
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMixed valence Sn doped (CH3NH3)3Bi2Br9 produced by mechanochemical synthesis†
Xiaohan
Jia
a,
Yuhan
Liu
 ab,
Prajna
Bhatt
a,
Robin S.
Perry
bc,
Ivan P.
Parkin
a and
Robert G.
Palgrave
*a
ab,
Prajna
Bhatt
a,
Robin S.
Perry
bc,
Ivan P.
Parkin
a and
Robert G.
Palgrave
*a
aDepartment of Chemistry, University College London, 20 Gordon Street, London, WC1H 0AJ, UK. E-mail: r.palgrave@ucl.ac.uk
bLondon Centre for Nanotechnology and Department of Physics and Astronomy, University College London, London, WC1E 6BT, UK
cISIS neutron spallation source, Rutherford Appleton Laboratory (RAL), Harwell Campus, Didcot, OX11 0QX, UK
First published on 20th January 2023
Abstract
Bismuth halides with formula A3Bi2X9, where A is an inorganic or organic cation, show desirable properties as solar absorbers and luminescent materials. Control of structural and electronic dimensionality of these compounds is important to yield materials with good light absorption and charge transport. Here we report mechanochemical reaction of (CH3NH3)3Bi2Br9 with SnBr2 at room temperature in air, yielding a material with strong absorption across the visible region. We attribute this to mixed valence doping of Sn(II) and Sn(IV) on the Bi site. X-Ray diffraction shows no secondary phases, even after heating at 200 °C to improve crystallinity. X-Ray photoelectron spectroscopy suggests the presence of Sn(II) and Sn(IV) states. A similar approach to dope Sn into the iodide analogue (CH3NH3)3Bi2I9 was unsuccessful.
Introduction
Halide perovskites with formula ABX3, where A and B are cations, and X is a halide, have recently been found as desirable solar absorbers.1–3 Compared with the most widely commercialised silicon solar cell, solution-processable halide perovskite solar cells have several advantages, including high optical absorption coefficient, defect tolerance, and tunable band gap.4 However, the most commonly used B-site metal, Pb, hinders commercialisation due to the high toxicity and the poor intrinsic air stability of many Pb(II) based perovskites.5 Thus, there is motivation to develop lead-free analogues. Bi(III) shares the same electronic configuration ([Xe] 4f14 5d10 6s2) with Pb(II), and shows excellent potential as a Pb alternative: it has low toxicity, and complex bismuth halides are characterised by good air stability.6–10 Bi(III) cannot fit into the ABX3 perovskite structure, but instead forms a series of structures with distorted BiX6 corner-or-edge-shared octahedra, leading to halide perovskite derivatives, A3Bi2X9 which adopt either 0D, 1D or 2D inorganic sublattices.11–14 The 2D layered structure is of special interest due to its vibronic properties and electronic strucutre,15,16 and remarkable luminescent efficiencies.14 The 2D structure is formed when X is bromide ion, while when X is iodide, the material adopts a 0D structure, with isolated [Bi2I9]3− clusters. While the use of iodide anions results in greater light absorption due to a smaller band gap, the 0D iodide structure is detrimental to carrier mobility. Likewise, the 2D A3Bi2Br9 may show higher mobility, but suffers from relatively large bandgap energies, which limit power conversion efficiency (PCE) of photovoltaic (PV) applications.17,18 Here we seek to find new methods to induce visible light absorption in 2D A3Bi2Br9 compounds, which may enhance PV properties, and also give greater control over their luminescent properties,19 or make them suitable for other optoelectronic applications.Intervalence charge transfer (IVCT) is defined by the electron transfer between two atoms of the same element that in the ground state have different oxidation states.20 IVCT is known for Sb(III)/Sb(V) as well as Sn(II)/Sn(IV) in halide materials.21 Although Sn(II) is a promising B-site candidate for lead-free perovskite solar absorber, its PV application is limited by the potential degradation of devices caused by the rapid oxidation of Sn(II) to Sn(IV).22 Where both Sn(II) and Sn(IV) are present in the same compound, light absorption may induce electron transfer between the two sites of different oxidation state. The activation energy of the transition between the symmetrical ground states typically lies in the visible and near infra-red regions of the electromagnetic spectrum, and so can cause a broad and intense optical absorption peak.23
We have recently shown that Cs3Bi2Br9 can be doped with mixed valence Sn through a solution phase synthesis.24 Here we show that a related hybrid material, MA3Bi2Br9 (MA = (CH3NH3)+) can similarly be doped with mixed valence Sn(II)/Sn(IV) by a very rapid room temperature, solvent free mechanochemical reaction carried out in air. Mechanochemical reactions have been used as a synthesis strategy for a long time in many areas such as to prepare polymeric materials,25 metal organic frameworks (MOFs)26,27 and halide perovskites.28 They have proven very successful for preparing conventional and vacancy ordered halide perovskites,29 including MA3Bi2Br9.30 Solvent-free mechanochemical reactions conducted by solid-state grinding, can be a sustainable synthesis approach.31,32 Here we report an ambient condition mechanochemical synthesis of a mixed valence compound, Sn:MA3Bi2Br9. We find that partial substitution of Bi(III) with equal amounts of Sn(II) and Sn(IV) dramatically increases the optical absorption in the visible region, which corresponds to the spontaneous colour change from yellow to black during the reaction. This may be a viable strategy for overcoming the principal challenge of such 2D A3B2X9 compounds, their large bandgaps.
