:15.9% to the C3–C5 bond population; (iii) the N2 non-bonding electron density is not formed until S5; (iv) the C1 pseudoradical center reappears at S6, integrating 0.15 e, with no energy cost but an energy release of 15.5 kcal mol−1 from S2 to S6; (v) formation of the second C1–O4 single bond takes place at a C–O distance of 1.78 Å and an initial population of 0.68 e, by sharing the C1 pseudoradical center and some O1 non-bonding electron density in a relationship of 42.6:50.0%; (vi) this reaction takes place through a non-concerted two-stage one-step mechanism,46 in which formation of the second C1–O4 bond takes place when the first one is almost completed, with a synchronicity index of S = 0.18 and a completion percentage of 97.4%; (vii) while geometries predict the first bond formation, Parr functions fail to explain the C5 selectivity; (viii) despite AY 18 having a pseudodiradical structure, the presence of only one C3 pseudoradical center in most of the reaction progress suggests a pmr-type 32CA reaction mechanism. This change of mechanism from a pdr- to a pmr-type is a consequence of the polarization of the pseudodiradical AY 18 caused by the presence of the carbonyl group of benzaldehyde 26.
A comparative analysis of two BET studies highlights the following observations: (i) the carbonyl group of benzaldehyde 26 is able to change the reactivity and/or mechanism of a pseudodiradical TAC and enable it to participate in a pmr-type 32CA reaction. While it has been observed that substituents can change the electronic structure and reactivity of TACs,4,10–12 this is the first time that the reactivity of a TAC has been modulated by the other reagent. Note that AY 18 behaves as a pseudodiradical TAC in the reaction with styrene 25; (ii) usually, pdr-type 32CA reactions have lower activation energies than pmr-type ones,3 however, in this case, the pmr-type 32CA reaction of benzaldehyde 26 has a lower activation energy than the pdr-type 32CA reaction of styrene 25 due to the higher polar character of the former which favors it over the latter; (iii) the higher polar character increases the asynchronicity of the single bond formation. While the reaction of styrene 25 is highly asynchronous, the one with benzaldehyde 26 has a non-concerted two-stage one-step mechanism;46 (iv) while Parr functions successfully explain the asynchronicity in the bond formation in the reaction of styrene 25, the prediction fails in the reaction of benzaldehyde 26; (v) it is interesting to note the orientation of the V(C1) and V(C3) monosynaptic basins of the pseudodiradical AY 18 at the beginning of these 32CA reactions; while in the reaction of styrene 25 the V(C1) and V(C3) are oriented towards the styrene moiety, in the reaction of benzaldehyde 26 the V(C1) monosynaptic basin is initially oriented in the opposite direction (see Fig. S6 in the ESI†).
4. Conclusions
The 32CA reactions of N-methyl AY 18 with benzaldehyde 26 and MFB 21, experimentally performed by Nyerges et al.14 and Moshkin et al.,15 were studied within MEDT at the ωB97X-D/6-311G(d) level, in order to characterize the reactivity of an experimental pseudodiradical TAC for the first time. To this end, the corresponding 32CA reaction with styrene 25 was also considered in order to determine the effect of the carbonyl substituent on the reactivity of AY 18 and the mechanism of the reaction.ELF topological analysis of AY 18 indicates that this TAC presents a pseudodiradical structure similar to that of the simplest unsubstituted AY 1 participating in pdr-type 32CA reactions. Analysis of its charge distribution indicates that its structure cannot be represented as the hypothetical Huisgen's zwitterionic 1,3-dipole. No relevant structural changes are observed between the aryl derivatives 21, 25, and 26.
Analysis of CDFT reactivity indices allows classifying AY 18 as a supernucleophile, while substitution at the moderately electrophilic styrene 25 turns benzaldehyde 26 and MFB 21 into strong electrophilic species. The electrophilic Parr functions at MFB 21 indicate that the aldehyde carbonyl group is more electrophilically activated than the carboxylate one.
