
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNanoscale domain imaging of Li-rich disordered rocksalt-type cathode materials with X-ray spectroscopic ptychography†
Hideshi
Uematsu
 *abc,
Nozomu
Ishiguro
*acd,
Masaki
Abe
abc,
Shuntaro
Takazawa
abc,
Jungmin
Kang
c,
Itsuki
Konuma
e,
Naoaki
Yabuuchi
e and
Yukio
Takahashi
*acdf
*abc,
Nozomu
Ishiguro
*acd,
Masaki
Abe
abc,
Shuntaro
Takazawa
abc,
Jungmin
Kang
c,
Itsuki
Konuma
e,
Naoaki
Yabuuchi
e and
Yukio
Takahashi
*acdf
aInternational Center for Synchrotron Radiation Innovation Smart (SRIS), Tohoku University, 2-1-1 Katahira, Aoba-ku, Sendai 980-8577, Japan. E-mail: hideshi.uematsu.r5@dc.tohoku.ac.jp; nozomu.ishiguro.c1@tohoku.ac.jp; ytakahashi@tohoku.ac.jp
bDepartment of Metallurgy, Materials Science and Materials Processing, Graduate School of Engineering, Tohoku University, 6-6-2 Aoba-yama, Aoba-ku, Sendai 980-8579, Japan
cRIKEN SPring-8 Center, 1-1-1, Koto, Sayo, Hyogo 679-5148, Japan
dInstitute of Multidisciplinary Research for Advanced Materials (IMRAM), Tohoku University, 2-1-1 Katahira, Aoba-ku, Sendai 980-8577, Japan
eDepartment of Chemistry and Life Science, Yokohama National University, 79-5 Tokiwadai, Hodogaya-ku Yokohama, Kanagawa 240-8501, Japan
fInstitute for Materials Research, Tohoku University, 2-1-1 Katahira, Aoba-ku, Sendai, Miyagi 980-8577, Japan
First published on 10th January 2023
Abstract
Lithium-rich disordered rocksalt-type cathode materials are promising for high-capacity and high-power lithium-ion batteries. Many of them are synthesized by mechanical milling and may have heterogeneous structures and chemical states at the nanoscale. In this study, we performed X-ray spectroscopic ptychography measurements of Li-rich disordered rocksalt-type oxide particles synthesized by mechanical milling before and after delithiation reaction at the vanadium K absorption edge, and visualized their structures and chemical state with a spatial resolution of ∼100 nm. We classified multiple domains with different chemical states via clustering analysis. A comparison of the domain distribution trends of the particles before and after the delithiation reaction revealed the presence of domains, suggesting that the delithiation reaction was suppressed.
1 Introduction
Lithium-ion batteries have become indispensable in our society for use in mobile devices, automobiles, and even for storing surplus electricity. Cathode active materials with high energy density and fast charge/discharge capability are required. Li-rich metastable cathode materials have recently been attracting attention for the development of the cathode materials that satisfy the requirements. In Li-rich disordered rocksalt (DRS)-type cathode materials (LixTM2-xO2, x > 1.1 TM: transition metal elements; e.g., V, Ti, Mn, Nb, Cr, Mo), Li and TM randomly occupy the same site.1–7 They achieve a higher reversible capacity than conventional layered rocksalt type cathode active materials such as commercial LiCoO2 and Li–Ni–Mn–Co–O2 compounds.1 Although such Li-rich DRS-type phases are often difficult to synthesize because of the need for sintering at high temperatures, they are synthesized by mechanical milling (MM).1,8 MM is a simple and powerful approach to obtaining metastable, amorphous, nanocomposite, and nanosized particles by mixing and crushing powders.9 It is expected that the reversible capacity and rate capability can be increased by reducing the particle size to the nanoscale by MM.4 Note that MM is a physical mixing process; hence, there are chemical and structural heterogeneities in the synthesized materials. Chemical and structural heterogeneities will consequently lead to a heterogeneous de-/lithiation reaction distribution, where reactions proceed only in a certain domain. Moreover, such a de-/lithiation reaction distribution will cause the capacity fade with local overcharge/overdischarge and structural degradation.10–13 Therefore, it is crucial to observe the heterogeneous structures at the nanoscale and identify the domains where the de-/lithiation reaction proceeds.X-ray spectroscopic microscopy using synchrotron radiation has been extensively used to visualize heterogeneous structures in bulk materials.14,15 In particular, X-ray spectroscopic ptychography16 is a promising tool for obtaining spatially resolved X-ray absorption fine structure (XAFS) spectra and phase images with a spatial resolution of 40–80 nm, independent of lens fabrication accuracy. Chemical state imagings of the gold nanoparticles,17 lithium iron phosphate cathode,18,19 Pt-supported ceria–zirconia (CZ) solid-solution catalyst,20,21 and zinc-oxide coated nanoporous alumina22 have been performed by analyzing spatially resolved XAFS spectra obtained by X-ray spectroscopic ptychography. In addition, X-ray spectroscopic ptychography measurements have been applied in the determination of crystal orientation in polycrystalline vanadium oxide using the linear polarization dependency of XAFS23 and the local coordination structure in manganese oxide in a nanoscale area using an extended X-ray absorption fine structure.24 X-ray spectroscopic microscopy measurements provide spatially resolved chemical state information from image data with more than 103 pixels. Machine learning methods can help reconstruct such huge complex data into lower-dimensional data that is easier for humans to understand. Machine learning methods are expected to mine hidden material information and promote better materials design. Thus, the detection of inert domains in LiCoO2 particles on electrodes,25 the analysis of oxygen diffusion pathways in CZ particles,20 and the classification of Ni K-edge XAFS spectra of Ni-rich cathode active material particles26 have been reported. Recently, we have reported the presence of multiple domains in spinel-type lithium nickel manganese oxide cathode particle by analyzing the correlation between the valence and the composition.27 The combination of X-ray spectroscopic ptychography and machine learning is expected to enable the visualization of nanoscale domains.
In this study, we selected 0.25Li2O−0.75LiVO2 (Li10/7V6/7O2) particles as an example of Li-rich DRS-type cathode material synthesized by MM. X-ray spectroscopic ptychography measurements were performed at the vanadium (V) K edge. The chemical state maps reflecting the V valence, the V site symmetry, and the composition were derived from the spatially resolved X-ray absorption near-edge structure (XANES) and phase images. Moreover, we visualized the domains with different chemical states by clustering analysis.
