
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRole of phosphatidylserine in amyloid-beta oligomerization at asymmetric phospholipid bilayers†
Jack
Robinson‡
 a,
Nirod Kumar
Sarangi‡
ab and
Tia E.
Keyes
*ab
a,
Nirod Kumar
Sarangi‡
ab and
Tia E.
Keyes
*ab
aSchool of Chemical Sciences, Dublin City University, Dublin 9, Ireland
bNational Center for Sensor Research, Dublin City University, Dublin 9, Ireland. E-mail: tia.keyes@dcu.ie
First published on 24th October 2022
Abstract
Amyloid-beta (Aβ1–42) aggregation triggers neurotoxicity and is linked to Alzheimer's disease. Aβ1–42 oligomers, rather than extended fibrils, adhere to the cell membrane, causing cell death. Phosphatidylserine (PS), an anionic phospholipid, is prevalent in neuronal membranes (< 20 molar percentage) and, while isolated to the cytoplasmic leaflet of the membrane in healthy cells, its exposure in apoptotic cells and migration to exoplasmic leaflet is triggered by oxidative damage to the membrane. It is widely believed that PS plays a crucial role in the Aβ peptide interaction in the membranes of neuronal cells. However, due to the complexity of the cell membrane, it can be challenging to address molecular level understanding of the PS-Aβ binding and oligomerization processes. Herein, we use microcavity supported lipid bilayers (MSLBs) to analyse PS and Aβ1–42 binding, oligomer formation, and membrane damage. MSLBs are a useful model to evaluate protein–membrane interactions because of their cell-like dual aspect fluidity, their addressability and compositional versatility. We used electrochemical impedance spectroscopy (EIS) and confocal fluorescence microscopy to compare the impact of Aβ1–42 on simple zwitterioinic membrane, dioleoylphosphatidylcholine (DOPC), with MSLBs comprised of transversally asymmetric binary DOPC and dioleoylphosphatidylserine (DOPS). Monomeric Aβ1–42 adsorbs weakly to the pristine zwitterionic DOPC membrane without aggregation. Using a membrane integrity test, with pyranine trapped within the cavities beneath the membrane, Aβ1–42 exposure did not result in pyranine leakage, indicating that DOPC membranes were intact. When 10 mol% DOPS was doped asymmetrically into the membrane's outer leaflet, oligomerization of Aβ1–42 monomer was evident in EIS and atomic force microscopy (AFM), and confocal imaging revealed that membrane damage, resulted in extensive pyranine leakage from the pores. The effects were time, and DOPS and Aβ1–42 concentration-dependent. Membrane pore formation was visible within 30 minutes, and oligomerization, membrane-oligomer multilayer, and Aβ1–42 fibril formation evident over 3 to 18 hours. In asymmetric membranes with DOPS localized to the lower leaflet, optothermally (laser induced) damage increased local DOPS concentrations at the distal leaflet, promoting Aβ1–42 aggregation.
Introduction
Alzheimer's disease (AD) is the most common form of dementia. Mainly associated with older adults, it is an irreversible brain disorder that causes progressive and irreversible loss of cognitive function.1 Amyloid-fibrils in AD are generated from amyloid-beta (Aβ), a naturally secreted peptide, found in various isoforms of different lengths. Among the isoforms, the 40 residue (Aβ1–40) constitutes the most common Aβ isoform in the brain, and the 42 residue (Aβ1–42) exhibits a considerable rise in some kinds of AD.2 Aβ has been detected in its oligomeric form in the cerebrospinal fluid and brain homogenates of Alzheimer's patients in the femto to picomolar range.3,4 There is substantial evidence indicating that the self-aggregation of monomeric Aβ to fibrils, particularly oligomers, plays an important role in the progression of AD.5–8 Although there is still debate over the amyloid model and the relative roles of tau and amyloid in disease progression, the recent FDA approval of Aducanumab suggests causality, although the drug is not currently approved by the European Medicines Agency.9 In AD, Aβ peptide is released from cells by the action of β- and γ-secretases on amyloid precursor protein (APP), where it undergoes amyloidogenesis; aggregation to oligomers, multimers, and fibrils in the extracellular space, eventually forming the plaques that are characteristic of the disease.10 Furthermore, pathogenic amyloid aggregates are not confined to AD but also play a major role in Parkinson's, Huntington's, and Diabetes.11 Aβ peptides are composed of 36 to 43 amino acids that spontaneously self-aggregate through hydrophobic-hydrophobic interactions, especially at higher concentrations (micro to submillimolar range).12–14 All amyloid aggregates can serve as cellular hallmarks for AD; however, soluble, small oligomers formed from Aβ1–42 have recently been shown to be more toxic than larger fibrils.15,16 Because AD currently has no cure and cannot