
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Using Koopmans’ theorem for constructing basis sets: approaching high Rydberg excited states of lithium with a compact Gaussian basis†
Jan
Šmydke

Department of Radiation and Chemical Physics, FZU - Institute of Physics of the Czech Academy of Sciences, Na Slovance 1999/2, 18200 Praha 8, Czech Republic. E-mail: jan.smydke@gmail.com
First published on 10th July 2023
Abstract
For accurate ab initio description of Rydberg excited states, this study suggests generating appropriate diffuse basis functions by cheap variational optimization of virtual orbitals of the corresponding ion core. By following this approach, dozens of converged correlated lithium Rydberg states, namely, all the states up to 24 2S, 25 2P, 14 2D, 16 2F and 16 2G, not yet achieved via other ab initio approaches, could be obtained at the EOM-CCSD level of theory with compact and mostly state-selective contracted Gaussian basis sets. Despite its small size and Gaussian character, the optimized basis leads to highly accurate excitation energies that differ merely in the order of meV from the reference state-of-the-art explicitly correlated Gaussian method and even surpass Full-CI results on the Slater basis by an order of magnitude.
1 Introduction
Investigation of Rydberg excited states of atomic and molecular systems is mostly the domain of the quantum defect theory (QDT),1 in which a motion of the highly excited electron is modeled by an effective one-electron approach. It is assumed that the average distance between the highly excited electron and the positively charged ion core is sufficiently large that the electron experiences a Coulombic field of the ion core analogous to the field in the hydrogen atom, though effectively shielded by the other electrons. The energy spectrum of such a model necessarily differs from the spectrum of hydrogen, introducing characteristic quantum defects δ of the principal quantum number n in the energy level formula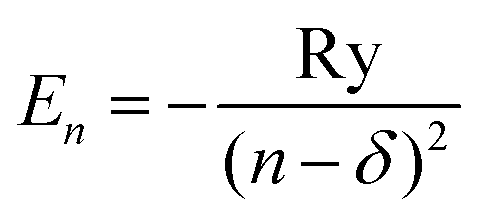 | (1) |
By contrast, the ab initio theories for excited states either suffer from overall poor accuracy of the computed energy spectrum or provide only the few lowest excited states in high precision. This stems from the inherent complexity of the ab initio methods, which deal with many-body systems and balance between scalability and an accurate description of the electronic correlation. Nevertheless, the need for a proper ab initio description of the Rydberg states as opposed to the quantum defect asymptotic theory comes also hand in hand with the fact that the Rydberg states often interact with valence excited states in a similar energy window, strongly affecting the spectroscopy and dynamics of the underlying chemical systems.2 In such cases, an efficient full many-body treatment is thus necessary.
The sharp contrast between the ability of the advanced quantum chemical methods to describe highly correlated ground electronic states even in difficult electronic structures on the one hand and their failure to reliably describe the higher excited states on the other hand grows out of the insufficiency of the commonly used basis sets to reach and mimic the diffuse and structurally more complicated excited state wave functions. The standard Gaussian basis sets (GTO) tend to be extensively optimized for the ground state to describe enough electronic correlation while keeping the number of basis functions low. The exponential-type basis functions (ETO) like the Slater-type orbitals (STO) and Coulomb–Sturmians (CS) are, of course, of higher quality than the Gaussians due to the correct cusp at nuclei and their natural diffuse characteristics. Despite the superiority of the ETOs and in particular of the CS functions, which constitute a complete orthogonal set, the ETOs (as well as the GTOs) are by no other means optimal for excited state description of many-electron systems.
Hence, a common way of building custom basis sets for excited states is to extend a standard GTO basis with a large set of primitive diffuse functions. The primitives are often put in the form of the even tempered Gaussians (ETG), in which the exponents are given by a simple formula
log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) ζk = logζ0 + klogα; logα < 0, k = 0…(N − 1) ζk = logζ0 + klogα; logα < 0, k = 0…(N − 1) | (2) |
The aim of the present study is a tailored optimization of the diffuse basis rather than a mere inclusion of a large number of functions. The intention is to systematically generate such a basis that would approximate at least a few Rydberg orbitals. Such orbitals could then serve as the optimal functions for a correlated treatment of the corresponding Rydberg states. The ultimate outcome, although beyond the scope of this work, might lead to a specific diffuse basis suitable for a complex scaling treatment of related resonance states, which is highly sensitive to the basis set quality.3 In order to employ standard quantum chemistry codes, this study exclusively uses Gaussian basis sets. Nevertheless, the investigated approach is universal and applicable to any basis set type.
This study focuses on systems that can be modeled as a closed-shell ion core with an odd electron moving around. For such systems, a simple trick can be used to describe the Rydberg orbitals that can be the target of the basis set optimization. As is well known from Koopmans’ theorem,4 the canonical restricted Hartree–Fock (RHF) virtual orbitals describe an electron captured by the system (i.e. the Rydberg electron captured by the ion core). Since the eigenvalues of the converged Fock operator (i.e. the one defined by the converged Hartree–Fock occupied orbitals) are stationary with respect to variations of the eigenvectors, the Rydberg orbitals can be found by a variational procedure. For the lithium atom (Li), which is the subject of this work, it means that by a variational minimization of the Li+ virtual orbital energies, while varying the basis set parameters and at the same time maintaining the Hartree–Fock energy minimal (in order to keep the converged Fock operator), one should end up with such Li+ virtual orbitals that could serve as the appropriate basis functions for the description of the Rydberg excited electron in the neutral Li atom.
