
Barrier heights, reaction energies and bond dissociation energies for RH + HO2 reactions with coupled-cluster theory, density functional theory and diffusion quantum Monte Carlo methods†
Xiaojun
Zhou
 *a,
Zhanli
Cao
b,
Fan
Wang
c and
Zhifan
Wang
d
*a,
Zhanli
Cao
b,
Fan
Wang
c and
Zhifan
Wang
d
aDepartment of Physics, Shaanxi University of Science and Technology, Xi’an, 710021, P. R. China. E-mail: xiaojunzhou82@163.com
bSchool of Science, Xi’an University of Posts and Telecommunications, Xi’an, 710121, P. R. China
cInstitute of Atomic and Molecular Physics, Key Laboratory of High Energy Density Physics and Technology, Ministry of Education, Sichuan University, Chengdu, P. R. China
dSchool of Electronic Engineering, Chengdu Technological University, Chengdu, P. R. China
First published on 15th November 2022
Abstract
Hydrogen abstraction reactions by the HO2 radical from hydrocarbon molecules are an important class of reactions in the autoignition of hydrocarbon fuels. Performance of DLPNO-CC and DFT methods using three hybrids and four double hybrids as well as FN-DMC with the single-Slater–Jastrow trial wavefunction on barrier heights and reaction energies of RH + HO2 reactions as well as bond dissociation energies of the involved X–H molecules is evaluated by comparison with the highly accurate CCSD(T)-F12b/CBS results in this study. Our results show that the DLPNO-CCSD(T)-F12 method can achieve highly accurate barrier heights, reaction energies and X–H bond energies for RH + HO2 reactions at a relatively low computational cost, and it is applicable to the H-abstraction reactions of larger molecules. Among all DFAs, MN15 and the employed double hybrids can achieve accurate barrier heights and reaction energies with MADs of less than or around 2 kJ mol−1, but their error on X–H bond energies is more pronounced. Only DSD-BLYP and DSD-PBEB95 can provide X–H bond energies with MADs less than 4 kJ mol−1. Considering dispersion correction in DFT calculations does not improve these barrier heights and reaction energies. The error of FN-DMC on barrier heights and reaction energies is slightly larger than that of MN15 and those of double hybrids, but it can achieve results within chemical accuracy for these reactions and the X–H bond energies.
1. Introduction
Hydrogen atom abstraction by the hydroperoxyl radical (HO2) from fuel molecules is a key reaction class during the autoignition processes at temperatures of 600–1200 K.1,2 Hydrogen peroxide (H2O2) and hydrocarbon radicals will be formed when HO2 abstracts a hydrogen atom from hydrocarbon fuels (RH): RH + HO2 → R + H2O2, and two highly reactive hydroxyl radicals (OH) are produced subsequently from the decomposition of H2O2: H2O2 + M → OH + OH + M. These reactions provide a channel for the transformation of the relatively unreactive HO2 radical to the chain propagating radical OH. It has been reported that the profiles of products and ignition delay times in the oxidation of various hydrocarbons are rather sensitive to the kinetics of the abstraction reaction RH + HO2 → R + H2O2.3,4 Besides, HO2 is an important sink for formaldehyde (HCHO) and trifluoroacetaldehyde (CF3CHO) in atmosphere chemistry.5 Nevertheless, there have been limited experimental measurements on rate constants of H-abstraction from hydrocarbons by the hydroperoxyl radical.6 Therefore, rate constants of these hydrogen atom abstraction reactions are mainly estimated or calculated with theoretical methods.7–9 To obtain reasonable reaction rate constants, highly accurate electronic structure approaches are important to achieve reliable barrier heights.Density functional theory (DFT)10 has emerged as the most popular method in electronic structure calculations because it represents an incomparable balance of efficiency and accuracy. However, accuracy of DFT results depends on the employed exchange–correlation (XC) functions. It has been shown that barrier heights of hydrogen abstraction reactions with hybrid XC functionals tend to be rather sensitive to the percentage of the Hartree–Fock (HF) exchange.11 The Minnesota XC functionals developed by the Truhlar group with about 50% of the HF exchange, such as M05-2X,12 M06-2X,13 and M08-HX,14 have been demonstrated to provide reliable barrier heights of 19 hydrogen transfer reactions (HTBH38/08).15 On the other hand, a study on the performance of different XC functionals on the 2017 GMTKN55 database shows that double hybrid density functionals (DHDF) are the most reliable and accurate functionals in calculating barrier heights, and they clearly outperform hybrid density functionals in most cases.16 For instance, mean absolute errors of B2GP-PLYP,17 DSD-BLYP,18 DSD-PBEP8619 and DSD-PBEB9520 on barrier heights for 19 hydrogen-transfer and 19 non-hydrogen-transfer reactions are around 1.0 kcal mol−1.16 London-dispersion interactions21 have been shown to be important for a quantitative description and qualitative understanding of barrier heights and reaction energies.22,23 However, barrier heights for the RH + HO2 reaction are not included in the above benchmark database. Performance of hybrid functionals and DHDFs as well as effects of London-dispersion corrections on barrier heights for RH + HO2 deserves further study.
The coupled-cluster (CC) approach24 at the singles and doubles (CCSD) level25 augmented by a perturbative treatment of triple excitations [CCSD(T)]26 is often regarded as the “gold standard” in quantum chemistry. It can provide highly accurate results for systems that are well-described using a single-reference wavefunction. However, CCSD(T) can only be applied to rather small molecules due to its high computational scaling and strong basis set dependence. Strong basis-set dependence can be alleviated with explicitly correlated CCSD(T)-F12x (x = a, b) methods.27,28 Computational cost of CCSD(T)-F12x is slightly larger than that of the standard CCSD(T) method when applying the same basis set, but highly accurate results can be achieved using a small basis set. This means that CCSD(T)-F12x methods can possibly be applied to larger systems than the standard CCSD(T) method.27 On the other hand, locally correlated coupled-cluster methods such as DLPNO-CCSD(T)29 have been developed to reduce computational cost by exploiting the short range nature of dynamical electron correlation. The performance of DLPNO-CCSD(T) relative to standard CCSD(T) has been studied extensively.30–33 Mean absolute deviations of DLPNO-CCSD(T) on seven sets of barrier heights in the GMTKN55 are less than 0.3 kcal mol−1 compared with standard CCSD(T) results.31 In addition, the DLPNO-CCSD(T1) method34 with an iterative treatment on triples, being slightly more computationally expensive, shows an improvement in accuracy over the DLPNO-CCSD(T) method, especially for open shell species.31