be prevented, models that can improve understanding of the mechanisms behind Aβ aggregation can provide insights into early detection strategies, understanding of possible preventative strategies and the development of therapy.16,17 Furthermore, given the prevalence of amyloid aggregation in numerous disease conditions beyond AD, tools to investigate Aβ aggregation are broadly important. The current understanding of amyloid action at the cellular interface is based on in-cellulo that have either targeted specific receptors or examined non-specific interactions with lipids within membranes in biophysical models.18,19 Numerous in vitro studies have revealed that the lipidic membrane in general, and negatively charged phospholipids like phosphatidylserine (PS) in particular, can promote Aβ oligomer formation at the bilayer interface.16,20–24 Aβ oligomers can develop in solution at high concentrations (μM to mM) due to self-assembly, while at low concentrations (pM to nM), oligomers form upon interaction with the lipidic interface. Of note, Aβ oligomers have the ability to disrupt cell membrane integrity and permeability.21,22,24–26To date, a number of models have been proposed to explain Aβ oligomer induced membrane toxicity, for example, (a) the poration model, proposes that amyloid forms stable transmembrane pores at the neuronal membrane, disrupting intracellular Ca2+ homeostasis; (b) the carpet model, hypothesizes that amyloid adsorption at the membrane interface destabilizes the membrane structure; and (c) the membrane dissolution model, hypothesizes amyloid precipitates serve as detergent, stripping off lipid from the bilayer.17,27,28 In all of these models, membrane disruption can result in oxidative stress, a key effector in AD progression that leads to cellular apoptosis.29–32 Under conditions of oxidative stress, phosphatidylserine, which is normally confined to the cytosolic leaflet of the lipid bilayer in the naturally asymmetric neuronal membrane, migrates to the extracellular leaflet. This is a primary step in the onset of apoptosis, and a number of studies have indicated that anionic lipids, particularly PS, influence Aβ oligomerization, fibrilization, and fibril growth.33–39 PS is particularly important as it is prevalent at the plasma membrane of neuronal cells, where Aβ peptide interaction occurs.40 The electrostatic attraction between positively charged Aβ and negatively charged PS is the primary driving force.41–43 And, while there is significant evidence, notably from in vitro models, that Aβ and PS interaction has a role in inducing the formation of oligomer scaffold,44–48 other membrane factors such as cholesterol content, membrane packing density, and lipid rafts also play a role.15,21,49–53 Kim et al. recently reported on the impact of different anionic phospholipids, such as dioleoyl phosphatidylserine (DOPS) or dioleoyl phosphatidylglycerol (DOPG), and mixed vesicles of DOPC/DOPS and DOPC/DOPG on the formation of Aβ fibrils. They observed that the liposome packing density (regulated by varying fatty acid on the PS in terms of length and unsaturation) and the DOPS/DOPG ratio, influenced the rate of fibrinogenesis and the length of the resulting fibrils.54 Although the concentration of PS in natural membranes was much greater than anticipated, the study showed the impact of membrane packing/fluidity and charge on amyloidgenisis.
Such biophysical models of the lipid membrane have provided deep insights into Aβ-cell membrane interactions because they permit a reductionist approach, separating the variables that affect oligomer growth at membranes. Aβ-membrane interactions have been studied using a diverse of models, including liposomes, black lipid membranes, and supported lipid bilayer membranes (SLBs).3,22,37,55–59 However, there are challenges in addressing some aspects of Aβ interaction. Liposomes for example, have a lack of control over compositional asymmetry, and are generally not addressable with surface-sensitive analytical tools. Substrate supported lipid bilayers (SLBs) and their variants address these issues, but they suffer different limitations, particularly in the study of dynamics of membrane/protein interactions that require lateral or transversal diffusion of lipid or components due to frictional lipid-substrate or protein/peptide-substrate interaction, which hinders diffusion and can even lead to protein/peptide denaturation.60–64 Although all of these models have provided crucial insights on Aβ-membrane interaction, the impact of varying PS concentrations within the physiological concentration limit and its asymmetric distribution across the bilayer on the recognition and assembly of Aβ, as well as the associated kinetics of oligomer formation under these conditions, has not yet been fully explored.