In principle, the presented idea of variational minimization of virtual orbitals for obtaining the Rydberg functions is not confined to closed-shell ion cores but could be used with any system for which the Koopmans’ theorem for electron affinity is valid. For open-shell ion cores, it is generally not correct to use the unrestricted (UHF) or an arbitrary restricted open-shell Hartree–Fock (ROHF) method variant. However, the canonical ROHF method as discussed by Plakhutin et al.5–8 guarantees the validity of Koopmans’ theorem for various open shell electronic systems, so the presented method is applicable for them too.
Although the suggested approach is based on the Hartree–Fock model only, it can be anticipated that due to the Rydberg character of the excited electron, the model can be satisfactory at least for higher excited states. In our earlier studies of helium (He) resonances,3,9 we used an analogous approach to obtain a basis for He excited states by appropriately modifying the Fock operator10 so that the virtual orbitals describe excited electrons rather than the captured ones. The quality of the optimized basis was outstanding, leading not only to good transition energies but also to a wide interval of resonance energy stability along the complex scaling parameter ϑ. Such a basis enabled an extensive non-Hermitian dynamical propagation of He under extremely intense laser radiation.
We are not aware of many other approaches for specific Gaussian basis set optimization regarding the Rydberg states. Durand and Volatron11 used electronically correlated transition energies computed using the configuration interaction method to find the optimal diffuse Gaussian exponents for the description of Rydberg states of water. Kaufmann et al.12 provide a universal ready-to-use formula for Gaussian exponents to describe either the atomic Rydberg functions or the low-lying continuum states. They basically tried an approximate one-to-one mapping between exponential-type functions and Gaussians in order to cover a portion of a complete bound or continuum spectrum. Despite using a less sophisticated principal idea, the method introduced in the present study offers more flexibility in the description of specific systems as opposed to using a fixed universal basis and is also less limited as to the number of achievable Rydberg states.
The rest of the article is organized as follows. Section 2 describes the basis set optimization and other computational details. Section 3 discusses important properties of the optimized basis and compares the resulting Rydberg excitation energies to other highly accurate values known in the literature. A summary of the results and conclusions are drawn in Section 4.
2 Computational details
The whole basis set optimization process can be summarized in the following. First, a standard basis set is included for a good description of the electronic correlation. That should suffice for the ground as well as for the Rydberg states since the part of the wave function, in which the correlation plays a role, is in all states spatially distributed similarly; only the Rydberg electron occupies distant areas where it does not significantly contribute to the dynamical electronic correlation. To describe the Rydberg electron attracted by a closed-shell ion core, diffuse functions are appended to the basis and the virtual orbital energies of the ion core are minimized. This is achieved by varying the diffuse functions while maintaining the optimal Hartree–Fock energy and avoiding near linear dependencies. In accord with Koopmans’ theorem, such optimal virtual orbitals are just the Rydberg orbitals. Since their shape is controlled solely by the Hartree–Fock field of the ion core, it is advantageous to improve the field quality before the actual virtual orbital optimization by an additional high-exponent function to mimic the wave-function cusp. Eventually, contracted basis functions are formed from the optimal Rydberg orbitals using their expansion coefficients (LCAO) in the primitive diffuse basis. The final basis set for productive computations of the Rydberg states then consists of the standard basis together with the high-exponent function and appropriate subsets of the contracted optimized Rydberg functions as discussed in Section 3.1.Practically, for lithium, the well established aug-ano-pVQZ13 basis of Neese and Valeev has been chosen as the standard basis set. To improve the Hartree–Fock wave function of the Li+ ion core, an additional ETG series of 19 high-exponent primitive S functions was used to minimize the Hartree–Fock energy. From the resulting 1S orbital, an auxiliary contracted function was formed, consisting of the ETG primitives only, which was used further instead of the high-exponent series. Independently for each angular momentum L ∈ {S,P,D,F,G}, a mostly diffuse ETG set of the given L was added to the basis and by varying its parameters, the energy of the first Li+ virtual orbital of that L symmetry was minimized. The orbital optimization led to ETG sets that spanned not only diffuse functions but also rather tight ones. The process thus had to avoid such parameters that caused linear dependence of the ETG functions with the aug-ano-pVQZ basis. The optimal ETG parameters were then fixed, and only the number (N) of the ETG primitive functions was further gradually increased until a satisfactorily large number of virtual orbital energies were no longer changing by more than 10−9 a.u. The numbers of ETG primitives reached 30 for S, 50 for P, 40 for D, 30 for F, and 25 for G ETG series. In the end, all the optimized virtual orbitals, namely, 25 S, 28 P, 22 D, 21 F and 14 G, were contracted in the diffuse ETG subspace and put together in a final huge basis denoted as [25S-28P-22D-21F-14G]. The basis set in this notation means that it contains the aug-ano-pVQZ plus the high-exponent contracted S function plus the given numbers of optimal contracted Rydberg functions.
During the basis set optimization process, the Hartree–Fock orbital energies were calculated using the MRCC14,15 program package, while the multidimensional minimization itself was driven by the mdoptcli16 utility, which uses procedures from the GNU Scientific Library (GSL).17 All the correlated computations using the coupled cluster (CCSD) and equation of motion coupled cluster (EOM-CCSD) methods employed the GAMESS 2021 R118 package, recompiled to allow a large number of primitive basis functions.