2 Experimental
2.1 Sample preparation
Li10/7V6/7O2, which is a solid solution sample, x = 0.25 in xLi2O −(1 − x)LiVO2, was synthesized by MM (denoted as as-prepared Li10/7V6/7O2). The details of the synthesis processes are presented in ESI.† To simulate the half charged state of Li10/7V6/7O2, chemical delithiation was performed on as-prepared Li10/7V6/7O2 using iodine in acetonitrile (denoted as delithiated Li10/7V6/7O2). We prepared LiVO2 (ESI†) and LiVO3 (ESI†) as standard compounds in spectroscopic ptychogrpahy. X-ray diffraction (XRD) patterns of as-prepared Li10/7V6/7O2 (Fig. S1, ESI†) showed peaks corresponding to those of Fm![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m space groups, but no peaks corresponding to those of layered rocksalt-type LiVO2 (space group Rm) are found. The reversible capacity of the Li10/7V6/7O2 electrode reaches > 350 mA h g−1 with good capacity retention (Fig. S2, ESI†). This capacity was superior to that of nanosized Li1.2V0.6Nb0.2O2 synthesized by MM6 and that of Li3PO4, where Li atoms occupy tetrahedral sites as in Li2O, integrated into LiMnO2.28
m space groups, but no peaks corresponding to those of layered rocksalt-type LiVO2 (space group Rm) are found. The reversible capacity of the Li10/7V6/7O2 electrode reaches > 350 mA h g−1 with good capacity retention (Fig. S2, ESI†). This capacity was superior to that of nanosized Li1.2V0.6Nb0.2O2 synthesized by MM6 and that of Li3PO4, where Li atoms occupy tetrahedral sites as in Li2O, integrated into LiMnO2.28
2.2 X-ray spectroscopic ptychography
The as-prepared Li10/7V6/7O2, delithiated Li10/7V6/7O2, LiVO2, and LiVO3 particles were dispersed in respective acetonitrile solutions, dropped on respective 500 nm-thick Si3N4 membranes (Norcada) in argon atmosphere glove box, and kept in an argon atmosphere until mounting on the piezoelectric stage inside the ptychography chamber in vacuum. X-ray spectroscopic ptychography experiments were performed at the BL29XUL beamline of SPring-8. The incident X-ray was monochromatized by the Si(111) double-crystal monochromator and focused using Kirzpatrick–Baez mirrors to a size of 300 nm (FHWM). Forty X-ray energies between 5420 and 5540 eV were selected with the smallest energy gap of 0.5 eV. The samples were placed in the focal plane and raster-scanned with a step width of 150 nm. The exposure time at each position was 1 s. Multiple diffraction patterns were collected using a pixel array detector (Dectris EIGER 1 M). The measurement of all energy points took 4–8 h per particle. Probe and projected sample images were reconstructed by an extended ptychographic iterative engine29 with the Kramers–Kronig relationship (KKR) constraint.30 The input of initial images of the sample were unity, and those of the probe were calculated on the basis of the experimental setup. Iterative calculations were performed for 800 cycles, and the KKR constraint was applied every 50 cycles after the 500th iteration.2.3 Curve-fitting analysis for spatially resolved XANES
To evaluate features of the spatially resolved XANES, such as the height of the edge jump and the height and width of the pre-edge peak, the curve-fitting analysis of V K-edge XANES was performed on each pixel using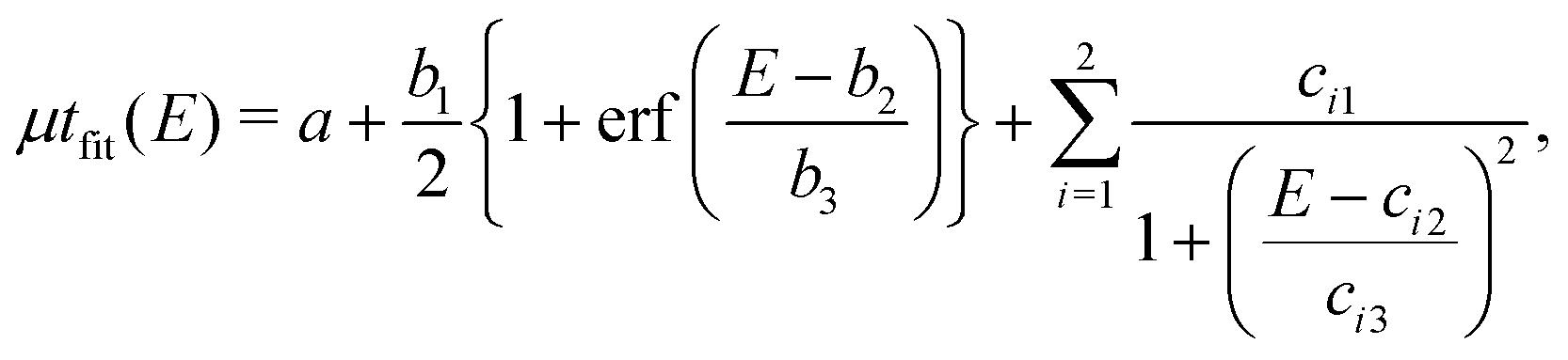 | (1) |
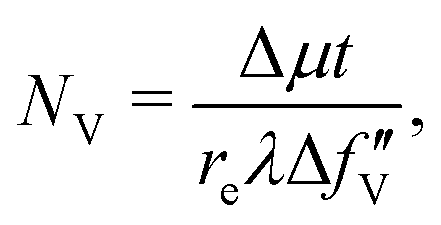 | (2) |
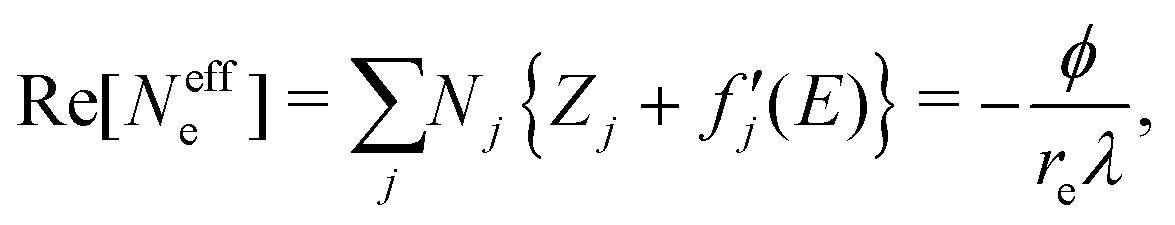 | (3) |
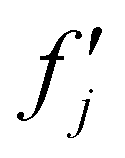 and
and 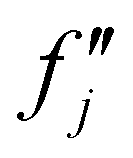 are the real and imaginary parts of the anomalous dispersion term of the atomic scattering factor, respectively, and