The computed activation Gibbs free energies of these 32CA reactions classified as polar FEDF processes are 10.5 (styrene 25), 8.2 (benzaldehyde 26), and 4.1 (MFB 21) kcal mol−1. These very low values agree with the highly reactive pseudodiradical structure and supernucleophilic character of AY 18, together with the experimental outcomes showing that the reaction with the strongest electrophile MFB 21 is the most favorable one. While the reaction of styrene 25 is exo steoreoselective, those of benzaldehyde 26 and MFB 21 are endo stereoselective; the latter one involving MFB 21 is also completely chemoselective towards the aldehyde group, in agreement with the experimental results and the analysis of Parr functions.
The BET study of the molecular mechanism of the 32CA reactions of AY 18 with styrene 25 and benzaldehyde 26 reveals that while the reaction of styrene 25 is highly asynchronous towards the terminal C4 ethylene carbon, that of benzaldehyde 26 follows a highly asynchronous two-stage one-step mechanism involving the carbonyl C5 carbon. However, while the 32CA reaction of AY 18 with styrene 25 is characterized as a pdr-type 32CA reaction, the one involving benzaldehyde 26 follows a pmr-type mechanism prompted by the presence of the carbonyl group.
The present MEDT study is the first theoretical analysis of an experimental pseudodiradical AY whose reactivity is modulated by the other reagent, showing that the mechanism of 32CA reactions can be modified not only by changing the electronic structure of TACs through appropriate substitution but also by the nature of their opposing ethylene derivative, which opens up new perspectives for future synthesis designs without the need for catalysts for their 32CA reactions.47
Author contributions
M. R.-G.: data curation, formal analysis, investigation, supervision, writing – original draft and writing – review & editing. L. R. D.: data curation, formal analysis, funding acquisition, investigation, and writing – review & editing. R. J.: data curation, investigation, and writing – review & editing.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
We acknowledge the financial support from the Ministry of Science and Innovation (MICINN) of the Spanish Government, project PID2019-110776GB-I00 (AEI/FEDER, UE). R. J. also acknowledges the University of Valencia for financial support for invited-researcher stays within the Vice-Rectorate for Research's “Talent Attraction” sub-programme, INV21-01-15.References
- (a) W. Carruthers, Some Modern Methods of Organic Synthesis, Cambridge University Press, Cambridge, UK, 2nd edn, 1978 Search PubMed; (b) A. Padwa, 1,3-Dipolar Cycloaddition Chemistry, Wiley-Interscience, New York, NY, USA, 1984, vol. 1 and 2 Search PubMed; (c) W. Carruthers, Cycloaddition Reactions in Organic Synthesis, Pergamon, Oxford, UK, 1990 Search PubMed; (d) A. Padwa and W. H. Pearson, Synthetic Applications of 1,3-Dipolar Cycloaddition Chemistry Toward Heterocycles and Natural Products, John Wiley & Sons, Inc., New York, NY, USA, 2002, vol. 59 CrossRef.
- L. R. Domingo, Molecules, 2016, 21, 1319 CrossRef PubMed.
- M. Ríos-Gutiérrez and L. R. Domingo, Eur. J. Org. Chem., 2019, 267–282 CrossRef.
- L. R. Domingo, E. Chamorro and P. Pérez, Lett. Org. Chem., 2010, 7, 432–439 CrossRef CAS.
- L. R. Domingo, M. Ríos-Gutiérrez and P. Pérez, J. Org. Chem., 2018, 83, 2182–2197 CrossRef CAS PubMed.
- (a) Y. N. Markitanov and V. M. Timoshenko, Chem. Heterocycl. Compd., 2021, 57, 1149–1154 CrossRef CAS; (b) N. S. Zimnitskiy, A. Yu Barkov, I. B. Kutyashev, V. Yu Korotaev and V. Ya Sosnovskikh, Chem. Heterocycl. Compd., 2021, 57, 743–750 CrossRef CAS; (c) I. B. Kutyashev, M. V. Ulitko, A. Yu Barkov, N. S. Zimnitskiy, V. Yu Korotaev and V. Ya Sosnovskikh, Chem. Heterocycl. Compd., 2021, 57, 751–763 CrossRef CAS.
- (a) S. Katahara, Y. Sugiyama, M. Yamane, Y. Komiya, T. Sato and N. Chida, Org. Lett., 2021, 23(8), 3058–3063 CrossRef CAS PubMed; (b) R. Narayan, M. Potowski, Z.-J. Jia, A. P. Antonchick and H. Waldmann, Acc. Chem. Res., 2014, 47, 1296–1310 CrossRef CAS PubMed.
