
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Recent progress in solid-state NMR of spin-½ low-γ nuclei applied to inorganic materials
Mark E.
Smith
 abc
abc
aVice-Chancellor and President's Office and Department of Chemistry, University of Southampton, Southampton, SO17 1BJ, UK. E-mail: m.e.smith@soton.ac.uk
bDepartment of Chemistry, Lancaster University, Bailrigg, Lancaster LA1 4YB, UK
cDepartment of Physics, University of Warwick, Coventry, CV4 7AL, UK
First published on 24th November 2022
Abstract
Significant technological and methodological advances in solid-state NMR techniques in recent years have increased the accessibility of nuclei with small magnetic moments (hereafter termed low-γ) underpinning an increased range of applications of such nuclei. These methodological advances are briefly summarised, including improvements in hardware and pulse sequences, as well as important developments in associated computational methods (e.g. first principles calculations, spectral simulation). Here spin-½ nuclei are the focus, with this Perspective complementing a very recent review that looked at half-integer spin low-γ quadrupolar nuclei. Reference is made to some of the original reports of such spin-½ nuclei, but recent progress in the relevant methodology and applications to inorganic materials (most within the last 10 years) of these nuclei are the focus. An overview of the current state-of-the-art of studying these nuclei is thereby provided for both NMR spectroscopists and materials researchers.
1. Introduction
In adopting a fully multinuclear approach to NMR there are many NMR-active nuclei to potentially observe across the Periodic Table. However the NMR characteristics of nuclei vary greatly, with one of the most important factors determining the observability of a particular isotope being the magnitude of the signal.2 For a given isotope the combination of external factors (i.e. the applied magnetic field, temperature) and intrinsic nuclear properties (i.e. gyromagnetic ratio (γ), nuclear spin (I) and natural abundance (NA, a percentage)) influence the magnitude of the magnetisation and hence the observed signal (see eqn. (2.19)–(2.22) in ref. 2) which provide a factor at fixed field and temperature of γ2(NA)I(I + 1). The size of the NMR signal also depends on the rate of precession of this magnetisation introducing a further factor of γ.2 The IUPAC recommended standardised comparison of the observability of a nucleus is provided by a combination of these factors termed the receptivity (RC) (eqn (1)),3| RC = γ3(NA)I(I + 1). | (1) |
| Isotope | ν o (MHz) @ 14.1 T | Natural abundance (NA) | Receptivity (RC) | Commonly used chemical shift referencing |
|---|---|---|---|---|
| 89Y | 29.502 | 100 | 0.322 | IUPAC reference9 Y(NO3)3 H2O/D2O |
| Secondary references 1 M YCl3 in H2O at 0 ppm and solid Y2Ti2O7 at 65 ppm10 | ||||
| 103Rh | 19.032 | 100 | 0.0856 | IUPAC reference9 Rh(acac)3, CDCl3, satd. |
| 0.5 M solution of the Rh(H2O)63+ ion 9915.8 ppm11 | ||||
| It has also been suggested to reference directly against 1H12 | ||||
| 107Ag | 24.282 | 51.82 | 0.0943 | As for 109Ag |
| 109Ag | 27.138 | 48.18 | 0.133 | IUPAC reference9 AgNO3 D2O (sat.) |
| 9.1 M AgNO3 (0.54 M Fe(NO3)3) −47.3 ppm | ||||
| 183W | 24.966 | 14.40 | 0.0287 | IUPAC reference9 1 M Na2WO4 D2O |
| H3PW12O40 in water −99 ppm13 |
In examining categories of nuclei across the Periodic Table one of the classes is made up of those nuclei with smaller gyromagnetic ratios. A convenient definition for such nuclei is those possessing smaller magnetic moment than 15N, since in the early days of commercial magic angle spinning (MAS) probes (i.e. up to the mid-1990s) there were no standard probes equipped on the so-called X-channel to tune to such low frequencies and were very much the preserve of the specialist, resulting in very few studies of such nuclei at that time. A useful shorthand for the nuclei with magnetic moments smaller than 15N is ‘low-γ’ which is now commonly used in the literature.5 It is convenient to split this group further into spin-½ and those with higher spin. The latter group experience the same interactions as for spin-½, but with the significant additional complication of the quadrupolar interaction.2,4,6,7 Low-γ non-integer spin quadrupolar nuclei were recently comprehensively reviewed.1
There are a range of other factors which complicate the observation of low-γ nuclei since the Larmor frequency (ν0 = γB0, B0 is the main static magnetic field applied) will be lower at given applied magnetic fields than for nuclei with higher magnetic moments. Lower frequencies tend to result in more acoustic ringing, longer longitudinal relaxation times (T1) and weaker internuclear interactions.2 Acoustic ringing can have a number of sources which have been described in detail,8 but tends to be worse at lower Larmor frequencies. Also given the elements in this category (Table 1) tend to be heavier, they can also exhibit quite large shift ranges and often have significant associated anisotropies which can make the lines cover a wide frequency range. This adds to the challenges of detection and sensitivity, with recent methods of tackling these issues discussed below. The last time low-γ spin-½ nuclei were collectively reviewed was around 20 years ago.2,5 There have been updates on particular aspects of these nuclei individually which are referenced below. This Perspective will therefore examine the methodological and technical advances that have enhanced the ability to observe and interpret the spectra from low-γ spin-½ nuclei. Their applications to probe inorganic materials problems are illustrated by recent examples taken from the last decade. To be completely comprehensive low-γ spin-½ nuclei would also include 57Fe and 169Tm. However the magnetic nature of both these isotopes means NMR reports from them are usually from internal field observation or heavily paramagnetically perturbed spectra which are quite different from conventional NMR approaches so are not discussed further here.
2. Recent advances in solid-state NMR methodology
The physical background and technological advances underpinning NMR since 2001/2 have been the subject of previous reviews14–17 and are briefly recapped here. The intrinsic sensitivity of any spectroscopic approach such as NMR depends on population differences determined by the Boltzmann factor which provide the signal. The local interactions provide information about the environment a nucleus is in, but in powder samples the anisotropy of these interactions causes line broadening which both diminishes sensitivity by spreading the signal over a greater frequency range and degrades the spectral resolution by increasing the linewidth of the individual components. Magic angle spinning (MAS) is the most common approach for solid-state NMR as it can average anisotropic line broadening, greatly increasing the spectral resolution.2,4 This is particularly effective for spin-½ nuclei in highly crystalline and ordered solids as there is no broadening from dispersion or the quadrupolar interaction.2,4,6As commercial spectrometers with MAS probes first became available for solid-state NMR these were typically equipped with wide-bore (89 mm) magnets of 4.7–9.4 T. Low-γ nuclei therefore resonated at low frequencies and there were no commercial probes available for such frequencies and the modest (by today's standards) magnetic field did not help observe the intrinsically weak signals. The low observation frequencies also introduced additional experimental complexities such as probe ringing.2,8 In the mid-1990s more wide-bore 11.7 and 14.1 T instruments became available meaning that observation of low-γ nuclei became much more feasible, which one starts to see in the reviews of around 2001/2.2,5 Fields have continued to rise as persistent magnetic fields of 18.8–20.0 T have become much more widely available, with the current more common state-of-the-art corresponding to wide-bore 20/21.1 T commercial instruments with now some extension to ≥21 T with persistent standard bore (57 mm) magnets and first reports of commercial 28.4 T instruments being installed. It should be noted that the advantages of the very high magnetic fields are less pronounced for spin-½ systems than for non-integer spin quadrupolar nuclei.1,2 The other major experimental advance is the increase in the MAS rate, but this is less important for smaller magnetic moment spin-½ nuclei. Choosing the optimal combination of applied magnetic field and MAS rate depends on a range of factors such as the nature of the sample (e.g. highly crystalline vs. glassy) and the local interactions experienced determining the extent of the line broadening. For low-γ spin-½ nuclei in samples without a high density of high-γ nuclei modest MAS rates can usually narrow the lines. This is borne out by looking at the literature on low-γ spin-½ nuclei since although some work using faster MAS certainly exists the use of relatively modest MAS rates by using larger rotors (e.g. ≥7 mm) to boost the sample volume and hence the signal size is still quite widely used. Higher applied magnetic fields will also give larger signals, but given chemical shift anisotropy (CSA) is the likely dominant line broadening mechanism2 in these heavy spin-½ nuclei then at higher magnetic fields the signal will be spread over a wider frequency range. Faster spinning will produce fewer spinning sidebands, but if that means using a smaller rotor this gain in signal could be offset by the smaller sample. The intrinsic linewidth under MAS also plays a role in determining the optimum conditions, with it being very low for highly crystalline materials, but can be very significant for glassy/amorphous materials. Hence compared to low-γ spin-½ quadrupolar nuclei where the highest field with fast spinning is often the best choice, a more nuanced compromise of the choice of applied magnetic field and spinning speed is necessary for the nuclei considered here. In the last 20 years there has been much progress in approaches to boost the signal. These have included using echo trains, hyperpolarisation methods, such as dynamic nuclear polarisation (DNP), where there can be some very impressive gains in signal observed and indirect detection which are described in more detail below. There are other very recent review articles that also provide useful background to recent methodological advances.1,18,19
Once a signal is observed then the spectrum itself or approaches which manipulate the spin provide information about the environment of the observed nucleus which in turn is providing information about the structure. Key is the translation of the experimentally measured data on the nucleus to the structural detail. The initial approach which is still used is effectively the fingerprinting approach of direct comparison of the spectral parameters with those resonances from a known structure. With real developments in the understanding of the theoretical underpinnings, first principles density functional theory (DFT) calculations of both the structure and the NMR interaction parameters, combined with the availability of significant computational power, new computational approaches for extracting structural information have become much more widely applied and added a new dimension to the NMR methodology. The reader is referred to several very helpful reviews for more details of how such computational work is being applied.20–22 The approaches are most directly applicable to extended crystalline solids. There are also examples of the computational approaches being adapted to calculation of the parameters with some atomic disorder (e.g. from solid solutions, see below examples from 89Y) and even from glasses. Combining simulation of the observed NMR spectra and calculation of the NMR interaction parameters from the structure has led to the powerful new approach termed ‘NMR crystallography’ which has already provided real new understanding of materials.23
2.1 Techniques for sensitivity enhancement
The sensitivity issues for low-γ, spin-½ nuclei start with the intrinsically small magnetic moments. However on top of this several other factors exacerbate accumulating a good signal-to-noise. There can be low natural abundance, although this is not much of a problem for the nuclei listed in Table 1. The nuclide of interest can be diluted in the sample. Also in the samples of interest there could be significant line broadening, which for heavier spin-½ nuclei is likely from chemical shift anisotropy. Low-γ, spin-½ nuclei can also suffer from long longitudinal (T1) relaxation times, which must be carefully accounted for. When MAS is applied to a highly crystalline sample with inhomogeneous anisotropic line broadening, for these nuclei the static powder pattern breaks up into a series of sharp signals – the isotropic resonance plus a set of spinning sidebands. This effectively concentrates the signal into these much sharper features and hence improves the signal-to-noise. In 2.1.1 other echo trains for increasing the sensitivity are examined. However given that inherent low signal sensitivity is one of the key weaknesses for NMR there has always been an impetus to enhance the signal by the transfer of signal from a stronger source or to use a strong signal to indirectly detect a weaker signal. For example this could be transfer of magnetisation from stronger sources as in cross-polarisation (CP) from nuclei with larger nuclear moments (2.1.2) or from the magnetisation of unpaired electrons in dynamic nuclear polarisation (DNP, 2.1.3), where under favourable conditions spectacular gains in signal are possible. Given low sensitivity is even more of an issue for low-γ nuclei there is an added premium in gaining signal from other sources. There is also the possibility of detecting the nuclei indirectly (2.1.4) or some combination of these techniques might be used.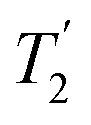 (the spin–spin relaxation time) and whose separation in the frequency domain is (2τ)−1. The peaks of the spikelets quite closely map out the lineshape that would be obtained by Fourier transformation of one half-echo. The gain in S/N comes from the concentration of the signal intensity into these narrow spikelets. The maximum possible enhancement will depend on the value of
(the spin–spin relaxation time) and whose separation in the frequency domain is (2τ)−1. The peaks of the spikelets quite closely map out the lineshape that would be obtained by Fourier transformation of one half-echo. The gain in S/N comes from the concentration of the signal intensity into these narrow spikelets. The maximum possible enhancement will depend on the value of 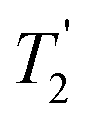 and it has been shown24 that the gain in S/N ratio is
and it has been shown24 that the gain in S/N ratio is 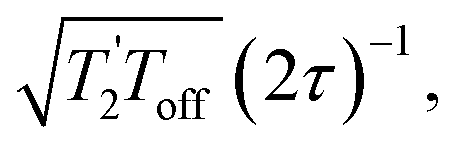 where Toff is the time for the free induction decay of the individual echo to become comparable to the noise level of the standard echo. This was first demonstrated for non-integer spin quadrupolar nuclei,25 with it being realised that the gain in sensitivity could help with the observation in particular of low-γ nuclei including spin-½ nuclei if they are experiencing a significant anisotropy.26 This approach works if the effective spin–spin relaxation time (i.e. the decay of the echo envelope,
where Toff is the time for the free induction decay of the individual echo to become comparable to the noise level of the standard echo. This was first demonstrated for non-integer spin quadrupolar nuclei,25 with it being realised that the gain in sensitivity could help with the observation in particular of low-γ nuclei including spin-½ nuclei if they are experiencing a significant anisotropy.26 This approach works if the effective spin–spin relaxation time (i.e. the decay of the echo envelope, 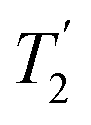 ) is much longer than the decay of the magnetisation caused by the local interactions. The ideal conditions for the CPMG sequence to work are when there is a large difference between the inhomogeneous and homogeneous broadening. Applications to spin-½ nuclei to enhance the sensitivity27,28 include reports on 109Ag with significant increases in sensitivity observed.28
) is much longer than the decay of the magnetisation caused by the local interactions. The ideal conditions for the CPMG sequence to work are when there is a large difference between the inhomogeneous and homogeneous broadening. Applications to spin-½ nuclei to enhance the sensitivity27,28 include reports on 109Ag with significant increases in sensitivity observed.28
 | ||
| Fig. 1 (a) CPMG echo sequence, with the two pulses (p1, p2) a combination that will create an echo (usually 90°, 180°) with the echo train repeated n times. In combination with MAS the repeat period is synchronised to an integer multiple of the MAS rotation period (τr). (b) The Wurst CPMG pulse sequence with the two WURST pulses identical and two-step phase cycling (φ1 = φreceiver = 0, π and φ2 = π/2) resulting in echo formation.34 (b) Reproduced from the ref. 34 with permission from Elsevier copyright 2013. | ||
Two other points to make here are that although this CPMG approach was initially applied to static samples it can be readily coupled with MAS, as long as the two echo processes are aligned. Second, if the static line covers a very wide frequency range such that the excitation bandwidth by the pulses causes an issue, the full spectrum can be collected piecewise by stepping the rf carrier frequency, which is referred to as Variable-Offset Cumulative Spectroscopy (VOCS).29,30 If one uses a VOCS experiment with different frequency offsets this can be combined with the CPMG sequence, with then the VOCS frequency steps set at an exact multiple of the spikelet separation. There are various considerations about the optimal pulses for such echo trains and typical practical conditions used can be found in the literature.26–28 In the classical configuration rectangular 90° and 180° pulses are used. However with modern digital spectrometers shaped and modulated pulses often result in better echoes, such as for example using chirp pulses.31 These concepts are the basis for broader more uniform excitation through for example WURST (wideband uniform-rate smooth truncation) pulses that can be used for such echoes and have been applied to spin-½ nuclei (Fig. 1(b)).32 The pulses are shaped with usually modulation of both the amplitude and phase, with the latter the more complex which results in the pulse creating an effective frequency sweep and the much broader excitation.33,34 Echoes can be formed through using different pulse lengths and/or sweep rates. Optimisation of such approaches has been considered in detail.33–35 No matter which approach is applied if the spectrum is split up into spikelets these points selectively sample the static envelop of the spectrum which is a less complete data set than the full static spectrum. Also if several distinct sites are present their apparent relative intensity will depend on the relative 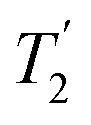 for each site.