Experimental section
Bismuth(III) oxide (Bi2O3, Fine Chemicals), caesium carbonate (Cs2(CO)3, 99%, Sigma-Aldrich), hydrobromic acid (HBr, 47 wt% in H2O, Sigma-Aldrich), hydroiodic acid (HI, 57 wt% in H2O, Sigma-Aldrich), methylamine solution (CH5N, 40 wt% in H2O, Sigma-Aldrich), tin(II) bromide (SnBr2, Sigma-Aldrich), tin(IV) bromide (SnBr4, Sigma-Aldrich), tin(II) iodide (SnI2, 99.99%, Sigma-Aldrich), tin(II) oxide (SnO, 99%, Sigma-Aldrich) were used as received without purification.Synthesis of MA3Bi2X9 (MA = CH3NH3+, X = Br or I) microcrystals
Microcrystalline MA3Bi2X9 was synthesised by firstly reacting the stoichiometric ratio of Bi2O3 and aqueous methylamine separately with excess 3 M hydrohalic acid, then mixing the two solutions. For example, to prepare 5 g of MA3Bi2Br9, pale yellow bismuth oxide powder (1.893 g, 4.26 mmol) was stirred and slowly dissolved in excess dilute hydrobromic acid (∼3 M, 23 mL) at 80 °C for 10 min to prepare the bismuth bromide precursor solution. Methylamine solution 40 wt% in H2O (0.989 g, MA![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Bi = 3:2), was added dropwise into the bismuth bromide solution in air. The clear reaction mixture was stirred at 80 °C for another 10 min to ensure the reaction to be completed. The bright yellow microcrystalline solid was precipitated by evaporating the solvent until sufficient product has crystallised, at which point it was isolated by filtration and dried at 60 °C in air.
:Bi = 3:2), was added dropwise into the bismuth bromide solution in air. The clear reaction mixture was stirred at 80 °C for another 10 min to ensure the reaction to be completed. The bright yellow microcrystalline solid was precipitated by evaporating the solvent until sufficient product has crystallised, at which point it was isolated by filtration and dried at 60 °C in air.
Synthesis of B-site Sn doped MA3Bi2Br9 by mechanochemical reaction
B-site mixed-valent Sn(II)/Sn(IV) doped MA3Bi2Br9 microcrystals (MA3(SnxBi(1−x))2Br9) were prepared by mechanochemical reaction (solid-state grinding) at room temperature in air from stoichiometric amounts of SnBr2 and the as-made MA3Bi2X9 microcrystals.For example, 30% Sn-doped MA3Bi2Br9 (MA3(Sn0.3Bi0.70)2Br9) was prepared by grinding SnBr2 powder (3 mmol) together with the as-synthesised yellow crystalline MA3Bi2Br9 (3.5 mmol) in an agate mortar until two solids were homogeneously mixed. No solvent was added to the reaction. The colour of the reactants turned black immediately upon grinding. Samples were made with Sn contents of x = 0, 0.01, 0.25, 0.3, 0.35, 0.4, 0.5, 0.6. All samples were stored in glass sample vials in air.
To further improve homogeneity of dopant and introduce crystallinity, half of each as-synthesised MA3(SnxBi(1−x))2Br9 sample (including x = 0, MA3Bi2Br9) were heated at 200 °C in furnace in air for two hours.
Characterisation details are given in the ESI.†
Results
As-produced yellow MA3Bi2Br9 microcrystals showed a powder diffraction pattern matching that reported by Ishihara et al.,33 and corresponding to a trigonal crystal structure with the space group P![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m1. We carried out Rietveld refinement using GSAS II,34 starting with the model of Ishihara et al. It was necessary to add a small amount of preferred orientation in the (100) direction, using the March–Dollase algorithm,35 to match the peak intensities well. No unidentified peaks were seen; a final RWP value of 4.01% was obtained. Refined lattice parameters for the as prepared undoped MA3Bi2Br9 phase were a = 8.2136(28) Å, c = 10.015(27) Å, these are 0.3% and 0.9% larger respectively than those reported by Ishihara et al. Further crystallisation of as-synthesised MA3Bi2Br9 was induced by heating at 200 °C in air. Diffraction peaks became sharper after annealing, but no changes to the lattice parameters were observed within error (Fig. 1 and Table S2, ESI†).
m1. We carried out Rietveld refinement using GSAS II,34 starting with the model of Ishihara et al. It was necessary to add a small amount of preferred orientation in the (100) direction, using the March–Dollase algorithm,35 to match the peak intensities well. No unidentified peaks were seen; a final RWP value of 4.01% was obtained. Refined lattice parameters for the as prepared undoped MA3Bi2Br9 phase were a = 8.2136(28) Å, c = 10.015(27) Å, these are 0.3% and 0.9% larger respectively than those reported by Ishihara et al. Further crystallisation of as-synthesised MA3Bi2Br9 was induced by heating at 200 °C in air. Diffraction peaks became sharper after annealing, but no changes to the lattice parameters were observed within error (Fig. 1 and Table S2, ESI†).