The diffusion Monte Carlo (DMC) method35–37 offers a good compromise between efficiency and accuracy in solving quantum mechanical problems stochastically. Key advantages of the DMC method are attractive scaling of N3 with the electron number and high parallel efficiency. DMC calculations have been carried out on super-computers with more than 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cores38 and it is applied to systems with up to around 1000 electrons.39 Furthermore, the DMC method can capture a large fraction of electron correlation37 and it is not sensitive to the employed basis sets.40 These properties enable its applicability to large molecules with high accuracy. In DMC calculations, important sampling is adopted to improve statistical efficiency by introducing a trial wavefunction. To circumvent the fermion sign problem, the fixed-node (FN) approximation41 is employed to constrain the DMC solution to have identical nodes of the trial wavefunction. Therefore, error of the FN-DMC energy depends on the node surface of the trial wavefunction. FN-DMC using the single-Slater–Jastrow trial wavefunction has been adopted extensively in a large variety of problems including reaction barrier heights of H + H2,42–44 organic molecules,45–47 and surface reaction.48–50 Some previous works show that mean absolute errors of FN-DMC on barrier heights of hydrogen-transfer reactions and non-hydrogen-transfer reactions are 1.0–1.5 kcal mol−1 using the single-Slater–Jastrow trial wavefunction.51,52
000 cores38 and it is applied to systems with up to around 1000 electrons.39 Furthermore, the DMC method can capture a large fraction of electron correlation37 and it is not sensitive to the employed basis sets.40 These properties enable its applicability to large molecules with high accuracy. In DMC calculations, important sampling is adopted to improve statistical efficiency by introducing a trial wavefunction. To circumvent the fermion sign problem, the fixed-node (FN) approximation41 is employed to constrain the DMC solution to have identical nodes of the trial wavefunction. Therefore, error of the FN-DMC energy depends on the node surface of the trial wavefunction. FN-DMC using the single-Slater–Jastrow trial wavefunction has been adopted extensively in a large variety of problems including reaction barrier heights of H + H2,42–44 organic molecules,45–47 and surface reaction.48–50 Some previous works show that mean absolute errors of FN-DMC on barrier heights of hydrogen-transfer reactions and non-hydrogen-transfer reactions are 1.0–1.5 kcal mol−1 using the single-Slater–Jastrow trial wavefunction.51,52
A wide array of different methods have been adopted to calculate the barrier heights of RH + HO2 reaction with variable accuracy and associated computational cost, such as B3LYP, BP86, TPSSh, BMK, B97K, RCCSD(T) and CCSD(T)-R12 methods, by Aguilera et al.7 In the present work, barrier heights and reaction energies for H-abstraction reactions by the HO2 radical from methane, ethane, propane, n-butane and iso-butane are studied by DLPNO-CCSD(T), DLPNO-CCSD(T1), and DLPNO-CCSD(T)-F12;53 B3LYP,54 M06-2X, and MN15;55 and B2GP-PLYP, DSD-BLYP, DSD-PBEP86, and DSD-PBEB95 as well as FN-DMC, and these reactions are listed as follows:
| CH4 + HO2 → CH3 + H2O2 | (R1) |
| C2H6 + HO2 → C2H5 + H2O2 | (R2) |
| C3H8 + HO2 → n-C3H7 + H2O2 | (R3) |
| C3H8 + HO2 → i-C3H7 + H2O2 | (R4) |
| n-C4H10 + HO2 → p-C4H9 + H2O2 | (R5) |
| n-C4H10 + HO2 → s-C4H9 + H2O2 | (R6) |
| i-C4H10 + HO2 → i-C4H9 + H2O2 | (R7) |
| i-C4H10 + HO2 → t-C4H9 + H2O2 | (R8) |
Note that the isomers of alkane radicals involved in (R3)–(R8) are given in Fig. 1. The purpose of this work is to evaluate the reliability of these methods on barrier heights and reaction energies of (R1)–(R8), and to provide a reasonable method for the study of H-abstraction of HO2 from large hydrocarbon molecules. This paper is organized in the following manner: computational details are described in Section II. The results of barrier heights and reaction energies for (R1)–(R8) as well as bond dissociation energies with these methods are presented in Section III. Summary and conclusions are given in Section IV.
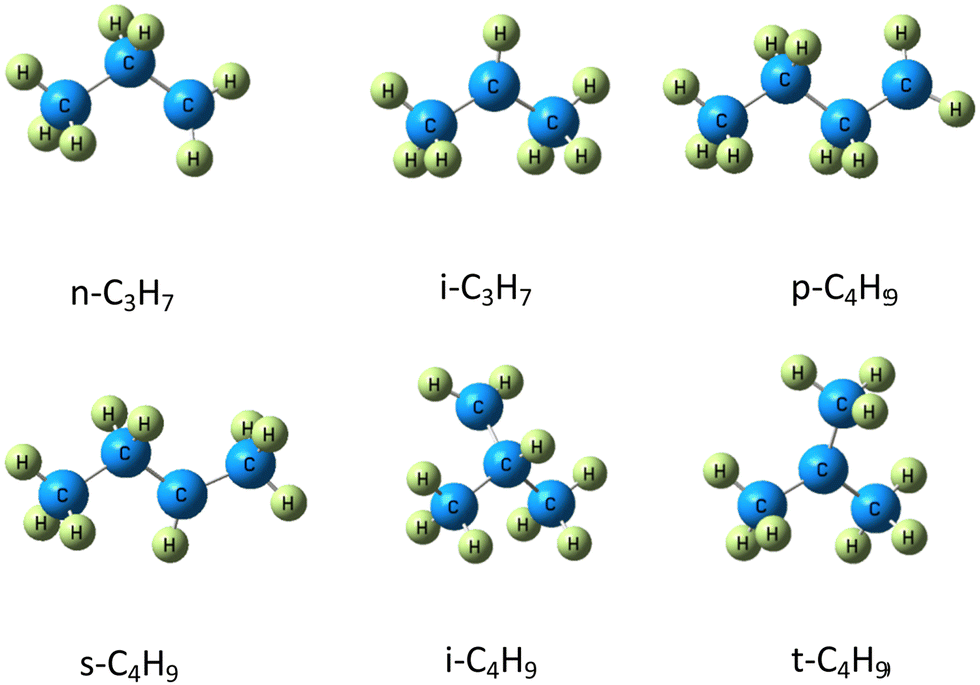 | ||
| Fig. 1 The set of radicals formed in reactions of R3–R8. | ||
2. Computational details
To obtain quantitative barrier heights, reliable geometries are required.56 The B3LYP method has been shown to be reasonable for geometry optimization.57 In this work, geometries and frequencies of reactants, products and transition states for (R1)–(R8) are obtained at the B3LYP/def2-TZVP level58 using the Gaussian 16 program,59 and their Cartesian coordinates are given in the ESI.† Stability of wavefunction at the B3LYP/def2-TZVP level is also tested, and our results show that all the wave functions are stable. Vibrational analyses are carried out to confirm that all of the reactants and products have no imaginary frequency and transition-state structures have only one imaginary frequency. In addition, the intrinsic reaction coordinate (IRC) calculations are performed to verify the transition-state connecting the desired reactants and products. To provide reliable barrier heights and reaction energies, accurate single point energies for reactants, products and transition states of (R1)–(R8) are calculated employing various electronic structure methods. Performances of DLPNO-CCSD(T), DLPNO-CCSD(T1), and DLPNO-CCSD(T)-F12; B3LYP, M06-2X, and MN15; and B2GP-PLYP, DSD-BLYP, DSD-PBEP86, and DSD-PBEB95 as well as FN-DMC methods on barrier heights and reaction energies are evaluated by comparing their results with those of CCSD(T)-F12b/CBS. Note that CCSD(T)-F12b/CBS results are derived from two-point extrapolation using cc-pVDZ-F12 and cc-pVTZ-F12 basis sets adopting the extrapolation scheme proposed in ref. 60. The following total energy expression is employed in this work:| E = E∞HF + ΔE∞CCSD-F12b + ΔE∞(T) | (1) |
| E∞HF = EXHF + aexp(−bX) | (2) |
| ΔE∞CCSD-F12b/(T) = (ΔEcorrlarge − ΔEcorrsmall)F + ΔEcorrsmall | (3) |
In this work, both DLPNO-CCSD(T) and DLPNO-CCSD(T1) calculations are carried out using the aug-cc-pVQZ basis set and the corresponding auxiliary basis sets.63,64 In addition, the explicitly correlated DLPNO-CCSD(T)-F12 method employing a perturbative F12 correction on top of the DLPNO-CCSD(T) correlation energy together with the cc-pVTZ-F1258,65 and aug-cc-pVTZ-F12/C basis sets as well as the near-complete auxiliary basis set cc-pVTZ-F12-CABS for F12 is also adopted in calculating the energies of stationary points on potential energy surfaces of these reactions. All DLPNO-CC calculations are carried out using the ORCA program package.66,67 Accuracy of DLPNO-CC is mainly controlled by thresholds for the PNO occupation number TCutPNO, the strong pair approximation cut-off TCutPairs, and the domain size parameter TCutMKN. In this work, all DLPNO calculations employ “TightPNO” setting68 with values of 10−7, 10−5, and 10−3 for the above three thresholds.