are the real and imaginary parts of the anomalous dispersion term of the atomic scattering factor, respectively, and 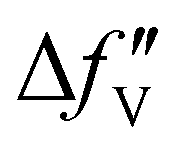 is the difference between the
is the difference between the 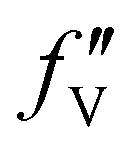 values before and after the edge jump. The values of
values before and after the edge jump. The values of 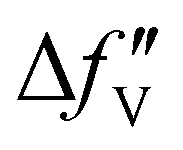 was estimated to be 3.53 from database references,31 and
was estimated to be 3.53 from database references,31 and 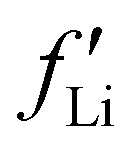 ,
, 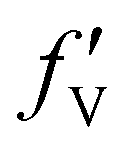 , and
, and 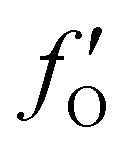 are 0.00278, −7.14, and 0.0757, respectively. Then, Re[Neffe]/NV is given as
are 0.00278, −7.14, and 0.0757, respectively. Then, Re[Neffe]/NV is given as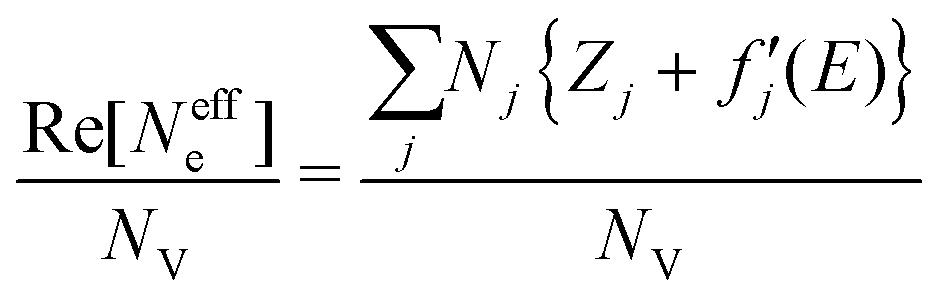 | (4) |
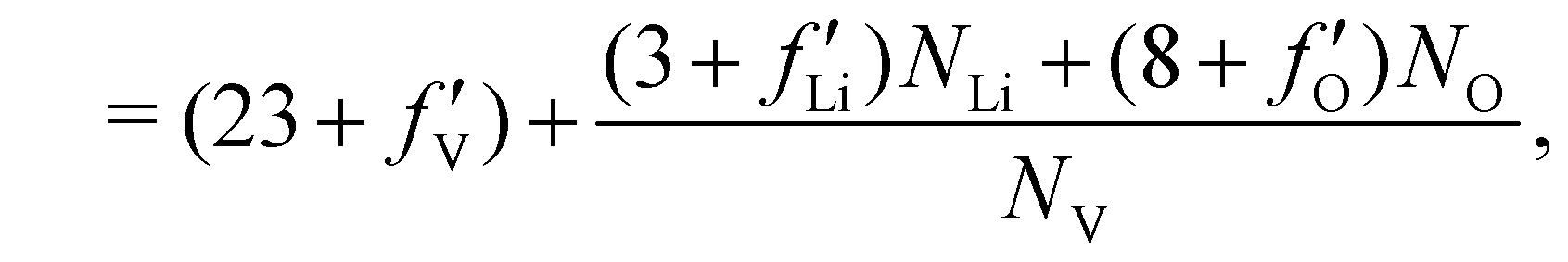 | (5) |
2.4 Clustering analysis
We performed clustering analysis by the k-means method32 based on the similarity between the pixels in three-dimensional data space with descriptors such as edge energy, PE/Δμt, and Re[Neffe]/NV for the data clustering. The number of clusters was set to 6, where the slope of the decrease in the within-cluster sum of squared error is gradual (Fig. S3, ESI†), as determined by the elbow method.333 Results and discussion
3.1 Reconstructed images and spatially resolved XANES
Fig. 1(a)–(c), respectively, show the field-emission scanning electron microscopy (SEM), reconstructed absorption, and reconstructed phase images of as-prepared Li10/7V6/7O2 particles P1–P3 and delithiated Li10/7V6/7O2 particles D1–D3. The pixel size of the reconstructed images is 15 nm. The absorption image at the energy above the V K-edge depends on the number of V atoms along the incident X-ray direction. On the other hand, the phase shift originates from the amount of electrons, which reflects the amount of all existing atoms, including the lighter Li and O. The full-period spatial resolutions of the reconstructed images were estimated to be better than 80–120 nm using the phase retrieval transfer function (Fig. S5, ESI†). The shape of the particles in the phase images (Fig. 1(c)) is in good agreement with that in the SEM images (Fig. 1(a)).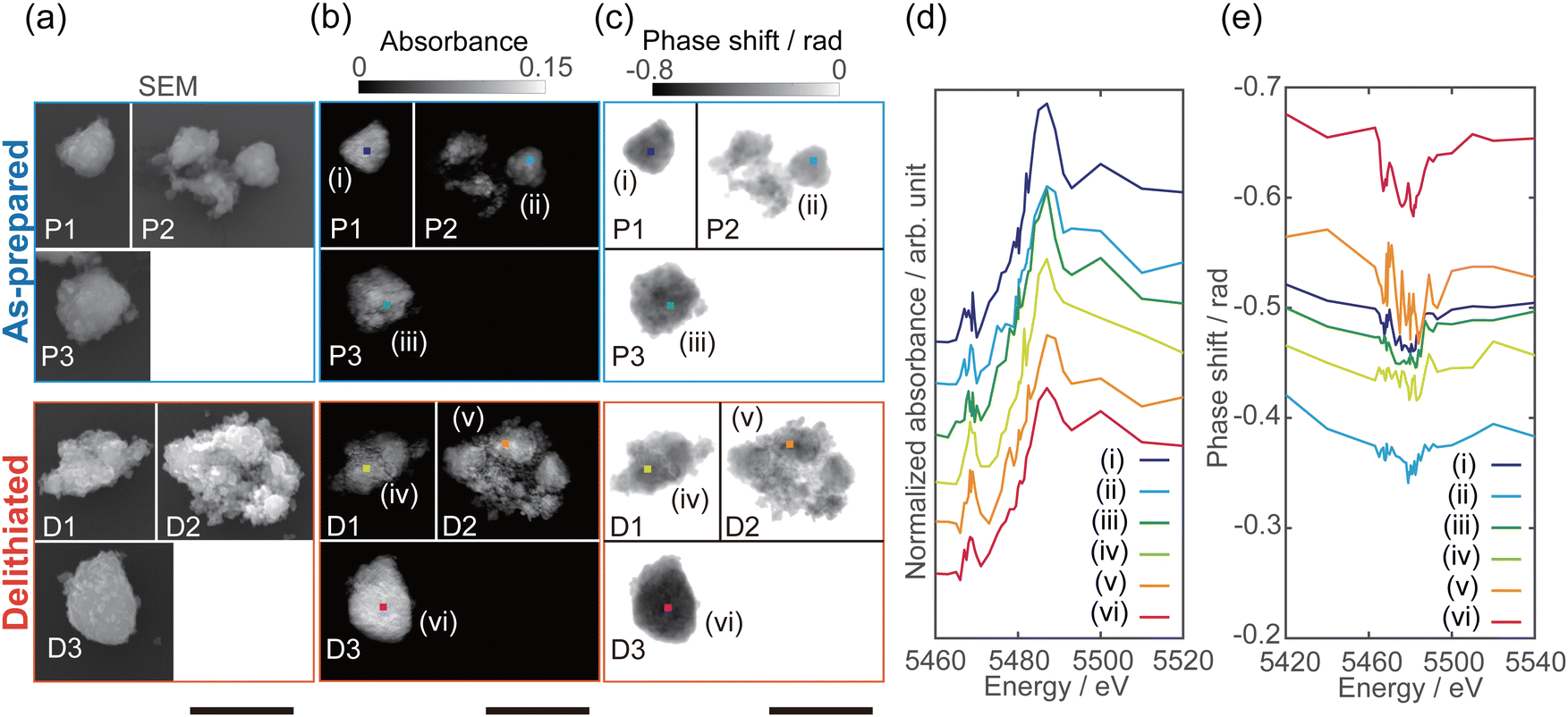 | ||
| Fig. 1 (a–c) Images of as-prepared Li10/7V6/7O2 particles P1–P3 and delithiated Li10/7V6/7O2 particles D1–D3. (a) SEM images. (b) Reconstructed absorption images at 5540 eV. (c) Reconstructed phase shift images at 5420 eV. The pixel size of reconstructed images is 15 nm. The scale bars are 2 μm. (d) XANES and (e) phase spectra of as-prepared and delithiated Li10/7V6/7O2 particles. Offset is added to the V K-edge XANES spectra in (d). The extracted size of both spectra (d and e) is 120 × 120 nm2. | ||
By stacking the reconstructed absorption and phase images, spatially resolved V K-edge XANES and phase spectra were successfully obtained, as shown in Fig. 1(d) and (e). The spatially resolved XANES spectra are in good agreement with the conventional XAFS spectra (Fig. S4, ESI†) of the pellet sample, where the absorption edge energy and pre-edge peak intensity were increased in delithiated Li10/7V6/7O2 compared with those in as-prepared Li10/7V6/7O2. Chaurand et al. have reported that the V K-edge energy increases with the V valence.34 The shift of the absorption edge should reflect the valence change with delithiation (ESI† Fig. S4). As shown in Fig. 1(d), the edge energy differed between particles, and there are regions that do not follow the trend of the edge energy in the conventional XAFS spectra.