3 Results and discussion
3.1 Properties of the optimized basis
It was found that the [7S-6P-5D] subset of the huge [25S-28P-22D-21F-14G] basis exhibits already converged CCSD ionization potential (IP) with respect to the basis set size and similarly also all the Li bound state excitation energies computed at the EOM-CCSD level (up to the state 8 2S). This is apparent in Table 1, which shows that by adding more of the optimized diffuse S, P or D functions (basis [10S-9P-8D]) or by including F and G functions (basis [7S-6P-5D-4F-3G]) to the [7S-6P-5D] basis, the IP as well as the excitation energy values were no longer affected. The IP value (5.3877 eV) was calculated as the difference between the CCSD energy of the Li+ cation and the neutral Li atom, underestimating the experimental19 value of 5.3917 eV by 4 meV. This is in accordance with the expectations as the CCSD method is not Full-CI equivalent in the description of the neutral 3-electron Li system. The [7S-6P-5D] basis can thus be considered as a minimal saturated set that can safely be extended to achieve higher Li Rydberg states.| State | [7S-6P-5D] | [7S-6P-5D-4F-3G] | [10S-9P-8D] |
|---|---|---|---|
| 2 2S ROHF | −7.4327268276 | −7.4327268276 | −7.4327268280 |
| 2 2S CCSD | −7.4744349730 | −7.4744349759 | −7.4744353492 |
| IP CCSD | 5.3877 | 5.3877 | 5.3877 |
| 2 2P | 1.8472 | 1.8472 | 1.8472 |
| 3 2S | 3.3704 | 3.3704 | 3.3704 |
| 3 2P | 3.8317 | 3.8317 | 3.8317 |
| 3 2D | 3.8754 | 3.8753 | 3.8754 |
| 4 2S | 4.3378 | 4.3378 | 4.3378 |
| 4 2P | 4.5186 | 4.5186 | 4.5186 |
| 4 2D | 4.5373 | 4.5373 | 4.5373 |
| 4 2F | 4.5379 | ||
| 5 2S | 4.7456 | 4.7456 | 4.7456 |
| 5 2P | 4.8341 | 4.8341 | 4.8341 |
| 5 2D | 4.8437 | 4.8437 | 4.8437 |
| 5 2F | 4.8440 | ||
| 5 2G | 4.8441 | ||
It should be stressed that the obtained diffuse basis was optimized for Li Rydberg states and not for Li+, although it was the virtual orbitals of the cation that determined the Li Rydberg functions. Therefore, Li+ excited states are not converged with respect to the basis. This can be seen from Table 2, where the not yet converged Li+ states are described by the very same basis sets as the converged states of the neutral Li in Table 1.
| State | [7S-6P-5D] | [7S-6P-5D-4F-3G] | [10S-9P-8D] |
|---|---|---|---|
| 1 1S RHF | −7.2364151179 | −7.2364151179 | −7.2364151179 |
| 1 1S CCSD | −7.2764420008 | −7.2764420020 | −7.2764423045 |
| 2 1S | 60.8448 | 60.8448 | 60.8446 |
| 2 1P | 62.2642 | 62.2642 | 62.2610 |
| 3 1S | 69.2199 | 69.2199 | 69.2194 |
| 3 1D | 69.7706 | 69.7706 | 69.7474 |
| 3 1P | 69.8285 | 69.8285 | 69.8204 |
| 4 1S | 72.0843 | 72.0843 | 72.0826 |
| 4 1D | 72.3562 | 72.3561 | 72.3458 |
| 4 1P | 72.3774 | 72.3774 | 72.3753 |
As anticipated above, to achieve higher Li Rydberg states of a particular angular momentum L, just more of the optimized Rydberg functions of the given L can be added to the minimal [7S-6P-5D] basis (schematically as [7S-6P-5D + kL]), since the resulting correlated states are no longer affected by the functions of other L. Truly, Table 3 shows that by gradually increasing the number of Rydberg S functions included to the minimal basis, the correlated bound states remain intact, and only new 2S states emerge as the basis grows. This, as well as a strong dominance of the EOM-CCSD R1 operator amplitude20 that excites to the appropriate Rydberg orbital, confirms the Rydberg character of the states (i.e. systems of an electron and a net positive charge of the ion core, ignoring the interactions between that electron and the individual other electrons of the ion core) and hence also suitability of the presented basis set optimization scheme. Analogous results were obtained also for 2P, 2F and 2G states. Only the 2D states computed with [7S-6P-5D+kD] basis exhibited unsaturated behavior, as shown in Table 4. Although the states below 7 2D (regardless of their symmetry) were unaffected by the additional D functions, none of the higher 2D states could achieve a converged excitation energy. This could mean that in such highly excited 2D states, the Rydberg excited electron is still extensively interacting with other electrons in the ion core, yet the provided basis is no longer capable of sufficient electronic correlation description over that large distance. In contrast to the other angular momentum states, the motion of the excited electron in 2D states thus may not be driven by a mere positive charge of the ion core as is typical for Rydberg states. For more discussion of the high 2D states’ behaviour, see also Fig. 1 and the last paragraph of this subsection.