DFT calculations are technically easy to perform, but it is non-trivial to choose the right density functional approximations (DFAs) for a specific problem. In this work, DFT calculations with various DFAs including the B3LYP, M06-2X, and MN15 hybrid functionals and B2GP-PLYP, DSD-BLYP, DSD-PBEP86 and DSD-PBEB95 double hybrid functionals are carried out to evaluate their performance on barrier heights and reaction energies of (R1)–(R8) reactions. The results of the above DFAs in conjunction with Ahlrichs’ type quadruple-zeta basis sets58 (def2-QZVP) are compared with CCSD(T)-F12b/CBS values. D3 with Becke–Johnson damping69 [D3(BJ)] is currently the most popular dispersion correction, and the results for B3LYP-D3(BJ), B2GP-PLYP-D3(BJ), DSD-PBEP86-D3(BJ) and DSD-PBEB95-D3(BJ) are also reported in this paper. It is worth mentioning that the MN15 calculation is carried out with the Gaussian 16 program package59 using the default ultrafine integration grid. Other DFA calculations are performed with the ORCA 4.2.1 program using the integration grid4 and final-grid6. All energies in the SCF steps have been converged to 10−7 au.
As for DMC calculations, the Slater–Jastrow trial wavefunction employed in this work takes the following forms:
ψT(![[x with combining right harpoon above (vector)]](https://www.rsc.org/images/entities/i_char_0078_20d1.gif) ) = Φs()eJ( ) = Φs()eJ(![[x with combining right harpoon above (vector)]](https://www.rsc.org/images/entities/char_0078_20d1.gif) ) ) | (4) |
) is a single-determinant wavefunction and J() is the Jastrow factor. The Slater determinant adopted in DMC calculations is usually obtained from a DFT calculation. Previous DMC calculations demonstrate that DMC results do not depend much on the employed XC functional.70 In this work, molecular orbitals in trial wavefunctions are obtained from B3LYP calculations with the scalar-relativistic energy-consistent Hartree–Fock pseudopotential71 (PP) developed by Burkatzki, Filippi, and Dolg (BFD) in conjunction with the cc-pVTZ basis sets, except for hydrogen, using the Gaussian 16 program package. The T-move scheme72 is employed to treat the nonlocal terms in PPs to ensure the stability of DMC calculations. Parameters in the Jastrow factor are optimized by the variance minimization method73 and energy minimization method74 based on variational Monte Carlo (VMC)37 calculations. The optimized trial wavefunctions are subsequently adopted in DMC calculations. To eliminate time-step bias, a quadratic extrapolation is carried out to obtain the final DMC energy at zero-time step from DMC energies at time steps of 0.02, 0.06 and 0.10 a.u. All QMC calculations are accomplished using the CASINO program package.36
3. Results and discussion
3.1 Barrier heights
CCSD(T) can achieve highly accurate results for single reference states, while its error may be somewhat larger for molecules with a pronounced multi-reference character. High-level coupled cluster theory has been shown to be necessary for some reactions.75 Unfortunately, post-CCSD(T) calculations are generally rather expensive and it can only be applied to very small molecules using a small basis set. To evaluate the performance of CCSD(T) on such types of reactions, CCSDT(Q) calculations with the cc-pVDZ basis set are performed on stationary points of R1 by using the MRCC program package76 interfaced through the CFOUR program package.77 Our results indicate that the difference between CCSDT(Q) and CCSD(T) results is less than 0.4 kJ mol−1 for the forward reaction, while their difference is about 1.4 kJ mol−1 for the reverse reaction. These results show that CCSD(T) can probably provide reliable results for these reactions. Furthermore, core-valence correlation and relativistic effects are not considered in this work. It should be noted that most of the core-valence correlation and relativistic effects will be cancelled out in calculating barrier heights and reaction energies. To further test this point, non-relativistic CCSD(T)/aug-cc-pVQZ and CCSD(T)/aug-cc-pCVQZ calculations as well as CCSD(T)/aug-cc-pVQZ calculations using the DHK Hamiltonian78 are carried out for R1. Our results indicate that the effects of core-valence correlation on barrier heights are less than 0.5 kJ mol−1, while relativistic effects on barrier heights are less than 0.2 kJ mol−1. These results indicate that the core-valence correlation and scalar relativistic effects are insignificant.| Barriera | CCSD(T)-F12a/cc-pVXZ-F12 | CCSD(T)-F12b/cc-pVXZ-F12 | Best estimateb | ||||
|---|---|---|---|---|---|---|---|
| DZ | TZ | DZ | TZ | CBS | |||
| a V‡f denotes forward barrier heights, and V‡r denotes reverse barrier heights. b Ref. 7. | |||||||
| R1 | V‡f | 100.76 | 99.37 | 101.68 | 100.08 | 99.45 | 100.4 |
| V‡r | 26.27 | 25.48 | 26.24 | 25.87 | 25.82 | — | |
| R2 | V‡f | 83.34 | 81.76 | 83.93 | 82.47 | 81.92 | 81.6 |
| V‡r | 24.34 | 23.60 | 24.18 | 23.98 | 24.02 | — | |
| R3 | V‡f | 81.17 | 79.62 | 81.72 | 80.35 | 79.87 | 82.0 |
| V‡r | 20.73 | 19.96 | 20.52 | 20.36 | 20.42 | — | |
| R4 | V‡f | 70.33 | 68.66 | 70.69 | 69.36 | 68.91 | 67.3 |
| V‡r | 22.46 | 21.76 | 22.21 | 22.15 | 22.26 | — | |
| R5 | V‡f | 80.83 | 79.33 | 81.35 | 80.02 | 79.58 | 81.4 |
| V‡r | 20.40 | 19.66 | 20.17 | 20.04 | 20.13 | — | |
| R6 | V‡f | 69.07 | 67.40 | 69.39 | 68.12 | 67.70 | 64.4 |
| V‡r | 19.96 | 19.20 | 19.66 | 19.61 | 19.71 | — | |
| R7 | V‡f | 81.72 | 80.21 | 82.24 | 80.95 | 80.54 | 79.0 |
| V‡r | 19.77 | 18.95 | 19.52 | 19.32 | 19.37 | — | |
| R8 | V‡f | 60.49 | 58.83 | 60.68 | 59.53 | 59.20 | 57.2 |
| V‡r | 19.26 | 18.62 | 18.98 | 19.02 | 19.16 | — | |
| MD | 0.80 | −0.35 | 0.94 | 0.20 | |||
| MAD | 0.80 | 0.35 | 0.98 | 0.27 | |||
According to the results in Table 1, CCSD(T)-F12a/cc-pVTZ-F12 underestimates these barrier heights, while CCSD(T)-F12a/cc-pVDZ-F12 overestimates these barrier heights compared with CCSD(T)-F12b/CBS results. In addition, CCSD(T)-F12a/cc-pVTZ-F12 results agree better with CCSD(T)-F12b/CBS results than those of CCSD(T)-F12a/cc-pVDZ-F12. On the other hand, barrier heights of CCSD(T)-F12b using cc-pVDZ-F12 or cc-pVTZ-F12 basis sets are higher than those of CCSD(T)-F12b/CBS in most cases. Barrier heights of CCSD(T)-F12b/cc-pVTZ-F12 are closer to those of CCSD(T)-F12b/CBS than CCSD(T)-F12a/cc-pVTZ-F12, while the mean deviation of CCSD(T)-F12b becomes larger than that of CCSD(T)-F12a when the cc-pVDZ-F12 basis set is employed. According to our results, CCSD(T)-F12a is more sensitive to the choice of basis sets than CCSD(T)-F12b on these barrier heights; however, both CCSD(T)-F12a and CCSD(T)-F12b methods using the cc-pVDZ-F12 basis set are able to provide reliable barrier heights with MADs of less than 1 kJ mol−1. It is worth mentioning here that MADs of these barrier heights for CCSD-F12a and CCSD-F12b methods are larger than 10 kJ mol−1. This means that triple excitations have a significant effect on these barrier heights.