In this context, microcavity supported lipid bilayers (MSLBs) in combination with electrochemical impedance spectroscopy (EIS) offer a useful approach. The microcavity pore provides aqueous reservoirs at each lipid leaflet interface, avoiding any loss of activity or assembly of peptide due to friction, and the layer by layer approach to fabrication enables facile and precise building of asymmetry into the bilayers structure with transversal fluidity that is expected to be cell-like.33,34 EIS is a sensitive, label-free technique that, by measuring changes to the admittance of bilayer membrane, can reveal details of molecular mechanism of Aβ activity in real time.
Exploiting these methods, herein, we study the role of DOPS content within DOPC on membrane association and assembly of Aβ1–42. At a fixed concentration of Aβ1–42 (1 μM) and using EIS, we established that 10 mol% DOPS is sufficient to facilitate Aβ oligomerization without damaging the membrane. At 5 mol% DOPS, no large-scale perturbation of membrane or oligomer formation is detected in the presence of Aβ1–42 (1 μM). However, at membranes comprising 20 mol% of DOPS, the activity of Aβ1–42 triggered the assembly processes leading to membrane damage. Atomic force microscopy (AFM) and confocal microscopy imaging were used to further identify large-scale oligomer formation and membrane damaging effect caused by Aβ1–42. As PS is localized to the cytoplasmic leaflet of the neuronal membrane in homeostatic cells and only migrates during membranal damage, e.g., under oxidative stress, we induced optothermal damage to mimic this process and observed that DOPS transverse diffusion stimulated Aβ1–42 assembly.
Experimental
Materials
1,2-Dioleoyl-sn-glycero-3-phosphocholine (DOPC) and 1,2-dioleoyl-sn-glycero-3-phospho-L-serine (DOPS) were purchased with maximum degree of purity (<99%) from Avanti Polar Lipids (Alabama, USA) and used without further purification. 1,2-Dioleoyl-sn-glycero-3-phosphoethamine labelled Atto655 (DOPE-Atto655) was purchased from ATTO-TEC GmbH (Siegen, Germany). HEPES salt and sodium azide were purchased from Sigma-Aldrich (Wicklow, Ireland). Amyloid beta (Aβ1–42) was purchased from Biolegend UK Ltd (London,UK). Gold on silicon wafers were purchased from Ams Biotechnology (Abingdon, UK). Polybead microspheres of different sizes were purchased from Polysciences Europe Gmbh (Baden-Württemberg, Germany). Aqueous solutions were prepared using MilliQ water (18 mΩ cm) (Milipore Corp. Bedford, USA). Polydimethylsiloxane silicon elastomer (PDMS) was purchased from Dow Corning Gmbh (Wiesbaden, Germany) and mixed following supplier's instructions. 1,1,1,3,3,3-Hexafluoroisopropanol (HFIP) and Thioflavin T (Tht) were purchased from Sigma-Aldrich (Wicklow, Ireland). Beta Amyloid (1–42) HiLyteTM Fluoro 555 (Aβ555) was purchased from Eurogentec (Seraing, Belgium).Methods
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :DOPS (80:20)//DOPC, DOPC:DOPS (90:10)//DOPC or DOPC:DOPS (95:5)//DOPC, lipid bilayers where the distal leaflet is composed of 80 mol% DOPC and 20 mol% of DOPS (or 90 mol% DOPC and 10 mol% DOPS, or 95 mol% DOPC and 5 mol% DOPS) while the proximal leaflet is composed of 100 mol% of DOPC to model an apoptotic cell membrane. The double forward slash ‘’//’’ indicates the two monolayers of different composition in the bilayer, the distal (away from substrate, at the bulk aqueous interface) leaflet and proximal leaflet (closest to substrate) of the designated membrane. Similarly, the other sets of asymmetric membranes such as DOPC//DOPC:DOPS (80:20), DOPC//DOPC:DOPS (90:10) or DOPC//DOPC:DOPS (95:5) contain 100 mol% DOPC at distal leaflet and proximal leaflet with varied DOPC:DOPS compositions including 80 mol% DOPC and 20 mol% of DOPS, 90 mol% DOPC and 10 mol% of DOPS, and 95 mol% DOPC and 5 mol% DOPS, as an analogue of a non-apoptotic cell membrane. Microcavity supported lipid bilayers (MSLBs) were assembled across aqueous buffer filled gold microcavity array using a combination of the Langmuir–Blodgett and vesicle fusion (LB-VF) methods. Successful formation of the MSLB across the gold microcavity array was confirmed using fluorescence life-time imaging microscopy (Fig. S1B–D, ESI†) by selectively labelling each layer with a different fluorophore so that each individual leaflet could be imaged separately and confirmed to form a continuous bilayer film across the array.