for each site.
 | ||
| Fig. 2 (a) Conventional cross-polarisation (CP) sequence between an abundant spin (I) and dilute spin (X), with the inset showing schematically the change in the energy level spacing from the applied magnetic field (B0) to the spinlock fields (B1s) to create the Hartmann–Hahn condition. (b) A PRESTO sequence creating polarisation transfer by recoupling (RC).41 (c) CP created using a WURST pulse in the BRAIN sequence.42 (d) The pulse sequence for dynamic nuclear polarisation (DNP) for indirect detection (i.e. via I) and a schematic showing the magnetisation pathways for both indirect and direct pathways. (e) An HMQC-type sequence that provides enhancement via indirect detection of the abundant spin I. | ||
As digital spectrometer technology has evolved more sophisticated pulse sequences and shapes have been implemented ameliorating the power issues on the hardware. An interesting approach is the PRESTO (phase-shifted recoupling effects a smooth transfer of order) sequence.41 PRESTO is effective at recoupling spins and thereby transferring magnetisation (Fig. 2(b)). As it does not require the conventional spinlock for polarisation transfer it greatly reduces the radiofrequency power requirements on the probe. The paper that initially demonstrated the PRESTO approach noted ‘The method is particularly attractive for nuclei with low gyromagnetic ratios or for those experiencing strong anisotropic spin interactions…’, with one of the examples chosen to illustrate the approach 19F–109Ag in AgSbF6.41 More recently developments for polarisation transfer have included pulse sequences that allow broader excitation at lower powers on both channels. BRoadband Adiabatic INversion (BRAIN) CP uses an adiabatic inversion pulse (i.e. like WURST) applied to the rare spin nuclide and a conventional spin-locking pulse on the abundant nucleus channel (Fig. 2(c)); both static42 and in combination with MAS have been reported.43 Under the frequency sweep created by the WURST pulse on the dilute spin there will be points where the Hartmann–Hahn matching condition is fulfilled with the conventional spin-locking pulse applied to the abundant spin for each frequency element of the powder pattern. This has the advantages that there is a much lower power requirement on the X-spin side and the transfer efficiency is far less frequency offset dependent than for the conventional hard pulse CP experiment.42 Another recent CP concept to boost the signal from dilute, weaker signals is based on the idea of solution and chemical exchange saturation transfer, but modified for dilute heteronuclei in solids. Although chemical exchange cannot occur as in a solution there is a group of highly abundant, more distant protons that are used to replenish the magnetisation of the more local protons that are in stronger dipolar coupling with the X nucleus and are undergoing the CP action. This has been termed PROgressive Saturation of the Proton Reservoir (PROSPR) and has been demonstrated on some low-γ quadrupolar nuclei.44
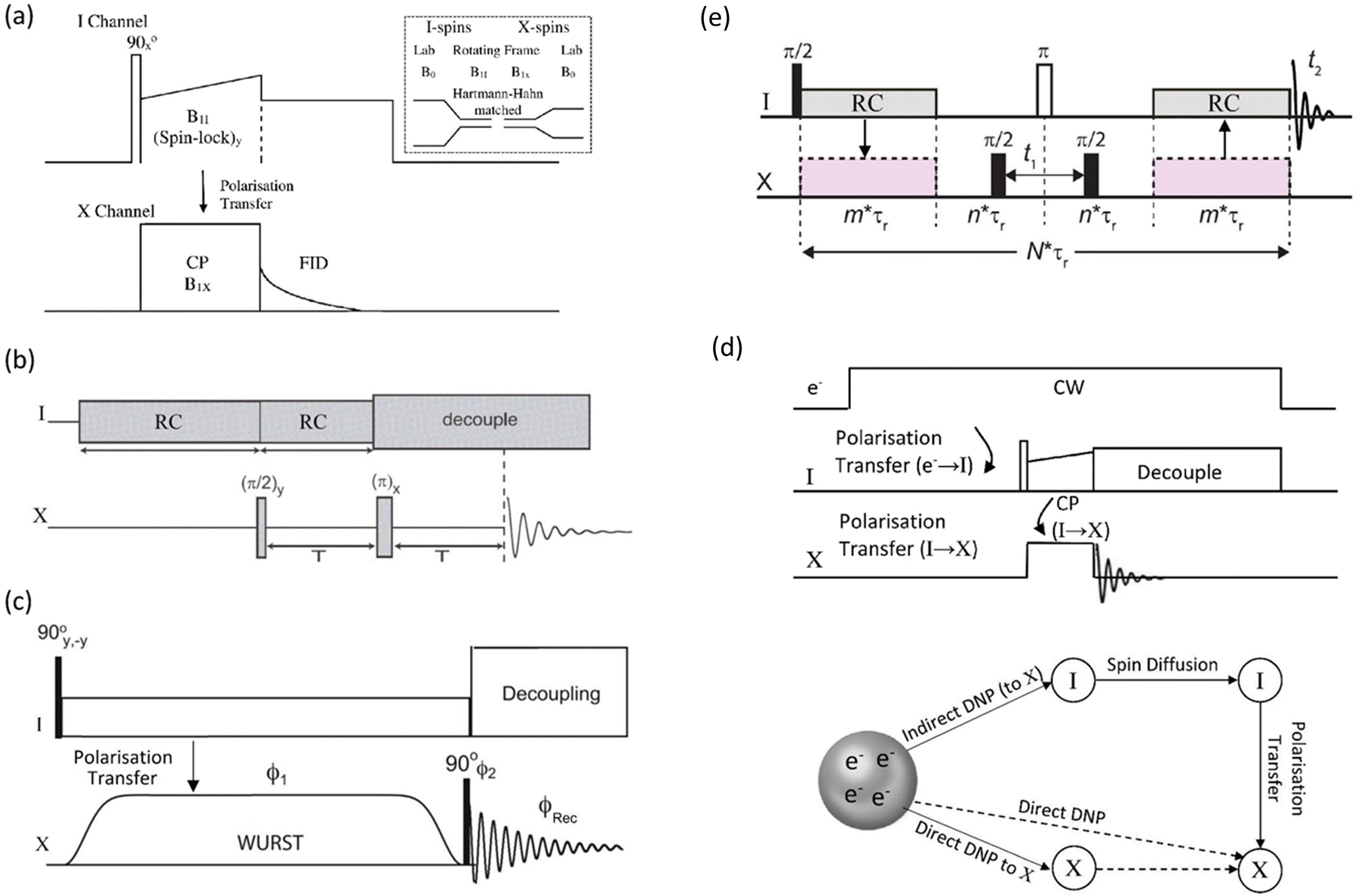
The application of DNP to enhance the signal from challenging nuclei has been summarised, including for 89Y.47 For 89Y there are several examples in the recent literature with a range of approaches to the polarisation. The common approach is indirect polarisation using an exogeneous free radical which polarises 1H with subsequent CP to 89Y. The ‘model’ sample used was a frozen solution of 0.5 M Y(NO3)3 with the 12 mM AMUPol biradical added. Very significant signal enhancement was observed with the frozen solution added and the different peaks to different local coordinations.52 The same experimental approach was then applied to understanding hydrated yttrium-doped barium zirconate. Two samples were examined with 20 and 30% yttrium and water contents of ∼7 and ∼9%. Without DNP only very weak signals were observed, whereas with microwave irradiation applied two 89Y signals are observed. Rather than via a biradical addition some samples can have intrinsic paramagnetic ions to act as the electron polarisation source effectively meaning the bulk sample is observed. Mixed samples of ceria with yttria and doped with gadolinia (general formula Ce1−x−yYxGdyO2−x/2−y/2 with x = 0.1, 0.25, and 0.4 and y = 0.001 and 0.005) have been examined with DNP. The nature of the field sweep used indicated that polarisation transfer occurs via the solid-effect mechanism and produced very significant gains in the signal and experimental time savings of four orders of magnitude (Fig. 3).53 In a different DNP study of Y2O3 in a frozen solution of Y(NO3)3 signal enhancements of >80 were observed using the trityl OX063 monoradical with polarisation transfer under MAS occurring via the cross-effect mechanism. In Gd2O3-doped Y2O3 (for T1 relaxation reduction) spectra could be obtained quickly (∼30 min).54
 | ||
| Fig. 3 89Y MAS NMR Hahn-echo spectra from co-precipitated ceria samples with various levels (10, 25 and 40%) of Y2O3 substitution and 0.1% Gd2O3 showing the signal enhancement (ε) via DNP when microwaves (μW) are applied, with the different yttrium coordinations [x]Y observed.53 Reproduced from ref. 53 with permission from the American Chemical Society, copyright 2021. | ||
 | ||
| Fig. 4 Demonstration of the signal enhancements produced by proton-detected compared to directly-detected solid-state NMR signals for (a) 109Ag in AgSO3CH3 and (b) 89Y in Y(NO3)3·6H2O. The CP condition/pulse sequence used for acquisition, sensitivity (S), total experiment time and gain in sensitivity from proton detection (ξ) are indicated – see ref. 55 for the definitions. The proton-detected NMR spectra were obtained from the positive projections of the corresponding 2D HETCOR spectrum.55 Reproduced from ref. 55 with permission from the American Chemical Society, copyright 2018. | ||
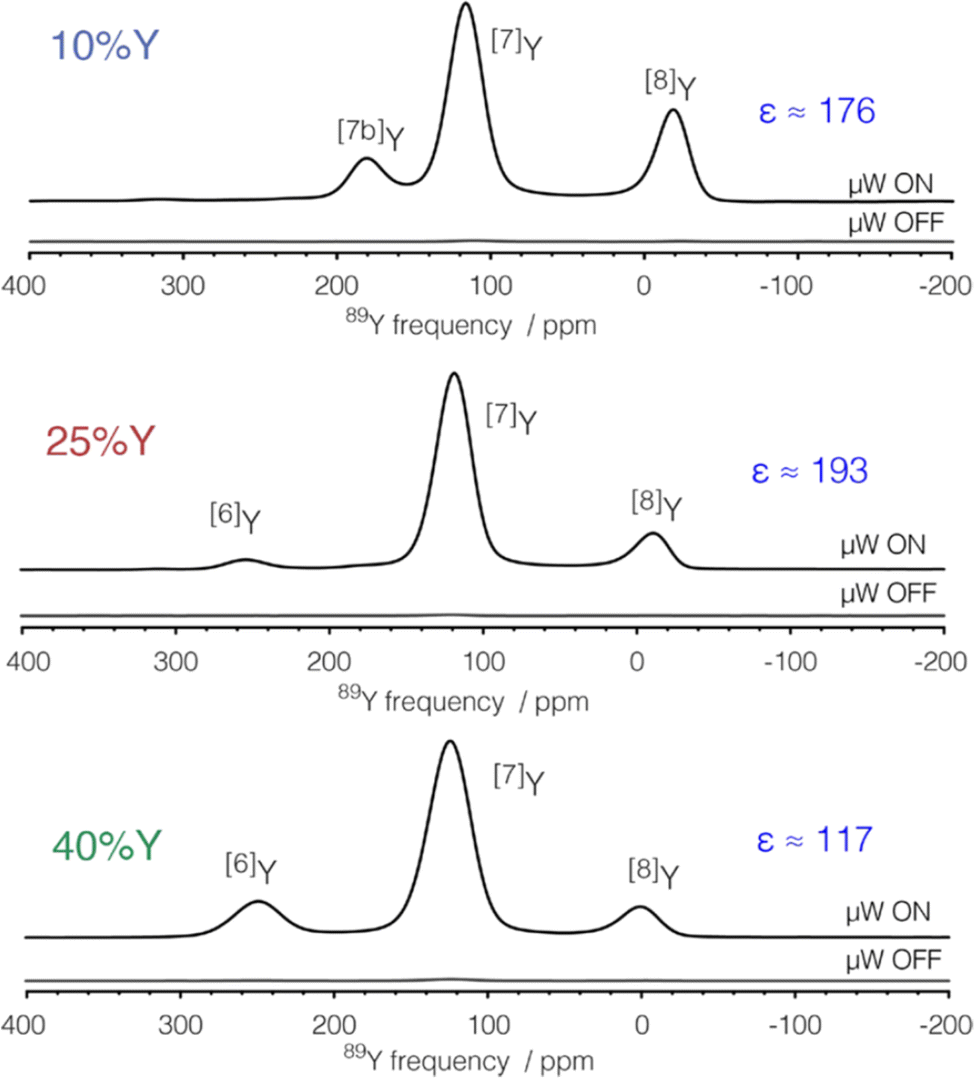
To gain even more in signal DNP has recently been combined with indirect detection, which has become possible with the availability of low-temperature fast MAS probes. A detailed theoretical and experimental study with DNP-enhanced 2D indirectly detected HETCOR (idHETCOR) experiments on four spin-½ nuclei (13C, 15N, 113Cd, 89Y), with 1.3 and 3.2 mm probes used.58 For the experiments on yttria nanoparticles (50 nm) impregnated with a 16 mM solution of TEKPol in TCE-d2, Fig. 5 shows the signals obtained with the two probes used. Two prominent yttrium signals are observed with differing numbers of hydroxyl nearest neighbours. The faster spinning and higher rf fields shows much better sensitivity in the 1.3 mm probe (Fig. 5(a) and (b), note the respective acquisition times (AT) of 1.5 minutes vs. 2.7 hours). Then with indirect detection for an idHETCOR a very significant gain in sensitivity was observed compared to a direct HETCOR of 42 (Fig. 5(c) and (d)).58
 | ||
| Fig. 5 The effect of signal enhancement for various NMR experiments on yttria nanoparticles (50 nm) impregnated with a 16 mM solution of TEKPol in TCE-d2, with a comparison of the signals obtained with two different NMR probes. The faster spinning and higher rf fields in the 1.3 mm probe show much better sensitivity (cf. (a) and (b)), noting the respective acquisition times (ATs). With indirect detection (idHETCOR) a very significant gain in sensitivity was observed compared to a direct HETCOR of 42 ((c) and (d)). A full list of experimental parameters can be found in Table S1 in ESI of ref. 58. Reproduced from ref. 58 with permission from Elsevier, copyright 2020. | ||
2.2 Computational approaches – structure, NMR parameters and lineshape calculations
In parallel with experimental advances, the role of complementary computational work in overall NMR methodologies for understanding materials has developed rapidly in recent years. Progress has been driven by the greater availability of the necessary compute power with better, more sophisticated code. This was recently described in a review of non-integer spin quadrupolar low-γ nuclei1 and is briefly recapitulated here. There are two distinct aspects of the computational work, i.e. (i) direct extraction of the NMR interaction parameters from the experimental data, and (ii) the use of first-principles calculations of both the structure and the NMR interaction parameters. To simulate spectra one can calculate the full evolution of the Hamiltonian, with SIMPSON being a widely used approach59 although others exist. Given they are full calculations of the Hamiltonian considerable computing resource is often necessary. The much more commonly adopted approach to extracting parameters from spectra is to use a simulation program based on formulae for frequencies derived directly from the Hamiltonian and a whole range of programmes are available.60–64 In using such programs they will be making some different approximations, for example perhaps the relevance of the high field limit (i.e. the Zeeman interaction dominates), ideal excitation, etc., which are likely to be highly valid for most cases, especially for the spin-½ nuclei being described here, but needs to always be considered.First principles calculations of structure and the resulting NMR interaction parameters from that structure have become much more straightforward as the theoretical underpinnings have allowed the development of more readily used and efficient code. Several relatively recent comprehensive reviews provide the reader with the detailed background and development of these approaches.20–22 A cluster is usually taken as an approximation for the extended solid or periodic boundary conditions are applied, being a good approximation for extended crystalline solids. Although these approaches were initially aimed at periodic systems there have been adaptations that allow parameters to be calculated when there is some atomic disorder (e.g. from solid solutions). A discrepancy between the calculated and observed NMR parameters suggests that the structural detail is not correct and the structure can be refined. Hence spectral simulation and NMR interaction parameters extracted from the calculated structure can be considered interactively. As a result the measured NMR parameters can more intimately and directly constrain the structure in what has become termed ‘NMR crystallography’.23,65,66 The literature clearly shows that such an approach is being increasingly used to give new structural insight, strengthening the role of NMR in understanding structure-function relations in materials.