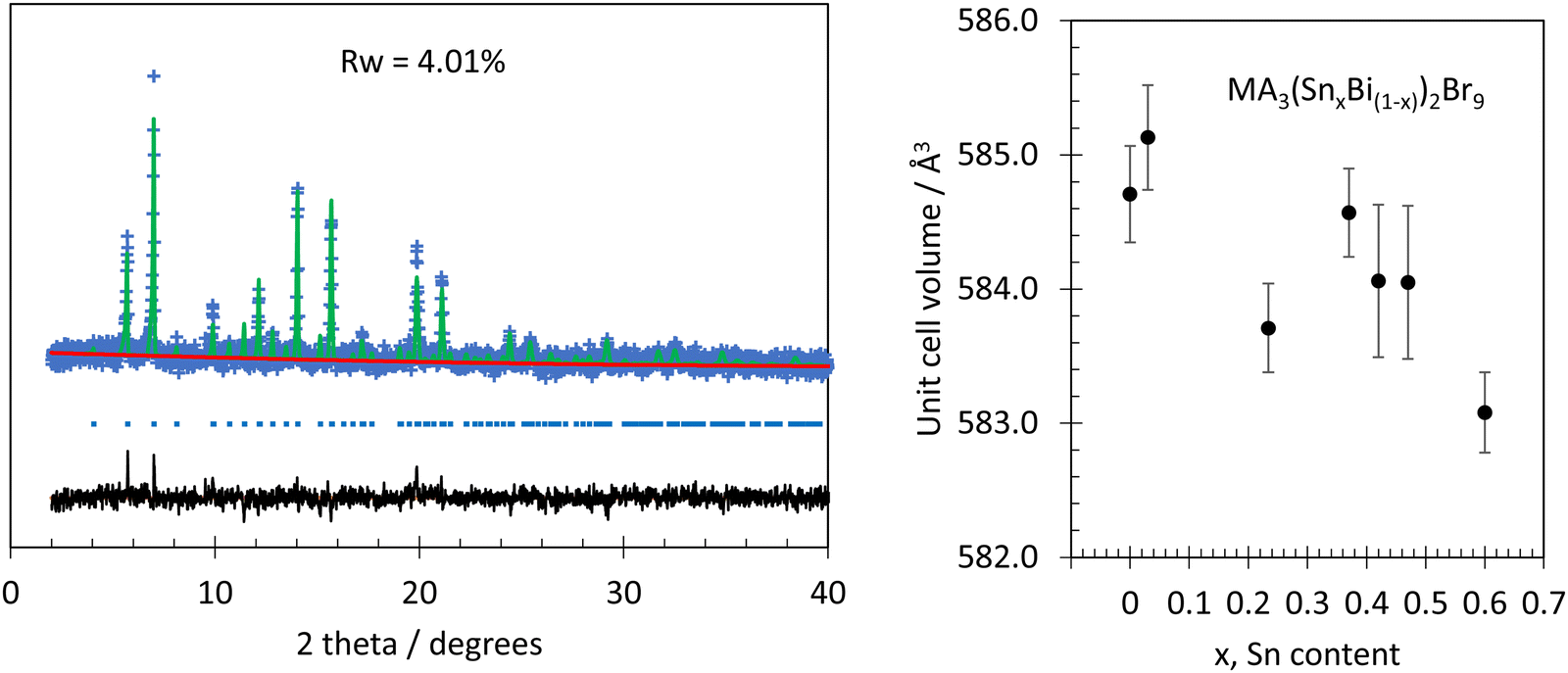 | ||
| Fig. 1 Left, powder XRD data (Mo Kα, blue points) from MA3Bi2Br9 annealed at 200 °C in air. The model (green line) was refined from the structure reported by Ishihara et al. The background is shown in red and the residual is in black. Blue tick marks show positions of reflections. Right: Variation in unit cell volume with analytical Sn content in MA3(SnxBi(1−x))2Br9. Error bars are plus and minus three times the standard deviation. | ||
During room temperature mechanochemical reaction between microcrystalline MA3Bi2Br9 and SnBr2, a colour change from bright yellow to black began to occur as soon as the two solids came into contact, and proceeded rapidly upon grinding to give a homogenous colour change after less than one minute (Fig. 2 and Fig. S1, ESI†). In contrast, no obvious colour change occurred when MA3Bi2Br9 was ground with SnBr4 instead of SnBr2 (Fig. S1, ESI†). The powder XRD patterns of mechanochemically doped MA3(SnxBi(1−x))2Br9 samples, after heating at 200 °C, were analysed by Rietveld refinement as described above. The models used did not include Sn dopants: only the lattice parameters, preferred orientation, and peak profile were refined, starting in each case from the model obtained from the undoped MA3Bi2Br9 sample. Final RWP values were below 6% for all samples, and no secondary phases were detected. The reactant, SnBr2, was confirmed to be crystalline by PXRD, and so the absence of SnBr2 peaks from the product after mechanosynthesis shows that it has been consumed by the reaction process. Only slight changes to the diffraction patterns were observed upon doping. There was a small decrease in lattice parameters with increasing Sn content, the cell volume falling from 584.70(12) Å3 in the undoped material, to 583.08(10) Å3 for the highest nominal doping level, x = 0.6. This trend is shown in Fig. 1. This decrease is mainly due to the contraction of the c parameter, i.e. a decrease in interlayer spacing, while the a parameter remains constant within error for all samples.
