
Interplay of conformational relaxation and hydrogen bond dynamics in the excited states of fluorescent Schiff base anions†
Souradip
Dasgupta
,
Arkaprava
Chowdhury
 ,
Dipak Kumar
Sahoo
and
Anindya
Datta
*
,
Dipak Kumar
Sahoo
and
Anindya
Datta
*
Department of Chemistry, Indian Institute of Technology Bombay, Powai, Mumbai 400 076, India. E-mail: anindya@chem.iitb.ac.in; Tel: +91 22 2576 7149
First published on 22nd November 2022
Abstract
Time resolved fluorescence spectroscopic investigation of four Schiff base anions has established that their excited state dynamics is governed by several solvent properties: polarity, viscosity and hydrogen bond donating ability. With viscous protic solvents like glycerol, fluorescence lifetimes of anions have been found to be markedly longer than those in ethanol, implying that conformational relaxation of molecules plays a key role in their nonradiative relaxation. Surprisingly, the lifetimes in less viscous aprotic solvents, like acetonitrile, are found to be even longer. The only plausible rationalization of this observation is in the light of hydrogen bond-assisted nonradiative phenomena that are operative in protic solvents. This contention draws support from a time evolution of the emission in the red end of the spectrum in low to moderately hydrogen bond donating protic solvents, with regard to an absence of such a rise time in aprotic solvents and strongly hydrogen bond donating solvents, viz., 2,2,2-trifluoroethanol. Rudimentary quantum chemical calculations provide a preliminary idea about the nature of excited state hydrogen bond redistribution involved in the process.
1. Introduction
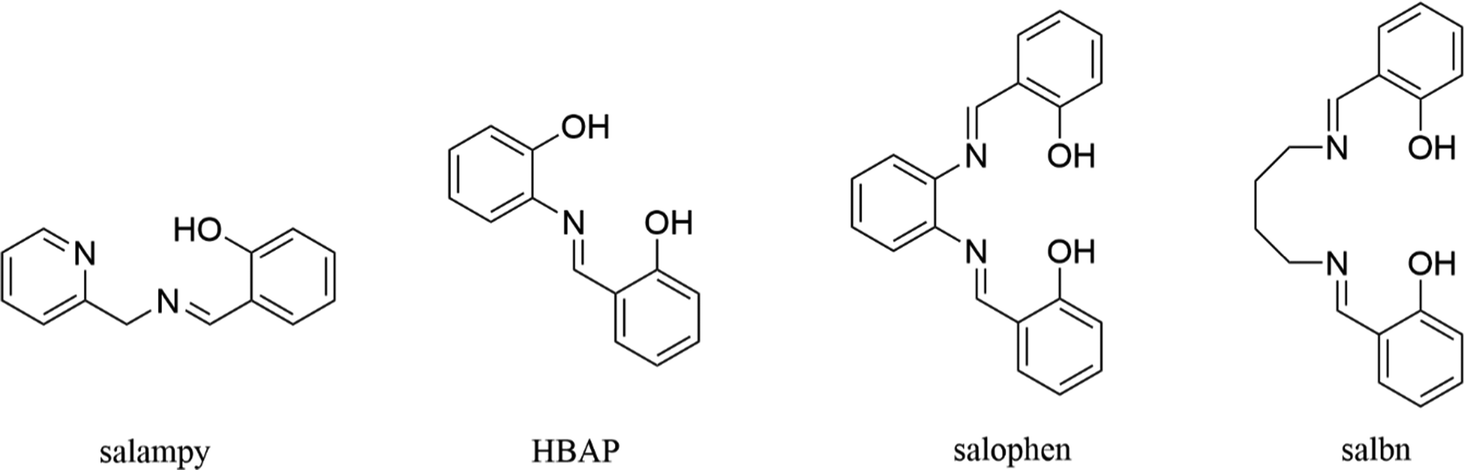
Fluorogenic molecules have attracted significant attention in recent times because of their importance in the fields of organic electronics and sensing of analytes.1–11 Excited state dynamics of such molecules are governed by highly efficient nonradiative processes, cessation of which by specific chemical or physical inputs renders the molecules strongly fluorescent.12 Schiff bases constitute a class of fluorogenic molecules that are attractive from several different perspectives. They are easy to synthesize and derivatise,13 making them excellent candidates as building blocks for novel functional materials.14–16 Generally, their fluorescence is very weak, as their flexible structures sustain efficient nonradiative pathways associated with the rotation of carbon–carbon and carbon–nitrogen bonds.17–23 On the other hand, they are excellent ligands that can bind strongly and selectively with metal ions. The consequent rigidification can lead to efficient suppression of the non-radiative pathways in principle, provided it is not overwhelmed by factors like the heavy atom effect. This forms the basis of the much explored applications of Schiff bases as fluorogenic “turn on” sensors of metal ions.24–26 The practical utility of Schiff bases as sensors in real biological media is limited by their well-known propensity for hydrolysis, the mechanism of which is an interesting problem in itself.27 The pursuit for Schiff bases that are immune to hydrolysis continues to be one of the Holy Grails of this field.28Over the last few years, our group has explored a family of Schiff bases (Scheme 1), with the mandate of understanding and optimizing the conditions for obtaining enhanced fluorescence from them, by complexation and aggregation.29–34 It has been established that these compounds exhibit selective enhancement of fluorescence with Zn(II) and Al(III) and that the Al(III) complexes emit more strongly than the Zn(II) complexes in solution. For salophen ((2,2′-((1,2-phenylenebis(azaneylylidene))bis(methaneylylidene))diphenol), Scheme 1), this has been explained in the light of opening of additional non-radiative channels in the dimeric Zn(II) complex, in which the ligands are π-stacked.30 This contention draws support from the marked decrease in the fluorescence of the Al(III) complex in its crystals, due to the π-stacking interactions induced between the ligand molecules in these complexes.29 Interestingly, fluorescence is restored in the solid state, upon frustrating the stacking by small chemical substitutions.31 Moreover, Zn(II) complexes with significantly stronger fluorescence are obtained with salampy ((2-(((pyridin-2-ylmethyl)imino)methyl)phenol), Scheme 1), a Schiff base, in which the ligands are not π-stacked.32 However, the Al(III) complex of this Schiff base has a larger fluorescence quantum yield (ϕf) and a longer fluorescence lifetime (τf), possibly because they are more rigid.35 Hence, a systematic understanding of the factors that determine the strength of fluorescence complexes has begun to emerge from this series of studies.
 | ||
| Scheme 1 Chemical structures of the Schiff bases. | ||
An interesting aspect of the photophysics of Schiff bases has come to light in the course of these investigations. The anions of all the four Schiff bases (Scheme 1), namely salophen, salampy, HBAP (2-((2-hydroxybenzylidene)amino)phenol) and salbn (2,2′-((butane-1,4-diylbis(azaneylylidene))bis(methaneylylidene))diphenol) have been found to be emissive to a significantly greater extent compared to the neutral molecules, but to a lesser extent than the metal complexes. The neutral Schiff bases typically have τf values of tens of picoseconds and the complexes have lifetimes in the nanosecond regime, unless π-stacking is at play. The anions typically have fluorescence lifetimes of 800–900 ps in methanolic solutions32–34 and their emission spectra exhibit a time dependent fluorescence Stokes shift (TDFSS) over timescales comparable to the solvation times reported earlier.36 This observation is in line with an earlier report on a Schiff base anion.19 The present work takes a closer look at the excited state dynamics involved. Time evolution of fluorescence of the anions of four Schiff bases (Scheme 1) have been investigated in protic and aprotic solvents, with a range of polarity and viscosity. The possible contributions of solvation and hydrogen bond dynamics have been weighed against each other.37–41 A clearer picture of the excited state dynamics of these anions has hence emerged.