3. Applications to inorganic materials
The nuclei examined here were first reviewed as a group in 1994 as part of an article looking at NMR of less commonly studied spin-½ nuclei.67 Their application collectively to inorganic materials was comprehensively reviewed around 2001.2,5 These works2,5 contained compilations of the NMR interaction parameters known at that time. There have been some subsequent updates on observations of individual nuclei, with some of the illustrative examples including inorganic materials. These are referenced below. Several of the advances described in Section 2 can be combined for example DNP and NMR crystallography. Some of the reviews of the developments of NMR techniques also make some reference to low-γ spin-½ nuclei.14 Some brief reference to low-γ spin-½ nuclei has been made in recent reviews of solid-state NMR applied to glasses68 and to oxide-based materials.69 Most of the illustrations of the application of the nuclei reported on here are taken from examples of the last few years, although to give some context for the reader who is not a long-standing afficionado of the solid-state NMR of these nuclei reference to some of the key early publications in the observation of these nuclei is provided. The present review updates of NMR parameters already collected in previous literature reviews.3.1. Yttrium-89
With 100% natural abundance yttrium is potentially highly attractive, with two of the pioneering studies70,71 showing from crystalline materials narrow resonances could be observed under MAS and despite the relatively few data points then available suggesting an extensive shift range in diamagnetic materials. Y2O3 provides a good illustration showing two sharp 89Y resonances with a shift difference of ca. 40 ppm corresponding to the two octahedral YO6 environments in the crystal structure. The intensity ratio of these two peaks at 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 is as expected from the known site occupancies. The high purity oxide and low-γ nature of 89Y result in the T1 values for both of these sites being just under four hours.72 This early work indicated that the 89Y chemical shift becomes more negative with an increasing number of nearest neighbour oxygen atoms, and to become less paramagnetic as the electronegativity of the coordinating group increases. In oxide-based materials yttrium shows typical coordinations YO5–YO9. The influence of different effects means that direct structural assignment to such a coordination based solely on the isotropic chemical shift is often ambiguous. The data below shows that δiso for 89Y moves to lower frequencies as the coordination number and/or with decreasing field strengths of the next-nearest neighbour cations, with these effects causing quite considerable overlap of the shifts associated with particular local coordinations. However in a given compound/solid solution the appearance of different peaks and their trend with composition gives good insight into changes of siting and structure. Now coupled with calculations greatly aid this situation of identification of the sites present. The applications reviewed below include direct phase identification in crystalline samples and structural siting within glasses since 2002. A particularly powerful application of 89Y has been to probe ordering within a wide range of solid solutions given its relatively large chemical shift range.22,73 This builds on the original application of 89Y NMR to solid solutions in a range of lanthanide-doped variants of a pyrochlore Y2(Sn/Ti)2O7.10 Recently a very helpful tutorial review has been published introducing from first principles the application of 89Y to such ordering problems along with a good overview of the systems studied to date by such an approach, complementing well what is presented here.74 A recent computational chemistry approach modelled the chemical shift in a wide range of molecular compounds and showed how δiso readily distinguishes the different ligands based on electronegativity arguments. This work is important in enabling 89Y NMR to probe surface-formed species on for example oxides and that the CSA can often be more discriminating than δiso.75
:1 is as expected from the known site occupancies. The high purity oxide and low-γ nature of 89Y result in the T1 values for both of these sites being just under four hours.72 This early work indicated that the 89Y chemical shift becomes more negative with an increasing number of nearest neighbour oxygen atoms, and to become less paramagnetic as the electronegativity of the coordinating group increases. In oxide-based materials yttrium shows typical coordinations YO5–YO9. The influence of different effects means that direct structural assignment to such a coordination based solely on the isotropic chemical shift is often ambiguous. The data below shows that δiso for 89Y moves to lower frequencies as the coordination number and/or with decreasing field strengths of the next-nearest neighbour cations, with these effects causing quite considerable overlap of the shifts associated with particular local coordinations. However in a given compound/solid solution the appearance of different peaks and their trend with composition gives good insight into changes of siting and structure. Now coupled with calculations greatly aid this situation of identification of the sites present. The applications reviewed below include direct phase identification in crystalline samples and structural siting within glasses since 2002. A particularly powerful application of 89Y has been to probe ordering within a wide range of solid solutions given its relatively large chemical shift range.22,73 This builds on the original application of 89Y NMR to solid solutions in a range of lanthanide-doped variants of a pyrochlore Y2(Sn/Ti)2O7.10 Recently a very helpful tutorial review has been published introducing from first principles the application of 89Y to such ordering problems along with a good overview of the systems studied to date by such an approach, complementing well what is presented here.74 A recent computational chemistry approach modelled the chemical shift in a wide range of molecular compounds and showed how δiso readily distinguishes the different ligands based on electronegativity arguments. This work is important in enabling 89Y NMR to probe surface-formed species on for example oxides and that the CSA can often be more discriminating than δiso.75
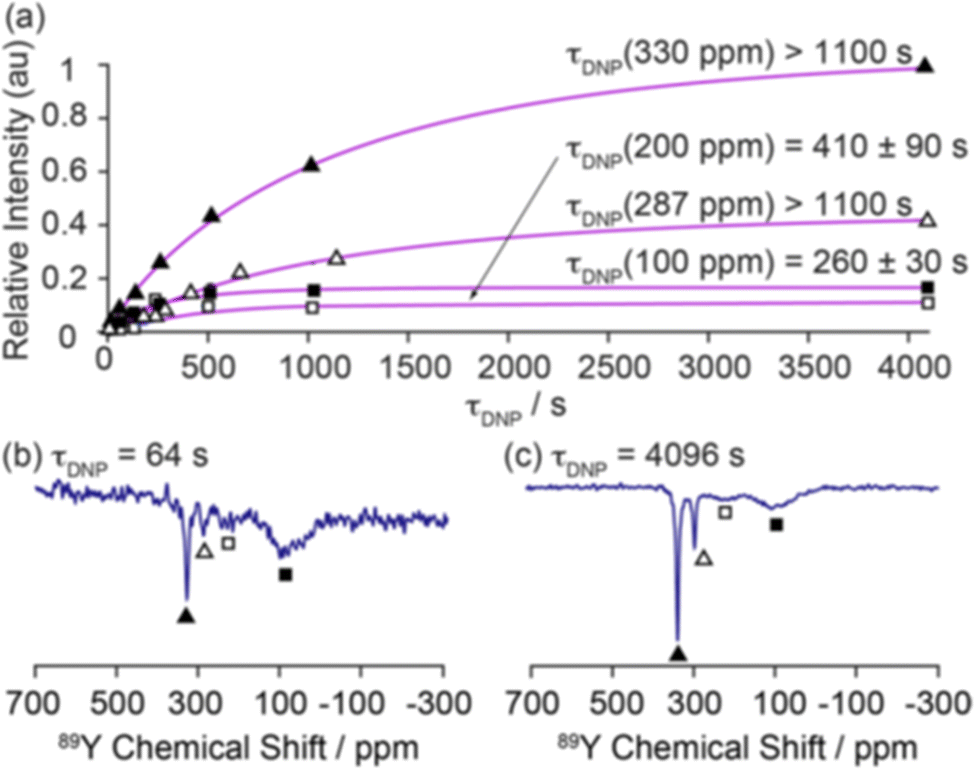 | ||
| Fig. 6 Demonstration of the magnetisation build up for 89Y under DNP for 0.1% Gd2O3-doped Y2O3 with a trityl polarising agent with (a) fitted build-up curves of the bulk (330 ppm, filled triangles; 287 ppm, hollow triangles) and surface sites (∼200 ppm, hollow squares; ∼100 ppm, filled squares). The 89Y MAS NMR spectra recorded at τ = (b) 64 and (c) 4096 s are shown to demonstrate the surface selectivity of the approach.54 Reproduced from ref. 54 with permission from the American Chemical Society, copyright 2019. | ||
Some other oxides to which Y2O3 is added are of real technological significance. ZrO2 and CeO2 occur in fluorite-structured forms with the metal in MO8 coordination. As the differently valent yttrium is added this creates oxygen vacancies. Some of the properties of these materials depend on the creation of these vacancies so that understanding their distribution can be very important. Yttria-stabilised zirconia is an important engineering ceramic with high toughness, is an important material for solid oxide fuel cells (SOFC) and as an oxygen sensor. As the yttrium-content was varied from 5.9 to 42.9 mol% three peaks of varying intensity were observed in the 89Y MAS NMR spectra at 80–90, 190–195 and 290–315 ppm which are attributed to YO8, YO7 and YO6 respectively, the latter two sites being associated with one and two oxygen vacancies respectively.77 The integrated intensity of these peaks could be extracted and compared to models of the vacancy distribution. The data shows a clear preference for vacancies associating with the zirconium site.77 An equivalent study has been carried out for yttria-doped ceria and again three peaks are observed, with a shift offset showing peaks occurring at 0, 120 and 250 ppm reflecting the different relative electronegativity of the elements in the system. In this case the changing intensity showed that there is a preference for the vacancies to be associated with yttrium. The intensity data along with observations of 17O suggest some pairing of yttrium centres occurs.78 A further study made a direct comparison of yttria-doped ZrO2 and CeO2, as well as looking at micro- and nanocrystalline variants. The vacancy preference differences between ZrO2 and CeO2 on doping with yttria were confirmed. When the samples were made nanocrystalline there was a marked tendency for the vacancy distribution to become much more random which was attributed to the energetics changing due to the changing balance of electrostatic and strain terms.79 Very recently DNP has been applied to examine yttria-doped ceria. Polarisation was driven from Gd3+ dopants giving large sensitivity enhancements homogeneously throughout the sample approaching 200. The data confirms that the vacancies in this system have a preference for the cerium. The much greater sensitivity allowed 89Y–89Y homonuclear dipolar correlation experiments to be undertaken and is the first such observation for 89Y. The experiments provide insight into the local distribution of yttrium and indicate there is no spatial clustering of the yttrium into domains.53 It is also possible to then have mixtures of ZrO2 and CeO2 as the underlying system and/or to dope with other oxides (e.g. Sc2O3, CaO), which then leads to multiple doping and the effect of these changes on the structural chemistry can likewise be investigated. For yttria-doped CeO2–ZrO2 the vacancy preference for the metal centres follows what is observed in the binary systems.80 Small additions of CaO, Sc2O3 and Y2O3 to a base 8 mol% yttria-stabilised composition showed CaO created more oxygen vacancies around the yttrium site, whereas they were decreased when Sc2O3 was added, which was explained as the larger Ca2+ preferring to remain as CaO8 whilst the smaller Sc3+ ion prefers to be ScO7.81
The early NMR work looking at atomic ordering in these pyrochlores used 119Sn MAS NMR of rare-earth (RE) doped Y2−xRExSn2O7 since 119Sn is a relatively high-γ spin-½ nucleus. Distinct paramagnetically shifted peaks could be seen, attributed to differing numbers of NNN REs.82 This was soon followed up by using 89Y MAS NMR to complement the 119Sn NMR in the stannates, but also opened up Y2Ti2O7 for investigation10 since the other two nuclei are even more challenging than 89Y at natural abundance. An early extension of 89Y NMR of Y2Ti2O7 was to examine low levels of calcium doping on the yttrium site at 5 and 10% to create ionic conduction pathways. On doping a shoulder on the main resonance to high frequency is observed.83 The most likely cause of the shoulder, given a shift in the direction of a lower coordination suggests that the yttrium coordination is lower and hence is associated with vacancies. Although there was not resolution of these separate peaks the estimated intensity suggests the vacancies are more likely forming near yttrium. Ashbrook and co-workers have built up an extensive body of work that has defined what NMR can do in examining ordering in inorganic solid solutions in crystals. For yttrium-containing pyrochlore systems there have been various dimensions of the ordering investigated with one case of A-site ordering in (Y,La)2Ti2O7,84 the majority of the work on Y2(Sn,Ti)2O7,85–88 with further reports on Y2(Zr,Sn)2O789 and Y2(Hf,Sn)2O7.90 The series of papers on Y2(Sn,Ti)2O785–88 shows nicely the evolution in the sophistication of the methodology.