000 Hz to 0.01 Hz with an AC modulation amplitude of 0.01 V at a potential bias of 0 V (vs. Ag/AgCl). All measurements were performed in a glass cell (approximate volume of 5 mL) in HEPES buffer at pH 7.4. The MSLB with the desired bilayer was initially analyzed prior to the addition of Aβ1–42 to ensure signal stability. Resuspended Aβ1–42 was then added into the glass cell with HEPES buffer at pH 7.4. The EIS response of the bilayer in contact with Aβ1–42 was measured every 30 minutes for 20 hours. Each measurement takes 10 minutes and was carried out at room temperature (20 ± 1 °C). An equivalent circuit model (ECM) was used to fit the measured AC impedance data using Z-View software to estimate the membrane resistivity and capacitance, as reported previously.61,66,75 The ECM model is shown in Fig. S2 (ESI†), where RS, RM, RC are resistance of the solution, membrane and cavity array respectively, CPEc + dl is the electrode double layer constant phase element and CPEM is the constant phase element of the membrane. Constant phase elements (CPE = 1/Q(jω)m) were used instead of pure capacitors to account for defects/inhomogeneities in the cavity array electrode and the bilayers. The ECM provided an excellent fit of the measured EIS data across the whole frequency range. When the impedance response was fitted, the only elements that changed significantly on incubation with Aβ1–42 were RM and CPEM, indicating that the model is valid. During the fit, the CPE exponent, m for membrane and cavity array was found to be 0.94 ± 0.02 and 0.5 ± 0.01 respectively.
:DOPS (90:10) and DOPC:DOPS (95:5). The detailed procedure is provided in ESI.† All AFM images were analyzed using Nanoscope 7.30 software, and the whole images were plane fitted with a 1st order polynomial.
000:1, prepared as previously reported.61,63 Confocal imaging was carried out using a Leica TSP DMi8 confocal microscope in sequential mode with “between frames”, where the first laser scans the entire spatial x-y region, followed by the second laser scanning the same region, and so on until all the lasers have scanned, and a 63× oil immersion objective lens was used to image the resulting arrays. By pre-sonicating the microcavity array in a 5 μM pyranine solution of HEPES, membrane impermeable probe pyranine was introduced into the pores of the array ahead of bilayer assembly. The pyranine was excited at 405 nm and emission detected between 440 and 560 nm.62 Aβ555 at the designated concentration was allowed to interact with the PDMS spanning membrane and was excited at 561 nm with a detection window of 570–630 nm. 20 μM Thioflavin T (tht) was dissolved in the contact solution from EIS studies (HEPES buffer used for the electrochemical cell) and excited at 405 nm with a detection window 430–580 nm.
:DOPS (80:20)//DOPC, DOPC:DOPS (90:10)//DOPC or DOPC:DOPS (95:5)//DOPC, lipid bilayers where the distal leaflet is composed of 80 mol% DOPC and 20 mol% of DOPS (or 90 mol% DOPC and 10 mol% DOPS, or 95 mol% DOPC and 5 mol% DOPS) while the proximal leaflet is composed of 100 mol% of DOPC to model an apoptotic cell membrane. The double forward slash ‘’//’’ indicates the two monolayers of different composition in the bilayer, the distal (away from substrate, at the bulk aqueous interface) leaflet and proximal leaflet (closest to substrate) of the designated membrane. Similarly, the other sets of asymmetric membranes such as DOPC//DOPC:DOPS (80:20), DOPC//DOPC:DOPS (90:10) or DOPC//DOPC:DOPS (95:5) contain 100 mol% DOPC at distal leaflet and proximal leaflet with varied DOPC:DOPS compositions including 80 mol% DOPC and 20 mol% of DOPS, 90 mol% DOPC and 10 mol% of DOPS, and 95 mol% DOPC and 5 mol% DOPS, as an analogue of a non-apoptotic cell membrane. Microcavity supported lipid bilayers (MSLBs) were assembled across aqueous buffer filled gold microcavity array using a combination of the Langmuir–Blodgett and vesicle fusion (LB-VF) methods. Successful formation of the MSLB across the gold microcavity array was confirmed using fluorescence life-time imaging microscopy (Fig. S1B–D, ESI†) by selectively labelling each layer with a different fluorophore so that each individual leaflet could be imaged separately and confirmed to form a continuous bilayer film across the array.