2. Materials and methods
2.1. Materials
All the chemicals and spectroscopy grade solvents (Spectrochem, Mumbai, India) are used as received without further purification unless mentioned. Deuterated ethanol (EtOD) and Coumarin-153 (C153) are procured from Sigma Aldrich and used for the experiments.2.2. Synthesis of Schiff bases and their anions
Salophen, salampy and HBAP are prepared and purified following our previous reports, using salicylaldehyde (SD Fine Chemicals, Mumbai, India) and respective aryl amines.30,32,34,42 Salbn is prepared by dropwise addition of salicylaldehyde (2 equiv.) to a methanolic solution of 1,4-butanediamine (1 equiv., Sigma Aldrich) at room temperature.43 The yellow precipitate obtained is recrystallized from methanol. The structures of the products are determined by 1H NMR spectra on a Bruker 400 MHz NMR spectrometer and by single X-ray diffraction. 1H NMR spectra and crystal structure (CCDC 176004) of salbn are presented subsequently (Fig. S1, ESI†). Detailed characterization of the other Schiff bases have been reported in our earlier reports.30,32,34 The Schiff base anions are prepared by adding the Schiff bases to a saturated alcoholic solution of potassium hydroxide (Merck, Mumbai). For the studies carried out in aprotic solvents, preparation of the anions in methanol is followed by vacuum evaporation of the solvent and addition of the corresponding aprotic solvent, such as acetonitrile (ACN) and N,N-dimethylformamide (DMF). Filtration of the same solution is performed to get rid of the KOH residue.2.3. Steady state fluorescence spectroscopy
Steady state spectra are recorded on a Shimadzu UV-NIR-3600 spectrophotometer and a Horiba Fluoromax 4 spectrofluorimeter. Quinine sulphate in 0.1 M H2SO4 (ϕf = 0.54) is used as a standard for measurement of fluorescence quantum yields (ϕf).442.4. Time-resolved fluorescence spectroscopy
Fluorescence lifetimes in the pico–nanosecond time regime are recorded on a DeltaFlex Time Correlated Single Photon Counting (TCSPC) spectrometer. Vertically polarized second harmonic output of a Mai-Tai Laser, with a repetition rate of 8 MHz, is used as the excitation source. Fluorescence decays are collected at magic angle (54.7°) polarization with respect to the excitation light. The full width at half maxima (FWHM) of the instrument response function (IRF), using a hybrid photomultipler detector (HPPD), is 74 ps. Decays are fitted to the sum of exponential functions (eqn (1)), with iterative reconvolution by a commercially available EzTime software package.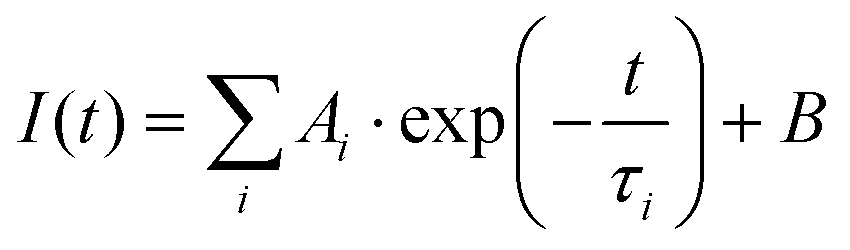 | (1) |
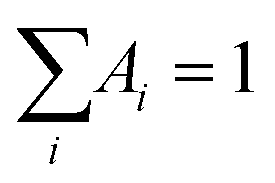 . B is the background signal. Fluorescence decays in the femto–picosecond domain are recorded using Femtosecond Optical Gating (FOG) technique with λex = 400 nm. The second harmonic, 400 nm of a modelocked 100 fs Ti:Sapphire (Tsunami, Spectra Physics, USA, 1 W, 80 MHz repetition rate) was used for the excitation. Details of the setup are discussed elsewhere.45
. B is the background signal. Fluorescence decays in the femto–picosecond domain are recorded using Femtosecond Optical Gating (FOG) technique with λex = 400 nm. The second harmonic, 400 nm of a modelocked 100 fs Ti:Sapphire (Tsunami, Spectra Physics, USA, 1 W, 80 MHz repetition rate) was used for the excitation. Details of the setup are discussed elsewhere.45
Time Resolved Emission Spectra (TRES) are constructed using steady state fluorescence intensities and temporal parameters from fluorescence transients (FOG decays for all the solvents except glycerol) at regular intervals across the range of fluorescence wavelengths of the Schiff base anions. Jacobian correction (i.e., I(![[v with combining macron]](https://www.rsc.org/images/entities/i_char_0076_0304.gif) ) = λ2I(λ)) is incorporated into the steady state spectral intensity.46 Each of the TRES is fitted to a modified log-normal function:
) = λ2I(λ)) is incorporated into the steady state spectral intensity.46 Each of the TRES is fitted to a modified log-normal function:
 | (2) |
![[small nu, Greek, tilde]](https://www.rsc.org/images/entities/i_char_e0e1.gif) 0 = spectral peak position, and Δ = bandwidth of the spectral band.
0 = spectral peak position, and Δ = bandwidth of the spectral band.
The apparent solvent correlation C(t) as obtained from the TRES function is defined as:
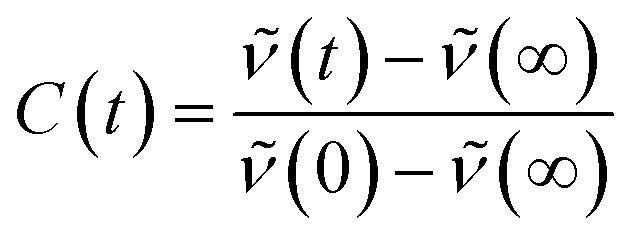 | (3) |
2.5. Quantum chemical calculations
The electronic structure calculations for the ground and excited states are performed with the Gaussian 09 program suite.47 The ground state geometry optimization of the salampy anion and its hydrogen bonded complexes with ethanol molecules are carried out using a density functional theory (DFT) method with the CAM-B3LYP/6-31G(d,p) level of theory. The final ground state optimized structure of the salampy−-ethanol hydrogen bonded complex obtained is used to carry out the excited state electronic structure calculations using the time-dependent density functional theory (TDDFT) method with the same CAM-B3LYP/6-31G(d,p) level of theory.37 Visualization of all the electronic structures and the measurements of the hydrogen bond lengths are done by Chemcraft software.3. Results and discussion