 | ||
| Fig. 2 (a) Diffuse reflectance spectrum from undoped MA3Bi2Br9 treated with the Kubelka Munk function (black points). The optical absorption is modelled using the Elliot model (green), consisting of an exciton peak (blue) and a continuum absorption (red) (b) Optical absorption spectra of MA3(SnxBi(1−x))2Br9, each curve is marked with the x value indicating the nominal Sn composition. (c) Photographs of doped and undoped samples. | ||
The UV-vis absorbance spectra of room temperature mechanochemically doped MA3(SnxBi(1−x))2Br9, taken from diffuse reflectance measurements using the Kubelka Munk function, is shown in Fig. 3. The undoped material, MA3Bi2Br9 shows an absorption spectrum characteristic of an exciton absorption close to the interband absorption edge. This is commonly observed for hybrid perovskites and related materials. Following Saba et al.,36 we model the spectrum close to the absorption edge using Elliot's theory.37 The continuum absorption is modelled using a Heaviside step function, and the exciton peak modelled using a Dirac delta function. The parameters of these two functions were adjusted to fit the observed spectrum (Fig. 3). The exciton peak is centred at 2.78 eV, while the band gap (taken as the mid point of the Heaviside function) is 2.91 eV, giving an exciton binding energy of 130 meV, a value typical of 2D layered perovskite-like materials.38
 | ||
| Fig. 3 Left, Sn 3d spectra from Sn doped MA3Bi2Br9 and Cs2SnBr6, a pure Sn(IV) compound. Right, Bi 4f spectra from Sn doped (top) and undoped (bottom) MA3Bi2Br9. Note BE values are referenced to Br 3d5/2 at 68.5 eV. | ||
Our measured exciton binding energy is significantly larger than the exciton binding energy seen in MAPbI3 (11 meV),39 although it is much smaller than that found in the all-inorganic analogues Cs3Bi2Br9 (940 meV)15 and Cs3Bi2I9 (300 meV)40
Our optical analysis is in contrast to some previous work, which did not use an exciton model, and instead modelled the absorption as a simple band-to-band transition, for example, Qi et al. who found a bandgap of 2.65 eV,41 and Leng et al. who found a bandgap of 2.5 eV for MA3Bi2Br9 nanoparticles.19
Broad absorption features are present in the visible regions in the doped MA3(SnxBi(1−x))2Br9 microcrystals, corresponding to the dramatic colour change seen in the samples (Fig. 2). The sample with x = 0.3 showed the greatest enhancement in absorption, above this level of doping the intensity of the additional absorption decreased, corresponding to the colour change from black to red with increasing nominal Sn doping. Any band gaps and exciton energies cannot be determined accurately in the doped samples, due to the strong absorption band that appears upon doping. We have previously shown that Sn doping of a related compound, Cs3Bi2Br9, leads to intervalence charge transfer (IVCT) absorption, due to the presence of mixed Sn oxidation states.24 To verify whether the strong optical absorption see here is due to IVCT, we carried out XPS analysis on the samples.
XPS was used to probe the composition and chemical environments of Sn in the doped system. The additional Sn peaks in XPS survey spectra for MA3(SnxBi(1−x))2Br9 compared with pure phase MA3Bi2Br9 shows that Sn was successfully detected in all doped samples. Survey spectra of the selected MA3(SnxBi(1−x))2Br9 before and after heating are presented in Fig. S4 (ESI†). No additional unexpected element except O was detected. The oxygen can be associated with oxides or oxyhalides such as BiOBr, although none were detected from XRD, which may be due to limited crystallinity of these products, or their presence only on the surface in amounts too small to detect with the XRD methodology used here.
The analytical ratio of Sn to Bi for each sample was calculated from the average of three XPS measurements by applying the equation 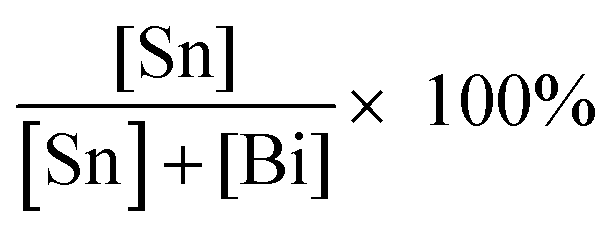 (Table S1, ESI†). For room temperature synthesised compounds, XPS values were within ±10 atomic% of nominal amounts, and the absolute errors between repeat measurements were ±5 atomic%. The surface sensitive nature of XPS may be the cause of the discrepancy with nominal values if the surface has a different composition to the bulk. For the heated samples, except for x = 0.30, all samples have the percentage error within ±2% to the theoretical ratios. This suggests that by annealing, the homogeneity of doped materials can be increased.