For (Y1−xLax)2Ti2O789Y MAS NMR data complemented neutron and X-ray diffraction as well as scanning electron microscopy and electron probe microanalysis. As lanthanum substitutes for yttrium a pyrochlore solid solution exists at x = 0 to 0.134, followed by a phase mixture of the pyrochlore with a monoclinic La2Ti2O7-type phase in the range x = 0.134 to 0.807, after which a solid solution in the monoclinic phase forms. The key is different peaks could be observed attributed to different combinations of NNN. In Y2Ti2O7 the data suggested that lanthanum substitutes randomly, whereas at high x in the monoclinic structure yttrium showed a preference for two of the sites.84 When substitution on the B site is examined in Y2Ti2−xSnxO7 a single phase pyrochlore solid solution exists for all x. There is a ∼15 ppm change in the chemical shift of 89Y observed per tin NNN substitution. The evolution of the intensity of each of these peaks closely follows that expected for the tin and titanium randomly occupying the B site.85 The use of first-principles DFT calculations allowed the assumptions of assignment of resonances to particular NNN configurations to be directly confirmed. The data also showed when the equilibrium atomic distribution had occurred.86 The understanding of the local environments in this solid solution was extended by applying to 89Y for the first time the two-dimensional (2D) CSA-amplified phase adjusted spinning sideband (PASS) experiments extracting CSA for 89Y which was compared to DFT calculations. The span Ω can be more reliably extracted than the skew κ and is shown to be a sensitive probe of the NNN environment.87 The most recent addition to work on this system takes the modelling a step further by examining a more comprehensive ensemble of configurations of the distribution of NNN. For example with say two Sn and four Ti NNN that corresponds to a single high level NNN combination, but there are three distinct configurations (Fig. 7). The NMR parameters will be largely determined by being 2 Sn and 4 Ti, but with subtle variations arising from the next level of configuration and by taking these into account spectral details were even more closely reproduced (Fig. 7).88
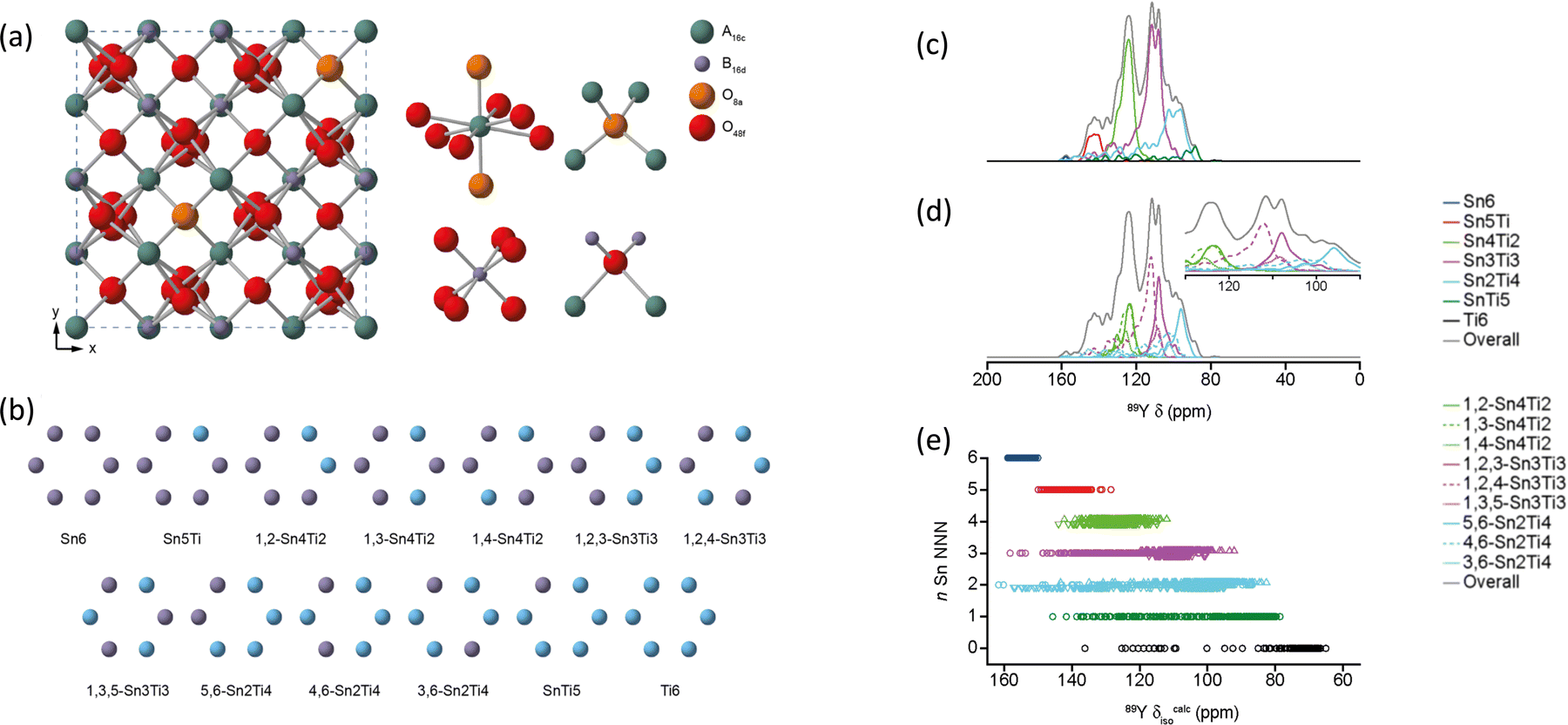 | ||
| Fig. 7 (a) Structure of a typical A2B2O7 pyrochlore, with expansions of the local environments of the Wyckoff 16c (A), 16d (B), and 8a and 48f(O) sites. (b) Possible arrangements on the six NNN B sites that surround the pyrochlore A and B sites for Y2(Sn,Ti)2O7. (c) 89Y MAS NMR spectra of Y2SnTiO7 showing the breakdown into different components assuming full tin/titanium disorder, with the isotropic chemical shift decreasing as the number of NNN tins decreases. (d) The fine detail is produced when the different ordering of the tin/titanium for a given number of NNN tin is also considered. (e) The DFT calculated shift ranges show significant overlaps of different NNN tins across the structural models generated.88 Reproduced from ref. 88 with permission from the American Chemical Society, copyright 2019. | ||
For Y2Zr2−xSnxO7 as the zirconium content increases the equilibrium phase changes from pyrochlore to defect fluorite. However for much of the compositional range two phases co-exist. The NMR data was able to provide clear quantitative evidence as to the phases present and details of the coordinations and NNN ordering. A more complex picture of the phase variation emerged and the distribution of zirconium between the two phases. Interestingly it was shown that the vacancies have a definite preference of associating with zirconium as opposed to tin.89 Similarly complex phase behaviour is observed in Y2Hf2−xSnxO7 with again pyrochlore (tin-rich) and defect-fluorite (hafnium-rich) structures present. Key to a real understanding was the full use of the NMR crystallography approach.90
| Compound | δ iso (ppm) | Local coordination and other comments | Ref. |
|---|---|---|---|
| YScO3 | 265 | YO8 | 91 |
| 263.1 | YO8, Ω = −221 ppm, κ = 0.33 | 92 | |
| YVO4 | −66 | YO8 | 98 |
| y-Y2Si2O7 | 170.8 | YO8 | 99 |
| 132.9 | YO8 | ||
| 95.1 | YO8 | ||
| 37.7 | YO8 | ||
| β-Y2Si2O7 | 207.3 | YO6 | 99 |
| 207.2 | YO6 | 100 | |
| γ-Y2Si2O7 | 198.9 | YO7 | 99 |
| δ-Y2Si2O7 | 121.1 | YO7 | 99 |
| X1-Y2SiO5 | 215.1 | YO7 | 99 |
| 73.9 | YO9 | ||
| X2-Y2SiO5 | 237.1 | YO6 | 99 |
| 149.5 | YO7 | ||
| G-(La,Y)2Si2O7 | 88–128 | YO8 | 101 |
| 95–101 | YO8 | ||
| YBO3 | 117.6, 113.8 | 2 × YO8 | 108 |
| 121.5, 118.1 | 2 × YO8 | 109 | |
| Y3BO6 | 15 peaks 297–168 | YO7, YO8 | 108 |
| YAl3(BO3)4 | 135 | YO6 | 111 |
| Y(OH)3 | 68 | YO9 | 112 |
Yttrium-doped BaZrO3 has been studied for several potential technological applications and there are several reports of 89Y NMR from such systems. The local yttrium environments have been probed (Bax(Zr0.8Y0.2)O3−δ) for four different values of x.93 Two signals are seen in the 89Y NMR data. One narrower peak at 420 ppm and the other much broader line in the range 100–300 ppm ascribed to YO5 and YO6 respectively. The much greater linewidth of the YO6 site is down to higher disorder around this site, although no definitive reason for this is suggested. Within the signal-to-noise recorded there is no evidence of any substitution on the A-site. The YO5 is associated with oxygen vacancies, with the intensity of this line correlated with the barium-content and indicates that maximising the barium-content is important. A study using DNP to enhance the 89Y signal in hydrated yttrium-substituted barium zirconate samples containing 20 and 30% yttrium and water contents of ∼7 and ∼9% respectively showed very significant gains in sensitivity, although using the indirect approach i.e. via the protons required long CP contact times as the 1H–89Y dipolar coupling is quite weak.52 It was noted52 that the low temperature of the DNP experiment (105 K) helps enable long contact times to be used and reduces the mobility and hence averaging of dipolar interactions. The increase in sensitivity allowed a 2D 1H–89Y CP MAS Frequency Switched Lee-Goldburg HETCOR spectrum to be collected. Two 89Y signals could be seen at ∼415 and 350 ppm, both assigned to octahedral coordination. The correlation experiment showed these two peaks were correlated to proton signals at 5.4 and 6.1 ppm respectively. Calculating possible local structural configurations and the resulting NMR parameters using DFT and GIPAW approaches gave direct evidence that the structural feature corresponding to the (415, 5.4) correlation is a proton bound to a yttrium – a ‘trapped’ site. The other correlation corresponds to a further distinct trapped proton site. This a very good example of how a leading-edge methodology is providing real insight into the local features that drive the properties that are of technological interest in such materials.
A further perovskite that has been studied using an approach that included 119Sn, 17O and 89Y NMR is BaSn1−xYxO3−x/2, with the 89Y NMR data collected for only x = 0.1, 0.3, and 0.5. As seen for the zirconate above two yttrium signals are resolved.94 One which shifts with composition from 300 to 343 ppm. As the first B-sublattice is known to be all tins, this effect is ascribed to changing Y cations in the second B-shell. A second site is seen, most clearly at high yttrium concentration and given it is a higher shift (∼450 ppm), as for the study by Lee et al. in a zirconate is thought to be a YO5 site.93 This latter peak disappears on hydration as the oxygen vacancy sites are occupied by hydroxyls. The experimental work was combined with computational outputs. There was some broad agreement with the trends e.g. relative shifts and changes with composition, but there were larger discrepancies between computed and observed values compared to the agreement in some other yttrium-containing systems which were not fully understood. Proton mobility remains high even at high yttrium doping which suggests there is quite strong order that prevents Y–O–Y formation as this more basic configuration would likely trap protons. The presence of magnetic ions complicated the spectrum through direct magnetic and relaxation effects, but 89Y MAS NMR has been reported from two double perovskites, Ba2YRuO695 and Ba2YReO6,96 which are geometrically frustrated antiferromagnets. 89Y MAS NMR showed a major peak at −5860 ppm for Ba2YRuO6, with the large shift a result of the direct magnetic interaction from the ruthenium, along with a second much smaller peak observed at −5100 ppm which are attributed to YO6 units with six and five ruthenium NNN respectively. The relative intensity suggests that there is strong yttrium/ruthenium ordering with only a few % disorder.95 For Ba2YReO6 only a single peak at −23290 ppm is observed suggesting almost perfect yttrium/rhenium ordering.96
Yttrium silicates are of technological interest as yttria is often used as a sintering aid in non-oxide ceramics (e.g. Si3N4, SiC), which then often results in yttrium-containing silicates or oxynitrides occurring as grain boundary phases. A study using 89Y MAS NMR reported data from the known polymorphs of Y2Si2O7 (y, α, β, γ, δ) and Y2SiO5 (X1, X2).99 This data expanded on the range of polymorphs and in some cases provided significantly better resolution than had previously been reported.71 The narrower lines obtained in this study allowed a fuller reconciliation with the crystal structures and in the case y-Y2Si2O7 indicated only a single yttrium resonance suggesting that the crystal structure determination needs re-examining. In α-Y2Si2O7 four yttrium sites were completely resolved in agreement with the structure. The two competing structures in the literature for δ-Y2Si2O7 could be distinguished as only one of these structures has a single crystallographic site for yttrium. As this data updates that appearing in the previous summary in the literature2 it is included in Table 2. There is a clear correlation with the mean Y–O distance across most of the dataset. The data for α-Y2Si2O7 clearly illustrates this with all four sites being YO8 coordinated, but a shift difference of >130 ppm across the sites resulting from the changing average Y–O bond length. There is also a trend of decreasing δiso with increasing coordination number of the yttrium, although there is quite strong overlap between the shift ranges, re-emphasising any assignment based on the shift alone has to be approached carefully (Table 2 and Fig. 8).