The emission spectra of all the anions are significantly Stokes shifted compared to their absorption spectra (Fig. 1, Table 1 and Fig. S2, Table ST1, ESI†). For example, salampy− exhibits absorption (λmaxabs) and emission (λmaxem) maxima at 383 and 492 nm, respectively in ethanol (EtOH). The large Stokes shift (109 nm for salampy−) indicates that the excited state processes are operational in the anions. For neutral Schiff bases, a similar observation is ascribed to the excited state intramolecular proton transfer (ESIPT) and the rapid non radiative depopulation of the cis-keto excited state associated with segmental motions of C–N bonds.17,20,21,48–54 ESIPT is precluded for anions, as they lack the proton that is essential for its occurrence. In polar aprotic solvents like N,N-dimethyl formamide (DMF) and acetonitrile (ACN), the emission spectra are slightly blue shifted (5–10 nm) compared to those in polar protic solvents (the Stokes shift for salampy− is 67 nm in DMF, but 84 nm in ACN, Table 1). However, in strongly hydrogen bonding solvents like 2,2,2-trifluoroethanol (TFE) the emission spectrum is further blue shifted (∼40 nm) as compared to methanol (MeOH). Similar observations are made for the other anions as well (Table ST1, ESI†). Hence, the emissive excited state is found to be stabilized to a greater extent in protic solvents with low to moderate hydrogen bond donating ability, than in aprotic solvents. This drastic difference in nature of the emissive excited state in TFE as compared to other protic solvents indicates the critical role of hydrogen bonding in the excited state processes of this class of anions. Viscosity also has a major role to play, as is indicated by the largest values of fluorescence quantum yields (ϕf) of all of the four anions in glycerol (Table 1 and Table ST1, Fig. S3, ESI†). This is likely to be due to the hindrance to the torsional motions associated with non-radiative channels, in solvents with high viscosity.55 The steady state spectra of salampy−, HBAP2− and salophen2− in methanol (MeOH) has been recorded afresh to reproduce our recent reports.33–35 Steady state spectra thus indicate the dependence of the excited state phenomena in the Schiff base anions on multiple parameters of the fluid medium in which it resides.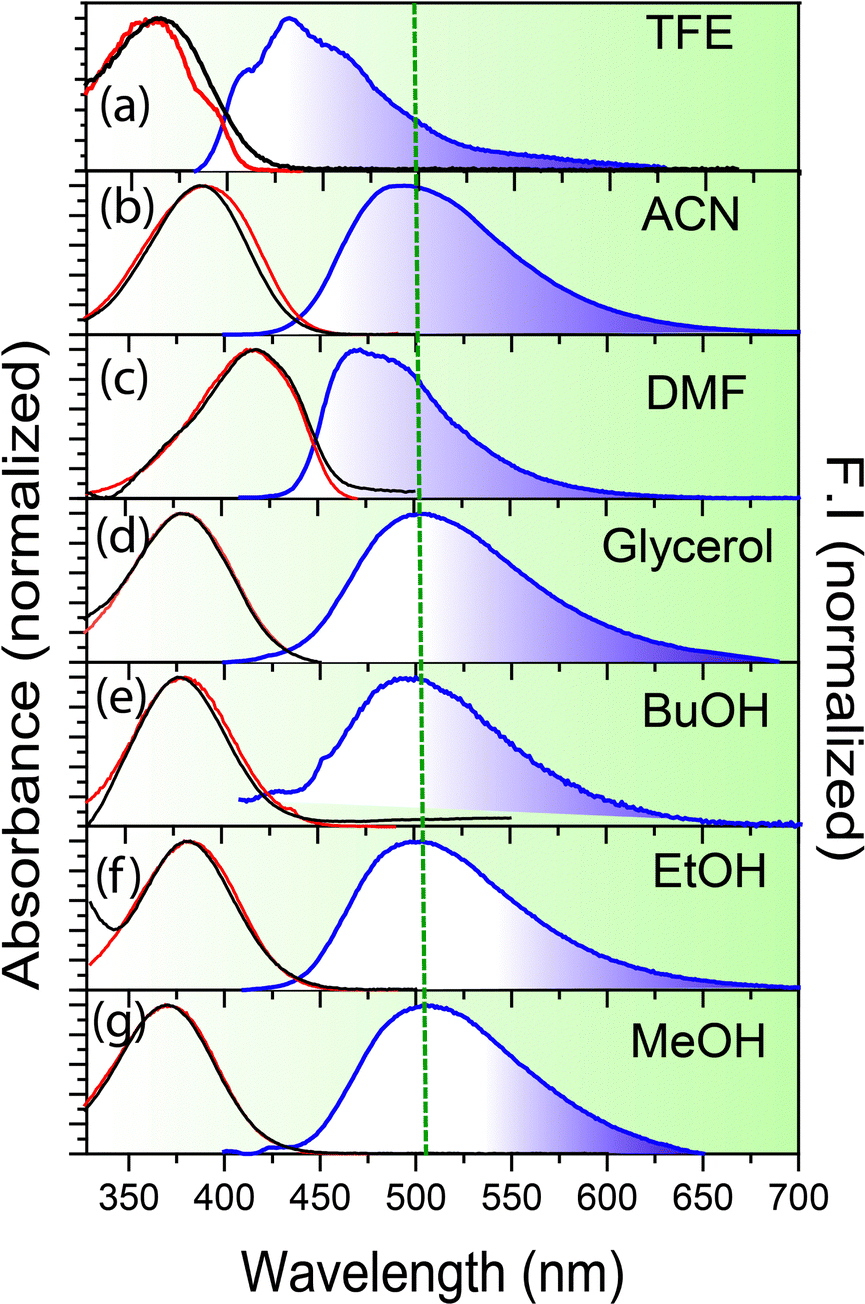 | ||
| Fig. 1 Absorption (black), fluorescence emission (blue) and excitation (red) of salampy− in (a) TFE (b) ACN (c) DMF (d) glycerol (e) BuOH (f) EtOH (g) MeOH. The emission spectra are recorded by exciting the samples at their corresponding λmaxabs. Excitation spectra are monitored at their respective λmaxem. | ||
| Solvent | Δf |
abs (cm−1) |
em (cm−1) |
Δλ (nm) | Δ (cm−1) |
ϕ f | τ f (ns) |
|---|---|---|---|---|---|---|---|
| ACN | 0.31 | 24![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 752 752 |
20492 |
84 | 4260 | 0.07 ± 0.01 | 3.3 |
| DMF | 0.27 | 24570 |
21097 |
67 | 3473 | 0.05 ± 0.01 | 4.4 |
| Glycerol | 0.27 | 26525 |
19920 |
125 | 6605 | 0.10 ± 0.05 | 1.3 |
| BuOH | 0.24 | 25974 |
19960 |
116 | 6014 | 0.03 ± 0.008 | 1.7 |
| EtOH | 0.29 | 26110 |
20325 |
109 | 5785 | 0.01 ± 0.006 | 1.0 |
| MeOH | 0.31 | 26596 |
19960 |
125 | 6636 | 0.03 ± 0.003 | 0.8 |
| TFE | 0.57 | 27269 |
23049 |
67 | 4220 | 0.01 ± 0.001 | — |
The plot thickens upon closer inspection of the time resolved fluorescence data in the femtosecond–picosecond and picosecond–nanosecond range when using femtosecond optical gating (FOG) and time correlated single photon count (TCSPC), respectively. The fluorescence decays, recorded at λmaxem, are slower in glycerol than in ethanol (Fig. 2a). This observation can be explained in light of hindrance to the segmental motion and consequently, conformational relaxation at higher viscosity32–34 and is in line with the larger ϕf values observed in glycerol. However, viscosity is not the sole factor that determines the excited state dynamics, as is borne out by a distinctly slower fluorescence decay of salampy− in ACN (Fig. 2a and Tables ST1, ST2, ESI†). Experiments with a library of solvents reveal that the slowest fluorescence decays are observed for solvents that are polar, but aprotic (Fig. S4, ESI†). Hence, it is inferred that hydrogen bonding interactions provide additional nonradiative decay channels for the anions in protic solvents. The dynamics of their excited states are thus experimentally indicated to be governed by potential energy surfaces that involve multidimensional reaction co-ordinates.56,57 This contention is fortified by the strong dependence of the fluorescence dynamics on the emission wavelength, observed in the nanosecond timescale for glycerol (Fig. 2b and Fig. S5, ESI†) and in the picosecond timescale for EtOH and butanol (BuOH) (Fig. 2c and Fig. S5, ESI†). Rise times at the red end of the emission spectra and corresponding fast decays at the blue end confirm the occurrence of an excited state relaxation (Table ST2, ESI†). The lack of rise times at the red of spectra in TFE is reminiscent of the fact that TFE is a strong hydrogen bonding solvent and it forms very strong hydrogen bond networks with the anions in the ground state itself, thus vindicating the role of hydrogen bond dynamics in the electronic excited state (Fig. 2e and Fig. S6, ESI†). The markedly slower dynamics in glycerol reaffirms the viscosity dependence and indicates the involvement of conformational relaxation in the process.