The difference between the barrier heights with CCSD(T)-F12b/CBS and those reported in ref. 7 is around or over 2 kJ mol−1 for R3–R8, and it reaches 3.3 kJ mol−1 for R6. It should be noted that the barrier heights for these reactions in ref. 7 are obtained by scaling the electron-correlation contributions to the electronic barrier height by an empirical factor of 1.053 in frozen-core CCSD(T)/cc-pVTZ calculations. Therefore, the results in ref. 7 may be subject to larger uncertainty for different size systems. According to CCSD(T)-F12b/CBS results, the forward barrier heights for those H transfer from the same carbon site, such as primary carbon sites (R2, R3, R5 and R7) or secondary carbon sites (R4 and R6), are rather close. This indicates that the system size has an insignificant effect on barrier heights for H transfer from the same type carbon site. Nevertheless, the carbon site of H transfer affects the forward barrier height to some extent. The barrier height for H transfer from a primary site (R5) is about 12 kJ mol−1 larger than that from a secondary site (R6), and it is about 21 kJ mol−1 higher than that from a tertiary carbon site (R8). On the other hand, the reverse barrier heights of these reactions are relatively insensitive to the carbon site.
| Reaction | Barriera | DLPNO- | B3LYP | M06-2X | MN15 | B2GP-PLYP | DSD- | DMC | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| CCSD(T) | CCSD(T1) | CCSD(T)-F12 | BLYP | PBEP86 | PBEB95 | |||||||
| a V‡f denotes forward barrier heights, and V‡r denotes reverse barrier heights. | ||||||||||||
| R1 | V‡f | 1.59 | 1.26 | 2.22 | −4.64 | −1.88 | 2.59 | 0.92 | −3.18 | −3.64 | 3.51 | 2.47 |
| V‡r | 2.43 | 2.05 | −0.13 | −17.66 | −7.15 | −1.55 | −2.72 | −2.18 | −2.22 | −2.18 | 1.51 | |
| R2 | V‡f | 2.34 | 1.92 | 2.09 | −5.77 | −3.10 | 1.17 | 0.84 | −3.51 | −3.47 | 3.35 | 5.06 |
| V‡r | 2.68 | 2.22 | 0 | −13.85 | −5.98 | 1.34 | −1.38 | −1.59 | −1.38 | −0.92 | 1.09 | |
| R3 | V‡f | 2.72 | 2.30 | 2.01 | −3.97 | −2.85 | 1.42 | 1.67 | −2.93 | −2.89 | 3.89 | 4.60 |
| V‡r | 2.72 | 2.22 | −0.04 | −11.67 | −5.65 | 1.55 | −0.54 | −1.21 | −0.92 | −0.38 | 1.51 | |
| R4 | V‡f | 3.01 | 2.55 | 2.18 | −5.86 | −3.56 | 0.71 | 0.92 | −3.64 | −3.26 | 3.39 | 4.48 |
| V‡r | 2.72 | 2.18 | 0.29 | −9.08 | −4.52 | 4.27 | 0.25 | −0.84 | −0.46 | 0.59 | 1.63 | |
| R5 | V‡f | 3.01 | 2.59 | 1.84 | −3.60 | −2.68 | 1.51 | 1.84 | −2.85 | −2.72 | 4.02 | 4.44 |
| V‡r | 2.68 | 2.18 | 0.08 | −11.25 | −5.31 | 1.84 | −0.33 | −1.09 | −0.75 | −0.21 | 1.26 | |
| R6 | V‡f | 3.47 | 3.05 | 2.18 | −3.77 | −3.31 | 0.71 | 1.97 | −2.93 | −2.55 | 4.14 | 5.02 |
| V‡r | 2.80 | 2.22 | 0.50 | −6.69 | −4.14 | 4.18 | 1.30 | −0.29 | 0.17 | 1.30 | 1.46 | |
| R7 | V‡f | 3.18 | 2.80 | 1.92 | −3.26 | −2.30 | 2.26 | 2.01 | −2.64 | −2.55 | 4.27 | 5.48 |
| V‡r | 2.76 | 2.22 | 0.17 | −10.63 | −4.90 | 2.13 | −0.25 | −1.13 | −0.75 | −0.13 | 4.02 | |
| R8 | V‡f | 3.60 | 3.14 | 2.26 | −6.32 | −3.26 | 1.38 | 0.67 | −3.93 | −3.22 | 3.35 | 3.85 |
| V‡r | 2.76 | 2.13 | 0.71 | −4.73 | −2.93 | 6.95 | 1.59 | −0.38 | 0.33 | 2.01 | 2.55 | |
| MD | 2.78 | 2.32 | 1.15 | −7.67 | −3.97 | 2.03 | 0.55 | −2.15 | −1.90 | 1.88 | 3.16 | |
| MAD | 2.78 | 2.32 | 1.17 | 7.67 | 3.97 | 2.23 | 1.20 | 2.15 | 1.96 | 2.35 | 3.16 | |
| MAD(all V‡f) | 2.87 | 2.45 | 2.09 | 4.65 | 2.87 | 1.47 | 1.35 | 3.20 | 3.04 | 3.74 | 4.42 | |
| MAD(all V‡r) | 2.69 | 2.18 | 0.24 | 10.69 | 5.07 | 2.98 | 1.05 | 1.09 | 0.87 | 0.96 | 1.89 | |
According to the results in Table 2, DLPNO-CC methods always overestimate these barrier heights. Deviations of the barrier heights with DLPNO-CCSD(T) are 1–4 kJ mol−1, and they are reduced by about 0.3–0.6 kJ mol−1 when DLPNO-CCSD(T1) is employed. This improvement is slightly more pronounced on reverse barrier heights than that on forward barrier heights. Computational cost of DLPNO-CCSD(T1) is about twice that of DLPNO-CCSD(T) using the aug-cc-pVQZ basis set for these systems; however, iterative treatment of perturbative triple excitations has a rather small effect on these barrier heights. Our results show that DLPNO-CCSD(T)-F12 with the cc-pVTZ-F12 basis set provides smaller deviations on barrier heights in most cases than those from the DLPNO-CCSD(T) with the aug-cc-pVQZ basis sets. This may be because both DLPNO-CCSD(T)-F12 and reference CCSD(T)-F12b/CBS results are from explicit correlation calculations. In fact, DLPNO-CCSD(T)-F12 improves reverse barrier heights pronouncedly with an MAD of 0.24 kJ mol−1, while its MAD on forward barrier heights is only slightly smaller than that of DLPNO-CCSD(T) or DLPNO-CCSD(T1). To further investigate the influence of the basis set, DLPNO-CCSD(T)-F12 calculations using the cc-pVQZ-F12 basis sets are also performed. Our results show that differences between results using the cc-pVTZ-F12 basis set and those with the cc-pVQZ-F12 basis set are less than 0.5 kJ mol−1 in most cases. This result indicates that basis set incompleteness error of DLPNO-CCSD(T)-F12 employing the cc-pVTZ-F12 basis set is negligible for barrier heights of these reactions.