The characteristic peak around 5466 eV, known as the pre-edge peak, is related to the electronic transition of the 1s electron to 3d orbitals in compounds of 3d-transition metals such as V, Mn, and Ti. The pre-edge peak intensity indicates the degree of d–p hybridization, allowing forbidden 1s → 3d transition; the degree of hybridization of 4p orbitals or ligand 2p orbitals with the 3d orbitals depends on the site symmetry (i.e., coordination structure).35,36 Chaurand et al. have reported that the pre-edge peak intensities in the V K-edge XANES spectra increase in the order of the tetrahedral structure (Na3VO4), the pyramidal structure (V2O5), and the octahedral structure (Ca3(V10O28)−17H2O).34 Nakajima et al. have proposed that the V atoms in the DRS-type Li–Nb–V–O2 cathode material reversibly migrate from octahedral to tetrahedral sites during charging, on the basis of the result of the operando XAFS measurement.2 In addition, Baur et al. have reported that formation of tetrahedrally coordinated V in the amorphous phase from the DRS-type Li2VO2F cathode during charging, based on the X-ray total scattering data.3 As shown in Fig. 1(d), the pre-edge peak intensities in the delithiated Li10/7V6/7O2 particles increase compared with those in the as-prepared Li10/7V6/7O2 particles, indicating an increase in the V valence and/or the amount of tetrahedrally coordinated V.
3.2 Chemical state mapping
Chemical state mapping was performed by the curve-fitting analysis of spatially resolved XANES spectra and the phase images. We succeeded in extracting the following three chemical state maps: edge energy, Re[Neffe]/NV, and normalized pre-edge peak intensity (PE/Δμt), which depend on the V valence, (Li + V + O)/V molar ratio, and V site symmetry, respectively. Fig. 2(a)–(c) show the derived chemical state maps of as-prepared and delithiated Li10/7V6/7O2 particles. ESI† S6 show the reconstructed images of LiVO2 and LiVO3, and their spatially resolved XANES and phase spectra. Fig. S7 (ESI†) show the chemical state maps of LiVO2 and LiVO3. As shown in Fig. 2(a)–(c), there are a gradient from the surface and a mottled appearance at the submicron order such as heterogeneous distributions, while such distributions are not observed in the chemical state maps in LiVO2 and LiVO3. The heterogeneous distributions within each as-prepared Li10/7V6/7O2 particle and within each delithiated Li10/7V6/7O2 particle are also seen in the histograms of chemical state maps for all measured particles (see ESI† S8).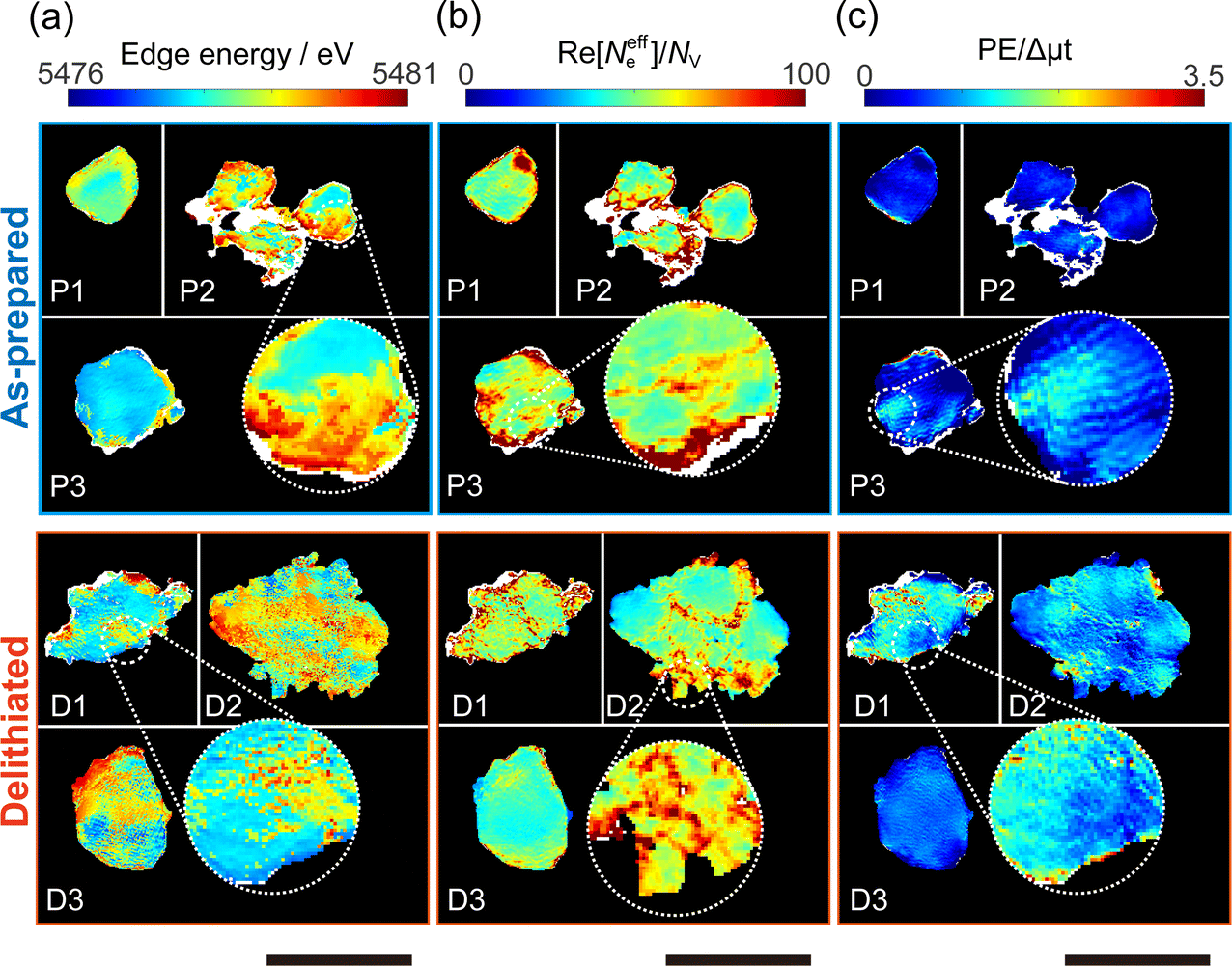 | ||
| Fig. 2 Chemical state maps of as-prepared Li10/7V6/7O2 particles P1–P3 and delithiated Li10/7V6/7O2 particles D1–D3. (a) Edge energy. (b) Re[Neffe]/NV (c) PE/Δμt. The pixel size of chemical state maps is 15 nm. The scale bars are 2 μm. Values in white areas are missing because fitting was not possible. | ||