(Table S1, ESI†). For room temperature synthesised compounds, XPS values were within ±10 atomic% of nominal amounts, and the absolute errors between repeat measurements were ±5 atomic%. The surface sensitive nature of XPS may be the cause of the discrepancy with nominal values if the surface has a different composition to the bulk. For the heated samples, except for x = 0.30, all samples have the percentage error within ±2% to the theoretical ratios. This suggests that by annealing, the homogeneity of doped materials can be increased.
The Sn 3d spectra from MA3(SnxBi(1−x))2Br9 are shown in Fig. 3. In common with other post transition metals, determination of Sn oxidation state by XPS can be challenging. An additional complication faced here is that binding energies are often referenced to adventitious C 1s peaks. This approach has faced recent criticism,42,43 and is especially difficult to implement in this case as our compounds contain carbon in the MA+ cation. Therefore to understand the X-ray photoemission spectra were taken from MA3(SnxBi(1−x))2Br9, and Cs2SnBr6, the latter a compound nominally containing only Sn(IV) which was synthesised as described previously.44 To avoid the need to calibrate the binding energy scale using C 1s, we calibrated to the Br 3d5/2 peak, which is present in both Cs2SnBr6 and MA3(SnxBi(1−x))2Br9. With the assumption that the bromide anion does not significantly change chemical environment in these compounds the Br 3d5/2 binding energy was set to 68.5 eV and the energy scale calibrated from there. The Sn 3d5/2 peak was found to be well modelled by a single Gaussian–Lorentzian peak over a Shirley background for Cs2SnBr6. The FWHM of the Sn 3d5/2 peak in Cs2SnBr6 was 1.38 eV. In Sn:MA3Bi2Br9, the Sn 3d5/2 spectrum consists of two maxima, and can be modelled well with two Gaussian Lorentzian components. The higher binding energy Sn 3d component in Sn:MA3Bi2Br9 has the same energy relative to Br 3d as found in Cs2SnBr6; we assign this as a Sn(IV) component. The lower binding energy component is assigned as a Sn(II) environment. The difference in energy between the two components is 1.4 eV, quite a large gap but the lower binding energy component remains too high in binding energy, and too broad, to be attributable to metallic Sn(0).45 Attempts to further confirm this assignment, by measuring a Sn(II) standard such as SnBr2 were hampered by oxidation on the surfaces of samples of SnBr2 available to us. The Sn(IV):Sn(II) ratio from this analysis was 0.86:1, showing there is a slight excess of Sn(II) compared to the expected case of equal quantities of Sn(IV) and Sn(II). We also examine the Auger parameter for Sn (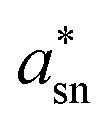 ) following the procedure of Felix et al.,46 which is described in detail in the SI. Felix et al. place Sn(IV) a* values between 926–928 eV, while Sn(II) values are between 927–929 eV, i.e. Sn(II) have higher
) following the procedure of Felix et al.,46 which is described in detail in the SI. Felix et al. place Sn(IV) a* values between 926–928 eV, while Sn(II) values are between 927–929 eV, i.e. Sn(II) have higher  values. We find a value of
values. We find a value of  for Cs2SnBr6 (pure Sn(IV)) to be 926.6 eV. Our MA3(Sn0.3Bi0.7)2Br9 sample showed
for Cs2SnBr6 (pure Sn(IV)) to be 926.6 eV. Our MA3(Sn0.3Bi0.7)2Br9 sample showed  = 927.7 eV, using the mid point of the Sn3d3/2 envelope, higher than the standard for Sn(IV), which would be expected for partial reduction of the Sn. Together, the core line and Auger data suggests Sn is present in a mixed valence state. The Bi 4f peaks are shown in Fig. 3, for doped (x = 0.3) and undoped samples. Again, the binding energy is referenced to the Br 3d peak. No change in Bi 4f peak position is observed after doping, and the Bi 4f peaks remain symmetrical in both cases, indicating that the Bi does not undergo significant chemical change during the doping.
= 927.7 eV, using the mid point of the Sn3d3/2 envelope, higher than the standard for Sn(IV), which would be expected for partial reduction of the Sn. Together, the core line and Auger data suggests Sn is present in a mixed valence state. The Bi 4f peaks are shown in Fig. 3, for doped (x = 0.3) and undoped samples. Again, the binding energy is referenced to the Br 3d peak. No change in Bi 4f peak position is observed after doping, and the Bi 4f peaks remain symmetrical in both cases, indicating that the Bi does not undergo significant chemical change during the doping.
Lastly we tested the hypothesis that the visible light absorption in the doped sample is due to formation of MASnBr3 as a secondary phase. It is known that MASnBr3 can be formed mechanochemically from SnBr2 and MABr in inert conditions.28 We attempted this reaction in air using the same procedure as used for synthesis of our doped MA3Bi2Br9. The result was a red powder, with XRD peaks matching cubic MASnBr3, and some small unidentified peaks that may be due to decomposition in air (Fig. S9 ESI†). The optical properties of this product are compared with the doped MA3Bi2Br9 samples in Fig. S9, ESI.† It is clear that the optical spectrum of MASnBr3 is quite different from our doped materials, having a band edge at 1.96 eV, determined by Tauc method, comparable with earlier experimental and computational reports for this material, which give band gaps close to 2.0 eV.28,47 In contrast the Sn:MA3Bi2Br9 samples have strong optical absorption down to around 1.4 eV. Given the lack of diffraction peaks from MASnBr3 in our Sn:MA3Bi2Br9 samples, and the difference in optical spectra, we can rule out the changes in optical spectra of the doped samples as being due to MASnBr3 contamination.