As for DFT results listed in Table 2, B3LYP always underestimates the barrier heights and its deviations are over 5 kJ mol−1 in most cases. We note that barrier heights of B3LYP in this work are 0.4–3.6 kJ mol−1 smaller than those reported in ref. 7, which is possibly due to the different basis sets and different geometry in single point energy calculations. MN15 overestimates almost all of the barrier heights, while M06-2X underestimates all barrier heights. Absolute deviations on barrier heights of MN15 are less than or close to those from M06-2X, except for the reverse barrier height of R8. In fact, deviations of barrier heights with MN15 do not exceed 2.5 kJ mol−1 for most of these reactions. Our results also show that MN15 provides significantly smaller absolute deviations on these barrier heights than B3LYP. It is interesting to find that both B3LYP and M06-2X give larger deviations on reverse barrier heights of R1–R7. As for the double hybrid functionals, DSD-BLYP and DSD-PBEP86 underestimate all barrier heights, while B2GP-PLYP and DSD-PBEB95 overestimate most of the barrier heights to some extent. In fact, barrier heights with DSD-BLYP and DSD-PBEP86 are rather close to each other. In addition, absolute deviation of DSD-BLYP, DSD-PBEP86 and DSD-PBEB95 for reverse barrier heights is always smaller than that of the corresponding forward barrier heights. Among the double hybrids, B2GP-PLYP is the best in calculating these barrier heights, where its MAD is less than 2.0 kJ mol−1. It can be seen that MADs of B2GP-PLYP are smaller than those of MN15, especially for reverse barrier heights. In fact, the MAD of MN15 for all reactions is close to those of the other three double hybrid functionals. Furthermore, both MN15 and the employed double hybrid functionals reach chemical accuracy on these barrier heights.
The work by Goerigk on GMTKN55 showed that MADs of barrier heights can usually be increased when London-dispersion corrections D3(BJ) are included in DFT treatments.16 In this work, D3(BJ) correction is also included in the B3LYP and well-performed B2GP-PLYP, DSD-PBEP86, and DSD-PBEB95 functionals. Deviations of those dispersion-corrected DFAs for all forward and reverse barrier heights are given in Table S1 of ESI.† Our results show that dispersion correction slightly improves the barrier heights of DSD-PBEB95 when they are overestimated. However, all barrier heights are further underestimated by B3LYP and DSD-PBEP86 when D3(BJ) correction is included. This is because the dispersion correction stabilizes the transition state more than it does on the reactants or products, and thus a functional that already underestimates a barrier height will do so even more with the dispersion correction. As a result, MADs of those B2GP-PLYP-D3(BJ), DSD-PBEP86-D3(BJ) and DSD-PBEB95-D3(BJ) are increased by less than 2 kJ mol−1 but the MAD of B3LYP-D3(BJ) is increased by 9 kJ mol−1 compared with those of the dispersion-uncorrected counterpart. This result may indicate that the effect of dispersion correction is less pronounced for the employed double hybrids. To further evaluate the effects of dispersion interaction, differences between barrier heights of R1–R8 by those dispersion-corrected and those of dispersion-uncorrected DFAs are presented in Table S2 of ESI.† These results show that the effect of dispersion correction increases with the system size for both the forward and reverse barrier heights. This indicates that dispersion becomes more important as the system size increases. In addition, dispersion correction is slightly more significant on the reverse barrier heights than on the forward barrier heights.
One can also see from Table 2 that all barrier heights of R1–R8 are overestimated by FN-DMC using the B3LYP wavefunction. Considering the fact that energies of FN-DMC using the T-move satisfy the variational principle, this means that the error of FN-DMC on TSs is somewhat larger than that for the reactants and products. Our FN-DMC results show that deviations of forward barrier heights for all reactions are larger than those for their corresponding reverse barrier heights, especially for R2 and R6. The MAD of FN-DMC for forward barrier heights is 4.4 kJ mol−1, while it is only 1.9 kJ mol−1 for reverse barrier heights. In addition, the MAD of the FN-DMC result for all these barrier heights is about 3.2 kJ mol−1. This indicates that FN-DMC with the single-Slater–Jastrow trial wavefunction is able to give reliable barrier heights of these H-abstraction reactions. This is consistent with our previous FN-DMC work on barrier heights of 19 H-transfer reactions where the error of FN-DMC was approaching chemical accuracy.51 Among all methods, DLPNO-CCSD(T)-F12 provides the smallest MADs on barrier heights of these H-abstraction reactions, followed by B2GP-PLYP. It can be seen that MADs of DLPNO-CCSD(T) or DLPNO-CCSD(T1) methods are close to or slightly larger than those of the present double hybrid functionals and MN15. FN-DMC outperforms M06-2X on barrier heights of R1–R8, which is also consistent with our previous results on 19 H-transfer reactions. In fact, all the employed methods except B3LYP reach chemical accuracy on these barrier heights.
3.2 Reaction energies and bond energies
Reaction energies (REs) and bond energies (BEs) are also highly important to understand the kinetics and thermodynamics of many chemical reactions. REs of R1–R8, and dissociation energies of the X–H bond in H2O2, CH4, C2H6, n-C3H8, i-C3H8, p-C4H10 and i-C4H10 molecules are discussed in this section. Reaction energies and dissociation energies can readily be obtained from the energies of the involved reactants and products. Reaction energies of R1–R8 and bond energies of X–H using DLPNO-CCSD(T), DLPNO-CCSD(T1), and DLPNO-CCSD(T)-F12; B3LYP, M06-2X, and MN15; and B2GP-PLYP, DSD-BLYP, DSD-PBEP86, and DSD-PBEB95 as well as FN-DMC are compared against reference values from CCSD(T)-F12b/CBS in this work. Deviations of REs and BEs for these methods are given in Tables 3 and 4, respectively, and their corresponding MADs are listed at the bottom of these tables. We note that the energy of the H atom is set to −0.5 a.u. in all DLPNO-CC and FN-DMC calculations. Reference values of REs and BEs from CCSD(T)-F12b/CBS as well as BEs obtained from a combination of experimental and theoretical atomization energies of the involved species in ref. 80 are also presented in the last column of tables for comparison. One can see that the differences between CCSD(T)-F12b/CBS results and those from ref. 80 are less than 1 kJ mol−1 in most cases. These results further confirm the reliability of the CCSD(T)-F12b/CBS method. According to the reference values in Table 4, the bond energy of C–H for hydrogen attached to a primary carbon is between 415 kJ mol−1 and 418 kJ mol−1, and that attached to a secondary carbon is about 404 kJ mol−1, and it is about 397 kJ mol−1 for tertiary carbon. These results indicate that the primary carbon radicals are the most active and the tertiary carbon radical is the most stable. This is consistent with the fact that barrier heights for H transfer from a primary carbon are the highest and those from a tertiary carbon are the lowest.| Reaction | DLPNO- | DSD- | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| CCSD(T) | CCSD(T1) | CCSD(T)-F12 | B3LYP | M06-2X | MN15 | B2GP-PLYP | BLYP | PBEP86 | PBEB95 | DMC | CCSD(T)-F12b | |