Now, we discuss the heterogeneous distribution of chemical state maps at the particle level. As shown in the enlarged view of the as-prepared Li10/7V6/7O2 particle P2 in Fig. 2(a), the V valence increases from the bulk to the surface of the particle toward V5+, suggesting that oxidation occurs from the surface to the bulk during synthesis. As for the delithiated Li10/7V6/7O2 particles, there were some regions remaining V3+, especially at the particle D1, even after the chemical delithiation. In the enlarged view of the delithiated Li10/7V6/7O2 particle D1 shown in Fig. 2(a), the region close to V5+ is localized, indicating that the delithiation reaction is inhibited within the particle and that the particle D1 has a lower delithiation reactivity than the other particles. As shown in Fig. 2(b), a mottled appearance is observed in Re[Neffe]/NV maps for the as-prepared Li10/7V6/7O2 particle P3 and also the delithiated Li10/7V6/7O2 particle D2, indicating the heterogeneous distribution of Li- or O-rich phases such as Li2O, implying that LiVO2 and Li2O were not completely mixed by MM. As shown in the enlarged image of the delithiated Li10/7V6/7O2 particle D1 in Fig. 2(c), the high PE/Δμt region is distributed around the region close to V5+ shown in Fig. 2(a), which may be due to the relationship between the delithiation reaction and the V site symmetry. Lee et al. have proposed that Li moves in the DRS phase through the octahedral–tetrahedral–octahedral sites diffusion pathway on an atomic scale (percolation theory).5 Taking into consideration this atomic-scale Li diffusion pathway, the V ions occupying tetrahedral sites are considered to be a barrier to Li diffusion. The distribution of PE/Δμt in the surrounding region close to V5+ may reflect the inhibition of Li diffusion.
3.3 Nanoscale domain imaging using clustering analysis
Domains with different characteristics, such as regions where it is difficult/easy for the delithiation reaction and impurity phases to proceed are expected to have different correlations between the chemical states. We performed clustering (grouping) analysis based on the similarity of chemical states among edge energy, Re[Neffe]/NV, and PE/Δμt, and grouped all pixels into 6 clusters. Fig. 3(a) shows the real-space distributions of 6 clusters, which are domain structures. We succeeded in visualizing domains with different correlations between V valence, composition, and V site symmetry. As far as we can see from Fig. 3(a), the domain sizes were estimated to be between 100–1000 nm. However, the images in Fig. 3(a) are generated projection images along incident X-rays (Fig. 2), and we cannot distinguish the overlapping domains. The domain structures are different between as-prepared and delithiated Li10/7V6/7O2 particles. Cluster 1 is more abundant in the as-prepared Li10/7V6/7O2 particles than in the delithiated Li10/7V6/7O2 particles and is also observed in the delithiated Li10/7V6/7O2 particle D1. Cluster 2 is partially observed in the as-prepared Li10/7V6/7O2 particle P3 and is abundant in the delithiated Li10/7V6/7O2 particle D1. Cluster 3 is distributed in delithiated Li10/7V6/7O2 particles and is rarely observed in as-prepared Li10/7V6/7O2 particles. Cluster 4 is observed around the surface of a particle, especially in the as-prepared Li10/7V6/7O2 particle P2. Clusters 5 and 6 are less abundant than other cluster, and are distributed around the surface of a particle.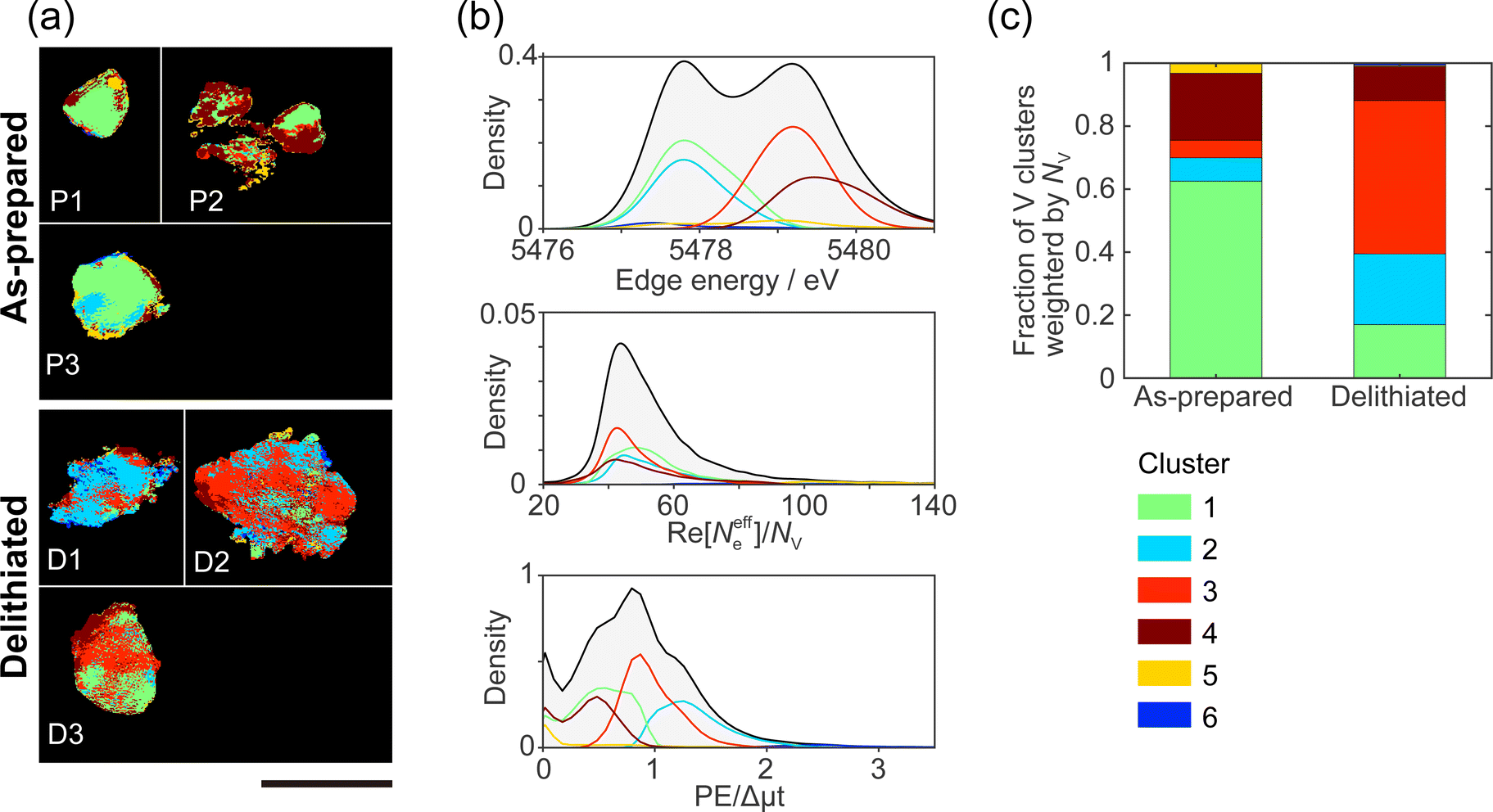 | ||
| Fig. 3 Results of clustering analysis. (a) Spatial distribution of clusters 1–6. The scale bar is 2 μm. (b) Projected probability density distribution of (top) edge energy, (center) Re[Neffe]/NV, and (bottom) PE/Dmt. (c) Fraction of clusters weighted by NV in as-prepared and delithiated Li10/7V6/7O2 particles. Each cluster is shown by the same color in (a), (b), and (c). | ||