The cause of the strong visible light absorption in the doped samples can now be discussed. It is known that mixed valence post transition metals can give rise to strong optical absorption, known as intervalence charge transfer (IVCT), for example in the classic mixed valence compound Cs2SbCl6, which contains both Sb(III) and Sb(V).48 IVCT manifests as a strong, broad absorption peak, the breadth, frequency and intensity all depend on the concentration of the mixed valence species.49 These are the characteristics of the new absorption feature we see in our doped samples. Together with the evidence for mixed valence Sn present in these samples from XPS, and the evidence of some, albeit slight, strucutral change upon doping from XRD (indicating that the Sn dopant has entered the lattice), and in the absence of detectable secondary phases that might explain the dramatic colour change in these samples, we attribute the emerging of the broad optical absorption peaks seen in Fig. 2 to the presence of IVCT resulting from the introduction of mixed-valence Sn(II) and Sn(IV), together replacing Bi(III).
The annealed MA3(SnxBi(1−x))2Br9 microcrystals, possess the same general trend in terms of sample colour and absorption enhancement in the visible region as shown in Fig. S3 (ESI†). From the spectra, it is clear that the interband transition is much more intense than the IVCT absorption in the annealed samples, likely due to increased crystallinity. We notice that the macroscopic visual appearance of annealed MA3(SnxBi(1−x))2Br9 samples remained as deep coloured, especially for x = 0.3 and 0.35. Hence, we interpret that the increase in crystallinity leads to an increase in the inter band transition, rather than a reduction in intensity of the IVCT. The stability of the samples was assessed after one year of storage in air under normal laboratory conditions. The colour of the samples appeared unchanged after this time, suggesting the samples are stable towards oxidation (or at least oxidation is limited to the surface region). However, attempts to dissolve the material, for example to spin coat into a thin film, caused rapid colour change from black to yellow, representing oxidation of Sn(II) and loss of IVCT.
The same investigations of mixed-valent Sn doping were carried out on the iodide analogue: MA3Bi2I9 to determine if mechanochemical synthesis is able to produce mixed valence doping in iodides. The Sn-doped MA3(SnxBi(1−x))2I9 samples were prepared by room temperature mechanochemical reaction in air where x = 0, 0.2, 0.3, 0.4, 0.5. Half of each sample was heated at 200 °C in the furnace in air for two hours to increase the crystallinity of as-made doped materials. No obvious colour change for both room temperature prepared and 200 °C heated MA3(SnxBi(1−x))2I9 (Fig. S5, ESI†). By increasing the doping level of Sn, no change in lattice parameter were observed by powder XRD, and the peak width slightly increased from low to high Sn-content (Fig. S6, ESI†). Impurity peaks from the starting material SnI2 were clear in powder XRD patterns in high Sn-containing samples. All results show the intended Sn doping was unsuccessful for the iodide analogue. Consistent with the sample colour, the UV-Vis absorption spectra for both room temperature and heated MA3(SnxBi(1−x))2I9 showed no significant change compared to undoped MA3Bi2I9 as shown in Fig. S7 (ESI†).
One of the reasons for the failure of mixed-valent Sn doping into MA3Bi2I9 may come from the structural difference between MA3Bi2Br9. Compared to 2D layered material MA3Bi2Br9, which has a trigonal crystal structure with the space group Pm1 crystal structure, the 0D MA3Bi2I9 lattice possesses monoclinic crystal structure with the space group C12/c1. Alternatively, there may be important chemical differences between I and Br which prevent Sn doping into its iodide. Further investigation on theoretical calculations using density functional theory (DFT) can be done to reveal reasons in electronic aspects.
Conclusion
We report the mixed valence doping the 2D hybrid layered perovskite, MA3Bi2Br9 using a room temperature mechanochemical synthesis. A spontaneous colour change from bright yellow to black was observed during the reaction. The strongest optical absorption of Sn-doped MA3(SnxBi(1−x))2Br9 is obtained when x = 0.3 We attribute this enhancement to mixed-valence doping of Sn(II) and Sn(IV) oxidation state on the Bi site, which introduces the intervalence charge transfer (IVCT) into MA3Bi2Br9. The IVCT process provides an additional energy transition in the visible region, resulting in a significant enhancement in light absorbance. Powder X-ray diffraction (PXRD) shows no secondary phases for MA3(SnxBi(1−x))2Br9 including x = 0, even after heating at 200 °C to induce crystallisation and homogeneity. X-Ray photoelectron spectroscopy (XPS) suggests the presence of Sn(II) and Sn(IV) oxidation states, while Bi remains Bi(III). MA3(SnxBi(1−x))2Br9 is air-stable for extended periods. In contrast, attempts to dope Sn into MA3Bi2I9 appear not to give a mixed valence product.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank Chantalle Krajewska for helpful discussions. Y. L. thanks the UCL-CSC scholarship scheme for PhD funding. We acknowledge the EPSRC National Facility for Photoelectron Spectroscopy (HarwellXPS), operated by Cardiff University and UCL under contract number PR16195.References
- A. Kojima, K. Teshima, Y. Shirai and T. Miyasaka, J. Am. Chem. Soc., 2009, 131, 6050–6051 CrossRef CAS PubMed.