 | ||
| Fig. 2 Fluorescence decays of salampy− using (a and b) TCSPC (nanosecond regime) and (c–e) FOG (femtosecond–picosecond regime) in (a) EtOH, glycerol and ACN at their corresponding emission maxima, (b) glycerol with λex = 400 nm, λem = 450 nm (blue end) and λem = 600 nm (red end) (c) in EtOH at λex = 400 nm, λem = 450 nm (blue end) and 600 nm (red end), (d) ACN with λex = 400 nm, λem = 450 nm (blue end) and 600 nm (red end), (e) in TFE at λex = 400 nm, λem = 450 nm (blue end), 550 nm (intermediate wavelength), 600 nm (red end). | ||
Fast decays at the blue end and corresponding rise times in the red end are commonly associated with solvation dynamics.46,56,58–62 Indeed, the time resolved emission spectra (TRES) of salampy− in EtOH (Fig. 3c) exhibit a TDFSS up to 50 ps. For BuOH and glycerol this TDFSS is slow and it saturates near the emission maxima in around 500 ps which is unlike that for EtOH (Fig. 3a, b and Fig. S7, ESI†). This can be attributed to the higher viscosity of BuOH and glycerol that tends to arrest different conformations of the probe in the excited state making their relaxation pathways slower. The decay of the apparent solvent correlation function, C(t) is fastest in ethanol followed by BuOH and glycerol (Fig. 3i and Fig. S8, Table ST3, ESI†), following the trend of increasing viscosity of the solvents. In deuterated ethanol (EtOD) the faster component is slightly slower as compared to EtOH because of the inertial effect on the rotation of the solvent molecules. However because of inhomogeneous broadening in liquids the slower component slows down only slightly on deuteration (Fig. 3i).63 Similar observations are obtained for the other Schiff base anions (Fig. S8, ESI†). Hence, a unified picture of viscosity dependent solvent relaxation, associated with TDFSS, is apparently obtained for the protic solvents other than TFE.
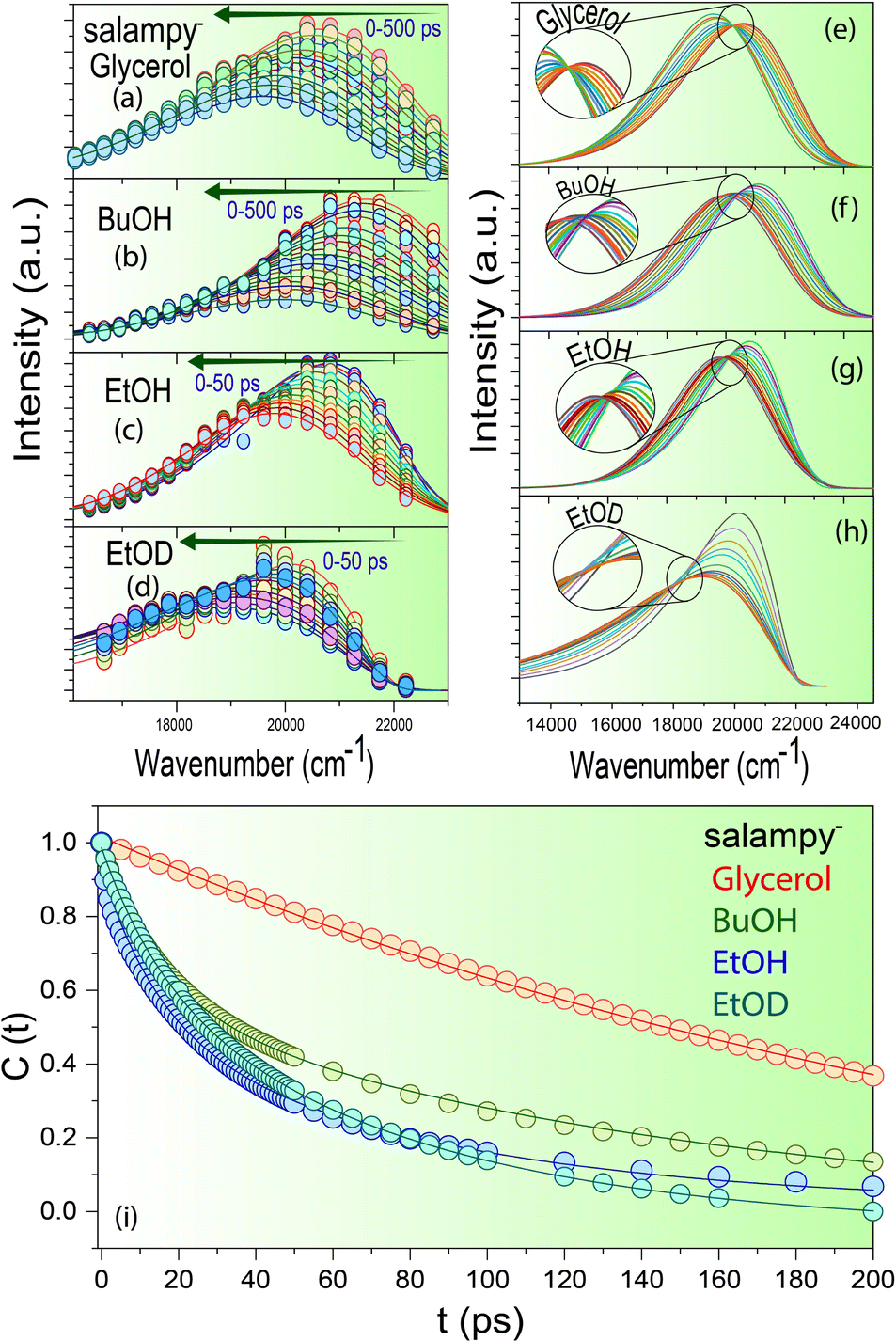 | ||
| Fig. 3 Time resolved emission spectra of salampy− in (a) glycerol (b) BuOH (c) EtOH (d) EtOD; time resolved area normalized emission spectra (TRANES) of salampy− in (e) glycerol (f) BuOH and (g) EtOH (h) EtOD; (i) apparent solvent correlation function (t) − (∞) of salampy− in glycerol, BuOH and EtOH. | ||
A twist in the tale is encountered in the form of an apparent absence of the emission wavelength dependence of the fluorescence decays of salampy− in polar aprotic solvents, namely ACN and DMF (Fig. 2d and Fig. S9, Table ST2, ESI†). There are previous reports where sub picosecond solvation is said to have been operative for C153 in ACN.64 Although that resolution is not achievable in the present set of experiments, it is evident that the magnitude of the rise at the red end obtained in solvents, viz., EtOH, BuOH and glycerol is perceivably much higher than that in polar aprotic solvents like DMF and ACN which prepares the basic foundation for us to believe in the difference of the nature of the operational excited state relaxation pathways. However, the absence of such a dependence in another polar protic solvent, viz., TFE further raises our interest. Experiments with C153, a well-known solvation probe reveal the presence of a 10 ps rise at the red end of the spectra owing to solvent relaxation in the excited state (Fig. S10 and Table ST4, ESI†). The decays of HBAP2− and salampy− in DMF (Fig. S9, ESI†) exhibit a weak, but perceivable wavelength dependence with the decays at the red end becoming faster. No rise time is observed at the red end of the spectra. This small wavelength dependence may be attributed to the conformational heterogeneity of the probes in these specific solvents. The absence of an apparent rise time indicates that dynamic solvation may be precluded in these liquids, the polarities of which are comparable to those of the protic solvents in which the rise times are observed (Table ST5, ESI†). In fact, it poses a serious question about the assignment of the observed relaxation to dynamic solvation, which is considered to arise out of non-specific, electrostatic interactions between solutes and solvent dipoles.46 In this situation, it is prudent to examine the possibility of a relaxation involving two states, rather than a continuous evolution by dynamic solvation. A convenient modality in this context is available in the form of time resolved area normalized emission spectra (TRANES), in which the presence of one or more isoemissive points is indicative of the involvement of discrete electronic states.65 Indeed, an isoemissive point, which is sometimes diffuse, is seen for each of the anions in hydrogen bonding polar protic solvents, viz., EtOH, BuOH, glycerol and EtOD (Fig. 3e–h and Fig. S11, ESI†), hence indicating relaxations involving two discrete states. The diffused nature of the isoemissive “points” may have two origins. The first one is trivial. TRES in EtOH and BuOH have been constructed from FOG data, for which the signal to noise ratio is not as good as TCSPC. The second possible issue is perhaps more important. Conformational heterogeneity of the excited state has been indicated for these anions. This would lead to a deviation from an ideal two state scenario.