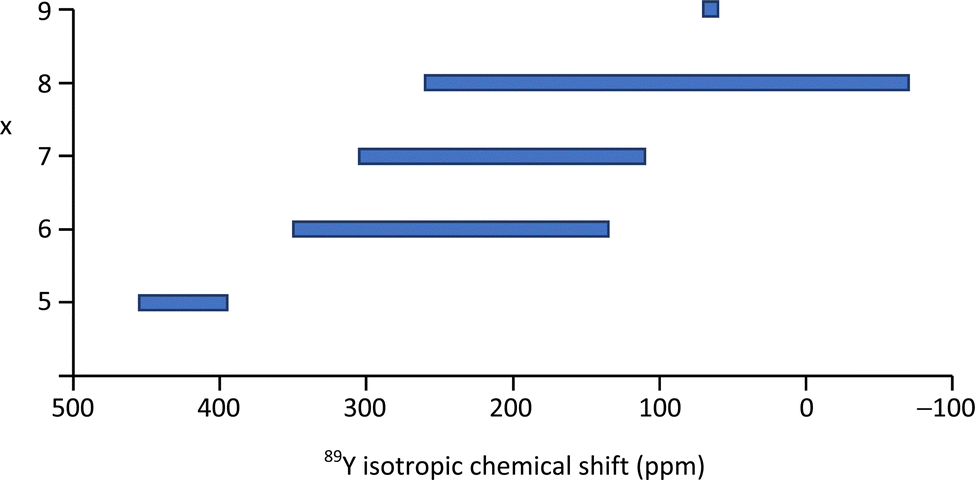 | ||
| Fig. 8 89Y isotropic chemical shift ranges associated with YOx local coordinations based on the values reported in the literature as given in Table 2 or summarised previously.2,5 | ||
The application of 89Y to crystalline silicates has been extended to examine solid solutions, in analogy to the work described above on pyrochlores and perovskites. An example is the solid solution Sc2Si2O7–β-Y2Si2O7.100 The data was collected using the CPMG approach for x = 0.5, 1.0, 1.5 and 2.0 in YxSc2−xSi2O7. The pure yttrium end member shows a single very sharp resonance (Fig. 9). The other spectra are much broader, with some asymmetry. Given that Sc3+ and Y3+ have very different ionic radii, as scandium enters the structure there must be some local distortion which results in the significant broadening observed from chemical shift dispersion. The asymmetry observed as scandium is added cannot be explained by the effect on the yttrium site of a random distribution of NNN substitution as the intensity does not vary as expected. It was suggested that it is more likely that the symmetry is being lowered with there being two crystallographically distinct sites. Small resonances are also observed from impurity X2-(Sc,Y)2SiO5. The CPMG sequence has been applied to 89Y in the solid solution (La,Y)2Si2O7.101 The phase diagram shows that different phases can co-exist and at 99% yttrium two signals are seen at 199 and 121 ppm, corresponding to γ- and δ-(La,Y)2Si2O7 polymorphs respectively. At 5% yttrium the γ has disappeared and the δ has become broader. This broadening results from the increasing range of NNN configurations possible. This system contains a further crystallographic phase G-(La,Y)2Si2O7, which as two distinct lanthanum/yttrium sites. Some of the spectra where G-(La,Y)2Si2O7 is present could be assigned to a single yttrium resonance showing that there is a strong preference for yttrium for one of the sites (RE2).101
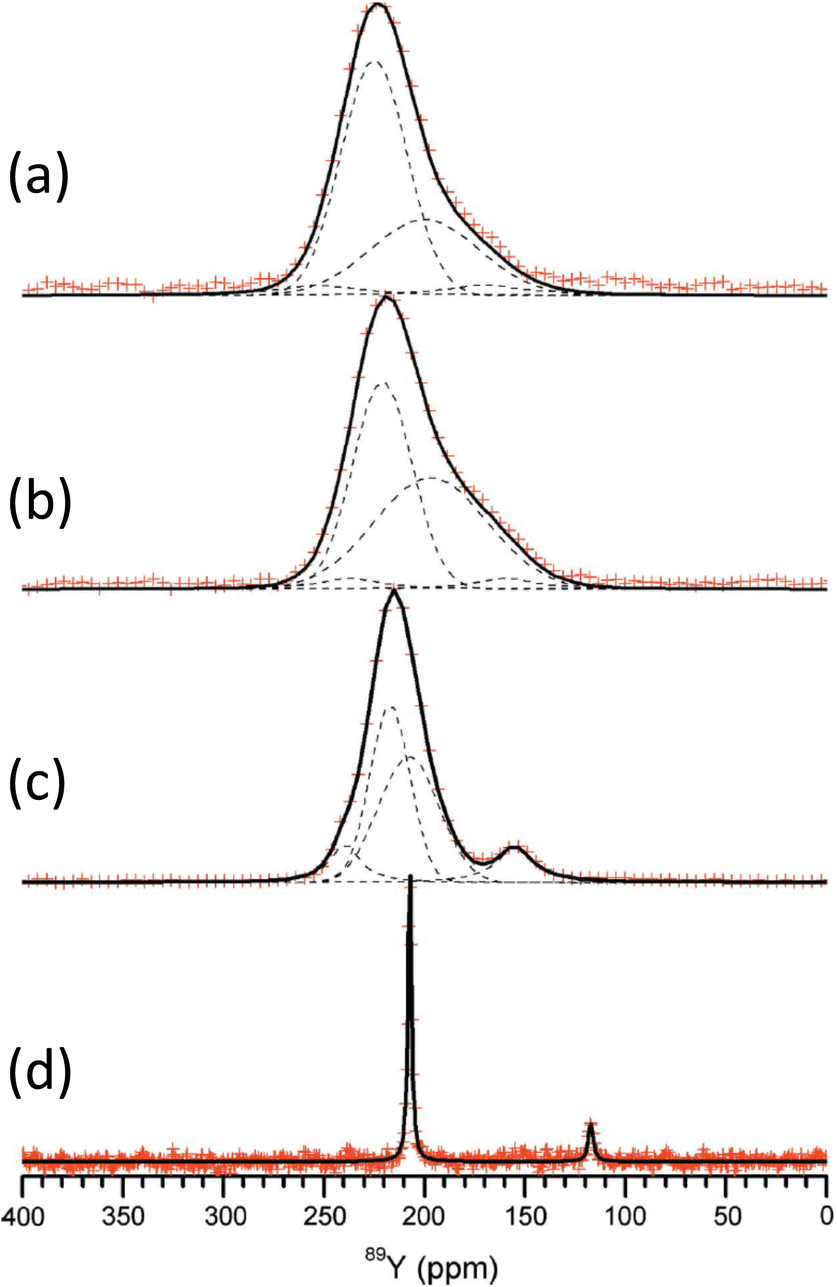 | ||
| Fig. 9 89Y CPMG MAS NMR spectra of YxSc2−xSi2O7 with (a) x = 0.5, (b) 1.0, (c) 1.5 and (d) 2.0. As scandium is added the appearance of more lines suggests inequivalent sites result from differing NNN yttrium and scandium.100 Reproduced from ref. 100 with permission from the International Union of Crystallography, copyright 2011. | ||
:1.109 Y3BO6 is a nominal composition, better represented by Y17.33(BO3)4(B2O5)2O16 with nine yttrium sites (YO8,YO7) in the structure110 and a complex 89Y MAS NMR spectrum was obtained with several peaks spanning the range 297–168 ppm.108 No assignment was given and this could usefully be revisited using NMR crystallography techniques. A further closely related phase YAl3(BO3)4 that crystallises in the huntite structure, containing YO6 showed a single 89Y MAS NMR resonance (Table 2).111 Applying 89Y MAS NMR to (Y2O3)x(Al2O3)0.4−x(B2O3)0.6 collected by a CPMG approach is probably the first such data reported from a glass showed quite broad resonances ∼100 ppm due to chemical shift dispersion. The addition of low levels of paramagnetic doping did not degrade the effectiveness of the CPMG approach, but considerably shortened the T1 allowing a good signal-to-noise to be collected relatively quickly, with an optimum level of doping suggested.102 With increasing yttria content the peak position changes monotonically from ∼70 ppm up to 120 ppm. By comparison with the model crystalline compounds the shifts are in much better agreement with the borates than those in the oxides or aluminates suggesting that yttrium bonding is dominated by borate-like environments. The crystallisation of this series of glasses was followed using 11B, 27Al and 89Y MAS NMR, with the yttrium showing the expected borate/aluminoborate phases formed.103 The second series of (Y2O3)0.2(Al2O3)x(B2O3)0.8−x (0.15 ≤ x ≤ 0.4) glasses was also studied by a combination of 11B, 27Al and 89Y MAS NMR. Across this series with increasing x, δiso for 89Y increases from 102 to 133 ppm. Comparison with the model crystalline compounds shows yttrium is becoming more associated with aluminium.104 As glasses from this series are crystallised the NMR very nicely shows how YBO3 and YAl3(BO3)4 form as the yttrium-containing phases (Fig. 10) and a model that has four (five with the unknown phase ‘X’) phases, with the equilibrium phases changing at around x = 0.3.105
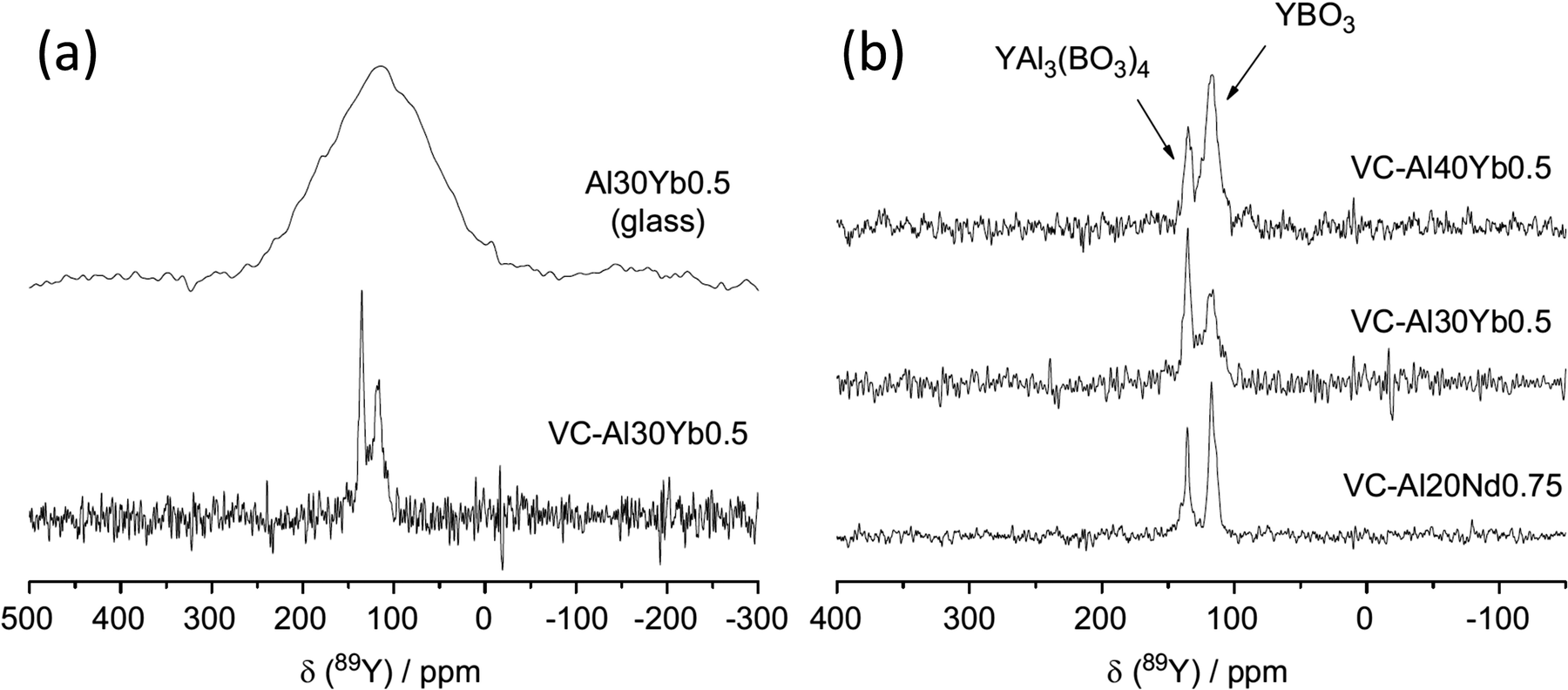 | ||
| Fig. 10 Application of 89Y MAS NMR to probe glass to ceramic transitions, with an example given for (a) (Y2O3)0.1(Al2O3)0.3(B2O3)0.6 (Al30) Yb-doped sample and (b) comparison of the major yttrium phases that form for different aluminium levels as well as a comparison of ytterbium to neodymium.105 Reproduced from ref. 105 with permission from Elsevier, copyright 2012. | ||
The concept of polyamorphism has been examined in Y2O3–Al2O3 glasses through three glasses in the compositional range 24 to 41 mol% Y2O3 using 27Al and 89Y MAS NMR in combination with microscopy and calorimetry.106 Polyamorphism is an interesting concept where there is a density driven ‘glass-in-glass’ phase separation. The 89Y MAS NMR data showed two resonances, a broader resonance covering the range 110–300 ppm and a narrower one at ∼75 ppm, with the relative integrated intensity and centre of gravity of the broader peak changing with composition. By comparison with the shift trends in crystalline yttrium aluminates the peak ∼220 ppm corresponds predominantly to YO8. Peaks at lower shift are most likely due to higher coordinations. Although the exact assignment was not possible the data showed distinct regions exist within the sample with differing average yttrium coordinations.106 In aluminosilicate glasses the structural incorporation of REs was investigated using 89Y as a proxy. A comprehensive study combined experimental data with DFT GIPAW calculations of the NMR parameters based on MD simulations of the glass structure.107 A comparison of direct 89Y MAS NMR and 27Al-89Y RAPT-CP-MAS NMR data showed relatively little difference in the spectra suggesting that aluminium is close to yttrium throughout the composition range. The computational work showed that yttrium exists in all coordinations five to eight, with six and seven dominant. It also showed that the CSA tends to be larger than in crystalline compounds. The chemical shift increases as silicon replaces aluminium and/or yttrium as the NNN.107
| Compound | δ iso (ppm) | Local coordination and other comments | Ref. |
|---|---|---|---|
| YF3 | −112 | YF9 | 113 |
| −107.0 | YF9 | 114 | |
| −109.6, −108.7 | YF9 | 116 and 117 | |
| β-NaYF4 | −78, −80 | YF9 | 113 |
| α-KY3F10 | −55 | YF8 | 113 |
| −50.8 | YF8 | 114 | |
| LiYF4 | −54 | YF8 | 113 |
| α-NaYF4 | −44 | YF8 | 113 |
| β-K2Y2F7 | −47.9, −49.9 | YF8 peaks in ratio 1:3 |
114 |
| KYF4 | −25, −32 | YF7 peaks in ratio 1:1 |
113 |
| −28.5, 2.1 | YF7 | 114 | |
| K2YF5 | −25 | YF7 | 113 |
| 29.7 | YF7 | 114 | |
| γ-K3YF6 | 20.9 | YF6 | 113 |
| r-YOF | 125.6 | YO4F4 | 116 |
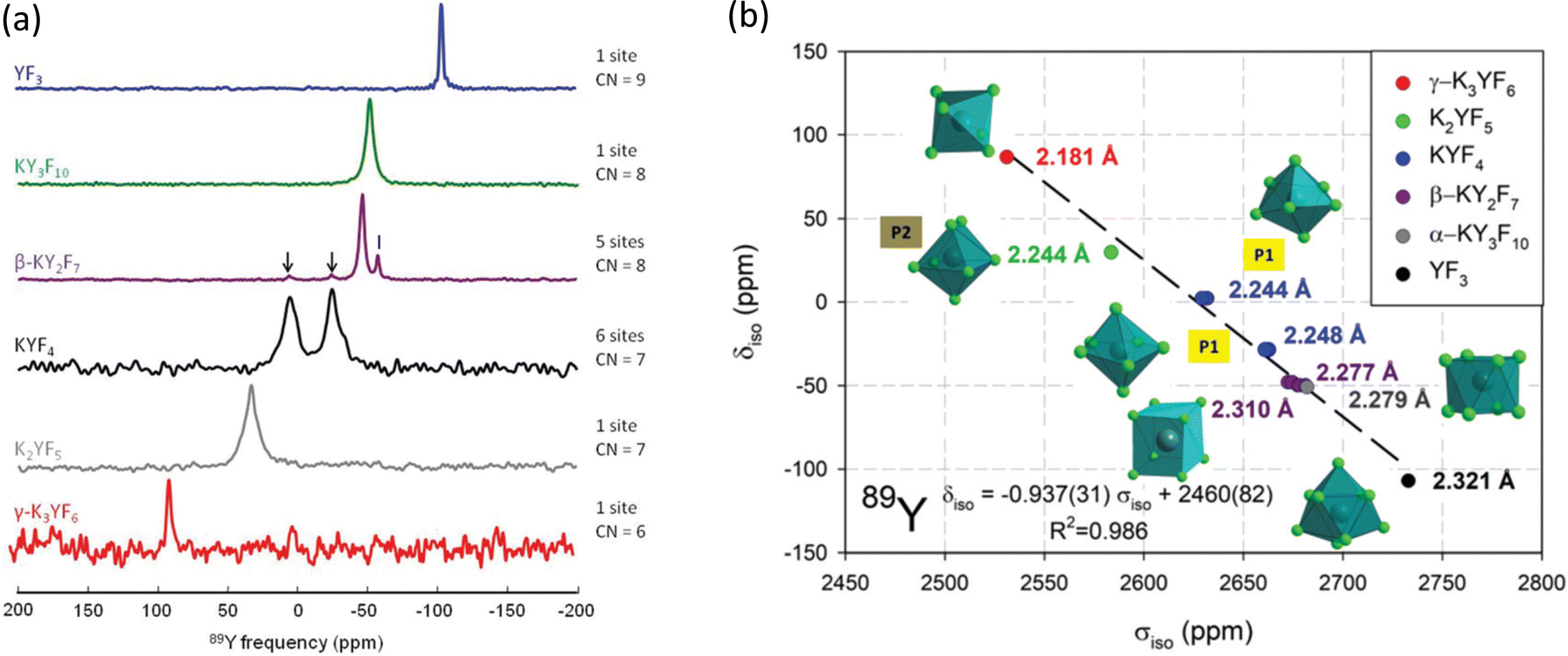 | ||
| Fig. 11 (a) 89Y VACP MAS (3 kHz) NMR spectra from various KF-YF3 binary compounds with the yttrium coordination number (CN) given. In β-KY2F7 known KYF4 impurities with I indicating an unidentified impurity. (b) Experimental 89Y δisoversus calculated 89Y σiso with the line showing the linear regression calculated on these values corresponding to the equation given.114 Reproduced from ref. 114 with permission from the Royal Society of Chemistry, copyright 2019. | ||
For yttrium fluoride nanoparticles of composition (H3O)YF10·xH2O 19F and 89Y showed that the nanoparticles did not consist of bulk YF3. Static and MAS NMR data was collected using VACP and showed two 89Y species with resonances at ∼55 and −36 ppm, with the intensity of the latter increasing with decreasing particle size suggesting it was a surface related species.115 Fluorolytic sol–gel formation of nanoscale yttrium fluorides/oxyfluorides from acetate precursors was followed by a combination of 19F and 89Y which included data from the crystalline oxyfluoride r-YOF.116 Yttrium was added to CaF2via a sol–gel route to form Ca1−xYxF2+x.117 To enhance the yttrium NMR signal 19F–89Y CP was used. At low yttrium contents a single resonance was observed at −33 ppm (which then shifts to more negative values with increasing yttrium content) attributed to yttrium occupying the YF8 cubic site in the host structure. At higher concentrations fluorine clustering occurs and a second signal at −50 to −60 ppm is observed, but associated with YF8 in a square antiprism. The trends in intensity qualitatively agree the expected changes in structure. Yttrium was also introduced into the closely related SrF2 to form a solid solution Sr1−xYxF2+x using mechanochemical and sol–gel approaches.118 The two synthetic approaches show very similar spectra and the same two yttrium species as for the calcium system, but with slightly different shift and opposite trends in the shift with changing composition which is related to the opposite trends in the lattice parameter as yttrium is added. However the two species and underlying structural explanations are the same for both systems.