| R1 | −0.84 | −0.79 | 2.34 | 13.01 | 5.27 | 4.14 | 3.64 | −1.00 | −1.42 | 5.69 | 0.96 | 73.64 |
| R2 | −0.33 | −0.29 | 2.09 | 8.08 | 2.89 | −0.17 | 2.22 | −1.92 | −2.09 | 4.27 | 3.97 | 57.91 |
| R3 | 0 | 0.08 | 2.05 | 7.70 | 2.80 | −0.13 | 2.22 | −1.72 | −1.97 | 4.27 | 3.10 | 59.45 |
| R4 | 0.29 | 0.38 | 1.88 | 3.22 | 0.96 | −3.56 | 0.67 | −2.80 | −2.80 | 2.80 | 2.85 | 46.65 |
| R5 | 0.33 | 0.42 | 1.76 | 7.66 | 2.64 | −0.33 | 2.18 | −1.76 | −1.97 | 4.23 | 3.18 | 59.45 |
| R6 | 0.67 | 0.84 | 1.67 | 2.93 | 0.84 | −3.47 | 0.67 | −2.64 | −2.72 | 2.85 | 3.56 | 47.99 |
| R7 | 0.42 | 0.59 | 1.76 | 7.36 | 2.59 | 0.13 | 2.26 | −1.51 | −1.80 | 4.39 | 1.46 | 61.17 |
| R8 | 0.84 | 1.00 | 1.55 | −1.59 | −0.33 | −5.56 | −0.92 | −3.56 | −3.56 | 1.34 | 1.30 | 40.04 |
| MAD | 0.47 | 0.55 | 1.89 | 6.44 | 2.29 | 2.19 | 1.85 | 2.11 | 2.29 | 3.73 | 2.55 | |
| Dissociation system | DLPNO- | B3LYP | M06-2X | MN15 | B2GP-PLYP | DSD- | DMC | CCSD(T)-F12b | Ref.a | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| CCSD(T) | CCSD(T1) | CCSD(T)-F12 | BLYP | PBEP86 | PBEB95 | ||||||||
| a Ref. 80. | |||||||||||||
| 1. H2O2 → HO2 + H | −0.25 | −0.08 | −4.44 | −18.50 | −8.20 | −8.85 | −8.21 | −1.81 | −3.52 | −5.43 | 1.26 | 387.77 | — |
| 2. CH4 → CH3 + H | −1.09 | −0.88 | −2.09 | −5.44 | −2.97 | −4.67 | −4.57 | −2.77 | −4.94 | 0.30 | 2.22 | 469.95 | 470.91 |
| 3. C2H6 → C2H5 + H | −0.63 | −0.42 | −2.38 | −10.46 | −5.35 | −9.02 | −6.07 | −3.77 | −5.65 | −1.20 | 5.19 | 455.39 | 456.27 |
| 4. n-C3H8 → n-C3H7 + H | −0.25 | 0.00 | −2.43 | −10.80 | −5.39 | −9.02 | −5.99 | −3.56 | −5.49 | −1.16 | 4.35 | 456.39 | — |
| 5. n-C3H8 → i-C3H7 + H | 0.00 | 0.25 | −2.59 | −15.32 | −7.28 | −12.45 | −7.58 | −4.61 | −6.36 | −2.67 | 4.06 | 444.42 | 444.01 |
| 6. p-C4H10 → p-C4H9 + H | 0.04 | 0.33 | −2.68 | −10.84 | −5.56 | −9.15 | −6.03 | −3.56 | −5.53 | −1.16 | 4.44 | 456.31 | — |
| 7. p-C4H10 → s-C4H9 + H | 0.42 | 0.71 | −2.76 | −15.57 | −7.36 | −12.33 | −7.54 | −4.44 | −6.24 | −2.58 | 4.81 | 445.26 | — |
| 8. i-C4H10 → i-C4H9 + H | 0.21 | 0.50 | −2.68 | −11.09 | −5.60 | −8.73 | −5.95 | −3.31 | −5.32 | −1.04 | 2.72 | 457.60 | — |
| 9. i-C4H10 → t-C4H9 + H | 0.59 | 0.92 | −2.85 | −20.09 | −8.53 | −14.42 | −9.13 | −5.40 | −7.08 | −4.09 | 2.51 | 436.64 | 433.88 |
| MAD | 0.39 | 0.45 | 2.77 | 13.12 | 6.25 | 9.85 | 6.79 | 3.69 | 5.57 | 2.18 | 3.51 | — | — |
According to the results in Table 3, REs from DSD-BLYP and DSD-PBEP86 methods are underestimated to some extent. This is consistent with the fact that forward barrier heights are underestimated more than the corresponding reverse barrier heights with these two functionals. On the other hand, DLPNO-CCSD(T)-F12, DSD-PBEB95 and FN-DMC overestimate the REs of these reactions. REs with M06-2X and B3LYP are also overestimated in most cases and these two functionals underestimate reverse barrier heights more than forward barrier heights. Our results show that B3LYP gives the largest MADs of 6.44 kJ mol−1 on these REs, and its deviation of R1 even reaches 13 kJ mol−1. This is consistent with the result that deviation of the reverse barrier height is significantly larger than that of the forward barrier height for R1. REs with DLPNO-CCSD(T) and DLPNO-CCSD(T1) agree the best with reference values and their MADs are less than or around 0.5 kJ mol−1. On the other hand, MADs of DLPNO-CCSD(T)-F12 and B2GP-PLYP on REs are somewhat larger. This is different from the results on barrier heights where the MAD of DLPNO-CCSD(T)-F12 and B2GP-PLYP is the smallest. These results indicate that DLPNO-CCSD(T) and DLPNO-CCSD(T1) can describe reactants and products better than DLPNO-CCSD(T)-F12 and B2GP-PLYP, while the transition states are calculated more accurately with DLPNO-CCSD(T)-F12 and B2GP-PLYP. Furthermore, deviations of REs with M06-2X, MN15 and FN-DMC are comparable to those of double hybrid functionals except for DSD-PBEB95. All these methods except for B3LYP can achieve chemical accuracy on REs.
The results in Table 4 show that bond energies of X–H are underestimated to some extent by DLPNO-CCSD(T)-F12 and all DFA methods. On the other hand, FN-DMC overestimates all these BEs. It should be noted that deviations on these BEs are reduced from tertiary carbon to primary carbon for butane with all DFAs. Similar to results on REs, DLPNO-CCSD(T) and DLPNO-CCSD(T1) give the smallest MADs of about 0.4 kJ mol−1, and B3LYP still provides the largest MADs of 13.1 kJ mol−1 for these BEs. The BE with the DLPNO-CCSD(T)-F12 method is underestimated by 2.8 kJ mol−1 on average. Bond energies with M06-2X are improved significantly compared with B3LYP and the MAD is reduced from 13.1 kJ mol−1 with B3LYP to 6.2 kJ mol−1 with M06-2X. In fact, MADs on these BEs with these DFAs are larger than the 5 kJ mol−1 except for DSD-PBEB95 and DSD-BLYP. B2GP-PLYP is the best XC functional among all DFAs on barrier heights and reaction energies; however, its deviation on X–H bond energies is sizeable. On the other hand, DSD-PBEB95 gives the smallest MADs on BEs among all DFAs, where its MAD is about 2 kJ mol−1. Deviations of DSD-BLYP on these BEs are slightly larger than those of DSD-PBEB95. These results indicate that the same functional behaves differently for different properties. FN-DMC is able to render reliable BEs for these X–H molecules and its MAD is smaller than that of most of the employed double hybrids, but it is still somewhat larger than that of DLPNO-CCSD(T)-F12. This is consistent with our previous findings51 that dissociation energies of involved molecules using FN-DMC are in excellent agreement with reference values. In summary, all DLPNO-CC methods, DSD-BLYP and DSD-PBEB95 as well as FN-DMC are able to provide the reliable BEs of those involved X–H molecules, and their accuracy is within chemical accuracy. However, the error of B3LYP, M06-2X, MN15, B2GP-PLYP and DSD-PBEP86 methods on BEs exceeds the chemical accuracy.
Deviations of dispersion-corrected B3LYP-D3(BJ), B2GP-PLYP-D3(BJ), DSD-PBEP86-D3(BJ) and DSD-PBEB95-D3(BJ) for those reaction energies and X–H bond energies are listed in Tables S3 and S4 of ESI.† Our results show that MADs of reaction energies are increased by 1–2 kJ mol−1 with B3LYP and DSD-PBEB95 when D3(BJ) correction is included. Nevertheless, MADs of B2GP-PLYP and DSD-PBEP86 with D3(BJ) correction are close to those of their dispersion-uncorrected counterparts. On the other hand, bond energies of these DFAs with D3(BJ) correction are rather close to those from their dispersion-uncorrected counterparts, except that dispersion correction decreases the MAD of B3LYP by about 2.5 kJ mol−1. These results show that dispersion correction is less important for these double hybrids.