- M. M. Lee, J. Teuscher, T. Miyasaka, T. N. Murakami and H. J. Snaith, Science, 2012, 338, 643–647 CrossRef CAS PubMed.
- H. J. Snaith, J. Phys. Chem. Lett., 2013, 4, 3623–3630 CrossRef CAS.
- H. S. Jung and N. G. Park, Small, 2015, 11, 10–25 CrossRef CAS.
- G. P. Nagabhushana, R. Shivaramaiah and A. Navrotsky, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 7717–7721 CrossRef CAS PubMed.
- W. J. Serfontein and R. Mekel, Res. Commun. Chem. Pathol. Pharmacol., 1979, 26, 391–411 CAS.
- A. M. Ganose, C. N. Savory and D. O. Scanlon, Chem. Commun., 2017, 53, 20–44 RSC.
- H. Tsai, W. Nie, J.-C. Blancon, C. C. Stoumpos, R. Asadpour, B. Harutyunyan, A. J. Neukirch, R. Verduzco, J. J. Crochet, S. Tretiak, L. Pedesseau, J. Even, M. A. Alam, G. Gupta, J. Lou, P. M. Ajayan, M. J. Bedzyk, M. G. Kanatzidis and A. D. Mohite, Nature, 2016, 536, 312–316 CrossRef CAS PubMed.
- J.-W. Lee, Z. Dai, T.-H. Han, C. Choi, S.-Y. Chang, S.-J. Lee, N. De Marco, H. Zhao, P. Sun, Y. Huang and Y. Yang, Nat. Commun., 2018, 9, 3021 CrossRef PubMed.
- I. C. Smith, E. T. Hoke, D. Solis-Ibarra, M. D. McGehee and H. I. Karunadasa, Angew. Chem., Int. Ed., 2014, 53, 11232–11235 CrossRef CAS PubMed.
- D. B. Mitzi, J. Chem. Soc., Dalton Trans., 2001, 1–12, 10.1039/B007070J.
- J. S. Manser, J. A. Christians and P. V. Kamat, Chem. Rev., 2016, 116, 12956–13008 CrossRef CAS PubMed.
- R. E. Brandt, V. Stevanović, D. S. Ginley and T. Buonassisi, MRS Commun., 2015, 5, 265–275 CrossRef CAS.
- Y. Shen, J. Yin, B. Cai, Z. Wang, Y. Dong, X. Xu and H. Zeng, Nanoscale Horiz., 2020, 5, 580–585 RSC.
- K. K. Bass, L. Estergreen, C. N. Savory, J. Buckeridge, D. O. Scanlon, P. I. Djurovich, S. E. Bradforth, M. E. Thompson and B. C. Melot, Inorg. Chem., 2017, 56, 42–45 CrossRef CAS PubMed.
- T. L. Hodgkins, C. N. Savory, K. K. Bass, B. L. Seckman, D. O. Scanlon, P. I. Djurovich, M. E. Thompson and B. C. Melot, Chem. Commun., 2019, 55, 3164–3167 RSC.
- D. Phuyal, S. M. Jain, B. Philippe, M. B. Johansson, M. Pazoki, J. Kullgren, K. O. Kvashnina, M. Klintenberg, E. M. J. Johansson, S. M. Butorin, O. Karis and H. Rensmo, J. Mater. Chem. A, 2018, 6, 9498–9505 RSC.
- M. Roy, S. Ghorui, Bhawna, J. Kangsabanik, R. Yadav, A. Alam and M. Aslam, J. Phys. Chem. C, 2020, 124, 19484–19491 CrossRef CAS.
- M. Leng, Z. Chen, Y. Yang, Z. Li, K. Zeng, K. Li, G. Niu, Y. He, Q. Zhou and J. Tang, Angew. Chem., Int. Ed., 2016, 55, 15012–15016 CrossRef CAS PubMed.
- J. W. Verhoeven, Pure Appl. Chem., 1996, 68, 2223–2286 CAS.
- K. Prassides, P. Day and A. K. Cheetham, J. Am. Chem. Soc., 1983, 105, 3366–3368 CrossRef CAS.
- I. I. Alkhatib, C. Garlisi, M. Pagliaro, K. Al-Ali and G. Palmisano, Catal. Today, 2020, 340, 209–224 CrossRef CAS.
- D. Chong, X. Wan and J. Zhang, J. Mater. Chem. C, 2017, 5, 6442–6449 RSC.
- C. J. Krajewska, R. Palgrave, S. R. Kavanagh, L. Zhang, D. J. Kubicki, D. O. Scanlon, K. Dey, K. Galkowski, C. P. Grey, S. D. Stranks and A. Walsh, Chem. Sci., 2021, 12, 14686–14699 RSC.
- D. A. Davis, A. Hamilton, J. L. Yang, L. D. Cremar, D. Van Gough, S. L. Potisek, M. T. Ong, P. V. Braun, T. J. Martinez, S. R. White, J. S. Moore and N. R. Sottos, Nature, 2009, 459, 68–72 CrossRef CAS PubMed.