More recently a collaboration between Eckert and Pöttgen has produced an extensive set of 89Y NMR data from intermetallics. Their first 89Y MAS NMR study examined YPd2Sn and YPdSn, also reporting 18 other related compounds for comparison. The shift range covered by the 20 compounds was ∼3000 ppm.121 There was only very modest sideband intensity for both YPdSn and YPd2Sn indicating a relatively small KSA. YPdSn has a much smaller linewidth than YPd2Sn which was possibly caused by local variations around the yttrium due Pd/Sn disorder.121 In the wider set of compounds reported three yttrium sites could be resolved in YPdSi. In the group of 20 compounds studied there was no obvious correlation to parameters that would normally be expected (e.g. electronegativity) since the density of states in the conduction band is generally not a reflection of the local environment given the band structure is delocalised. Full electronic structure calculations provide better insight. 89Y NMR has also been applied to the series YNiX and YIrX (X = Si, Ge, Sn, Pb), compounds which crystallise in an orthorhombic TiNiSi-type structure.122 There were some attempts to rationalise the shift changes observed with the s-electron density of states and additional quantum mechanical calculations. Although there was a general increase in the shift with the s density of states other structural factors must be complicating things. For YIrSi and YIrGe a sizeable KSA was observed which was related to the reverse T/X site occupancy in these two compounds.122 For the compounds Y2RuB6, Y2ReB6, Y2RhSi3, YRh2Si, and YPdSi, the first four have two and the last one three yttrium sites. 89Y MAS NMR was able to readily resolve these sites with shift differences >1500 ppm.123 A further report concerned the ThCr2Si2-structured silicides YT2Si2 (T = Co, Ni, Cu, Ru, Rh, Pd).124 Again a large range of shifts was observed and in one case, YRu2Si2 a large KSA was found which could be attributed to local structural distortion.124 The Pauli paramagnet YIrSb showed a single 89Y resonance that was shifted by ∼600 ppm from the isotypic YIrSn which is a result of the increased electron withdrawing power of antimony compared to tin reducing the s-electron spin density at the yttrium site.125 In the YCrB4-structured borides YTB4 (T = Mo, W, Re) the 89Y MAS NMR data shows very much smaller Knight shifts than for the compounds above. For YMoB4 and YWB4 the shifts are in the range for non-magnetic, insulating materials whereas YReB4 is a metal and therefore the Knight shift makes an important contribution.126 Recently a computational study has drawn together many of these experimental observations applying an ab initio DFT approach to calculating the 89Y shielding parameters. The calculated shift variation of ∼2500 ppm matches well with the experimentally observed shift variation, with some outliers. The calculations show that although the spin-contact (s-electron) term dominates other terms cannot be neglected including the chemical shift.127
Metal hydrides have several potential technological applications, such as for hydrogen storage and thin films can display varying optical properties. For yttrium hydride as hydrogen is added to yttrium metal the stoichiometry strongly influences the metallic character of the system. Addition of oxygen to form YHxOy can create photochromic materials.1121H–89Y CP-MAS showed a relatively broad peak 372–190 ppm, which was made up of at least 7 separate components. There are interesting changes in the spectrum on illumination, such as the increase of the Knight-shifted peak. A 2D 89Y–1H HETCOR spectrum confirmed the presence of the numerous local yttrium coordinations.112
3.2 Rhodium-103
103Rh is 100% naturally abundant like 89Y, but its smaller γ typically leads to lower sensitivity and longer relaxation times than 89Y meaning it is typically more difficult to observe. Its NMR was extensively reviewed by Carlton in 2008.12 Rhodium also possesses a very large chemical shift range of ∼12000 ppm largely determined from solution-state studies. Solid-state NMR reports made up only a very small fraction of those in the review by Carlton. The examples included rhodium nanoparticles.128 From insulating solids 1H–103Rh CP on two crystalline salts of polynuclear Rh(III) clusters were reported. One was a mesitylate salt of a dimer and the other the perchlorate salt of a trimer. To boost the signal-to-noise a 7.5 ppm MAS probe was used coupled with relatively long contact times (>10 ms) for the CP. For the mesitylate sample δiso = 10131 ppm, with slow spinning (∼1 kHz) showing a set of spinning sidebands (Fig. 12) allowing the CSA components to be extracted, with a span (Ω) of 758 ppm and skew of −0.33. For the perchlorate salt two sets of spinning sidebands were clearly resolved with an approximate intensity ratio of 1:2, with careful inspection of the more intense set of sidebands showing evidence of two partially resolved peaks, consistent with the presence of three inequivalent rhodium positions, with the δiso of 9401, 9359, and 9354 ppm. The first line has a span of 1720 ppm and skew of −0.64 and the latter two have a single set of parameters reported with a span of 1620 ppm and skew of −0.58.37 Despite these elegant results there has been no data building on this work. This is most likely due to the very large shift range observed making the CSA sizeable for everything apart from the most symmetric sites, as well as driving a significant intrinsic linewidth from chemical shift dispersion in samples with disorder/imperfections. These factors coupled with the low intrinsic sensitivity result in the low number of 103Rh NMR observations from solids. The recent successful development of indirect proton-detected 103Rh NMR data was reported from a solid rhodium complex, where two distinct peaks at chemical shifts of −8300 and −8375 ppm were observed.55 The shift difference is much larger than any expected scalar or residual dipolar coupling and was therefore attributed to different solid forms.
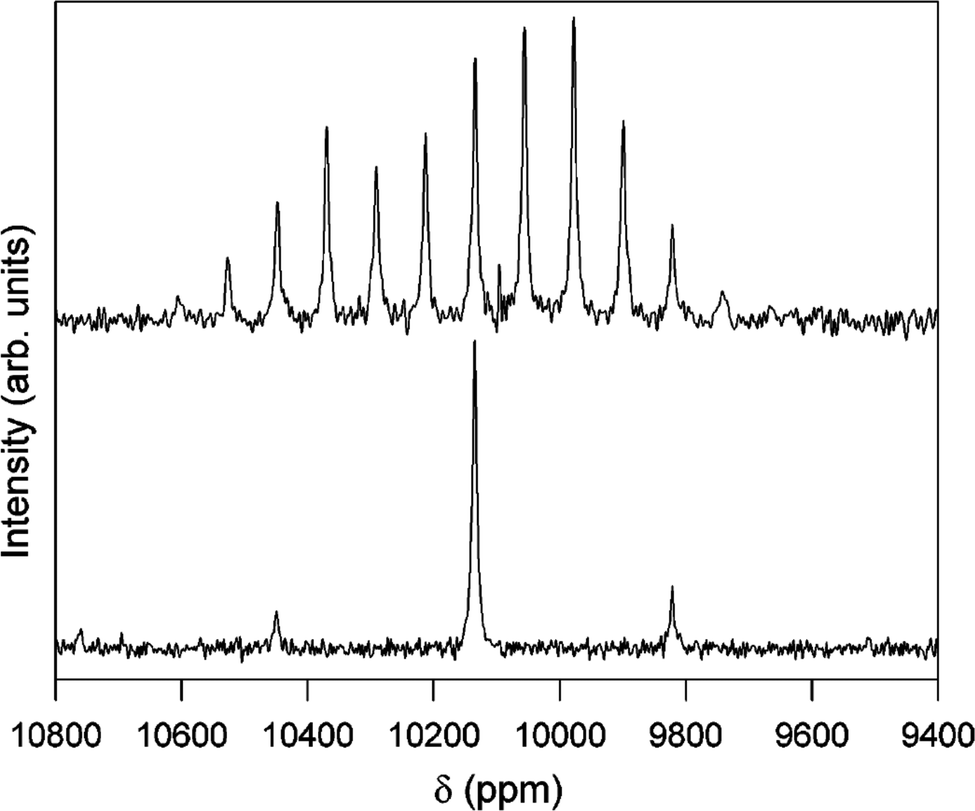 | ||
| Fig. 12
1H–103Rh CP-MAS NMR spectra of [[(H2O)4Rh(μ2-OH)2Rh(H2O)4]‚(dmtos)4]·8H2O comparing spinning rates of 1.0 (top) and 4.0 kHz (bottom). Both spectra acquired with 8 ms contact time, 5 s relaxation delay for 12000 (top) and 15800 (bottom) acquisitions.37 Reproduced from the ref. 37 with permission from the American Chemical Society, copyright 2006. | ||
3.3 Silver-109
Silver had two isotopes suitable for NMR spectroscopy, 107Ag and 109Ag, with almost equal natural abundance. However 109Ag has a 41% greater receptivity and forms the vast majority of NMR reports on silver.2 Early work covered fast-ion conducting glasses (where static lines were observed due to silver ion motion) and complexes of biological significance. As expected for a relatively heavy metal a sizable chemical shift range is observed and often spectra show very significant CSA. Merwin and Sebald38 showed that 1H–109Ag CP could significantly improve the sensitivity, but relatively long contact times (>10 ms) were necessary and that silver lactate was a good set-up compound. A body of work 2003–2007 by Penner and co-workers added a significant amount of new 109Ag NMR data from model compounds and extensively reviewed129,130 the development of it as a nucleus up to that point. An alternative set-up compound for CP was suggested in AgSO3CH3 which could also act as secondary chemical shift reference at 87.2 ppm.39 The literature up to around 2006 is very well covered through the four articles,2,5,129,130 with the data given here from studies since then. The key themes that have become more common for studies using 109Ag NMR are the state of silver in microporous materials, with some sporadic reports on other systems e.g. chromatographic media, quantum dots, silver cluster formation and some continuing work on fast-ion conducting glasses.The 109Ag MAS NMR spectrum reported from Ag2PO3F showed three resonances at 190, 38, and −10 ppm with an approximate intensity ratio of 2:1:1. There are three crystallographically distinct sites with significantly different local coordinations. The relative intensities allow the 190 ppm peak to be assigned to the Ag1 site which is also consistent with this site having the lowest coordination number. The differences in the local structures around the other two sites did not allow an assignment to the Ag2 and Ag3 sites on the basis of the information available.131 For the samples AgOP(OR)2 (R = CH3, C2H5, C4H9) using CP-MAS NMR allowed all the parameters of the CSA to be collected. With two peaks in the isotropic position J(109Ag,31P) couplings of ∼1300 Hz could be seen, with this magnitude indicating that the silver and phosphorus are directly bonded.132 The contribution that computational work now plays in NMR is illustrated by a study applying a full-potential linearised augmented plane wave (FP-LAPW) approach to calculate a range of properties including the NMR response for the compounds AgXC7H10N2 (X = Cl, Br, I).133 The approach was tested by calculating δiso (via the shielding) for AgNO3 which gave −72.13 ppm, compared to the experimental value of −76.0 ppm.129 The full CSA tensor was calculated for each of the three compounds, with the shift trends showing that as the electronegativity of the attached halogen decreases leads to a larger chemical shift.133
A study of 3–6 wt% silver-containing BEA zeolites after different treatments used a range of different probe techniques, but included 109Ag MAS NMR. For a 3% silver-containing sample 109Ag MAS NMR showed a single silver site at 21.5 ppm, but at 6% the single peak had shifted to −33.2 ppm, with the shift explained as changes in the paramagnetic character of small silver clusters. On reduction of the 6% sample a large shift to the Knight-shifted position of a silver nanoparticle at 5272 ppm is observed.137 For two small pore zeolites (Cha, Rho) silver loadings were varied between 14 and 26 wt% silver. On reduction extensive formation of metallic silver was observed. The differences in lineshape suggested those particles formed in Cha(2) are quite similar to that of silver metal whereas in Rho(4) the significantly narrower resonance observed suggests a lower aggregation of silver, which was corroborated by UV-Vis spectra.138 For silver-exchanged sodium zeolite Y, the as-prepared sample showed a single peak 42 ppm corresponding to Ag+ associated with the lattice. On reduction all the silver is transformed to the metal at 5570 ppm. As it is used for catalytic oxidation of ammonia the spectrum then shows a mixture of the two species.139 The mesoporous framework material silver 4,4′-bipyridine nitrate can capture perchlorate to form silver 4,4′-bipyridine perchlorate and the structural transformation can be followed by 109Ag NMR. For the initial compound 109Ag CP MAS NMR shows a single isotropic resonance at 438.5 ppm and a significant CSA. J-coupling to two equivalent 14N (I = 1) nuclei shows the expected quintet fine structure. As chlorate is added a second signal that increases in intensity with chlorate concentration is formed at 341 ppm with different CSA.140
Silver-ion chromatography can separate unsaturated compounds such as light olefin/paraffin mixtures. Tuning the selectivity of the stationary phase towards olefins occurs by varying the ratio of silver ion:mixed ligands. 109Ag NMR was carried out using indirect detection based around the 1H–109Ag dipolar couplings for proton-detected HETCOR spectra. For other samples as the silver coordination changed strong shift differences of ∼370 ppm were observed.145 Fast-ion conducting glasses in the series 40AgI–(60 − x)AgPO3–xAg2WO4 (0 ≤ x ≤ 25 mol%) with interesting optical properties which strongly depend on composition have been studied with a range of techniques including 31P and 109Ag NMR. For 109Ag both static and MAS NMR data was collected, with the MAS NMR spectra showing a single sharp peak ∼490 ppm, which did not change position with composition. This indicates that the silver is present as Ag+, the location of which is independent of composition consistent with the fact that it depends on the O/I ratio, which in constant across this series.146
3.4 Tungsten-183