| State | [7S-6P-5D] | [16S-6P-5D] | [25S-6P-5D] |
|---|---|---|---|
| 2 2S ROHF | −7.4327268276 | −7.4327268280 | −7.4327268280 |
| 2 2S CCSD | −7.4744349730 | −7.4744349806 | −7.4744349805 |
| IP CCSD | 5.3877 | 5.3877 | 5.3877 |
| 7 2P | 5.1070 | 5.1070 | 5.1070 |
| 7 2D | 5.1097 | 5.1097 | 5.1097 |
| 8 2S | 5.1540 | 5.1540 | |
| 9 2S | 5.2116 | 5.2116 | |
| 10 2S | 5.2610 | 5.2610 | |
| 11 2S | 5.2990 | 5.2990 | |
| 12 2S | 5.3264 | 5.3264 | |
| 13 2S | 5.3457 | 5.3457 | |
| 14 2S | 5.3592 | 5.3592 | |
| 15 2S | 5.3684 | 5.3684 | |
| 16 2S | 5.3747 | 5.3747 | |
| 17 2S | 5.3790 |
| State | [7S-6P-5D] | [7S-6P-10D] | [7S-6P-14D] |
|---|---|---|---|
| 2 2S ROHF | −7.4327268276 | −7.4327268276 | −7.4327268276 |
| 2 2S CCSD | −7.4744349730 | −7.4744350378 | −7.4744350426 |
| IP CCSD | 5.3877 | 5.3877 | 5.3877 |
| 7 2P | 5.1070 | 5.1070 | 5.1070 |
| 7 2D | 5.1097 | 5.1106 | 5.1088 |
| 8 2D | 5.1745 | 5.1655 | |
| 9 2D | 5.2165 | 5.1750 | |
| 10 2D | 5.2582 | 5.2159 | |
| 11 2D | 5.2719 | 5.2618 | |
| 12 2D | 5.2828 | 5.2733 | |
| 13 2D | 5.3004 | ||
| 14 2D | 5.3559 |
 | ||
| Fig. 1 Obtained EOM-CCSD excitation energy levels of 2 Li for angular momenta S up to G. Only states starting from n = 4 and higher are shown for better resolution of the unconverged high 2D states (see text). To help distinguish states of the same n, the states with n = 5 are grouped by the red oval. The level of the CCSD ionization limit is depicted in blue. | ||
Moreover, for all states with the principal quantum number n larger than 7, the optimized Rydberg functions are state selective. That means, only a single specific Rydberg function needs to be added to the [7S-6P-5D] basis to achieve the appropriate correlated Rydberg state, reducing the necessary basis set size dramatically. Except for 2D states, again, where the differences between the state selective basis and the [7S-6P-14D] basis reached even 0.1 eV, all the other L states exhibited negligible errors, from 3 × 10−9 eV for state 14 2G to 2 × 10−6 eV for state 10 2S.
Fig. 1 shows the EOM-CCSD excitation energy diagram for all the computed bound 2S to 2G angular momentum states starting from n = 4. One can clearly see the convergence of the 2S and 2P energy level sequences towards the CCSD ionization limit. Except for the 2D states, which could not yet achieve converged values with respect to the basis set, as discussed in Table 4 and still commented further below, the states with higher angular momenta exhibit the same trend, yet the number of the computed energy levels is smaller. The diagram also nicely illustrates the diminishing dependence of the energy levels on the angular momentum with higher n. From n = 5 the 2P, 2D, 2F and 2G states are already almost aligned. Only the 2S states keep their energy levels different even for higher n. Finally, as for the unconverged 2D states above n = 7, the figure markedly appears as if some levels were just missing while others were put in odd positions compared to the other angular momentum states of the same n. Such behaviour could suggest a poor numerical convergence of the EOM-CCSD procedure for the higher 2D states. However, the energies converged smoothly even for a tight convergence criterion and with no significant effect on the results. Moreover, all five components of the 2D degenerate states led to the same numerical value, despite belonging to different irreducible representations of the D2h computational symmetry group. The behaviour could also be due to poorly optimized Rydberg basis functions. However, no particular issues have been experienced with optimizing the D Rydberg orbitals. Therefore, it truly seems most likely that the highly excited D electron still non-negligibly interacts with the ion core S electrons holding a weaker Rydberg character than the states in other angular momenta. In effect, such states would require an even larger set of diffuse basis functions for the saturated description of their electronic correlation, as was already suggested.
3.2 Li bound excited states
In this section, the bound Li Rydberg states computed by the EOM-CCSD method using the optimized basis are presented and compared with the best non-relativistic results known in the literature. The most appropriate data come from the systematic studies of Li 2S, 2P and 2D states that employ the explicitly correlated Gaussians (ECG) and consider also the effect of the finite nuclear mass as well as the leading relativistic and QED corrections.21–23 These state-of-the-art studies provide highly accurate results for most Li excited states that had ever been computed. In this work, the comparison is made only to the non-relativistic ECG results, which practically reach the estimates of the exact non-relativistic energies.24–28 Another comparison is made to the Hylleraas-CI (Hy-CI) study,29 which provides considerably fewer 2S, 2P and 2D states. Nevertheless, the same study also presents Full-CI energies in optimal Slater-type-orbital (STO) basis that fully cover the principal quantum number n = 7, covering all the states from 2 2S to 7 2S and up to 72 I. The results for the individual angular momenta are compared in Tables 5 (2S), 6 (2P), 7 (2D), 8 (2F) and 9 (2G). The state energies are presented in a.u., while the excitation energies with respect to the ground 2 2S state are in eV rounded to 10−4 eV, for convenience. When appropriate, values corresponding to the estimates of the exact non-relativistic energies are labeled with a related bibliographic reference.| State | [25S-6P-5D] | ECG21 | STO Full-CI29 | Hy-CI29 | ||||
|---|---|---|---|---|---|---|---|---|
| E | EE | EE | ΔEE | EE | ΔEE | EE | ΔEE | |
| 2 2S | −7.4744350 | (−7.4780603)*,24 | (−7.477192) | (−7.478058969) | ||||