000 Hz to 0.01 Hz with an AC modulation amplitude of 0.01 V at a potential bias of 0 V (vs. Ag/AgCl). All measurements were performed in a glass cell (approximate volume of 5 mL) in HEPES buffer at pH 7.4. The MSLB with the desired bilayer was initially analyzed prior to the addition of Aβ1–42 to ensure signal stability. Resuspended Aβ1–42 was then added into the glass cell with HEPES buffer at pH 7.4. The EIS response of the bilayer in contact with Aβ1–42 was measured every 30 minutes for 20 hours. Each measurement takes 10 minutes and was carried out at room temperature (20 ± 1 °C). An equivalent circuit model (ECM) was used to fit the measured AC impedance data using Z-View software to estimate the membrane resistivity and capacitance, as reported previously.61,66,75 The ECM model is shown in Fig. S2 (ESI†), where RS, RM, RC are resistance of the solution, membrane and cavity array respectively, CPEc + dl is the electrode double layer constant phase element and CPEM is the constant phase element of the membrane. Constant phase elements (CPE = 1/Q(jω)m) were used instead of pure capacitors to account for defects/inhomogeneities in the cavity array electrode and the bilayers. The ECM provided an excellent fit of the measured EIS data across the whole frequency range. When the impedance response was fitted, the only elements that changed significantly on incubation with Aβ1–42 were RM and CPEM, indicating that the model is valid. During the fit, the CPE exponent, m for membrane and cavity array was found to be 0.94 ± 0.02 and 0.5 ± 0.01 respectively.
:DOPS (90:10) and DOPC:DOPS (95:5). The detailed procedure is provided in ESI.† All AFM images were analyzed using Nanoscope 7.30 software, and the whole images were plane fitted with a 1st order polynomial.
000:1, prepared as previously reported.61,63 Confocal imaging was carried out using a Leica TSP DMi8 confocal microscope in sequential mode with “between frames”, where the first laser scans the entire spatial x-y region, followed by the second laser scanning the same region, and so on until all the lasers have scanned, and a 63× oil immersion objective lens was used to image the resulting arrays. By pre-sonicating the microcavity array in a 5 μM pyranine solution of HEPES, membrane impermeable probe pyranine was introduced into the pores of the array ahead of bilayer assembly. The pyranine was excited at 405 nm and emission detected between 440 and 560 nm.62 Aβ555 at the designated concentration was allowed to interact with the PDMS spanning membrane and was excited at 561 nm with a detection window of 570–630 nm. 20 μM Thioflavin T (tht) was dissolved in the contact solution from EIS studies (HEPES buffer used for the electrochemical cell) and excited at 405 nm with a detection window 430–580 nm.
Results and discussion
Effect of Aβ1–42 on a symmetric zwitterionic DOPC membrane
In a recent report, on supercritical angle fluorescence measurements of Aβ oligomerization at a DOPC/DOPS (65/35) membrane, Seeger et al.55 hypothesized the following steps: (1) Aβ monomer adsorbs at the membrane, facilitated by long-range electrostatic interactions with anionic lipids; (2) Aβ monomers diffuse along the membrane and self-aggregate, concomitantly inducing lipid clustering that generate nanopores within the membrane, causing additional membrane strain; (3) further aggregation of Aβ removes some of the lipids that are tightly bound to the oligomer scaffold; and (4) the oligomer of Aβ is ejected from the membrane as a plaque leaving a porated/damaged membrane as shown schematically in Fig. 1.17,26–28,55 | ||
| Fig. 1 Hypothesized mechanism of amyloid-beta activity within a lipid bilayer proposed by Seeger et al.55 The majority of the membrane is made up of zwitterionic lipids (blue), and the amyloid beta peptide monomer (red) may adsorb to the membrane. Negatively charged lipids (green) attract amyloid beta peptides, causing the monomers to assemble within the membrane; however, some monomers may pull lipids from the membrane. Within the membrane, the monomers continue to aggregate and form oligomers. Amyloid-beta fibrils fall out of the membrane as a plaque, causing damage to the membrane. | ||
A central tenet of this and other hypotheses is that the membrane becomes porous on amyloid aggregation. Electrochemical impedance spectroscopy (EIS) is a highly sensitive method for exploring such effects because it directly addresses the admittance of the bilayer and was thus used here to distinguish membrane resistance and capacitance changes induced by the Aβ peptide following adsorption, and oligomerization. First to establish the electrochemical characteristics of the membrane prior to exposure to Aβ1–42, non-Faradaic EIS measurements were carried out on symmetric DOPC bilayers. The DOPC membrane was prepared by LB-VF method and spanned across the gold microcavity array, which serves as the working electrode in a three electrode cell. Representative non-Faradaic Nyquist plots of DOPC MSLB in the absence and presence of 1 μM Aβ1–42 in the contact buffer at different time intervals are shown in Fig. S3 (ESI†). The corresponding data was modeled using an ECM, reported previously (cf. Fig. S2, ESI†) to extract the membrane resistance and capacitance values. The absolute values of membrane resistance and capacitance across multiple replicates are respectively 40–60 MΩ cm2 and 0.6–0.9 μF cm−2. As the microcavity pore arrays are fabrication across a ∼1 cm × 1.5 cm flat gold substrate, and show some batch to batch variation in electrode area due to cavity packing and thiol modification, instead of normalizing with respect to the electroactive area, the mean (N = 3) of relative resistance/capacitance changes with and without the presence of Aβ1–42 are reported in Fig. 2. The relative membrane resistance (ΔRM) is defined as Rtime=tM − Rtime=0M; where Rtime=tM is the resistance at time, t and Rtime=0M is the resistance at time, 0 i.e., after bilayer preparation. Similarly, ΔCPEM are defined as, CPEtime=tM − CPEtime=0M.