4. Conclusion
H-abstraction reaction by HO2 from fuel molecules is a key reaction class during the autoignition processes at low-to-intermediate temperatures. The barrier heights of RH + HO2 reactions are calculated using DLPNO-CC and DFT methods as well as FN-DMC with the single-Slater–Jastrow trial wavefunction. Performance of different methods on these barrier heights is evaluated by comparison with CCSD(T)-F12b results extrapolated to the complete basis set limit. Our results show that DLPNO-CCSD(T)-F12 is in best agreement with CCSD(T)-F12b/CBS results among all the DLPNO-CC methods. Accuracy of DLPNO-CCSD(T1) on these barrier heights is similar to that of DLPNO-CCSD(T), which indicates that iterative treatment of triple excitations has a negligible effect on these barrier heights. Among all DFAs, B3LYP significantly underestimates barrier heights and B2GP-PLYP is the best in calculating these barrier heights, whose MAD is comparable to that of the highly accurate DLPNO-CCSD(T)-F12 method. MADs of MN15 and DSD-BLYP, DSD-PBEP86, and DSD-PBEB95 are around 2–3 kJ mol−1. Dispersion interactions will reduce these barrier heights and barrier heights are not improved when D3(BJ) correction is included in DFAs. Errors of FN-DMC on these barrier heights are somewhat larger than those from MN15 and double hybrid functionals, but it is smaller than that of M06-2X. In addition, barrier heights with all these methods except for B3LYP are within chemical accuracy for these reactions.Both reaction energies and bond energies of DLPNO-CCSD(T) and DLPNO-CCSD(T1) are in better agreement with CCSD(T)-F12b/CBS results than those from DLPNO-CCSD(T)-F12. All DFAs except for B3LYP can reach chemical accuracy on reaction energies, while only DSD-BLYP and DSD-PBEB95 can provide X–H bond energies within chemical accuracy. Among all employed DFAs, B2GP-PLYP gives the smallest MAD for reaction energies, but its MAD for X–H bond energies reaches 6.8 kJ mol−1. On the other hand, DSD-PBEB95 gives the smallest MAD of 2.2 kJ mol−1 for bond energies, but its MAD for reaction energies is the largest among these double hybrids. Dispersion correction shows a minor effect on reaction energies and bond energies in most cases. The error of FN-DMC is comparable to those of double hybrids on reaction energy, while it is smaller for X–H bond energies than that of most of the employed DFAs. According to our results, the DLPNO-CCSD(T)-F12 method can achieve highly accurate barrier heights, reaction energies and X–H bond energies for these RH + HO2 reactions at a relatively low computational cost, and it is applicable to H-abstraction reactions of larger molecules. On the other hand, MN15 and the investigated double hybrids can provide barrier heights and reaction energies for these reactions within chemical accuracy, while their error on X–H bond energies is rather pronounced except for DSD-BLYP and DSD-PBEB95. The error of FN-DMC is larger than those of MN15 and double hybrids, but its results on barrier heights, reaction energies and X–H bond energies are always within chemical accuracy. FN-DMC can also achieve reliable results for RH + HO2 reactions.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Natural Science Foundation of Shaanxi Province of China (grant no. 2021JQ-539), the Natural Science Foundation of Shaanxi Provincial Department of Education (grant no. 22JK0298) and the National Natural Science Foundation of China (grant no. 22103061).References
- P. G. Ashmore, Reaction kinetics, The Chemical Society, London, 1975 Search PubMed.
- F. Battin-Leclerc, O. Herbinet, P. A. Glaude, R. Fournet, Z. Zhou, L. Deng, H. Guo, M. Xie and F. Qi, Angew. Chem., 2010, 122, 3237–3240 CrossRef.
- Y. Mao, L. Yu, Z. Wu, W. Tao, S. Wang, C. Ruan, L. Zhu and X. Lu, Combust. Flame, 2019, 203, 157–169 CrossRef CAS.
- H. J. Curran, Proc. Combust. Inst., 2019, 37, 57–81 CrossRef CAS.
- B. Long, Y. Xia and D. G. Truhlar, J. Am. Chem. Soc., 2022, 144, 19910–19920 CrossRef CAS.
- S. E. Rawadieh, I. S. Altarawneh, M. A. Batiha, L. A. Al-Makhadmeh, M. H. Almatarneh and M. Altarawneh, Energ. Fuel., 2019, 33, 11781–11794 CrossRef CAS.
- J. Aguilera-Iparraguirre, H. J. Curran, W. Klopper and J. M. Simmie, J. Phys. Chem. A, 2008, 112, 7047–7054 CrossRef CAS PubMed.
- M. K. Altarawneh, B. Z. Dlugogorski, E. M. Kennedy and J. C. Mackie, Combust. Flame, 2013, 160, 9–16 CrossRef CAS.
- M. K. Altarawneh and B. Z. Dlugogorski, Combust. Flame, 2015, 162, 1406–1416 CrossRef CAS.
- W. Kohn, Rev. Mod. Phys., 1999, 71, 1253–1266 CrossRef CAS.
- Y. Zhao, N. Gonzalez-Garcıa and D. G. Truhlar, J. Phys. Chem. A, 2005, 109, 2012–2018 CrossRef CAS.
- Y. Zhao, N. E. Schultz and D. G. Truhlar, J. Chem. Theory Comput., 2006, 2, 36–382 Search PubMed.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215–241 Search PubMed.
- Y. Zhao and D. G. Truhlar, J. Chem. Theory Comput., 2008, 4, 1849–1868 CrossRef CAS PubMed.
- R. Peverati and D. G. Truhlar, Philos. Trans. R. Soc., A, 2014, 372, 20120476 CrossRef.
- L. Goerigk, A. Hansen, C. Bauer, S. Ehrlich, A. Najibi and S. Grimme, Phys. Chem. Chem. Phys., 2017, 19, 32184–32215 RSC.
- A. Karton, A. Tarnopolsky, J. F. Lamere, G. C. Schatz and J. M. L. Martin, J. Phys. Chem. A, 2008, 112, 12868–12886 CrossRef CAS.
- S. Kozuch, D. Gruzman and J. M. L. Martin, J. Phys. Chem. C, 2010, 114, 20801–20808 CrossRef CAS.
- S. Kozuch and J. M. L. Martin, Phys. Chem. Chem. Phys., 2011, 13, 20104–20107 RSC.
- S. Kozuch and J. M. L. Martin, J. Comput. Chem., 2013, 34, 2327–2344 CAS.
- S. Grimme, A. Hansen, J. G. Brandenburg and C. Bannwarth, Chem. Rev., 2016, 116, 5105–5154 CrossRef CAS PubMed.
- L. Goerigk and N. Mehta, Aust. J. Chem., 2019, 72, 563–573 CrossRef CAS.
- L. Goerigk, J. Chem. Theory Comput., 2014, 10, 968–980 CrossRef CAS.
- R. J. Bartlett and M. Musiał, Rev. Mod. Phys., 2007, 79, 291–352 CrossRef CAS.
- G. D. Purvis and R. J. Bartlett, J. Chem. Phys., 1982, 76, 1910–1918 CrossRef CAS.
- K. Raghavachari, G. W. Trucks and J. A. Pople, Chem. Phys. Lett., 1989, 157, 479–483 CrossRef CAS.
- G. Knizia, T. B. Adler and H. J. Werner, J. Chem. Phys., 2009, 130, 054104 Search PubMed.
- D. Feller, K. A. Peterson and J. G. Hill, J. Chem. Phys., 2010, 133, 184102 CrossRef PubMed.
- C. Riplinger and F. Neese, J. Chem. Phys., 2013, 138, 034106 CrossRef PubMed.
- E. Paulechka and A. Kazakov, J. Chem. Theory Comput., 2018, 14, 5920–5932 CrossRef CAS PubMed.
- D. G. Liakos, Y. Guo and F. Neese, J. Phys. Chem. A, 2020, 124, 90–100 CrossRef CAS PubMed.
- S. Mallick, B. Roy and P. Kumar, Comput. Theor. Chem., 2020, 1187, 112934 CrossRef CAS.