With tungsten having the lowest receptivity and being the heaviest of the nuclei reported here, these facts combine to make this the most difficult of them to observe. This is reflected in the continuing scarcity of solid-state NMR observations of 183W, which is well illustrated by the paucity of citations to the pioneering observations of this nucleus by MAS147 and 1H CP.40 There has been a recent addition to the literature of 183W via the report of NMR data from some polyoxometallates which have a range of potential technological applications.13 High field (18.8 T) was applied along with MAS at 10 kHz and CPMG with SPINAL-64 1H decoupling for most samples. 183W MAS NMR spectra were reported for K33H7P8W48O184 (crown-type), K8γ-SiW10O36 and NaK6PW11O39 (Keggin-type), K6P2W18O62 (Dawson-type) and (TBA)4SiW12O40. In all of these polyoxometalate units the tungsten is present as WO6, with these units clustered in various ways to form the complex. The isotropic chemical shifts were reported (Table 4). Despite the relatively poor signal-to-noise two distinct tungsten sites are observable in K33H7P8W48O184 in the ratio 2:1 and three equally populated sites for K8γ-SiW10O36 (Fig. 13). Similarly for K6P2W18O62, the two anticipated signals are seen, whereas although six are expected for NaK6PW11O39 only two could be seen. The CSA for 183W in these complexes ranges up to ∼1500 ppm.13 The proton-detected approach has also been applied to 183W, demonstrated on (NH4)2WS4,55 with the observed shift at 3648 ppm agreeing with the original CP observation.40
| Compound | δ iso (ppm) (±1) |
|---|---|
| K33H7P8W48O184 | −170, −202 |
| K8γ-SiW10O36 | −95, −125, −164 |
| NaK6PW11O39 | −136, −183 |
| K6P2W18O62 | −118, −168 |
| (TBA)4SiW12O40 | −145 |
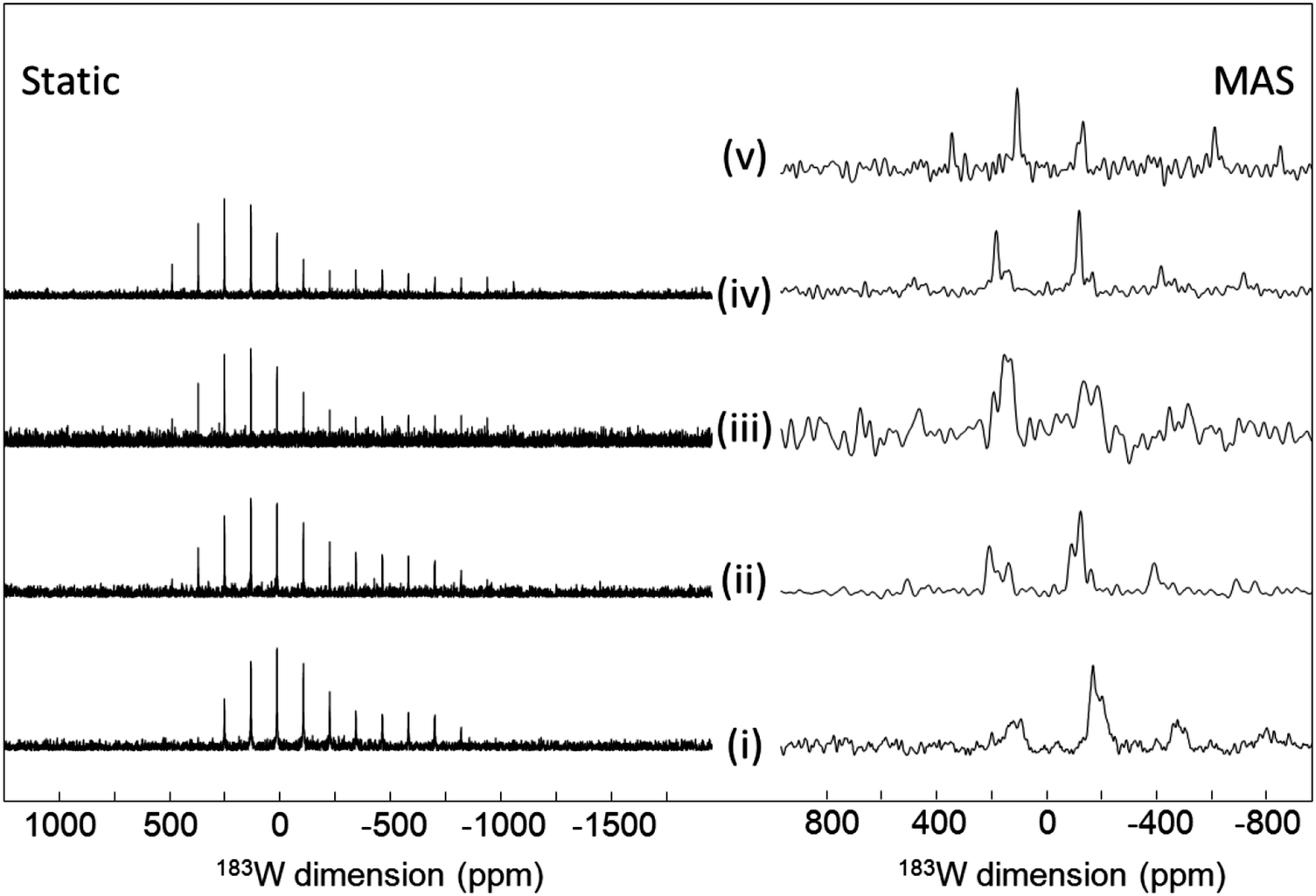 | ||
| Fig. 13 183W NMR spectra of (i) K33H7P8W48O184, (ii) K8SiW10O36, (iii) K6P2W18O62, (iv) NaK6PW11O39 and (v) (TBA)4SiW12O40. collected static (left) and MAS (right).13 For details of the collection conditions see the ESI of ref. 13. Reproduced from the ref. 13 with permission from the John Wiley and Sons Ltd, copyright 2017. | ||
4. Summary and outlook
A summary is presented of the progress made over the last 20 years in the application of solid-state NMR of spin-½ low-γ nuclei to the study of inorganic materials up to approximately spring 2022. Progress is illustrated in the period since the last overview of this set of nuclei collectively was provided around 20 years ago,2,5 as well as using more recent summaries of individual nuclei. Key differences that have emerged in the period are the better sensitivity available now and the complementary computational work. For sensitivity the much higher magnetic fields now typically available start things off in a much better place. It should be noted though that for nuclei where chemical shift-based mechanisms are important there can be a downside of higher magnetic fields with the CSA spreading the intensity over a greater frequency range and if disorder is present the intrinsic width under MAS will be greater. A range of other signal enhancement techniques have been successfully applied such as CPMG echo sequences, DNP and indirect detection. All show advantage and should help further develop these nuclei. DFT calculations of both structure and then the corresponding NMR parameters are widely impacting on solid-state NMR which is no different for this group of nuclei. The development of these four nuclei over the last 20 years has proceeded at significantly different rates. It is clear that 89Y has made the most progress with a significant body of work building on the foundations of the literature in early 2001. Between 2000–2006 a substantial database of 109Ag shifts was reported by Penner.129,130 Up to this point static work on fast-ion silver based materials had been the most common solid-state NMR application of 109Ag. Since then the use of 109Ag has been dominated by using the large shift difference between silver in Ag0 and Ag+ states to identify the nature of the silver present and the formation of silver nanoparticles in a range of settings. For 103Rh and 183W there still are very few solid-state NMR reports probably largely as a result of sensitivity limitations. As the methods for signal enhancement become more routine there will be increasing impact on the observation of such nuclei. Also as the leading-edge of individual techniques (e.g. ultrafast MAS, ultrahigh field, higher temperature DNP) develop so will their combinations which will open up new applications to such nuclei. This Perspective clearly illustrates that NMR continues to make real strides with a multinuclear approach becoming more fully realised.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The author gratefully acknowledges the support of the Universities of Southampton, Lancaster and Warwick of his research over the last decade. Copyright holders who gave permission for the use of figures from their publications are thanked. NMR infrastructure at Warwick funded through a variety of sources including EPSRC, the HEFCE and the University of Warwick, as well as through the Science City Advanced Materials project supported by Advantage West Midlands (AWM) and part funded by the European Regional Development Fund (ERDF) has been important in making low-γ nuclei more accessible. Current support from EPSRC is acknowledged via EP/T014911/1 for the provision of ultrahigh solid-state NMR infrastructure.References
- M. E. Smith, Magn. Reson. Chem., 2021, 59, 864–907 CrossRef CAS.
- K. J. D. MacKenzie and M. E. Smith, Multinuclear Solid-State NMR of Inorganic Materials, Pergamon, 2002 Search PubMed.
- R. K. Harris, E. D. Becker, S. M. C. De Menezes, R. Goodfellow and P. Granger, Pure Appl. Chem., 2001, 73, 1795–1818 CAS.
- M. J. Duer, Solid-State NMR Spectroscopy Principles and Applications, Blackwell Science, 2002 Search PubMed.
- M. E. Smith, in Annual Reports on NMR Spectroscopy, ed. G. A. Webb, 2001, vol. 43, pp. 121–175 Search PubMed.
- M. E. Smith and E. R. H. van Eck, Prog. Nucl. Magn. Reson. Spectrosc., 1999, 34, 159–201 CrossRef CAS.
- R. E. Wasylishen, S. E. Ashbrook and S. Wimperis, NMR of Quadrupolar Nuclei in Solid Materials, John Wiley & Sons, 2012 Search PubMed.
- I. P. Gerothanassis, Prog. Nucl. Magn. Reson. Spectrosc., 1987, 19, 267–329 CrossRef CAS.
- R. K. Harris, E. D. Becker, S. M. C. De Menezes, P. Granger, R. E. Hoffman and K. W. Zilm, Pure Appl. Chem., 2008, 80, 59–84 CrossRef CAS.
- C. P. Grey, M. E. Smith, A. K. Cheetham, C. M. Dobson and R. Dupree, J. Am. Chem. Soc., 1990, 112, 4670–4675 CrossRef CAS.
- M. C. Read, J. Glaser, M. Sandstrom and I. Toth, Inorg. Chem., 1992, 31, 4155–4159 CrossRef CAS.
- L. Carlton, in Annual Reports on Nmr Spectroscopy, ed. G. A. Webb, Elsevier Academic Press Inc, San Diego, 2008, vol. 63, pp. 49–178 Search PubMed.
- M. Haouas, J. Trebosc, C. Roch-Marchal, E. Cadot, F. Taulelle and C. Martineau-Corcos, Magn. Reson. Chem., 2017, 55, 902–908 CrossRef CAS.
- J. V. Hanna and M. E. Smith, Solid State Nucl. Magn. Reson., 2010, 38, 1–18 CrossRef CAS PubMed.
- C. Leroy and D. L. Bryce, Prog. Nucl. Magn. Reson. Spectrosc., 2018, 109, 160–199 CrossRef CAS.
- B. F. Chmelka, J. Magn. Reson., 2019, 306, 91–97 CrossRef CAS PubMed.
- S. E. Ashbrook, J. M. Griffin and K. E. Johnston, in Annual Review of Analytical Chemistry, ed. P. W. Bohn and J. E. Pemberton, Annual Reviews, Palo Alto, 2018, vol. 11, pp. 485–508 Search PubMed.
- D. L. Bryce, in Annual Reports on NMR Spectroscopy, ed. G. A. Webb, Elsevier, 2022, ch. 1, vol. 107, pp. 1–46 Search PubMed.
- F. A. Perras and A. L. Paterson, in Comprehensive Inorganic Chemistry III, ed. J. Reedijk and K. R. Poeppelmeier, Elsevier, 2021 DOI:10.1016/B978-0-12-823144-9.00015-7.
- T. Charpentier, Solid State Nucl. Magn. Reson., 2011, 40, 1–20 CrossRef CAS.
- C. Bonhomme, C. Gervais, F. Babonneau, C. Coelho, F. Pourpoint, T. Azais, S. E. Ashbrook, J. M. Griffin, J. R. Yates, F. Mauri and C. J. Pickard, Chem. Rev., 2012, 112, 5733–5779 CrossRef CAS PubMed.
- S. E. Ashbrook and D. M. Dawson, Acc. Chem. Res., 2013, 46, 1964–1974 CrossRef CAS PubMed.
- R. K. Harris, R. E. Wasylishen and M. J. Duer, NMR Crystallography, John Wiley & Sons, 2012 Search PubMed.
- R. Lefort, J. W. Wiench, M. Pruski and J. P. Amoureux, J. Chem. Phys., 2002, 116, 2493–2501 CrossRef CAS.
- F. H. Larsen, H. J. Jakobsen, P. D. Ellis and N. C. Nielsen, J. Phys. Chem. A, 1997, 101, 8597–8606 CrossRef CAS.
- F. H. Larsen, J. Skibsted, H. J. Jakobsen and N. C. Nielsen, J. Am. Chem. Soc., 2000, 122, 7080–7086 CrossRef CAS.
- R. Siegel, T. T. Nakashima and R. E. Wasylishen, J. Phys. Chem. B, 2004, 108, 2218–2226 CrossRef CAS.
- I. Hung, A. J. Rossini and R. W. Schurko, J. Phys. Chem. A, 2004, 108, 7112–7120 CrossRef CAS.
- D. Massiot, I. Farnan, N. Gautier, D. Trumeau, A. Trokiner and J. P. Coutures, Solid State Nucl. Magn. Reson., 1995, 4, 241–248 CrossRef CAS.
- A. J. Pell, R. J. Clement, C. P. Grey, L. Emsley and G. Pintacuda, J. Chem. Phys., 2013, 138, 114201 CrossRef PubMed.
- E. Kupce and R. Freeman, J. Magn. Reson., Ser. A, 1995, 115, 273–276 CrossRef CAS.
- A. W. MacGregor, L. A. O'Dell and R. W. Schurko, J. Magn. Reson., 2011, 208, 103–113 CrossRef CAS.
- R. W. Schurko, Acc. Chem. Res., 2013, 46, 1985–1995 CrossRef CAS PubMed.
- L. A. O'Dell, Solid State Nucl. Magn. Reson., 2013, 55–56, 28–41 CrossRef PubMed.
- I. Hung and Z. H. Gan, J. Magn. Reson., 2010, 204, 256–265 CrossRef CAS PubMed.
- L. H. Merwin and A. Sebald, J. Magn. Reson., 1990, 88, 167–171 CAS.
- B. L. Phillips, J. R. Houston, J. Feng and W. H. Casey, J. Am. Chem. Soc., 2006, 128, 3912–3913 CrossRef CAS.
- L. H. Merwin and A. Sebald, J. Magn. Reson., 1992, 97, 628–631 CAS.
- G. H. Penner and W. L. Li, Solid State Nucl. Magn. Reson., 2003, 23, 168–173 CrossRef CAS PubMed.
- L. H. Merwin and A. Sebald, Solid State Nucl. Magn. Reson., 1992, 1, 45–47 CrossRef CAS.
- X. Zhao, W. Hoffbauer, J. Gunne and M. H. Levitt, Solid State Nucl. Magn. Reson., 2004, 26, 57–64 CrossRef CAS PubMed.
- K. J. Harris, A. Lupulescu, B. E. G. Lucier, L. Frydman and R. W. Schurko, J. Magn. Reson., 2012, 224, 38–47 CrossRef CAS.
- S. Wi, R. W. Schurko and L. Frydman, Solid State Nucl. Magn. Reson., 2018, 94, 31–53 CrossRef CAS PubMed.
- M. J. Jaroszewicz, A. R. Altenhof, R. W. Schurko and L. Frydman, J. Am. Chem. Soc., 2021, 143, 19778–19784 CrossRef CAS.
- A. G. M. Rankin, J. Trebosc, F. Pourpoint, J. P. Amoureux and O. Lafon, Solid State Nucl. Magn. Reson., 2019, 101, 116–143 CrossRef CAS PubMed.
- P. Berruyer, L. Emsley and A. Lesage, Emagres, 2018, 7, 93–104 CAS.
- F. A. Perras, T. Kobayashi and M. Pruski, Emagres, 2018, 7, 35–50 CAS.
- A. J. Rossini, J. Phys. Chem. Lett., 2018, 9, 5150–5159 CrossRef CAS.
- M. Rosay, L. Tometich, S. Pawsey, R. Bader, R. Schauwecker, M. Blank, P. M. Borchard, S. R. Cauffman, K. L. Felch, R. T. Weber, R. J. Temkin, R. G. Griffin and W. E. Maas, Phys. Chem. Chem. Phys., 2010, 12, 5850–5860 RSC.
- M. Ha and V. K. Michaelis, in Modern Magnetic Resonance, ed. G. A. Webb, Springer, 2018, pp. 1183–1206 Search PubMed.
- Q. Z. Ni, E. Daviso, T. V. Can, E. Markhasin, S. K. Jawla, T. M. Swager, R. J. Temkin, J. Herzfeld and R. G. Griffin, Acc. Chem. Res., 2013, 46, 1933–1941 CrossRef CAS PubMed.
- F. Blanc, L. Sperrin, D. Lee, R. Dervisoglu, Y. Yamazaki, S. M. Haile, G. De Paepe and C. P. Grey, J. Phys. Chem. Lett., 2014, 5, 2431–2436 CrossRef CAS.
- D. Jardon-Alvarez, N. Kahn, L. Houben and M. Leskes, J. Phys. Chem. Lett., 2021, 12, 2964–2969 CrossRef CAS.
- N. J. Brownbill, D. Lee, G. De Paepe and F. Blanc, J. Phys. Chem. Lett., 2019, 10, 3501–3508 CrossRef CAS PubMed.
- A. Venkatesh, M. J. Ryan, A. Biswas, K. C. Boteju, A. D. Sadow and A. J. Rossini, J. Phys. Chem. A, 2018, 122, 5635–5643 CrossRef CAS PubMed.
- A. Venkatesh, D. Gioffre, B. A. Atterberry, L. Rochlitz, S. L. Carnahan, Z. R. Wang, G. Menzildjian, A. Lesage, C. Coperet and A. J. Rossini, J. Am. Chem. Soc., 2022, 144, 13511–13525 CrossRef CAS PubMed.
- F. A. Perras, T. W. Goh and W. Y. Huang, Solid State Nucl. Magn. Reson., 2022, 120, 101807 CrossRef CAS.
- Z. R. Wang, M. P. Hanrahan, T. Kobayashi, F. A. Perras, Y. H. Chen, F. Engelke, C. Reiter, A. Purea, A. J. Rossini and M. Pruski, Solid State Nucl. Magn. Reson., 2020, 109, 101685 CrossRef CAS PubMed.
- M. Bak, J. T. Rasmussen and N. C. Nielsen, J. Magn. Reson., 2000, 147, 296–330 CrossRef CAS.
- D. Massiot, F. Fayon, M. Capron, I. King, S. Le Calve, B. Alonso, J. O. Durand, B. Bujoli, Z. H. Gan and G. Hoatson, Magn. Reson. Chem., 2002, 40, 70–76 CrossRef CAS.
- K. Eichele, WSolids1, version 1.21.3, Universität Tübingen, 2015.
- T. F. Kemp and M. E. Smith, Solid State Nucl. Magn. Reson., 2009, 35, 243–252 CrossRef CAS.