| 3 2S | −7.3505733 | 3.3704 | 3.3732*,24 | −0.0027 | 3.3727 | −0.0022 | 3.3733 | −0.0028 |
| 4 2S | −7.3150230 | 4.3378 | 4.3410*,25 | −0.0032 | 4.3406 | −0.0027 | 4.3413 | −0.0035 |
| 5 2S | −7.3000352 | 4.7456 | 4.7486*,25 | −0.0030 | 4.7486 | −0.0030 | 4.7496 | −0.0040 |
| 6 2S | −7.2923000 | 4.9561 | 4.9579*,25 | −0.0018 | 4.9595 | −0.0033 | 4.9612 | −0.0050 |
| 7 2S | −7.2878069 | 5.0784 | 5.0795*,25 | −0.0011 | 5.1047 | −0.0263 | 5.0878 | −0.0094 |
| 8 2S | −7.2850298 | 5.1540 | 5.1563 | −0.0023 | 5.2109 | −0.0569 | 5.1611 | −0.0071 |
| 9 2S | −7.2829112 | 5.2116 | 5.2079 | 0.0037 | ||||
| 10 2S | −7.2810981 | 5.2610 | 5.2442 | 0.0167 | ||||
| 11 2S | −7.2797011 | 5.2990 | 5.2708 | 0.0282 | ||||
| 12 2S | −7.2786917 | 5.3264 | 5.2907 | 0.0357 | ||||
| 13 2S | −7.2779819 | 5.3457 | 5.3062 | 0.0396 | ||||
| 14 2S | −7.2774894 | 5.3592 | ||||||
| 15 2S | −7.2771501 | 5.3684 | ||||||
| 16 2S | −7.2769175 | 5.3747 | ||||||
| 17 2S | −7.2767584 | 5.3790 | ||||||
| 18 2S | −7.2766498 | 5.3820 | ||||||
| 19 2S | −7.2765758 | 5.3840 | ||||||
| 20 2S | −7.2765254 | 5.3854 | ||||||
| 21 2S | −7.2764911 | 5.3863 | ||||||
| 22 2S | −7.2764676 | 5.3870 | ||||||
| 23 2S | −7.2764516 | 5.3874 | ||||||
| 24 2S | −7.2764405 | 5.3877 | ||||||
Table 5 shows the 2S states computed with the [25S-6P-5D] basis. We can see that the ground state energy of the present study is still more than 3 millihartree above the highly precise computations. This is well understandable due to the Gaussian character of the basis, lack of any explicit electronic correlation, only the CCSD level of theory describing the three-electron system and also the relatively small basis size. Nevertheless, when we compare the excitation energies, we can see that the present Gaussian basis results are consistently only a few meV off the ECG values up to the state 8 2S. From the state 9 2S, the differences increase (even change the sign) and one might speculate that the error could reach up to tens of meV for the highest computed state 24 2S. Such a sudden drop in accuracy may be put down to the higher D orbital space insufficiency, as discussed with the Table 4 and Fig. 1, since the D functions contribute to the 2S states correlation energy via double excitations. On the other hand, there is an apparent convergence of the excitation energies to the CCSD ionization limit, which is just 4 meV below the experimental value. By taking this into account, the error estimate for the highest achieved excitation energies with respect to the accurate non-relativistic values may still fall in the interval of only a few meV. The results are also consistent within a few meV with the Hy-CI and with the STO Full-CI excitation energies except that the latter deviates from the precise ECG values for its highest 7 2S and 8 2S states by an order of magnitude more than the Gaussian basis results of the present study.
In Table 6, we can see that the 2P results in the basis [7S-24P-5D] are remarkably close to the ECG values within meV accuracy, while the STO Full-CI excitation energies differ from the present calculations by an order of magnitude more. It can also be noticed that the present Gaussian excitation energies are all closer to the ECG results and with a very consistent difference compared to the Hy-CI values. From this trend and from the apparent convergence towards the IP limit like in the 2S states, we might speculate that the accuracy of the computed excitation energy of the highest achieved 25 2P state could also be within a few meV.
| State | [7S-24P-5D] | ECG22 | STO Full-CI29 | Hy-CI29 | ||||
|---|---|---|---|---|---|---|---|---|
| E | EE | EE | ΔEE | EE | ΔEE | EE | ΔEE | |
| 2 2S | −7.4744358 | (−7.4780603)*,24,26 | (−7.477192) | (−7.478058969) | ||||
| 2 2P | −7.4065515 | 1.8472 | 1.8478*,27 | −0.0005 | 1.8660 | −0.0187 | 1.8479 | −0.0007 |
| 3 2P | −7.3336219 | 3.8317 | 3.8343 | −0.0026 | 3.8513 | −0.0196 | 3.8353 | −0.0036 |
| 4 2P | −7.3083789 | 4.5186 | 4.5217 | −0.0031 | 4.5391 | −0.0205 | 4.5238 | −0.0052 |
| 5 2P | −7.2967853 | 4.8341 | 4.8374 | −0.0033 | 4.8542 | −0.0201 | 4.8415 | −0.0074 |
| 6 2P | −7.2905206 | 5.0046 | 5.0080 | −0.0034 | 5.0245 | −0.0199 | 5.0094 | −0.0048 |
| 7 2P | −7.2867570 | 5.1070 | 5.1104 | −0.0034 | 5.1278 | −0.0208 | 5.1224 | −0.0154 |
| 8 2P | −7.2843201 | 5.1733 | 5.1767 | −0.0034 | ||||
| 9 2P | −7.2826463 | 5.2188 | 5.2221 | −0.0033 | ||||
| 10 2P | −7.2814198 | 5.2522 | 5.2545 | −0.0023 | ||||
| 11 2P | −7.2804485 | 5.2787 | ||||||
| 12 2P | −7.2796394 | 5.3007 | ||||||
| 13 2P | −7.2789788 | 5.3186 | ||||||
| 14 2P | −7.2785302 | 5.3309 | ||||||
| 15 2P | −7.2782993 | 5.3371 | ||||||
| 16 2P | −7.2779292 | 5.3472 | ||||||
| 17 2P | −7.2775941 | 5.3563 | ||||||
| 18 2P | −7.2773332 | 5.3634 | ||||||
| 19 2P | −7.2771260 | 5.3691 | ||||||
| 20 2P | −7.2769625 | 5.3735 | ||||||
| 21 2P | −7.2768372 | 5.3769 | ||||||
| 22 2P | −7.2767405 | 5.3796 | ||||||
| 23 2P | −7.2766658 | 5.3816 | ||||||
| 24 2P | −7.2766082 | 5.3832 | ||||||
| 25 2P | −7.2765638 | 5.3844 | ||||||
Similarly in Table 7, the 2D states obtained from the [7S-6P-14D] basis are closest to the ECG results with consistent differences in meV, while the differences from the Hy-CI values are slightly less regular. The STO Full-CI excitation energies, again, differ from the present results by an order of magnitude more than the ECG values. The excitation to the state 7 2D (the highest 2D state achieved by all three comparative studies) exhibits sudden deviation from the three reference results. This, again, reflects the insufficiency of the developed basis for a proper correlated description of the higher 2D states as discussed in Table 4 and Fig. 1.