Fig. 3(b) shows the probability density distributions of the chemical states. The three-dimensional scattering plots and two-dimensional probability density are shown in ESI† Fig. S9. As mentioned in Section 3.1, the edge energy depends on the V valence and increases with the V valence. The high PE/Δμt reflects a tetrahedral structure, and the low PE/Δμt reflects an octahedral structure. The high Re[Neffe]/NV reflects Li- and/or O-rich composition. In cluster 1, the V valence tends to be V3+ compared with those in clusters 3–5. Cluster 1 tends to be an octahedral structure and to be Li- and O-rich compared with clusters 3 and 4. In cluster 2, the V valence is close to V3+, as in cluster 1, but cluster 2 tends to be a tetrahedral structure. Cluster 2 tends to be Li- and O-poor compared with clusters 3 and 4. In cluster 3, the V valence tends to be V5+. Cluster 3 tends to be a tetrahedral structure. Cluster 3 is relatively Li- or O-poor compared with clusters 1 and 2. In cluster 4, the V valence tends to be V5+. Cluster 4 tends to be an octahedral structure. Clusters 5 and 6 are mostly located at the outer edges of the particles, and Re[Neffe]/NV values in clusters 5 and 6 are almost two times larger than those in clusters 1–4. The tendency of the chemical states for each cluster is also seen in the XANES spectra averaged for each cluster (ESI† Fig. S9(e)).
Fig. 3(c) shows the fractions of the clusters in the as-prepared and delithiated Li10/7V6/7O2 particles. The fractions of the clusters are weighted by NV. Cluster 1 is predominant for the as-prepared Li10/7V6/7O2 particles and a minority for the delithiated Li10/7V6/7O2 particles. On the other hand, cluster 3 is a minority for the as-prepared Li10/7V6/7O2 particles and predominant for the delithiated Li10/7V6/7O2 particles. The reversal of the fraction of clusters 1 and 3 could be due to the change of cluster 1 into cluster 3 with delithiation. Konuma et al. have reported that octahedrally coordinated V3+ reversibly migrates to tetrahedrally coordinated V5+ during charge/discharging process in Li8/7Ti2/7V4/7O2 DRS-type cathode, and also the migration of V ion is related to the invariant volume in electrode.7 The change in the fractions of cluster 1 and 3 can be related to the previous study. X-ray spectroscopic ptychography will provide information between macroscopic and atomic structures by visualizing the domain structure. The fraction of cluster 2 is higher in the delithiated Li10/7V6/7O2 particles than in the as-prepared Li10/7V6/7O2 particles. There are two possible origins of cluster 2. The first possibility is that the tetrahedral site is occupied by V3+ in the MM stage, and it remains without undergoing the delithiation reaction. This may be because V ions remain in the tetrahedral site when moving from one octahedral site to the neighboring octahedral site during the formation of the DRS-type structure in MM synthesis. The second possibility is that cluster 2 is generated from other clusters during reactions associated with the delithiation reaction, e.g., disproportionation reaction,37i.e., 2V4+ → V3+ + V5+, and/or the structural changes to an amorphous phase.3 Most charging and discharging are caused by a redox of V3+/V5+. Thus, cluster 2 that keeps V3+ in delithiated Li10/7V6/7O2 particles will not contribute to the capacity during charging. The domain belonging to cluster 2 will become a barrier to Li diffusion. To improve the capacity and rate capability of cathode materials, it is important to avoid producing the domain belonging to cluster 2 during MM synthesis and charging/discharging processes, for example, doping additives such as Nb stabilizing the atomic framework of DRS phase2 might be effective.