- I. Sandler, J. Chen, M. Taylor, S. Sharma and J. Ho, J. Phys. Chem. A, 2021, 125, 1553–1563 CrossRef CAS.
- Y. Guo, C. Riplinger, U. Becker, D. G. Liakos, Y. Minenkov, L. Cavallo and F. Neese, J. Chem. Phys., 2018, 148, 011101 CrossRef PubMed.
- W. M. C. Foulkes, L. Mitas, R. J. Needs and G. Rajagopal, Rev. Mod. Phys., 2001, 73, 33–83 CrossRef CAS.
- R. J. Needs, M. D. Towler, N. D. Drummond and P. L. Ríos, J. Phys.: Condens. Matter, 2010, 22, 023201 CrossRef CAS PubMed.
- B. M. Austin, D. Y. Zubarev and W. A. Lester Jr., Chem. Rev., 2012, 112, 263–288 CAS.
- M. J. Gillan, M. D. Towler and D. Alfé, Petascale computing opens up new vistas for quantum Monte Carlo, Psi-k Scientific Highlight of the Month, 2011, https://www.psi-k.org/newsletters/News_103/Highlight_103.pdf.
- L. K. Wagner and D. M. Ceperley, Rep. Prog. Phys., 2016, 79, 094501 CrossRef PubMed.
- M. Dubecký, L. Mitas and P. Jurečka, Chem. Rev., 2016, 116, 5188–5215 CrossRef.
- P. J. Reynolds, D. M. Ceperley, B. J. Alder and W. A. Lester Jr, J. Chem. Phys., 1982, 77, 5593–5603 CrossRef CAS.
- R. N. Barnett, P. J. Reynolds and W. A. Lester Jr, J. Chem. Phys., 1985, 82, 2700–2707 CrossRef CAS.
- P. J. Reynolds, R. N. Barnett, B. L. Hammond and W. A. Lester Jr, J. Stat. Phys., 1986, 43, 1017–1026 CrossRef.
- J. B. Anderson, J. Chem. Phys., 2016, 144, 166101 CrossRef PubMed.
- M. Barborini and L. Guidoni, J. Chem. Phys., 2012, 137, 224309 CrossRef.
- F. Fracchia, C. Filippi and C. Amovilli, J. Comput. Chem., 2014, 35, 30–38 CrossRef CAS.
- S. Pakhira, B. S. Lengeling, O. Olatunji-Ojo, M. Caffarel, M. Frenklach and W. A. Lester Jr, J. Phys. Chem. A, 2015, 119, 4214–4223 CrossRef CAS PubMed.
- Y. Kanai and N. Takeuchi, J. Chem. Phys., 2009, 131, 214708 CrossRef.
- P. E. Hoggan and A. Boufergune, Int. J. Quantum Chem., 2014, 114, 1150–1156 CrossRef CAS.
- Y. Wu, L. K. Wagner and N. R. Aluru, J. Chem. Phys., 2016, 144, 164118 CrossRef PubMed.
- X. Zhou and F. Wang, J. Comput. Chem., 2017, 38, 798–806 CrossRef CAS PubMed.
- K. Krongchon, B. Busemeyer and L. K. Wagner, J. Chem. Phys., 2017, 146, 124129 CrossRef.
- F. Pavošević, C. Peng, P. Pinski, C. Riplinger, F. Neese and E. F. Valeev, J. Chem. Phys., 2017, 146, 174108 Search PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- S. Y. Haoyu, X. He, S. L. Li and D. G. Truhlar, Chem. Sci., 2016, 7, 5032–5051 RSC.
- B. Long, J. L. Bao and D. G. Truhlar, J. Am. Chem. Soc., 2019, 141, 611–617 CrossRef CAS.
- Y. Xu, S. Xi, F. Wang and X. Li, J. Phys. Chem. A, 2019, 123, 3949–3958 CrossRef CAS.
- D. Feller, J. Comp. Chem., 1996, 17, 1571–1586 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Peterssonet, et al., Gaussian 16, Revision A.03, Gaussian, Inc., Wallingford, CT, 2016 Search PubMed.
- A. Tajti, P. G. Szalay, A. G. Csaszar, M. Kállay, J. Gauss, E. F. Valeev, B. A. Flowers, J. Vázquez and J. F. Stanton, J. Chem. Phys., 2004, 121, 11599 CrossRef CAS.
- J. G. Hill, K. A. Peterson, G. Knizia and H.-J. Werner, J. Chem. Phys., 2009, 131, 194105 CrossRef.
- H.-J. Werner, P. J. Knowles, R. D. Amos, A. Bernhardsson, A. Berning, P. Celani, D. L. Cooper, M. J. O. Deegan, A. J. Dobbyn, F. Eckert, C. Hampel, G. Hetzer, T. Korona, R. Lindh, A. W. Lloyd, S. J. McNicholas, F. R. Manby, W. Meyer, M. E. Mura, A. Nicklass, P. Palmieri, R. Pitzer, G. Rauhut, M. Schtuz, U. Schumann, H. Stoll, A. J. Stone, R. Tarroni and T. Thorsteinsson, MOLPRO, University of Birmingham, 1st edn, 2006 Search PubMed.
- T. H. Dunning Jr, J. Chem. Phys., 1989, 90, 1007–1023 CrossRef.
- R. A. Kendall, T. H. Dunning Jr and R. J. Harrison, J. Chem. Phys., 1992, 96, 6796–6806 CrossRef CAS.
- K. L. Schuchardt, B. T. Didier, T. Elsethagen, L. Sun, V. Gurumoorthi, J. Chase, J. Li and T. L. Windus, J. Chem. Inf. Model., 2007, 47, 1045–1052 CrossRef CAS.
- F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 73–78 CAS.
- F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2017, 8, e1327 Search PubMed.
- D. G. Liakos, M. Sparta, M. K. Kesharwani, J. M. L. Martin and F. Neese, J. Chem. Theory Comput., 2015, 11, 1525–1539 CrossRef CAS.
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS.
- K. Doblhoff-Dier, J. Meyer, P. E. Hoggan, G. J. Kroes and L. K. Wagner, J. Chem. Theory Comput., 2016, 12, 2583–2597 CrossRef CAS.
- M. Burkatzki, C. Filippi and M. Dolg, J. Chem. Phys., 2007, 126, 234105 CrossRef CAS PubMed.
- M. Casula, Phys. Rev. B: Condens. Matter Mater. Phys., 2006, 74, 161102 CrossRef.
- P. R. C. Kent, R. J. Needs and G. Rajagopal, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 12344–12351 CrossRef CAS.
- J. Toulouse and C. J. Umrigar, J. Chem. Phys., 2007, 126, 084102 CrossRef PubMed.
- B. Long, J. L. Bao and D. G. Truhlar, J. Am. Chem. Soc., 2016, 138, 14409–14422 CrossRef CAS.
- Z. Rolik, L. Szegedy, I. Ladjánszki, B. Ladóczki and M. Kállay, J. Chem. Phys., 2013, 139, 094105 CrossRef.
- J. F. Stanton, J. Gauss, L. Cheng, M. E. Harding and P. G. Szalay, CFOUR, A Quantum-Chemical Program Package, with contributions from A. A. Auer, R. J. Bartlett, U. Benedikt, C. Berger, D. E. Bernholdt, and Y. J. Bomble et al., https://www.cfour.de/ (accessed Jan 10, 2019).
- G. Jansen and B. A. Hess, Phys. Rev. A, 1989, 39, 6016–6017 CrossRef.
- S. Kanchanakungwankul, J. Zheng, I. M. Alecu, B. J. Lynch, Y. Zhao and D. G. Truhlar, https://comp.chem.umn.edu/freqscale.
- R. Peverati and D. G. Truhlar, https://comp.chem.umn.edu/db/dbs/mgae109.html.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2cp04463c |
| This journal is © the Owner Societies 2023 |