 | ||
| Fig. 2 Top representative temporal electrochemical data from DOPC bilayers suspended in a 1 μm diameter gold cavity array showing the relative change in membrane (A) resistance and (B) capacitance versus time before (grey) and after (orange) the addition of Aβ1–42(1 μM) in HEPES buffer at pH 7.4 over 20 hours. EIS measurements were performed in HEPES electrolytes (pH 7.4) at 20 °C using gold microcavity suspended bilayer as working electrode, Ag/AgCl (1 M KCl) as reference electrode and Pt coil as counter electrode. Measurements were taken at an AC perturbation amplitude of 10 mV with DC bias potentials 0 V (vs. Ag/AgCl), over the frequency range from 0.01 to 104 Hz. Equilibration of DOPC lipid bilayer was achieved within 2 hours after bilayer preparation (data from 0–2 h are excluded for clarity), and the impedance change remained constant for over 18 hours. The data recorded are the mean of N = 3 with error bars representing standard error. (C) Topographic tapping mode AFM images (5 μm × 5 μm) of DOPC bilayer without and with 1 μM Aβ1–42 at different time intervals. The depression induced by Aβ1–42 are indicated by the white arrow in the 15 h panel and the corresponding line profile shown to its right. For both EIS and AFM studies, the bilayer is prepared by the LB-VF method. The AFM are acquired on a freshly cleaved mica and imaged under the HEPES buffer. | ||
To evaluate membrane stability, impedance spectra were collected every 30 minutes for 20 hours. Data showed an immediate and systematic increase in resistance and decrease in capacitance of DOPC membrane in the electrochemical cell that equilibrates in under 2 hours of bilayer preparation, beyond which the signal remains constant for up to 18 hours. The initial equilibration data (from 0 to 2 h) are not included in the plot for clarity. Fig. 2A and B (grey) shows representative temporal stability data over 2–18 hours for DOPC MSLB showing relative resistance and capacitance after initial equilibration. For peptide binding studies, 1 μM Aβ1–42 in its monomeric form (cf. fluorescence correlation spectroscopy data in Fig. S4, ESI†) was added to the membrane contact buffer following membrane's electrochemical equilibration, and impedance sampled for at least 18 hours (i.e., within the stability window of pristine membrane). The corresponding relative changes in resistance and capacitance are shown in Fig. 2A and B (orange) respectively. After a modest fluctuation of membrane resistance within 1–2 hours of Aβ1–42 incubation (cf. the time window from 2–4 hours in Fig. 2A, orange), the resistance remains indistinguishable from that of pristine membrane for at least 10 hours. Beyond 10 hours, resistance starts to decrease. The capacitance, on the other hand, decreases systematically over the full experimental window after exposure to Aβ1–42. This may suggest weak and slow physisorption of Aβ1–42 at the membrane over time, but with limited impact on overall membrane organization. Our results are consistent with a previous study in which Aβ1–42 monomers were observed to weakly bind to zwitterionic DOPC bilayer.20 It is worth noting that in the absence of Aβ1–42, a modest decrease in capacitance of the pristine DOPC membrane (grey, Fig. 2B) occurs over the course of 18 h, which may be attributed to the impact of HEPES buffer, which has been reported to cause the membrane to rigidify.61,79 Nonetheless, the capacitance changes are greater in the presence of Aβ1–42. Decoupling the contributions of both effects is challenging, therefore, to gain more insight, we carried out atomic force microscopy (AFM) studies at a flat mica SLB platform to visualize any nano to sub-microscopic changes caused by Aβ1–42 to the DOPC membrane within the time range of EIS observations (Fig. 2C). In line with EIS, the membrane remains quite homogeneous in the presence of 1 μM Aβ1–42 peptide beyond 10 h. Some modest heterogeneity is evident after 15 h (cf. white arrow mark, Fig. 2C, right panel) consistent with the time frame of the EIS resistance decrease.