4 Conclusions
We have successfully visualized the chemical state distributions of the V valence, V site symmetry, and composition of Li-rich DRS-type vanadium oxide particles as synthesized by MM and their delithiated particles, using X-ray spectroscopic ptychography. The clustering analysis resulted in the division of chemical state maps into 6 clusters. The real-space distribution of the 6 clusters is the heterogeneous domain structure within and between particles, even in the synthesis stage. There were domains where the delithiation reaction proceeds or is inhibited in the particles. It is necessary for the delithiation reaction to occur in all particles to maximize the charge–discharge capacity.In this study, the resolution of X-ray spectroscopic ptychography was ∼100 nm. The spatial resolution depends mainly on the performance of the synchrotron source. We measured chemically delithiated particles to investigate the domain structure. Although the kinetics of electrochemical delithiation is expected to be more complex than that of chemical delithiation because of the presence of the other battery components, such as the electrolyte, binder, and conductive additives, the structural (thermodynamical) changes are by either electrochemical or chemical delithiation is considered similar.38 Still, operando measurement under working or simulated conditions in the actual battery is required to investigate the proceeding of the delithiation reaction in the electrode. Furthermore, operando measurements coupling with the impedance method that provides electrical signals of surface degradation, cracks, etc., will be helpful to investigate relationships between the impedance and microstructure changes. However, there are two large challenges to affording operando measurement. First is the improvement of the signal-to-noise ratio of spatially resolved XAFS spectra. The transmitted X-ray is attenuated to 1/10 in the 500 μm electrolyte at 5500 eV, and scattering from the electrolyte should also be considerable. Therefore, the total cell thickness, including the X-ray window, is preferably restrained to less than 50 μm. The second is measurement time. X-ray spectroscopic ptychography measurement takes 4–8 hours to obtain spatially resolved XANES region spectra of a μm size particle.27 Thus, it can only visualize a stable or steady state of samples presently. Therefore it is necessary to shorten the measurement time to measure more dynamic aspects of electrochemical reactions in various conditions. To overcome the above challenges, low-emittance beam with higher brightness X-rays, high dynamic-range detector to capture high brightness X-rays, and sparse measurement to reduce energy points39 are required. In the synchrotron radiation facilities with a low-emittance storage ring, which have been built or are under construction at several locations around the world, higher brightness will be provided. In the near future, we believe that X-ray spectroscopic ptychography will contribute to the design of functional materials with complex and heterogeneous structures at the nanoscale.
Author contributions
H. U.: conceptualization, methodology, software, investigation, visualization, writing – original draft preparation. N. I.: conceptualization, methodology, software, investigation, project administration, writing – reviewing and editing. M. A., S. T. and J. K. methodology, investigation. I. K.: investigation, resources, writing – reviewing and editing. N. Y.: resources, supervision, writing – reviewing and editing. Y. T.: conceptualization, methodology, project administration, writing – reviewing and editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Japan Society for the Promotion of Science (JSPS) KAKENHI (Grant No. JP18H05253, JP19H05814, JP20K15375, JP20K20523 and JP22J21779), Iketani Science and Technology Foundation (ISTF) (Grant No. 0331102-A), and Ministry of Education, Culture, Sports, Science and Technology of Japan (MEXT) program: “Data Creation and Utilization-Type Material Research and Development” project (Grant No. JPMXP1122712807). It was also supported in part by the “Dynamic Alliance for Open Innovation Bridging Human, Environment and Materials” project from the MEXT. The conventional transmission XAFS measurement was performed on BL37XU at SPring-8 with the approval of the Japan Synchrotron Radiation Research Institute (JASRI) (Proposal No. 2021A1483 and 2021B1566).Notes and references
- R. J. Clément, Z. Lun and G. Ceder, Energy Environ. Sci., 2020, 13, 345–373 RSC.
- M. Nakajima and N. Yabuuchi, Chem. Mater., 2017, 29, 6927–6935 CrossRef.
- C. Baur, C. Baur, M. E. Lǎcǎtuşu, M. Fichtner, M. Fichtner and R. E. Johnsen, ACS Appl. Mater. Interfaces, 2020, 12, 27010–27016 CrossRef PubMed.
- N. Yabuuchi, Curr. Opin. Electrochem., 2022, 34, 100978 CrossRef.
- J. Lee, A. Urban, X. Li, D. Su, G. Hautier and G. Ceder, Science, 2014, 343, 519–522 CrossRef PubMed.
- R. Qi, B. D. Campéon, I. Konuma, Y. Sato, Y. Kaneda, M. Kondo and N. Yabuuchi, Electrochemistry, 2022, 90, 4–9 CrossRef.
- I. Konuma, D. Goonetilleke, N. Sharma, T. Miyuki, S. Hiroi, K. Ohara, Y. Yamakawa, Y. Morino, H. B. Rajendra, T. Ishigaki and N. Yabuuchi, Nat. Mater., 2022 DOI:10.1038/s41563-022-01421-z.
- J. Chable, C. Baur, J. H. Chang, S. Wenzel, J. M. García-Lastra and T. Vegge, J. Phys. Chem. C, 2020, 124, 2229–2237 CrossRef CAS.
- T. Prasad Yadav, R. Manohar Yadav and D. Pratap Singh, Nanosci. Nanotechnol., 2012, 2, 22–48 CrossRef.
- J. Liu, M. Kunz, K. Chen, N. Tamura and T. J. Richardson, J. Phys. Chem. Lett., 2010, 1, 2120–2123 CrossRef CAS.
- Y. Yang, R. Xu, K. Zhang, S.-J. Lee, L. Mu, P. Liu, C. K. Waters, S. Spence, Z. Xu, C. Wei, D. J. Kautz, Q. Yuan, Y. Dong, Y.-S. Yu, X. Xiao, H.-K. Lee, P. Pianetta, P. Cloetens, J.-S. Lee, K. Zhao, F. Lin and Y. Liu, Adv. Energy Mater., 2019, 9, 1900674 CrossRef.
- C. Tan, A. S. Leach, T. M. Heenan, H. Parks, R. Jervis, J. N. Weker, D. J. Brett and P. R. Shearing, Cell Rep. Phys. Sci., 2021, 2, 100647 CrossRef CAS.