At this point, it is imperative to ask if the Schiff base anions under study are good solvation probes at all. This question is best answered by constructing Lippert–Mataga plots for the anions, with a view to estimate the values of Δμ, the change in magnitude of the dipole moment brought about by photoexcitation (Fig. 4, Table 1 and ST1, ESI†).44 This simple exercise turns out to be rather rewarding. For each of the anions, two distinct data sets with linear variation are obtained, one for polar aprotic solvents and the other for polar protic solvents (Fig. 4). Moreover, both the lines are practically parallel. In fact, some of them exhibit a slightly negative slope. Considering the slight deviation on either side to be within the experimental error limits the slopes are almost zero, implying Δμ = 0. In other words, photoexcitation does not lead to the creation of a dipole and hence, the anions are not good solvation probes. This realization necessitates a second look at the excited state relaxation observed in protic solvents (except TFE), as it can no longer be ascribed to solvation. However, the rise times at the red ends for solvents, viz., EtOH, glycerol, butanol necessarily means that a relaxation pathway is operative and more often than not these pathways are associated with a photoinduced electron transfer and consequent change in dipole moment in the electronically excited state. It is better to mention here that it is not imperative to assume that a change in magnitude of the dipole moment can only bring about relaxation, the direction of dipole moment in the photoexcited state also has an equal role to play and the Lippert–Mataga model does not account for that change (Fig. 5a).66,67 At the same time, it is important to recognize an additional degree of stabilization of the emissive excited states in protic solvents, which takes place in tens of picoseconds to a few nanoseconds, depending on the viscosity of the solvent. This extra degree of stabilization leaves its mark in the Lippert–Mataga plots as well. For solvents of similar polarity like MeOH and ACN, the difference in Δ is 1850 ± 500 cm−1 (Fig. 4) which is comparable to the magnitude of TDFSS of the probes in polar protic solvents (Table ST6, ESI†). This agreement between the two values is in line with the fact that the total amount of TDFSS essentially amounts to the energy stabilization brought about by excited state relaxation. In the present experiment, the extra stabilization is observed only in the protic solvents that have moderate hydrogen bond donating abilities and dynamic solvation is ruled out. The most likely candidate for the process that brings about the relaxation would be the reorganization of the solute–solvent hydrogen bonds post excitation. It is prudent to discuss the curious case of TFE at this point (Fig. 2 and Fig. S6, ESI†). Owing to its very strong hydrogen bond donating ability TFE forms very strong hydrogen bond networks with the anions in the ground state itself. So, the possibility of relaxation of the locally excited state via repositioning of hydrogen bonds is impeded and hence there is no rise time at the red end of the emission spectrum. However, the presence of a 10 ps rise at the red end of C153, a well-known solvation probe, is observed in TFE. Absence of such a rise in the Schiff base anions (Fig. 2e and Fig. S6, ESI†) indicates the absence of dynamic solvation of the excited state of the anions in this solvent and bolsters the contention about the predominant role of hydrogen bond dynamics. The slight wavelength dependence arises mostly because of conformational reorganization which led to a slight wavelength dependence in solvents like ACN, DMF as well. Dynamics of such hydrogen bond reorganization has been elucidated for several other molecular systems like fluorenone and coumarin 102, using ultrafast spectroscopic techniques and quantum chemical calculations.68,69
 | ||
| Fig. 4 Lippert–Mataga plots of (a) salampy− (b) HBAP2− (c) salophen2− (d) salbn2−. | ||
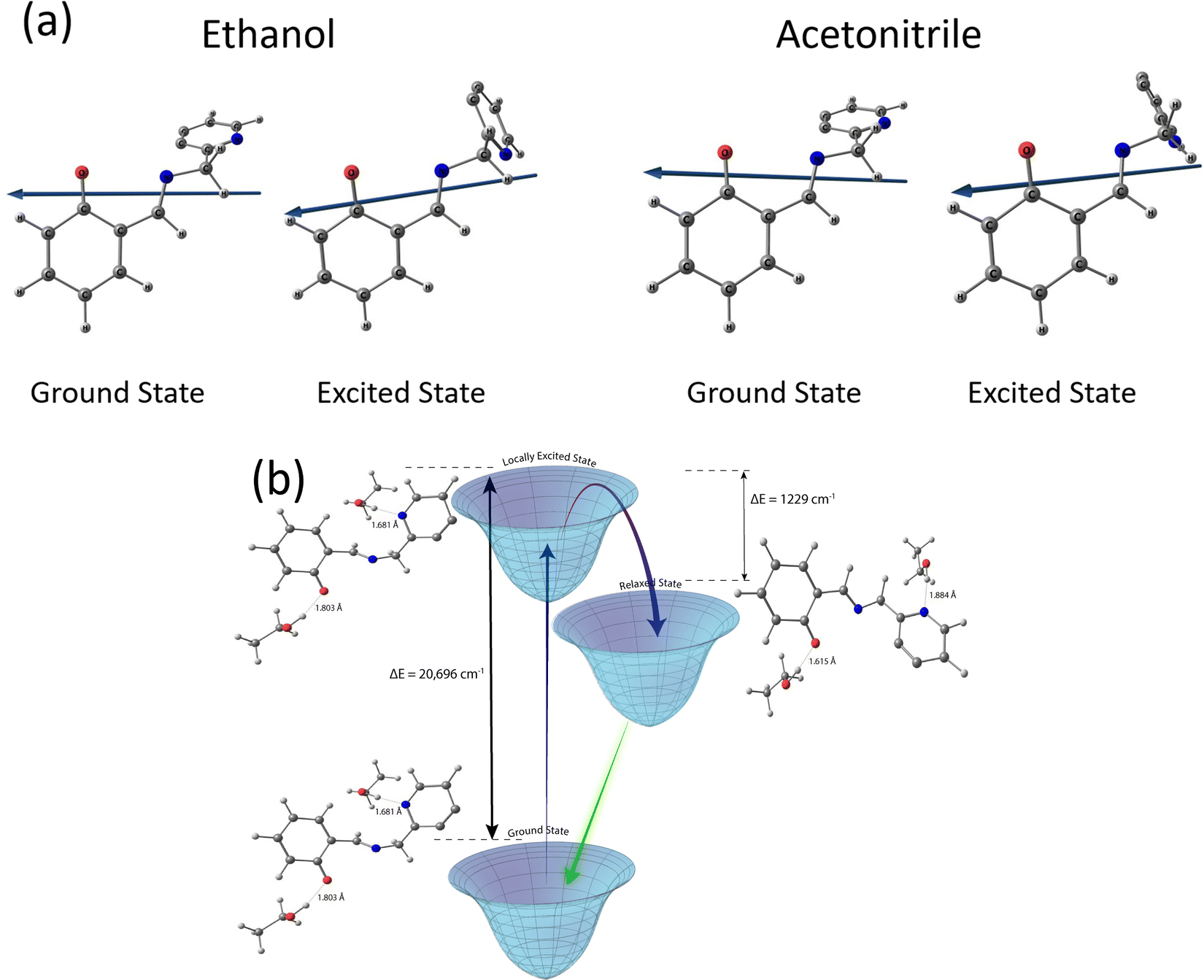 | ||
| Fig. 5 (a) Energy optimized structure in the ground and electronic excited state depicting the direction of dipole moment. (b) Quantum chemical calculations depicting the energy of the ground state (GS), locally excited state (LE) and relaxed state (RS). | ||
Taking a leaf out of these literature reports, elementary quantum chemical calculations have been performed to obtain a preliminary idea about the energetics of the ground and excited states of salampy− in EtOH (Fig. 5 and Table ST7, ESI†). These calculations indicate that the excited state relaxation involves the lengthening of the hydrogen bond between the phenyl O atom in the anion and the hydroxyl H atom of a solvent EtOH molecule, along with a shortening of the hydrogen bond between the pyridinium N atom of the anion and a solvent molecule. This redistribution of hydrogen bonds is estimated to be associated with a stabilization of the excited state by 1229 cm−1. This estimate is in excellent agreement with the experimentally observed TDFSS of 1000 cm−1 in EtOH (Table ST6, ESI†) and also with the amount of stabilization indicated by the difference in the Δv values between protic and aprotic solvents of the same polarity, in the Lippert–Mataga plots (Fig. 4). It should be reiterated that the results obtained here might not be quantitative. A more detailed study considering the solvent molecules explicitly is likely to portray a more holistic picture. However, the agreement between the experimental and computational results is indeed encouraging. The extra stabilization by hydrogen bond reorganization in the excited states decreases the energy gap between the excited and ground states. It appears that this is the driving force, in keeping with the energy gap law,70 for an increase in the rate of radiationless transition and consequently, smaller fluorescence lifetimes for the anions in protic solvents. The lifetimes are longer in aprotic solvents of the same polarities, due to the absence of such an extra amount of stabilization.