- F. Bellina and R. Rossi, Tetrahedron, 2006, 62, 7213–7256 CrossRef CAS.
- L. R. Domingo and J. A. Sáez, J. Org. Chem., 2011, 76, 373–379 CrossRef CAS PubMed.
- M. Ríos-Gutiérrez, A. Barakat and L. R. Domingo, Organics, 2022, 3, 122–136 CrossRef.
- L. R. Domingo, M. J. Aurell and P. Pérez, Tetrahedron, 2014, 70, 4519–4525 CrossRef CAS.
- L. R. Domingo, K. Kula, M. Ríos-Gutiérrez and R. Jasiński, J. Org. Chem., 2021, 86, 12644–12653 CrossRef CAS PubMed.
- (a) R. Grigg and S. Thianpatanagul, J. Chem. Soc., Chem. Commun., 1984, 180–181 RSC; (b) R. Grigg, J. Idle, P. McMeekin and D. Vipond, J. Chem. Soc., Chem. Commun., 1987, 49–51 RSC; (c) O. Tsuge, S. Kanemasa, M. Ohe and S. Takenaka, Bull. Chem. Soc. Jpn., 1987, 60, 4079–4089 CrossRef CAS; (d) M. Joucla and J. Mortier, Bull. Soc. Chim. Fr., 1988, 125, 579–583 Search PubMed.
- M. Nyerges, I. Fejes, A. Virányi, P. W. Groundwater and L. A. Töke, Synthesis, 2001, 1479–1482 CAS.
- V. S. Moshkin, E. M. Buev and V. Y. Sosnovskikh, Tetrahedron Lett., 2015, 56, 5278–5281 CrossRef CAS.
- A. G. Meyer and J. H. Ryan, Molecules, 2016, 21, 935 CrossRef PubMed.
- A. D. Becke and K. E. Edgecombe, J. Chem. Phys., 1990, 92, 5397–5403 CrossRef CAS.
- J.-D. Chai and M. Head-Gordon, Phys. Chem. Chem. Phys., 2008, 10, 6615–6620 RSC.
- M. J. Hehre, L. Radom, P. V. R. Schleyer and J. Pople, Ab initio Molecular Orbital Theory, Wiley, New York, NY, USA, 1986 Search PubMed.
- (a) H. B. Schlegel, J. Comput. Chem., 1982, 3, 214–218 CrossRef CAS; (b) H. B. Schlegel, in Modern Electronic Structure Theory, ed. D. R. Yarkony, World Scientific Publishing, Singapore, 1994 Search PubMed.
- K. Fukui, J. Phys. Chem., 1970, 74, 4161–4163 CrossRef CAS.
- (a) C. González and H. B. Schlegel, J. Phys. Chem., 1990, 94, 5523–5527 CrossRef; (b) C. González and H. B. Schlegel, J. Chem. Phys., 1991, 95, 5853–5860 CrossRef.
- (a) J. Tomasi and M. Persico, Chem. Rev., 1994, 94, 2027–2094 CrossRef CAS; (b) B. Y. Simkin and I. I. Sheikhet, Quantum Chemical and Statistical Theory of Solutions–Computational Approach, Ellis Horwood, London, UK, 1995 Search PubMed.
- (a) M. Cossi, V. Barone, R. Cammi and J. Tomasi, Chem. Phys. Lett., 1996, 255, 327–335 CrossRef CAS; (b) E. Cances, B. Mennucci and J. Tomasi, J. Chem. Phys., 1997, 107, 3032–3041 CrossRef CAS; (c) V. Barone, M. Cossi and J. Tomasi, J. Comput. Chem., 1998, 19, 404–417 CrossRef CAS.
- L. R. Domingo, RSC Adv., 2014, 4, 32415–32428 RSC.
- (a) A. E. Reed, R. B. Weinstock and F. Weinhold, J. Chem. Phys., 1985, 83, 735–746 CrossRef CAS; (b) A. E. Reed, L. A. Curtiss and F. Weinhold, Chem. Rev., 1988, 88, 899–926 CAS.