To confirm that Aβ1–42 adsorbs at the DOPC membrane without damaging or porating it, a confocal fluorescence microscopy-based leakage study was carried out on an analogous PDMS cavity array with DOPC MSLBs. Prior to bilayer formation, the cavities were filled with 5 μM pyranine, a fluorophore that is impermeable to the bilayer and thus expected to remain trapped in the cavity while the membrane is intact.62 This experiment also serves to confirm that the bilayer is effectively spanned across the pore array. Fluorescence imaging was then performed after ∼3 h incubation of DOPC MSLB with 1 μM Aβ555 at 20 ± 1 °C. The reflectance image in Fig. 3A clearly distinguishes between cavities that are filled (dark circular regions) with buffer/pyranine and a small number of cavities that are not filled (marked by an arrow) thus not bilayer sealed (cf. arrow, Fig. 3B). However, notably in the case of MSLB over gold support, where the dimension of pores is smaller and bilayer is supported by modification of the intervening top surface with OH terminate SAM, we observed continuous aqueous filling and well spanned membrane across the substrate (Fig. S1B–D, ESI†). The observation of occasional unfilled/unspanned pores in PDMS is attributed to the larger size of these pores (∼2 μm diameter, compared to 1 μm in gold) and to the hydrophobicity of the substrate that can sometimes mitigate against complete aqueous filling where plasma treatment may not have reached all pores. The confocal fluorescence images of a bilayer doped with DOPE-Atto655 (0.05 mol%) at the distal leaflet of the membrane (Fig. 3B) confirms the bilayer is spanned over the buffer/pyranine filled cavities. Fig. 3C shows the fluorescent image collected from Aβ555 (green), and the overlayed (Fig. 3D) image confirms it is weakly associated with the membrane above the pyranine (side on image shown in Fig. S7, ESI†). The pyranine signal (shown in yellow), from the filled pores is clearly visible (Fig. 3D) both from within the pores and is particular intense from the pore walls where the bright emission is attributed to the accumulated signal along the vertical walls of the pores in the confocal z-axis. This is attributed to weak adsorption of the pyranine to the plasma treated, hydroxylated PDMS surface. The propensity for adsorption of pyranine at hydroxylated surfaces has been noted previously at polymer and graphene.96–98
 | ||
| Fig. 3 (A) Reflectance image of DOPC MSLB spanning over pyranine containing buffer filled PDMS substrate. The dark grey circular features in the reflectance image are the cavities that filled with buffer. The occasional unfilled cavity is easily distinguished in reflectance, as indicated by an arrow and appear as bright spots. (B) A confocal fluorescence image of -Atto655 labelled DOPC MSLB across the same region as the reflectance image, showing the bilayer spanned over pyranine/buffer filled cavities. The pores where buffer failed to fill or where bilayer fail to span are also clear, as in these unfilled pores, the intense emission from the pore edges are due to emission from along vertical walls of the pores captured in the confocal volume due to the lipid lining the pore. (C) A confocal fluorescence image of the DOPC MSLB in contact with Aβ555 after 3 hours of incubation, showing homogenous weak emission from Aβ555 at the bilayer. The overlayed image of panels B (red) and C (green) along with signal obtained from pyranine (yellow) are shown in panel D. The emission is evident from the pores but is exceptionally bright at the pore walls due to weak adsorption of the pyranine at the hydroxylated PDMS. The red channel in panel B is the emission from DOPE-Atto655 (0.05 mol%) which was doped at the upper leaflet of DOPC bilayer with excitation wavelength, λex = 640 nm and detection window 650–780 nm; the yellow channel is the emission from pyranine (5 μM) with excitation at 405 nm and detection window 440–560 nm; and the green channel is the emission from Aβ555 (1 μM) with excitation at 561 nm and detection window 570–630 nm. In each panel, the scale bar was 10 μm. | ||
After 3 hours incubation with Aβ555, there was no change in bilayer emission image or pyranine emission intensity or any leakage evident of the pyranine, confirming, consistent with EIS and AFM data that the bilayer is intact and not damaged by the amyloid under these conditions.