- S. Kumar, S. Jain, M. Nehra, N. Dilbaghi, G. Marrazza and K. H. Kim, Coord. Chem. Rev., 2020, 420, 213407 CrossRef CAS.
- X. L. Li, C. Q. Yang, B. Sun, S. L. Cai, Z. M. Chen, Y. Q. Lv, J. Zhang and Y. Liu, J. Mater. Chem. A, 2020, 8, 16045–16060 RSC.
- C. A. López, C. Abia, J. Gainza, P. Kayser, N. N. Nemes, O. J. Dura, J. L. Martínez, M. T. Fernández-Díaz, C. Álvarez-Galván and J. A. Alonso, Mater. Adv., 2021, 2, 3620–3628 RSC.
- F. Palazon, Y. El Ajjouri, P. Sebastia-Luna, S. Lauciello, L. Manna and H. J. Bolink, J. Mater. Chem. C, 2019, 7, 11406–11410 RSC.
- S.-Y. Kim, Y. Yun, S. Shin, J. H. Lee, Y.-W. Heo and S. Lee, Scr. Mater., 2019, 166, 107–111 CrossRef CAS.
- T. Friscic, C. Mottillo and H. M. Titi, Angew. Chem., Int. Ed., 2020, 59, 1018–1029 CrossRef CAS.
- S. L. James, C. J. Adams, C. Bolm, D. Braga, P. Collier, T. Friscic, F. Grepioni, K. D. M. Harris, G. Hyett, W. Jones, A. Krebs, J. Mack, L. Maini, A. G. Orpen, I. P. Parkin, W. C. Shearouse, J. W. Steed and D. C. Waddell, Chem. Soc. Rev., 2012, 41, 413–447 RSC.
- H. Ishihara, K. Watanabe, A. Iwata, K. Yamada, Y. Kinoshita, T. Okuda, V. G. Krishnan, S. Q. Dou and A. Weiss, Z. Naturforsch., A: Phys. Sci., 1992, 47, 65–74 CrossRef CAS.
- B. H. Toby and R. B. Von Dreele, J. Appl. Crystallogr., 2013, 46, 544–549 CrossRef CAS.
- W. A. Dollase, J. Appl. Crystallogr., 1986, 19, 267–272 CrossRef CAS.
- M. Saba, M. Cadelano, D. Marongiu, F. Chen, V. Sarritzu, N. Sestu, C. Figus, M. Aresti, R. Piras, A. Geddo Lehmann, C. Cannas, A. Musinu, F. Quochi, A. Mura and G. Bongiovanni, Nat. Commun., 2014, 5, 5049 CrossRef CAS PubMed.
- R. J. Elliott, Phys. Rev., 1957, 108, 1384–1389 CrossRef CAS.
- R. L. Z. Hoye, J. Hidalgo, R. A. Jagt, J.-P. Correa-Baena, T. Fix and J. L. MacManus-Driscoll, Adv. Energy Mater., 2022, 12, 2100499 CrossRef CAS.
- Y. Yang, M. Yang, K. Zhu, J. C. Johnson, J. J. Berry, J. van de Lagemaat and M. C. Beard, Nat. Commun., 2016, 7, 12613 CrossRef CAS PubMed.
- S. Rieger, B. J. Bohn, M. Döblinger, A. F. Richter, Y. Tong, K. Wang, P. Müller-Buschbaum, L. Polavarapu, L. Leppert, J. K. Stolarczyk and J. Feldmann, Phys. Rev. B, 2019, 100, 201404 CrossRef CAS.
- Q. Li, L. Yin, Z. Chen, K. Deng, S. Luo, B. Zou, Z. Wang, J. Tang and Z. Quan, Inorg. Chem., 2019, 58, 1621–1626 CrossRef CAS PubMed.
- G. Greczynski and L. Hultman, Sci. Rep., 2021, 11, 11195 CrossRef CAS PubMed.
- G. Greczynski and L. Hultman, Prog. Mater. Sci., 2020, 107, 100591 CrossRef CAS.
- M. M. S. Karim, A. M. Ganose, L. Pieters, W. W. Winnie Leung, J. Wade, L. Zhang, D. O. Scanlon and R. G. Palgrave, Chem. Mater., 2019, 31, 9430–9444 CrossRef CAS PubMed.
- D. Wang, A. C. Miller and M. R. Notis, Surf. Interface Anal., 1996, 24, 127–132 CrossRef CAS.
- R. Félix, N. Llobera-Vila, C. Hartmann, C. Klimm, M. Hartig, R. G. Wilks and M. Bär, RSC Adv., 2018, 8, 67–73 RSC.
- A. Shukla, V. K. Sharma, S. K. Gupta and A. S. Verma, Mater. Chem. Phys., 2020, 253, 123389 CrossRef CAS.
- P. Day, N. S. Hush and R. J. H. Clark, Philos. Trans. R. Soc., A, 2008, 366, 5–14 CrossRef CAS PubMed.
- L. Atkinson and P. Day, J. Chem. Soc. A, 1969, 2423–2431, 10.1039/J19690002423.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2cp04487k |
| This journal is © the Owner Societies 2023 |