4. Conclusion
To conclude, the combination of conformational relaxation and hydrogen bond redistribution is found to play a significant role in determining the course of evolution of the excited state of Schiff base anions in protic solvents, viz., EtOH, BuOH and glycerol. The critical role of hydrogen bond dynamics is further evident from the absence of a rise time in strongly hydrogen bond donating solvents like TFE. The observed time evolution of the emission spectra is not due to solvation, as photoexcitation does not lead to a significant amount of dipole creation in these anions. Besides, such time evolution is not observed in polar aprotic solvents. The nature of the hydrogen bond redistribution in the excited state has been further bolstered by quantum chemical calculations, which indicate the shortening of the hydrogen bond between one of the N atoms of the anion and a solvent molecule, along with the lengthening of that between the O atom of the anion and a solvent molecule, in the excited state. The profound effect of these subtle changes in the state of solute–solvent hydrogen bonding, on the excited state dynamics of the anions, is illuminating. This could possibly open up future applications of this class of fluorophores in sensing minute changes in complex microenvironments of biological relevance.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
S. D. acknowledges DST-INSPIRE and IRCC, IIT Bombay for a Senior Research Fellowship. A. C. acknowledges the Ministry of Education, India for a Prime Minister's Research Fellowship. D. K. S. acknowledges IIT Bombay for an institute postdoctoral fellowship. A. D. thanks CSIR for a generous research grant and Prof. Sukhendu Nath for a fruitful discussion. The Central Instrumentation Facility, Department of Chemistry is thankfully acknowledged. The TCSPC experiments were performed in the central facility for Time Resolved Fluorescence Spectroscopy and Microscopy, IIT Bombay. The FOG experiments have been done on a Fund for Improvement of S&T Infrastructure (FIST)-funded facility of the Department of Chemistry.References
- S. Wang, W. Tan, W. Lang, H. Qian, S. Guo, L. Zhu and J. Ge, Fluorogenic and Mitochondria-Localizable Probe Enables Selective Labeling and Imaging of Nitroreductase, Anal. Chem., 2022, 94, 7272–7277 CrossRef CAS PubMed.
- L. Gutiérrez-Arzaluz, I. Nadinov, G. Healing, J. Czaban-Jóźwiak, J. Jia, Z. Huang, Y. Zhao, O. Shekhah, K. S. Schanze, M. Eddaoudi and O. F. Mohammed, Ultrafast Aggregation-Induced Tunable Emission Enhancement in a Benzothiadiazole-Based Fluorescent Metal–Organic Framework Linker, J. Phys. Chem. B, 2021, 125, 13298–13308 CrossRef.
- T. K. Heiss, R. S. Dorn, A. J. Ferreira, A. C. Love and J. A. Prescher, Fluorogenic Cyclopropenones for Multicomponent, Real-Time Imaging, J. Am. Chem. Soc., 2022, 144, 7871–7880 CrossRef CAS PubMed.
- G. Lukinavičius, G. Y. Mitronova, S. Schnorrenberg, A. N. Butkevich, H. Barthel, V. N. Belov and S. W. Hell, Fluorescent dyes and probes for super-resolution microscopy of microtubules and tracheoles in living cells and tissues, Chem. Sci., 2018, 9, 3324–3334 RSC.
- A. S. Klymchenko, Solvatochromic and Fluorogenic Dyes as Environment-Sensitive Probes: Design and Biological Applications, Acc. Chem. Res., 2017, 50, 366–375 CrossRef CAS.
- J. Mohanty, N. Thakur, S. Dutta Choudhury, N. Barooah, H. Pal and A. C. Bhasikuttan, Recognition-Mediated Light-Up of Thiazole Orange with Cucurbit[8]uril: Exchange and Release by Chemical Stimuli, J. Phys. Chem. B, 2012, 116, 130–135 CrossRef CAS PubMed.
- H. Liu, J. Chen, Y. Fu, Z. Zhao and B. Z. Tang, Achieving High Electroluminescence Efficiency and High Color Rendering Index for All-Fluorescent White OLEDs Based on an Out-of-Phase Sensitizing System, Adv. Funct. Mater., 2021, 31, 2103273 CrossRef CAS.
- I. Takashima, Y. Inoue, N. Matsumoto, A. Takagi and K. Okuda, A fluorogenic probe using a catalytic reaction for the detection of trace intracellular zinc, Chem. Commun., 2020, 56, 13327–13330 RSC.
- K. Santra, I. Geraskin, M. Nilsen-Hamilton, G. A. Kraus and J. W. Petrich, Characterization of the Photophysical Behavior of DFHBI Derivatives: Fluorogenic Molecules that Illuminate the Spinach RNA Aptamer, J. Phys. Chem. B, 2019, 123, 2536–2545 CrossRef CAS.
- M. Shen, T. Ding, J. Luo, C. Tan, K. Mahmood, Z. Wang, D. Zhang, R. Mishra, M. D. Lew and B. Sadtler, Competing Activation and Deactivation Mechanisms in Photodoped Bismuth Oxybromide Nanoplates Probed by Single-Molecule Fluorescence Imaging, J. Phys. Chem. Lett., 2020, 11, 5219–5227 CrossRef CAS PubMed.
- B. Samanta, J. Seikowski and C. Höbartner, Fluorogenic Labeling of 5-Formylpyrimidine Nucleotides in DNA and RNA, Angew. Chem., Int. Ed., 2016, 55, 1912–1916 CrossRef CAS.
- P. H. Doan, D. R. G. Pitter, A. Kocher, J. N. Wilson and T. Goodson, Two-Photon Spectroscopy as a New Sensitive Method for Determining the DNA Binding Mode of Fluorescent Nuclear Dyes, J. Am. Chem. Soc., 2015, 137, 9198–9201 CrossRef CAS PubMed.
- G. Sivaraman, M. Iniya, T. Anand, N. G. Kotla, O. Sunnapu, S. Singaravadivel, A. Gulyani and D. Chellappa, Chemically diverse small molecule fluorescent chemosensors for copper ion, Coord. Chem. Rev., 2018, 357, 50–104 CrossRef CAS.
- C. Kang, K. Yang, Z. Zhang, A. K. Usadi, D. C. Calabro, L. S. Baugh, Y. Wang, J. Jiang, X. Zou, Z. Huang and D. Zhao, Growing single crystals of two-dimensional covalent organic frameworks enabled by intermediate tracing study, Nat. Commun., 2022, 13, 1–9 Search PubMed.
- A. Foroughnia, A. D. Khalaji, E. Kolvari and N. Koukabi, Synthesis of new chitosan Schiff base and its Fe2O3 nanocomposite: Evaluation of methyl orange removal and antibacterial activity, Int. J. Biol. Macromol., 2021, 177, 83–91 CrossRef CAS.