| State | [7S-6P-14D] | ECG23 | STO Full-CI29 | Hy-CI29 | ||||
|---|---|---|---|---|---|---|---|---|
| E | EE | EE | ΔEE | EE | ΔEE | EE | ΔEE | |
| 2 2S | −7.4744350 | (−7.4780603)*,24,26 | (−7.477192) | (−7.478058969) | ||||
| 3 2D | −7.3320162 | 3.8754 | 3.8786*,27 | −0.0032 | 3.8937 | −0.0183 | 3.8789 | −0.0035 |
| 4 2D | −7.3076902 | 4.5373 | 4.5408 | −0.0034 | 4.5560 | −0.0187 | 4.5402 | −0.0028 |
| 5 2D | −7.2964306 | 4.8437 | 4.8472 | −0.0035 | 4.8624 | −0.0187 | 4.8482 | −0.0045 |
| 6 2D | −7.2903160 | 5.0101 | 5.0137 | −0.0036 | 5.0288 | −0.0187 | 5.0167 | −0.0066 |
| 7 2D | −7.2866912 | 5.1088 | 5.1140 | −0.0053 | 5.1291 | −0.0204 | 5.1226 | −0.0138 |
| 8 2D | −7.2846058 | 5.1655 | ||||||
| 9 2D | −7.2842566 | 5.1750 | ||||||
| 10 2D | −7.2827529 | 5.2159 | ||||||
| 11 2D | −7.2810660 | 5.2618 | ||||||
| 12 2D | −7.2806438 | 5.2733 | ||||||
| 13 2D | −7.2796484 | 5.3004 | ||||||
| 14 2D | −7.2776090 | 5.3559 | ||||||
As for the 2F states (Table 8), computed in the [7S-6P-5D-13F] basis, there are not many studies to compare with. The estimates of the exact non-relativistic values are taken from a relatively old review article by King.28 The differences between the estimates and the present results are within a few meV, which is still on a par with the precise ECG studies of Adamowicz et al.21–23 for the 2S, 2P and 2D states. The STO Full-CI results also differ by only a few meV, although with the opposite sign. The reason why the 2F excitation energies are this close to the STO Full-CI results in contrast to the 2S, 2P or 2D states may be due to a true Rydberg character of the 2F states.
| State | [7S-6P-5D-13F] | Exact non-rel. est.28 | STO Full-CI29 | |||
|---|---|---|---|---|---|---|
| E | EE | EE | ΔEE | EE | ΔEE | |
| 2 2S | −7.4744350 | (−7.4780603)*,24 | (−7.477192) | |||
| 4 2F | −7.3076701 | 4.5379 | 4.5414*,28 | −0.0035 | 4.5329 | 0.0050 |
| 5 2F | −7.2964194 | 4.8440 | 4.8475*,28 | −0.0035 | 4.8396 | 0.0045 |
| 6 2F | −7.2903080 | 5.0103 | 5.0064 | 0.0040 | ||
| 7 2F | −7.2866230 | 5.1106 | 5.1100 | 0.0006 | ||
| 8 2F | −7.2842313 | 5.1757 | ||||
| 9 2F | −7.2825915 | 5.2203 | ||||
| 10 2F | −7.2814185 | 5.2522 | ||||
| 11 2F | −7.2805499 | 5.2759 | ||||
| 12 2F | −7.2798861 | 5.2939 | ||||
| 13 2F | −7.2793602 | 5.3082 | ||||
| 14 2F | −7.2789311 | 5.3199 | ||||
| 15 2F | −7.2785961 | 5.3290 | ||||
| 16 2F | −7.2783444 | 5.3359 | ||||
The only reference data for the 2G states in Table 9 that we can compare to are those from the STO Full-CI computations. Similarly like for the 2F states, the results differ in the order of meV, although there are only three excited states available for comparison.