- C. H. Lin, Z. Ju, X. Zheng, X. Zhang, N. Zmich, X. Liu, K. J. Takeuchi, A. C. Marschilok, E. S. Takeuchi, M. Ge, G. Yu and Y. c K. Chen-Wiegart, Carbon, 2022, 188, 114–125 CrossRef CAS.
- M. Wolf, B. M. May and J. Cabana, Chem. Mater., 2017, 29, 3347–3362 CrossRef.
- I. L. Buurmans and B. M. Weckhuysen, Nat. Chem., 2012, 4, 873–886 CrossRef CAS PubMed.
- S. G. Urquhart, ACS Omega, 2022, 7, 11521–11529 CrossRef CAS PubMed.
- R. Hoppe, J. Reinhardt, G. Hofmann, J. Patommel, J.-D. Grunwaldt, C. D. Damsgaard, G. Wellenreuther, G. Falkenberg and C. G. Schroer, Appl. Phys. Lett., 2013, 102, 203104 CrossRef.
- D. A. Shapiro, Y. S. Yu, T. Tyliszczak, J. Cabana, R. Celestre, W. Chao, K. Kaznatcheev, A. L. Kilcoyne, F. Maia, S. Marchesini, Y. S. Meng, T. Warwick, L. L. Yang and H. A. Padmore, Nat. Photonics, 2014, 8, 765–769 CrossRef CAS.
- Y. S. Yu, M. Farmand, C. Kim, Y. Liu, C. P. Grey, F. C. Strobridge, T. Tyliszczak, R. Celestre, P. Denes, J. Joseph, H. Krishnan, F. R. Maia, A. L. Kilcoyne, S. Marchesini, T. P. C. Leite, T. Warwick, H. Padmore, J. Cabana and D. A. Shapiro, Nat. Commun., 2018, 9, 1–7 CrossRef PubMed.
- M. Hirose, N. Ishiguro, K. Shimomura, N. Burdet, H. Matsui, M. Tada and Y. Takahashi, Angew. Chem., Int. Ed., 2018, 57, 1474–1479 CrossRef CAS PubMed.
- M. Hirose, N. Ishiguro, K. Shimomura, D. N. Nguyen, H. Matsui, H. C. Dam, M. Tada and Y. Takahashi, Commun. Chem., 2019, 2, 1–7 CrossRef CAS.
- H. Yuan, H. Yuan, T. Casagrande, D. Shapiro, Y. S. Yu, B. Enders, J. R. Lee, A. Van Buuren, M. M. Biener, S. A. Gammon, T. F. Baumann and A. P. Hitchcock, ACS Appl. Nano Mater., 2021, 4, 621–632 CrossRef CAS.
- Z. Gao, M. Holler, M. Odstrcil, A. Menzel, M. Guizar-Sicairos and J. Ihli, Chem. Commun., 2020, 56, 13373–13376 RSC.
- M. Hirose, K. Shimomura, T. Higashino, N. Ishigurob and Y. Takahashia, J. Synchrotron Radiat., 2020, 27, 455–461 CrossRef PubMed.
- K. Zhang, F. Ren, X. Wang, E. Hu, Y. Xu, X. Q. Yang, H. Li, L. Chen, P. Pianetta, A. Mehta, X. Yu and Y. Liu, Nano Lett., 2017, 17, 7782–7788 CrossRef CAS PubMed.
- G. Qian, J. Zhang, S. Q. Chu, J. Li, K. Zhang, Q. Yuan, Z. F. Ma, P. Pianetta, L. Li, K. Jung and Y. Liu, ACS Energy Lett., 2021, 6, 687–693 CrossRef CAS.
- H. Uematsu, N. Ishiguro, M. Abe, S. Takazawa, J. Kang, E. Hosono, N. D. Nguyen, H. C. Dam, M. Okubo and Y. Takahashi, J. Phys. Chem. Lett., 2021, 12, 5781–5788 CrossRef CAS PubMed.
- M. Sawamura, S. Kobayakawa, J. Kikkawa, N. Sharma, D. Goonetilleke, A. Rawal, N. Shimada, K. Yamamoto, R. Yamamoto, Y. Zhou, Y. Uchimoto, K. Nakanishi, K. Mitsuhara, K. Ohara, J. Park, H. R. Byon, H. Koga, M. Okoshi, T. Ohta and N. Yabuuchi, ACS Cent. Sci., 2020, 6, 2326–2338 CrossRef CAS PubMed.
- A. Maiden and J. Rodenburg, Ultramicroscopy, 2009, 109, 1256–1262 CrossRef CAS PubMed.
- M. Hirose, K. Shimomura, N. Burdet and Y. Takahashi, Opt. Express, 2017, 25, 8593 CrossRef CAS PubMed.
- B. Henke, E. Gullikson and J. Davis, At. Data Nucl. Data Tables, 1993, 54, 181–342 CrossRef.
- K. P. Murphy, Machine Learning: A Probabilistic Perspective, The MIT Press, 2012 Search PubMed.
- R. Tibshirani, G. Walther and T. Hastie, J. Phys. Chem. B, 2001, 63, 411–423 Search PubMed.
- P. Chaurand, J. Rose, V. Briois, M. Salome, O. Proux, V. Nassif, L. Olivi, J. Susini, J. L. Hazemann and J. Y. Bottero, J. Phys. Chem. B, 2007, 111, 5101–5110 CrossRef PubMed.
- J. Wong, F. W. Lytle, R. P. Messmer and D. H. Maylotte, Phys. Rev. B: Condens. Matter Mater. Phys., 1984, 30, 5596–5610 CrossRef.
- T. Yamamoto, X-Ray Spectrom., 2008, 37, 572–584 CrossRef.
- L. de Picciotto, M. Thackeray, W. David, P. Bruce and J. Goodenough, Mater. Res. Bull., 1984, 19, 1497–1506 CrossRef.
- E. Salagre, S. Quílez, R. de Benito, M. Jaafar, H. P. van der Meulen, E. Vasco, R. Cid, E. J. Fuller, A. A. Talin, P. Segovia, E. G. Michel and C. Polop, Sci. Rep., 2021, 11, 1–12 CrossRef PubMed.
- N. Ishiguro and Y. Takahashi, J. Appl. Crystallogr., 2022, 55, 929–943 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Sample synthesis, electrochemical analysis, XRD and conventional XAFS of powder samples, spatial-resolution analysis of reconstructed images and chemical state maps of standard samples, histogram of chemical state maps, within-cluster sum of squared errors against the number of clusters, and average values and the probability density maps of chemical state maps in each cluster. See DOI: https://doi.org/10.1039/d2cp04087e |
| This journal is © the Owner Societies 2023 |