- Z. Zeng, X. Peng, J. Zheng and C. Xu, Heteroatom-Doped Nickel Oxide Hybrids Derived from Metal–Organic Frameworks Based on Novel Schiff Base Ligands toward High-Performance Electrochromism, ACS Appl. Mater. Interfaces, 2021, 13, 4133–4145 CrossRef CAS.
- S. Mitra and N. Tamai, Dynamics of photochromism in salicylideneaniline: A femtosecond spectroscopic study, Phys. Chem. Chem. Phys., 2003, 5, 4647 RSC.
- S. Mitra and N. Tamai, Femtosecond spectroscopic study on photochromic salicylideneaniline, Chem. Phys. Lett., 1998, 282, 391–397 CrossRef CAS.
- M. Ziółek, M. Gil, J. A. Organero and A. Douhal, What is the difference between the dynamics of anion- and keto-type of photochromic salicylaldehyde azine?, Phys. Chem. Chem. Phys., 2010, 12, 2107 RSC.
- W. Rodríguez-Córdoba, J. S. Zugazagoitia, E. Collado-Fregoso and J. Peon, Excited state intramolecular proton transfer in Schiff bases. Decay of the locally excited enol state observed by femtosecond resolved fluorescence, J. Phys. Chem. A, 2007, 111, 6241–6247 CrossRef.
- M. Ziółek, J. Kubicki, A. Maciejewski, R. Naskrecki and A. Grabowska, Enol-keto tautomerism of aromatic photochromic Schiff base N,N’-bis(salicylidene)-p-phenylenediamine: ground state equilibrium and excited state deactivation studied by solvatochromic measurements on ultrafast time scale, J. Chem. Phys., 2006, 124, 124518 CrossRef.
- L. Spörkel, G. Cui and W. Thiel, Photodynamics of Schiff Base Salicylideneaniline: Trajectory Surface-Hopping Simulations, J. Phys. Chem. A, 2013, 117, 4574–4583 Search PubMed.
- J. Dobkowski, P. Wnuk, J. Buczyńska, M. Pszona, G. Orzanowska, D. Frath, G. Ulrich, J. Massue, S. Mosquera-Vázquez, E. Vauthey, C. Radzewicz, R. Ziessel and J. Waluk, Substituent and Solvent Effects on the Excited State Deactivation Channels in Anils and Boranils, Chem. – Eur. J., 2015, 21, 1312–1327 CAS.
- J. S. Zugazagoitia, M. Maya, C. Damián-Zea, P. Navarro, H. I. Beltrán and J. Peon, Excited-State Dynamics and Two-Photon Absorption Cross Sections of Fluorescent Diphenyl-Tin IV Derivatives with Schiff Bases: A Comparative Study of the Effect of Chelation from the Ultrafast to the Steady-State Time Scale, J. Phys. Chem. A, 2010, 114, 704–714 CrossRef CAS PubMed.
- Y. Dong, R. Fan, W. Chen, P. Wang and Y. Yang, A simple quinolone Schiff-base containing CHEF based fluorescence ‘turn-on’ chemosensor for distinguishing Zn 2+ and Hg 2+ with high sensitivity, selectivity and reversibility, Dalton Trans., 2017, 46, 6769–6775 RSC.
- J. Wang, Y. Li, N. G. Patel, G. Zhang, D. Zhou and Y. Pang, A single molecular probe for multi-analyte (Cr 3+, Al 3+ and Fe 3+) detection in aqueous medium and its biological application, Chem. Commun., 2014, 50, 12258–12261 RSC.
- Y. Xiong, X. Li, M. Li, H. Qin, C. Chen, D. Wang, X. Wang, X. Zheng, Y. Liu, X. Liang and G. Qing, What Is Hidden Behind Schiff Base Hydrolysis? Dynamic Covalent Chemistry for the Precise Capture of Sialylated Glycans, J. Am. Chem. Soc., 2020, 142, 7627–7637 CrossRef CAS.
- X. Qiu, J. Huang, N. Wang, K. Zhao, J. Cui and J. Hao, Facile Synthesis of Water-Soluble Rhodamine-Based Polymeric Chemosensors via Schiff Base Reaction for Fe3+ Detection and Living Cell Imaging, Front. Chem., 2022, 10 DOI:10.3389/fchem.2022.845627.
- T. Khan and A. Datta, Impact of Molecular Arrangement and Torsional Motion on the Fluorescence of Salophen and Its Metal Complexes, J. Phys. Chem. C, 2017, 121, 2410–2417 CrossRef CAS.
- T. Khan, S. Vaidya, D. S. Mhatre and A. Datta, The Prospect of Salophen in Fluorescence Lifetime Sensing of Al 3 +, J. Phys. Chem. B, 2016, 120, 10319–10326 CrossRef CAS PubMed.
- T. Khan and A. Datta, Enhanced fluorescence with nanosecond dynamics in the solid state of metal ion complexes of alkoxy salophens, Phys. Chem. Chem. Phys., 2017, 19, 30120–30127 RSC.
- S. Dasgupta, S. Banerjee, S. Das and A. Datta, From fluorogens to fluorophores by elucidation and suppression of ultrafast excited state processes of a Schiff base, Phys. Chem. Chem. Phys., 2021, 23, 19494–19502 RSC.
- S. Dasgupta, A. Chowdhury and A. Datta, Time evolution of the solvated and conformationally relaxed emissive excited state of the anionic form of salophen, a Schiff base, J. Indian Chem. Soc., 2021, 98, 100122 CrossRef CAS.
- A. Chowdhury, S. Dasgupta and A. Datta, Deprotonation-induced enhancement in fluorescence of 2-((2-hydroxybenzylidene)amino)phenol, a Schiff base, Chem. Phys. Impact, 2021, 3, 100057 CrossRef.
- R. R. Maar and J. B. Gilroy, Group 13 Complexes of Chelating N 2 O 2 n − Ligands as Hybrid Molecular Materials, Chem. – Eur. J., 2018, 24, 12449–12457 CrossRef CAS.
- M. L. Horng, J. A. Gardecki, A. Papazyan and M. Maroncelli, Subpicosecond Measurements of Polar Solvation Dynamics: Coumarin 153 Revisited, J. Phys. Chem., 1995, 99, 17311–17337 CrossRef CAS.
- B. A. Capistran, S. H. Yuwono, M. Moemeni, S. Maity, A. Vahdani, B. Borhan, J. E. Jackson, P. Piecuch, M. Dantus and G. J. Blanchard, Excited-State Dynamics of a Substituted Fluorene Derivative. The Central Role of Hydrogen Bonding Interactions with the Solvent, J. Phys. Chem. B, 2021, 125, 12242–12253 CrossRef CAS PubMed.
- D. A. Horke, H. M. Watts, A. D. Smith, E. Jager, E. Springate, O. Alexander, C. Cacho, R. T. Chapman and R. S. Minns, Hydrogen Bonds in Excited State Proton Transfer, Phys. Rev. Lett., 2016, 117, 1–5 CrossRef.
- G. J. Zhao and K. L. Han, Hydrogen bonding in the electronic excited state, Acc. Chem. Res., 2012, 45, 404–413 CrossRef CAS PubMed.
- K. J. Gaffney, P. H. Davis, I. R. Piletic, N. E. Levinger and M. D. Fayer, Hydrogen bond dissociation and reformation in methanol oligomers following hydroxyl stretch relaxation, J. Phys. Chem. A, 2002, 106, 12012–12023 CrossRef CAS.
- D. Mandal, T. Tahara and S. R. Meech, Excited-State Dynamics in the Green Fluorescent Protein Chromophore, J. Phys. B: At., Mol. Opt. Phys., 2004, 108, 1102–1108 CAS.
- S. Dasgupta, A. Chowdhury and A. Datta, Time evolution of the solvated and conformationally relaxed emissive excited state of the anionic form of salophen, a Schiff base, J. Indian Chem. Soc., 2021, 98, 100122 CrossRef CAS.