| State | [7S-6P-5D-12G] | STO Full-CI29 | ||
|---|---|---|---|---|
| E | EE | EE | ΔEE | |
| 2 2S | −7.4744350 | (−7.477192) | ||
| 5 2G | −7.2964303 | 4.8437 | 4.8371 | 0.0066 |
| 6 2G | −7.2903074 | 5.0104 | 5.0041 | 0.0062 |
| 7 2G | −7.2866226 | 5.1106 | 5.1045 | 0.0061 |
| 8 2G | −7.2842310 | 5.1757 | ||
| 9 2G | −7.2825914 | 5.2203 | ||
| 10 2G | −7.2814184 | 5.2522 | ||
| 11 2G | −7.2805500 | 5.2759 | ||
| 12 2G | −7.2798892 | 5.2938 | ||
| 13 2G | −7.2793766 | 5.3078 | ||
| 14 2G | −7.2789649 | 5.3190 | ||
| 15 2G | −7.2785953 | 5.3291 | ||
| 16 2G | −7.2782391 | 5.3388 | ||
4 Conclusions
For an ab initio quantum chemical description of Li Rydberg states, an appropriate diffuse Gaussian basis was developed by a variational optimization of virtual orbitals of the corresponding Li+ ion core. The approach is based on Koopmans’ theorem, which says that a virtual orbital of a closed shell system describes a captured electron, which actually means a Rydberg state. The resulting basis consists of the standard aug-ano-pVQZ set for sufficient description of the electronic correlation, an extra high-exponent contracted function to achieve a high-quality Hartree–Fock field and a set of optimal diffuse Rydberg functions.At the EOM-CCSD level of theory, a minimal subset of the optimized basis could be found, for which the ionization potential and the excitation energies were converged with respect to the number of the Rydberg functions used. Higher Rydberg states could be effectively achieved by a state-selective inclusion of the corresponding Rydberg function, dramatically reducing the demands on the basis set size.
Dozens of states at high accuracy could be achieved by the present approach, namely, up to the states 24 2S, 25 2P, 14 2D, 16 2F and 16 2G, that is many more than by competitive ab initio methods. Compared to the state-of-the-art ECG approach, the computed excitation energies consistently differed mostly in the order of meV. Only the 2D states above 6 2D could not achieve convergence, which might be due to their supposedly weak Rydberg character. The results were also comparable to Hylleraas-CI energies; however, the differences were less regular than with the ECG values. Nevertheless, the presented excitation energies even surpassed accuracy of Full-CI results computed in an optimal STO basis by an order of magnitude.
Regardless of the excellent quality of the presented basis, plenty of room for improvement remains. The number of primitive functions is still too large for practical use in standard quantum chemistry codes, which typically impose various restrictions on the basis set size. The optimization process could also be more sophisticated, involve directly more virtual orbitals and the basis could use more flexible parameterization than ETG, e.g., ExTG.3 On the other hand, not all chemical systems would require this high accuracy for the excited states, so the optimization criteria might appropriately loosen.
Although the study was performed on the lithium atom, the approach is universal for any ion core + electron system including such with an open-shell ion core, as long as the appropriate canonical RHF or ROHF5–8 orbitals are used. This method could also be applied to molecules, where, however, still more research needs to be done on several fronts to assess its suitability or the need for more development. For example, the question of multiple Rydberg centers in a single molecule, the question of strong coupling of low-lying states with the ion core or the question of states with otherwise complicated character. Also, performing Rydberg basis optimization specifically for a given molecule should lead to smaller yet superior molecular basis sets than using the basis optimized just for an isolated atom. All these questions, however, would require more dedicated studies. Similarly, the method is not limited to Gaussian basis sets but can be applied to any basis set types. Once the presented tailored basis set generation process becomes sufficiently tuned so that it is feasible for common quantum chemistry codes, it could promise an affordable highly accurate approach to ab initio state-selective investigation of molecular Rydberg states and possibly also of their related resonances.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The work was financially supported by the Grant Agency of the Czech Republic (Grant 20-21179S). Computational resources were supplied by the project “e-Infrastruktura CZ” (e-INFRA CZ LM2018140) supported by the Ministry of Education, Youth and Sports of the Czech Republic.References
- M. J. Seaton, Quantum defect theory, Rep. Prog. Phys., 1983, 46(2), 167–257, DOI:10.1088/0034-4885/46/2/002.
- H. Reisler and A. I. Krylov, Interacting Rydberg and valence states in radicals and molecules: experimental and theoretical studies, Int. Rev. Phys. Chem., 2009, 28(2), 267–308 Search PubMed.
- P. R. Kaprálová-Žd’ánská and J. Šmydke, Gaussian basis sets for highly excited and resonance states of helium, J. Chem. Phys., 2013, 138, 024105 CrossRef PubMed.
- A. Szabo and N. S. Ostlund, Modern Quantum Chemistry, Dover Publications, New York, Mineola, NY, 1996 Search PubMed.
- B. N. Plakhutin, E. V. Gorelik and N. N. Breslavskaya, Koopmans’ theorem in the ROHF method: Canonical form for the Hartree-Fock Hamiltonian, J. Chem. Phys., 2006, 125(20), 204110 CrossRef CAS PubMed.
- E. R. Davidson and B. N. Plakhutin, Koopmans's theorem in the restricted open-shell Hartree–Fock method. II. The second canonical set for orbitals and orbital energies, J. Chem. Phys., 2010, 132(18), 184110 CrossRef.
- B. N. Plakhutin and E. R. Davidson, Canonical form of the Hartree-Fock orbitals in open-shell systems, J. Chem. Phys., 2014, 140(1), 014102 CrossRef PubMed.
- B. N. Plakhutin, Koopmans’ theorem in the Hartree-Fock method. General formulation, J. Chem. Phys., 2018, 148(9), 094101 CrossRef.
- P. R. Kaprálová-Žd’ánská, J. Šmydke and S. Civiš, Excitation of helium Rydberg states and doubly excited resonances in strong extreme ultraviolet fields: full-dimensional quantum dynamics using exponentially tempered Gaussian basis sets, J. Chem. Phys., 2013, 139, 104314 CrossRef PubMed.
- W. J. Hunt and W. A. Goddard III, Excited States of H2O Using Improved Virtual Orbitals, Chem. Phys. Lett., 1969, 3, 414 CrossRef CAS.