- (a) R. G. Parr and W. Yang, Density Functional Theory of Atoms and Molecules, Oxford University Press, New York, NY, USA, 1989 Search PubMed; (b) L. R. Domingo, M. Ríos-Gutiérrez and P. Pérez, Molecules, 2016, 21, 748 CrossRef PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson and H. Nakatsuji, et al., Gaussian 16, Revision A.03, Gaussian, Inc., Wallingford, CT, USA, 2016 Search PubMed.
- S. Noury, X. Krokidis, F. Fuster and B. Silvi, Comput. Chem., 1999, 23, 597–604 CrossRef CAS.
- R. Dennington, T. A. Keith and J. M. Millam, GaussView, Semichem Inc., Shawnee Mission, KS, USA, 6th edn, 2016 Search PubMed.
- B. Silvi and A. Savin, Nature, 1994, 371, 683–686 CrossRef CAS.
- R. Huisgen, Proc. Chem. Soc., 1961, 357–396 Search PubMed.
- L. R. Domingo, M. J. Aurell, P. Pérez and J. A. Sáez, RSC Adv., 2012, 2, 1334–1342 RSC.
- L. R. Domingo and M. Ríos-Gutiérrez, Application of Reactivity Indices in the Study of Polar Diels–Alder Reactions, in Conceptual Density Functional Theory: Towards a New Chemical Reactivity Theory, ed. S. Liu, Wiley-VCH GmbH, 2022, vol. 2, pp. 481–502 Search PubMed.
- R. G. Parr and R. G. Pearson, J. Am. Chem. Soc., 1983, 105, 7512–7516 CrossRef CAS.
- L. R. Domingo, M. Ríos-Gutiérrez and P. Pérez, RSC Adv., 2020, 10, 15394–15405 RSC.
- R. G. Parr and R. G. Pearson, J. Am. Chem. Soc., 1983, 105, 7512–7516 CrossRef CAS.
- R. G. Parr, Lv Szentpaly and S. Liu, J. Am. Chem. Soc., 1999, 121, 1922–1924 CrossRef CAS.
- L. R. Domingo, E. Chamorro and P. Pérez, J. Org. Chem., 2008, 73, 4615–4624 CrossRef CAS.
- L. R. Domingo, P. Pérez and J. A. Sáez, RSC Adv., 2013, 3, 1486–1494 RSC.
- L. R. Domingo and M. Ríos-Gutiérrez, Org. Biomol. Chem., 2019, 17, 6478–6488 RSC.
- M. G. Evans and M. Polanyi, Trans. Faraday Soc., 1935, 31, 875–894 RSC.
- G. S. Hammond, J. Am. Chem. Soc., 1955, 77, 334–338 CrossRef CAS.
- L. R. Domingo, M. Ríos-Gutiérrez and P. Pérez, Molecules, 2020, 25, 2535 CrossRef CAS PubMed.
- X. Krokidis, S. Noury and B. Silvi, J. Phys. Chem. A, 1997, 101, 7277–7282 CrossRef CAS.
- L. R. Domingo, J. A. Sáez, R. J. Zaragozá and M. Arnó, J. Org. Chem., 2008, 73(22), 8791–8799 CrossRef CAS PubMed.
- L. R. Domingo, M. Ríos-Gutiérrez and P. Pérez, J. Org. Chem., 2018, 83, 10959–10973 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Fig. S1 shows the ELF topological analysis of N-phenyl and N-benzyl AYs 23 and 24. Table S1 includes ωB97X-D/6-311G(d) total energies of the stationary points involved in the 32CA reactions of AY 18 with styrene 25, benzaldehyde 26, and MFB 21, in the gas phase and in benzene. Table S2 includes ωB97X-D/6-311G(d) absolute thermodynamic data of the three reactions. Table S3 gathers C1–C(O)4 and C3–C5 distances at the ωB97X-D/6-311G(d) optimized geometries of the TSs in the gas phase and in solvent. Table S4 includes the BET data along the exo stereoisomeric path of the 32CA reaction between AY 18 and styrene 25. Table S5 includes the BET data along the endo stereoisomeric path of the 32CA reaction between AY 18 and benzaldehyde 26. Fig. S2 shows the ELF topological analysis of S1 associated with the exo and endo steroisomeric reaction paths of the 32CA reactions of AY 18 with styrene 25 and benzaldeyde 26. See DOI: https://doi.org/10.1039/d2cp05032c |
| This journal is © the Owner Societies 2023 |