- F. A. Perras, C. M. Widdifield and D. L. Bryce, Solid State Nucl. Magn. Reson., 2012, 45–46, 36–44 CrossRef CAS.
- S. G. J. van Meerten, W. M. J. Franssen and A. P. M. Kentgens, J. Magn. Reson., 2019, 301, 56–66 CrossRef CAS PubMed.
- C. Martineau, J. Senker and F. Taulelle, in Annual Reports on NMR Spectroscopy, ed. G. A. Webb, 2014, vol. 82, pp. 1–57 Search PubMed.
- S. E. Ashbrook and D. McKay, Chem. Commun., 2016, 52, 7186–7204 RSC.
- A. Sebald, NMR Basic Principles and Progress, 1994, 31, 91–131 CAS.
- R. Youngman, Materials, 2018, 11, 476–499 CrossRef.
- O. B. Lapina, A. A. Shubin and V. V. Terskikh, in Modern Magnetic Resonance, ed. G. A. Webb, Springer, 2018, pp. 1125–1160 Search PubMed.
- A. R. Thompson and E. Oldfield, J. Chem. Soc., Chem. Commun., 1987, 27–29 RSC.
- R. Dupree and M. E. Smith, Chem. Phys. Lett., 1988, 148, 41–44 CrossRef CAS.
- T. Harazono and T. Watanabe, Bull. Chem. Soc. Jpn., 1997, 70, 2383–2388 CrossRef CAS.
- M. E. Smith, in NMR Crystallography, ed. R. K. Harris, R. E. Wasylishen and M. J. Duer, J. Wiley and Sons Ltd, 2009, ch. 23, pp. 341–353 Search PubMed.
- J. Xu, S. J. Jiang and Y. P. Du, ChemPhysChem, 2020, 21, 825–836 CrossRef CAS PubMed.
- L. Latsch, E. Lam and C. Coperet, Chem. Sci., 2020, 11, 6724–6735 RSC.
- Z. M. Liu, L. X. Liang, D. Xiao, Y. Ji, Z. C. Zhao, J. Xu and G. J. Hou, Phys. Chem. Chem. Phys., 2021, 23, 27244–27252 RSC.
- K. Kawata, H. Maekawa, T. Nemoto and T. Yamamura, Solid State Ion., 2006, 177, 1687–1690 CrossRef CAS.
- N. J. Kim and J. F. Stebbins, Chem. Mater., 2007, 19, 5742–5747 CrossRef CAS.
- P. Jain, H. J. Avila-Paredes, C. Gapuz, S. Sen and S. Kim, J. Phys. Chem. C, 2009, 113, 6553–6560 CrossRef CAS.
- H. Maekawa, K. Kawata, Y. P. Xiong, N. Sakai and H. Yokokawa, Solid State Ion., 2009, 180, 314–319 CrossRef CAS.
- R. J. Darby, I. Farnan and R. V. Kumar, Ionics, 2009, 15, 183–190 CrossRef CAS.
- C. P. Grey, C. M. Dobson, A. K. Cheetham and R. J. B. Jakeman, J. Am. Chem. Soc., 1989, 111, 505–511 CrossRef CAS.
- N. Kim and C. P. Grey, Dalton Trans., 2004, 3048–3052 RSC.
- E. J. Harvey, S. E. Ashbrook, R. L. Gregory and S. A. T. Redfern, J. Mater. Chem., 2006, 16, 4665–4674 RSC.
- S. E. Ashbrook, K. R. Whittle, G. R. Lumpkin and I. Farnan, J. Phys. Chem. B, 2006, 110, 10358–10364 CrossRef CAS.
- S. W. Reader, M. R. Mitchell, K. E. Johnston, C. J. Pickard, K. R. Whittle and S. E. Ashbrook, J. Phys. Chem. C, 2009, 113, 18874–18883 CrossRef CAS.
- M. R. Mitchell, D. Carnevale, R. Orr, K. R. Whittle and S. E. Ashbrook, J. Phys. Chem. C, 2012, 116, 4273–4286 CrossRef CAS.
- R. F. Moran, D. McKay, P. C. Tornstrom, A. Aziz, A. Fernandes, R. Grau-Crespo and S. E. Ashbrook, J. Am. Chem. Soc., 2019, 141, 17838–17846 CrossRef CAS PubMed.
- S. E. Ashbrook, M. R. Mitchell, S. Sneddon, R. F. Moran, M. de los Reyes, G. R. Lumpkin and K. R. Whittle, Phys. Chem. Chem. Phys., 2015, 17, 9049–9059 RSC.
- R. F. Moran, A. Fernandes, D. M. Dawson, S. Sneddon, A. S. Gandy, N. Reeves-McLaren, K. R. Whittle and S. E. Ashbrook, J. Phys. Chem. C, 2020, 124, 17073–17084 CrossRef CAS.
- S. Balamurugan, U. C. Rodewald, T. Harmening, L. van Wullen, D. Mohr, H. Deters, H. Eckert and R. Pottgen, Z. Naturforsch., B: J. Chem. Sci., 2010, 65, 1199–1205 CrossRef CAS.
- K. E. Johnston, M. R. Mitchell, F. Blanc, P. Lightfoot and S. E. Ashbrook, J. Phys. Chem. C, 2013, 117, 2252–2265 CrossRef CAS.
- J. H. Lee, Z. H. Gan and O. H. Han, J. Am. Ceram. Soc., 2014, 97, 3749–3754 CrossRef CAS.
- L. Buannic, F. Blanc, D. S. Middlemiss and C. P. Grey, J. Am. Chem. Soc., 2012, 134, 14483–14498 CrossRef CAS PubMed.
- T. Aharen, J. E. Greedan, F. Ning, T. Imai, V. Michaelis, S. Kroeker, H. D. Zhou, C. R. Wiebe and L. M. D. Cranswick, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 80, 134423 CrossRef.
- T. Aharen, J. E. Greedan, C. A. Bridges, A. A. Aczel, J. Rodriguez, G. MacDougall, G. M. Luke, V. K. Michaelis, S. Kroeker, C. R. Wiebe, H. D. Zhou and L. M. D. Cranswick, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 81, 064436 CrossRef.
- R. J. McCarty and J. F. Stebbins, Solid State Nucl. Magn. Reson., 2016, 79, 11–22 CrossRef CAS PubMed.
- R. Amantea, P. Ghigna, P. Mustarelli and V. Tartara, J. Solid State Chem., 2005, 178, 1692–1696 CrossRef CAS.
- A. I. Becerro, A. Escudero, P. Florian, D. Massiot and M. D. Alba, J. Solid State Chem., 2004, 177, 2783–2789 CrossRef CAS.
- M. Allix, M. D. Alba, P. Florian, A. J. Fernandez-Carrion, M. R. Suchomel, A. Escudero, E. Suard and A. I. Becerro, J. Appl. Crystallogr., 2011, 44, 846–852 CrossRef CAS.
- A. J. Fernandez-Carrion, M. Allix, P. Florian, M. R. Suchomel and A. I. Becerro, J. Phys. Chem. C, 2012, 116, 21523–21535 CrossRef CAS.
- H. Deters, A. S. S. de Camargo, C. N. Santos, C. R. Ferrari, A. C. Hernandes, A. Ibanez, M. T. Rinke and H. Eckert, J. Phys. Chem. C, 2009, 113, 16216–16225 CrossRef CAS.
- H. Deters, A. S. S. de Camargo, C. N. Santos and H. Eckert, J. Phys. Chem. C, 2010, 114, 14618–14626 CrossRef CAS.
- H. Deters, J. F. de Lima, C. J. Magon, A. S. S. de Camargo and H. Eckert, Phys. Chem. Chem. Phys., 2011, 13, 16071–16083 RSC.
- H. Deters and H. Eckert, Solid State Nucl. Magn. Reson., 2012, 41, 48–59 CrossRef CAS.
- N. K. Nasikas, S. Sen and G. N. Papatheodorou, Chem. Mater., 2011, 23, 2860–2868 CrossRef CAS.
- A. Jaworski, T. Charpentier, B. Stevensson and M. Eden, J. Phys. Chem. C, 2017, 121, 18815–18829 CrossRef CAS.
- M. T. Cohen-Adad, O. Aloui-Lebbou, C. Goutaudier, G. Panczer, C. Dujardin, C. Pedrini, P. Florian, D. Massiot, F. Gerard and C. Kappenstein, J. Solid State Chem., 2000, 154, 204–213 CrossRef CAS.
- B. Fuchs, F. Schroder, G. Heymann, R. Siegel, J. Senker, T. Justel and H. Huppertz, Z. Anorg. Allg. Chem., 2021, 647, 2035–2046 CrossRef CAS.
- J. H. Lin, S. Zhou, L. Q. Yang, G. Q. Yao, M. Z. Su and L. P. You, J. Solid State Chem., 1997, 134, 158–163 CrossRef CAS.
- O. Aloui-Lebbou, C. Goutaudier, S. Kubota, C. Dujardin, M. T. Cohen-Adad, C. Pedrini, P. Florian and D. Massiot, Opt. Mater., 2001, 16, 77–86 CrossRef CAS.
- C. V. Chandran, H. Schreuders, B. Dam, J. W. G. Janssen, J. Bart, A. P. M. Kentgens and P. J. M. van Bentum, J. Phys. Chem. C, 2014, 118, 22935–22942 CrossRef CAS.
- C. Bessada, A. Rakhmatullin, A. L. Rollet and D. Zanghi, J. Fluor. Chem., 2009, 130, 45–52 CrossRef CAS.
- J. Dabachi, M. Body, J. Dittmer, A. Rakhmatullin, F. Fayon and C. Legein, Dalton Trans., 2019, 48, 587–601 RSC.
- B. E. G. Lucier, K. E. Johnston, D. C. Arnold, J. L. Lemyre, A. Beaupre, M. Blanchette, A. M. Ritcey and R. W. Schurko, J. Phys. Chem. C, 2014, 118, 1213–1228 CrossRef CAS.
- G. Scholz, M. Dreger, R. Bertram and E. Kemnitz, Dalton Trans., 2015, 44, 13522–13529 RSC.
- T. Krahl, G. Scholz and E. Kemnitz, J. Phys. Chem. C, 2014, 118, 21066–21074 CrossRef CAS.
- B. Ritter, T. Krahl, G. Scholz and E. Kemnitz, J. Phys. Chem. C, 2016, 120, 8992–8999 CrossRef CAS.
- J. Roger, V. Babizhetskyy, S. Cordier, J. Bauer, K. Hiebl, L. Le Polles, S. E. Ashbrook, J. F. Halet and R. Guerin, J. Solid State Chem., 2005, 178, 1851–1863 CrossRef CAS.
- O. J. Zogal, P. Florian, D. Massiot, S. Paluch, N. Shitsevalova and D. F. Borshchevsky, Solid State Commun., 2009, 149, 693–696 CAS.
- C. Hoting, H. Eckert, T. Langer, I. Schellenberg and R. Pottgen, J. Solid State Chem., 2012, 190, 216–220 CrossRef.
- C. Hoting, H. Eckert, F. Haarmann, F. Winter and R. Pottgen, Dalton Trans., 2014, 43, 7860–7867 RSC.
- C. Hoting, H. Eckert, F. Haarmann and R. Pottgen, Z. Anorg. Allg. Chem., 2014, 640, 1303–1308 CrossRef.
- C. Hoting, H. Eckert, S. F. Matar, U. C. Rodewald and R. Pottgen, Z. Naturforsch., B: J. Chem. Sci., 2014, 69, 305–312 CrossRef.
- C. Benndorf, L. Heletta, T. Block, H. Eckert and R. Pottgen, Z. Anorg. Allg. Chem., 2017, 643, 294–298 CrossRef CAS.
- C. Benndorf, M. de Oliveira, C. Doerenkamp, F. Haarmann, T. Fickenscher, J. Kosters, H. Eckert and R. Pottgen, Dalton Trans., 2019, 48, 1118–1128 CAS.
- L. Kalantari, P. Blaha, K. H. Khoo and R. Laskowski, J. Phys. Chem. C, 2017, 121, 28454–28461 CAS.
- P. A. Vuissoz, T. Yonezawa, D. L. Yang, J. Kiwi and J. J. vanderKlink, Chem. Phys. Lett., 1997, 264, 366–370 CrossRef CAS.
- G. H. Penner and W. L. Li, Inorg. Chem., 2004, 43, 5588–5597 CrossRef CAS.
- G. H. Penner and X. L. Liu, Prog. Nucl. Magn. Reson. Spectrosc., 2006, 49, 151–167 CrossRef CAS.
- M. Weil, M. Puchberger, E. Fuglein, E. J. Baran, J. Vannahme, H. J. Jakobsen and J. Skibsted, Inorg. Chem., 2007, 46, 801–808 CrossRef CAS PubMed.
- F. Chen and R. E. Wasylishen, Magn. Reson. Chem., 2010, 48, 270–275 CrossRef CAS PubMed.
- M. Samsami, B. Azadegan, H. A. R. Aliabad and F. Amiri-Shookoh, J. Mol. Model., 2022, 28, 136 CrossRef CAS.
- G. E. Pavlovskaya, C. F. Horton-Garcia, C. Dybowski, D. R. Corbin and T. Meersmann, J. Phys. Chem. B, 2004, 108, 1584–1589 CrossRef CAS.
- Y. Liu, F. Chen, S. M. Kuznicki, R. E. Wasylishen and Z. H. Xu, J. Nanosci. Nanotechnol., 2009, 9, 2768–2771 CrossRef CAS PubMed.
- Y. Liu, F. Chen, R. E. Wasylishen, Z. H. Xu, J. Sawada and S. M. Kuznicki, J. Nanosci. Nanotechnol., 2012, 12, 6420–6427 CrossRef CAS PubMed.
- N. Popovych, P. Kyriienko, S. Soloviev, R. Baran, Y. Millot and S. Dzwigaj, Phys. Chem. Chem. Phys., 2016, 18, 29458–29465 RSC.
- C. W. Lopes, J. Martinez-Ortigosa, K. Gora-Marek, K. Tarach, J. A. Vidal-Moya, A. E. Palomares, G. Agostini, T. Blasco and F. Rey, J. Mater. Chem. A, 2021, 9, 27448–27458 RSC.
- J. Martinez-Ortigosa, C. W. Lopes, G. Agostini, A. E. Palomares, T. Blasco and F. Rey, Microporous Mesoporous Mater., 2021, 323, 111230 CrossRef CAS.
- I. R. Colinas, K. K. Inglis, F. Blanc and S. R. J. Oliver, Dalton Trans., 2017, 46, 5320–5325 RSC.
- L. E. Marbella and J. E. Millstone, Chem. Mater., 2015, 27, 2721–2739 CrossRef CAS.
- C. W. Liu, Y. R. Lin, C. S. Fang, C. Latouche, S. Kahlal and J. Y. Saillard, Inorg. Chem., 2013, 52, 2070–2077 CrossRef CAS PubMed.
- S. Uchida, T. Okunaga, Y. Harada, S. Magira, Y. Noda, T. Mizuno and T. Tachikawa, Nanoscale, 2019, 11, 5460–5466 RSC.
- K. Son and Z. Jang, J. Korean Phys. Soc., 2013, 62, 292–296 CrossRef CAS.
- H. Nan, C. Zhang, A. Venkatesh, A. J. Rossini and J. L. Anderson, J. Chromatogr. A, 2017, 1523, 316–320 CrossRef CAS PubMed.
- M. Blais-Roberge, S. H. Santagneli, S. H. Messaddeq, M. Rioux, Y. Ledemi, H. Eckert and Y. Messaddeq, J. Phys. Chem. C, 2017, 121, 13823–13832 CrossRef CAS.
- C. T. G. Knight, G. L. Turner, R. J. Kirkpatrick and E. Oldfield, J. Am. Chem. Soc., 1986, 108, 7426–7427 CrossRef CAS.
| This journal is © the Owner Societies 2023 |