- G. Durand and F. Volatron, An improved basis set for H2O. Test on Rydberg excited states, J. Chem. Phys., 1984, 80(5), 1937–1942 CrossRef CAS.
- K. Kaufmann, W. Baumeister and M. Jungen, Universal Gaussian basis sets for an optimum representation of Rydberg and continuum wavefunctions, J. Phys. B: At., Mol. Opt. Phys., 1989, 22(14), 2223 CrossRef CAS.
- F. Neese and E. F. Valeev, Revisiting the Atomic Natural Orbital Approach for Basis Sets: Robust Systematic Basis Sets for Explicitly Correlated and Conventional Correlated ab initio Methods?, J. Chem. Theory Comput., 2011, 7(1), 33–43 CrossRef CAS PubMed.
- MRCC, a quantum chemical program suite written by M. Kallay, P. R. Nagy, D. Mester, L. Gyevi-Nagy, J. Csoka, P. B. Szabo, Z. Rolik, G. Samu, J. Csontos, B. Hegely, A. Ganyecz, I. Ladjanszki, L. Szegedy, B. Ladoczki, K. Petrov, M. Farkas, P. D. Mezei and R. A. Horvath. Available from: https://www.mrcc.hu.
- M. Kallay, P. R. Nagy, D. Mester, Z. Rolik, G. Samu and J. Csontos, et al., The MRCC program system: Accurate quantum chemistry from water to proteins, J. Chem. Phys., 2020, 152, 074107, DOI:10.1063/1.5142048.
- mdoptcli; Multidimensional Optimizer for Command Line. Available from: https://sourceforge.net/projects/mdoptcli.
- M. Galassi, J. Davies, J. Theiler, B. Cough, G. Jungman and P. Alken, et al., GNU Scientific Library Reference Manual, Network Theory Ltd, 3rd edn, 2009. Available from: http://www.gnu.org/software/gsl/ Search PubMed.
- G. M. J. Barca, C. Bertoni, L. Carrington, D. Datta, N. De Silva and J. E. Deustua, et al., Recent developments in the general atomic and molecular electronic structure system, J. Chem. Phys., 2020, 152(15), 154102, DOI:10.1063/5.0005188.
- C. J. Lorenzen and K. Niemax, Quantum Defects of the n2P1/2,3/2 Levels in 39K I and 85Rb I, Phys. Scr., 1983, 27(4), 300, DOI:10.1088/0031-8949/27/4/012.
- J. F. Stanton and R. J. Bartlett, The equation of motion coupled-cluster method. A systematic biorthogonal approach to molecular excitation energies, transition probabilities, and excited state properties, J. Chem. Phys., 1993, 98(9), 7029–7039 CrossRef CAS.
- A. Bralin, S. Bubin, M. Stanke and L. Adamowicz, The 2S Rydberg series of the lithium atom. Calculations with all-electron explicitly correlated Gaussian functions, Chem. Phys. Lett., 2019, 730, 497–505 CrossRef CAS . Available from: https://www.sciencedirect.com/science/article/pii/S0009261419305251.
- S. Bubin and L. Adamowicz, Explicitly correlated Gaussian calculations of the 2Po Rydberg spectrum of the lithium atom, J. Chem. Phys., 2012, 136(13), 134305, DOI:10.1063/1.3698584.
- K. L. Sharkey, S. Bubin and L. Adamowicz, Lower Rydberg 2D states of the lithium atom: finite-nuclear-mass calculations with explicitly correlated Gaussian functions, Phys. Rev. A: At., Mol., Opt. Phys., 2011, 83, 012506, DOI:10.1103/PhysRevA.83.012506.
- M. Puchalski and K. Pachucki, Relativistic, QED, and finite nuclear mass corrections for lowlying states of Li and Be+, Phys. Rev. A: At., Mol., Opt. Phys., 2008, 78, 052511, DOI:10.1103/PhysRevA.78.052511.
- J. S. Sims and S. A. Hagstrom, Hylleraas-configuration-interaction study of the 2 2S ground state of neutral lithium and the first five excited 2S states, Phys. Rev. A: At., Mol., Opt. Phys., 2009, 80, 052507, DOI:10.1103/PhysRevA.80.052507.
- S. Bubin, J. Komasa, M. Stanke and L. Adamowicz, Isotope shift in the electron affinity of lithium, J. Chem. Phys., 2009, 131(23), 234112, DOI:10.1063/1.3275804.
- L. M. Wang, Z. C. Yan, H. X. Qiao and G. W. F. Drake, Variational energies and the Fermi contact term for the low-lying states of lithium: basis-set completeness, Phys. Rev. A: At., Mol., Opt. Phys., 2012, 85, 052513, DOI:10.1103/PhysRevA.85.052513.
- F. W. King, B. Bederson and H. Walther, High-Precision Calculations for the Ground and Excited States of The Lithium Atom, in Advances In Atomic, Molecular, and Optical Physics, ed. B. Bederson and H. Walther, Academic Press, 1999, vol. 40, pp. 57–112. Available from: https://www.sciencedirect.com/science/article/pii/S1049250X08601111 Search PubMed.
- M. B. Ruiz, J. T. Margraf and A. M. Frolov, Hylleraas-configuration-interaction analysis of the lowlying states in the three-electron Li atom and Be+ ion, Phys. Rev. A: At., Mol., Opt. Phys., 2013, 88, 012505, DOI:10.1103/PhysRevA.88.012505.
Footnote |
| † Electronic supplementary information (ESI) available: Basis set parameters developed in this work. See DOI: https://doi.org/10.1039/d2cp04633d |
| This journal is © the Owner Societies 2023 |