- A. R. Kennedy and J. Reglinski, N, N ′-Bis(salicylidene)-1-4-butanediamine, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2001, 57, o1027–o1028 CrossRef CAS.
- J. R. Lakowicz, Principles of fluorescence spectroscopy, Springer, 2006 Search PubMed.
- T. N. Burai, T. K. Mukherjee, P. Lahiri, D. Panda and A. Datta, Early events associated with the excited state proton transfer in 2-(2′-pyridyl)benzimidazole, J. Chem. Phys., 2009, 131, 034504 CrossRef PubMed.
- M. Maroncelli and G. R. Fleming, Picosecond solvation dynamics of coumarin 153: The importance of molecular aspects of solvation, J. Chem. Phys., 1987, 86, 6221–6239 CrossRef CAS.
- M. J. Frisch and H. B. Schlegel, et al., Gaussian 16, Revision C.01, Gaussian, Inc., Wallin, 2016 Search PubMed.
- M. Devi, P. Sharma, A. Kumar, S. Kaur, M. Kumar and V. Bhalla, ESIPT Active Assemblies for ‘On-On’ Detection, Cell Imaging and Hampering Cellular Activity of 2,6-Dichloro-4-nitroaniline, Chem. – Asian J., 2022, 17, e202101219 CrossRef CAS.
- C.-H. Wang, Z.-Y. Liu, C.-H. Huang, C.-T. Chen, F.-Y. Meng, Y.-C. Liao, Y.-H. Liu, C.-C. Chang, E. Y. Li and P.-T. Chou, Chapter Open for the Excited-State Intramolecular Thiol Proton Transfer in the Room-Temperature Solution, J. Am. Chem. Soc., 2021, 143, 12715–12724 CrossRef CAS PubMed.
- X. Liu, X. Liu, Y. Shen and B. Gu, A Simple Water-Soluble ESIPT Fluorescent Probe for Fluoride Ion with Large Stokes Shift in Living Cells, ACS Omega, 2020, 5, 21684–21688 CrossRef CAS PubMed.
- Z.-Y. Liu, J.-W. Hu, T.-H. Huang, K.-Y. Chen and P.-T. Chou, Excited-state intramolecular proton transfer in the kinetic-control regime, Phys. Chem. Chem. Phys., 2020, 22, 22271–22278 RSC.
- D. Jacquemin, M. Khelladi, A. De Nicola and G. Ulrich, Turning ESIPT-Based triazine fluorophores into dual emitters: From theory to experiment, Dyes Pigm., 2019, 163, 475–482 CrossRef CAS.
- Z.-Y. Liu, J.-W. Hu, C.-L. Chen, Y.-A. Chen, K.-Y. Chen and P.-T. Chou, Correlation among Hydrogen Bond, Excited-State Intramolecular Proton-Transfer Kinetics and Thermodynamics for −OH Type Proton-Donor Molecules, J. Phys. Chem. C, 2018, 122, 21833–21840 CrossRef CAS.
- P. Jagadesan, T. Whittemore, T. Beirl, C. Turro and P. L. McGrier, Excited-State Intramolecular Proton-Transfer Properties of Three Tris(N -Salicylideneaniline)-Based Chromophores with Extended Conjugation, Chem. – Eur. J., 2017, 23, 917–925 CrossRef CAS PubMed.
- K. E. Steege, J. Wang, K. E. Uhrich and E. W. Castner, Local Polarity and Microviscosity in the Hydrophobic Cores of Amphiphilic Star-like and Scorpion-like Macromolecules, Macromolecules, 2007, 40, 3739–3748 CrossRef CAS.
- Y. Tamoto, H. Segawa and H. Shirota, Solvation Dynamics in Aqueous Anionic and Cationic Micelle Solutions: Sodium Alkyl Sulfate and Alkyltrimethylammonium Bromide, Langmuir, 2005, 21, 3757–3764 CrossRef CAS.
- Y. Mei and W. Yang, Excited-State Potential Energy Surfaces, Conical Intersections, and Analytical Gradients from Ground-State Density Functional Theory, J. Phys. Chem. Lett., 2019, 10, 2538–2545 CrossRef CAS PubMed.
- S. Nandi, P. Mondal, R. Chowdhury, A. Saha, S. Ghosh and K. Bhattacharyya, Amyloid beta peptides inside a reconstituted cell-like liposomal system: aggregation, FRET, fluorescence oscillations and solvation dynamics, Phys. Chem. Chem. Phys., 2016, 18, 30444–30451 RSC.
- D. Sardana, K. Yadav, H. Shweta, N. S. Clovis, P. Alam and S. Sen, Origin of Slow Solvation Dynamics in DNA: DAPI in Minor Groove of Dickerson-Drew DNA, J. Phys. Chem. B, 2019, 123, 10202–10216 CrossRef CAS.
- S. S. Hossain, S. Paul and A. Samanta, Complete Solvation Dynamics of Coumarin 153 in Tetraalkylammonium Bromide-Based Deep Eutectic Solvents, J. Phys. Chem. B, 2020, 124, 2473–2481 CrossRef CAS.
- B. M. Luther, J. R. Kimmel and N. E. Levinger, Dynamics of polar solvation in acetonitrile–benzene binary mixtures: Role of dipolar and quadrupolar contributions to solvation, J. Chem. Phys., 2002, 116, 3370–3377 CrossRef CAS.
- N. Bera, M. Ghosh, S. Nandi and N. Sarkar, Femtosecond solvation dynamics study of hydrophobic and hydrophilic probes in various room temperature ionic liquids (RTILs) containing microemulsions, Chem. Phys. Lett., 2021, 767, 138356 CrossRef CAS.
- S. H. Lee, J. H. Lee and T. Joo, Deuterium isotope effect on the solvation dynamics of a dye molecule in methanol and acetonitrile, J. Chem. Phys., 1999, 110, 10969–10977 CrossRef CAS.
- M. L. Horng, J. A. Gardecki, A. Papazyan and M. Maroncelli, Subpicosecond Measurements of Polar Solvation Dynamics: Coumarin 153 Revisited, J. Phys. Chem., 1995, 99, 17311–17337 CrossRef CAS.
- A. S. R. Koti, M. M. G. Krishna and N. Periasamy, Time-Resolved Area-Normalized Emission Spectroscopy (TRANES): A Novel Method for Confirming Emission from Two Excited States, J. Phys. Chem. A, 2001, 105, 1767–1771 CrossRef CAS.
- M. M. Lindic, M. Zajonz, C. Gers-Panther, T. J. J. Müller and M. Schmitt, The excited state dipole moment of 2-[(4-methoxyphenyl)ethynyl]-3-(1-methyl-1H-indol-3-yl)-quinoxaline from thermochromic shifts, Spectrochim. Acta, Part A, 2020, 228, 117574 CrossRef CAS PubMed.
- T. Abe, Estimation of Angles between the Ground- and Excited-State Dipole Moments from Solvent Spectral Frequency Shifts, Bull. Chem. Soc. Jpn., 1991, 64, 3224–3228 CrossRef CAS.
- G. Zhao and K. Han, Ultrafast Hydrogen Bond Strengthening of the Photoexcited Fluorenone in Alcohols for Facilitating the Fluorescence Quenching †, J. Phys. Chem. A, 2007, 111, 9218–9223 CrossRef CAS PubMed.
- R. Ghosh, A. K. Mora, S. Nath and D. K. Palit, Ultrafast Dynamics of Hydrogen Bond Breaking and Making in the Excited State of Fluoren-9-one: Time-Resolved Visible Pump-IR Probe Spectroscopic Study, J. Phys. Chem. B, 2017, 121, 1068–1080 CrossRef CAS PubMed.
- R. Englman and J. Jortner, The energy gap law for radiationless transitions in large molecules, Mol. Phys., 1970, 18, 145–164 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2cp05007b |
| This journal is © the Owner Societies 2023 |