Effect of Aβ1–42 on an asymmetric negatively charged DOPC![[thin space (1/6-em)]](https://www.rsc.org/images/entities/h3_char_2009.gif) :DOPS (90:10)//DOPC membrane
:DOPS (90:10)//DOPC membrane
PS concentrations in plasma membranes range from 5 to 10% (mol/mol) and can reach up to 20 (molar%) in neuronal cell membranes.40,80 In healthy cells, PS is localized to the cytoplasmic leaflet. When the membrane is damaged, e.g. by oxidative stress, the PS translocates to the exterior leaflet, where it serves as an “eat me’’ signal for cellular apoptosis,81–85 a hallmark biomarker of AD. Typically Aβ is found in healthy brains at low femto to picomolar levels, but is elevated in AD.86,87
Using EIS, we therefore set up an onset monomeric Aβ1–42 working concentration in the pico to subnanomolar range at an asymmetric MSLB comprised of DOPC at the proximal leaflet and DOPC:DOPS (90:10) at the distal leaflet. This range of Aβ concentration is not expected to lead to oligomer formation, emulating a healthy brain cell.3,86,88,89 Previously, μM Aβ1–42 concentrations have been used to instigate oligomer/fibril formation, especially in zwitterionic membranes or in aqueous solution55,57,74,90–92 whereas pM-sub nM have been used to mimic oligomer formation when cholesterol and/or charged lipids are present.50,73 Because Aβ oligomerization is a slower process than its detergent-like behaviour, which is a fast process, EIS was employed to tap the later process. Furthermore, working below the critical micelle concentration of Aβ1–42 and incubating for a short period of time (30 minutes) allows us to capture the entire range of concentrations within our overall experimental time window (18 hours) and prevent difficulties that could result in aggregation-induced membrane damage.93
Fig. 4A shows the relative resistance changes to an asymmetric DOPC:DOPS (90:10)//DOPC membrane on titration of different concentrations of Aβ1–42. Fig. 4B shows the corresponding relative capacitance plot. Each data point was collected after 30 minutes of incubation at the designated Aβ1–42 concentrations. Over the concentration range 25–200 pM, no change in membrane resistance or capacitance was evident. However, from 400 pM of Aβ1–42, the membrane resistance proceeded to decrease, suggesting that oligomerization commences around 400 pM, (notwithstanding the limit of detection in our experimental set-up for detecting oligomerization). Since oligomerization and fibril formation at the bilayer interface is a slow process94 even at elevated (μM) amyloid concentrations, the decrease in resistance after 30 min incubation is most likely due to partial membrane insertion and the first step in oligomerization. Beyond 800 pM Aβ1–42, dramatic decreases in membrane resistance are evident, indicating that significant interaction of Aβ1–42 with the anionic membrane. The bilayer capacitance remains unchanged across the range 400 to 800 pM of Aβ1–42, but not to 1 nM. Together, the data suggests some insertion of monomeric Aβ1–42 into the membrane, leading to increased membrane admittance without changes to membrane thickness. 10 nM Aβ1–42 decreases resistance to ∼2.7 MΩ and capacitance modestly, indicating enhanced admittance and bilayer thickness. Above 10 nM Aβ1–42, membrane resistance drops as capacitance increases indicating membrane damage. The data reveal a significant change in bilayer impedance at higher Aβ1–42 concentrations, with the capacitance increasing to ∼0.8 μF sm−1 and the resistance decreasing to −6.84 MΩ, suggestive of pore formation and likely indicating that oligomerization of amyloid is underway from 400 pM. Given the dynamic and complex nature of the Aβ1–42-membrane interaction, extracting association data from the resistance curve was not possible as we cannot assume that the process had reached equilibrium at the 30 minute time point at which the data was collected. Therefore, to dynamically follow the oligomerization process at the DOPC:DOPS asymmetric membrane, we extended the incubation period of Aβ1–42 with membrane at different fixed concentrations of 100 nM, 1 μM and 2 μM.
 | ||
| Fig. 4 Concentration-dependent titration curve for the effect of Aβ1–42 on the impedance of the DOPC:DOPS (90:10) asymmetric membrane. (A) Represents the relative resistance change, whilst (B) represents the capacitance change of the membrane after incubation with different concentrations of Aβ1–42. A 30 minute incubation period following membrane equilibration was established for each concentration of Aβ1–42, followed by the addition of next concentration. Any change in membrane impedance caused by Aβ1–42 is normalized with respect to the pristine equilibrated membrane impedance (before any amyloid addition). The results shown are the averages of at least three independent experiments (N ≥ 3). | ||
Fig. 5A and B show representative plots of relative change to resistance and capacitance of an asymmetric DOPC:DOPS (90:10) membrane over time. As noted before, the initial 2 hours equilibration window is excluded from the plot. The data (Fig. 5A and B) show that the membrane is stable for 18 hours, similar to the pristine zwitterionic DOPC bilayer, (cf.Fig. 2) defining the experimental window.
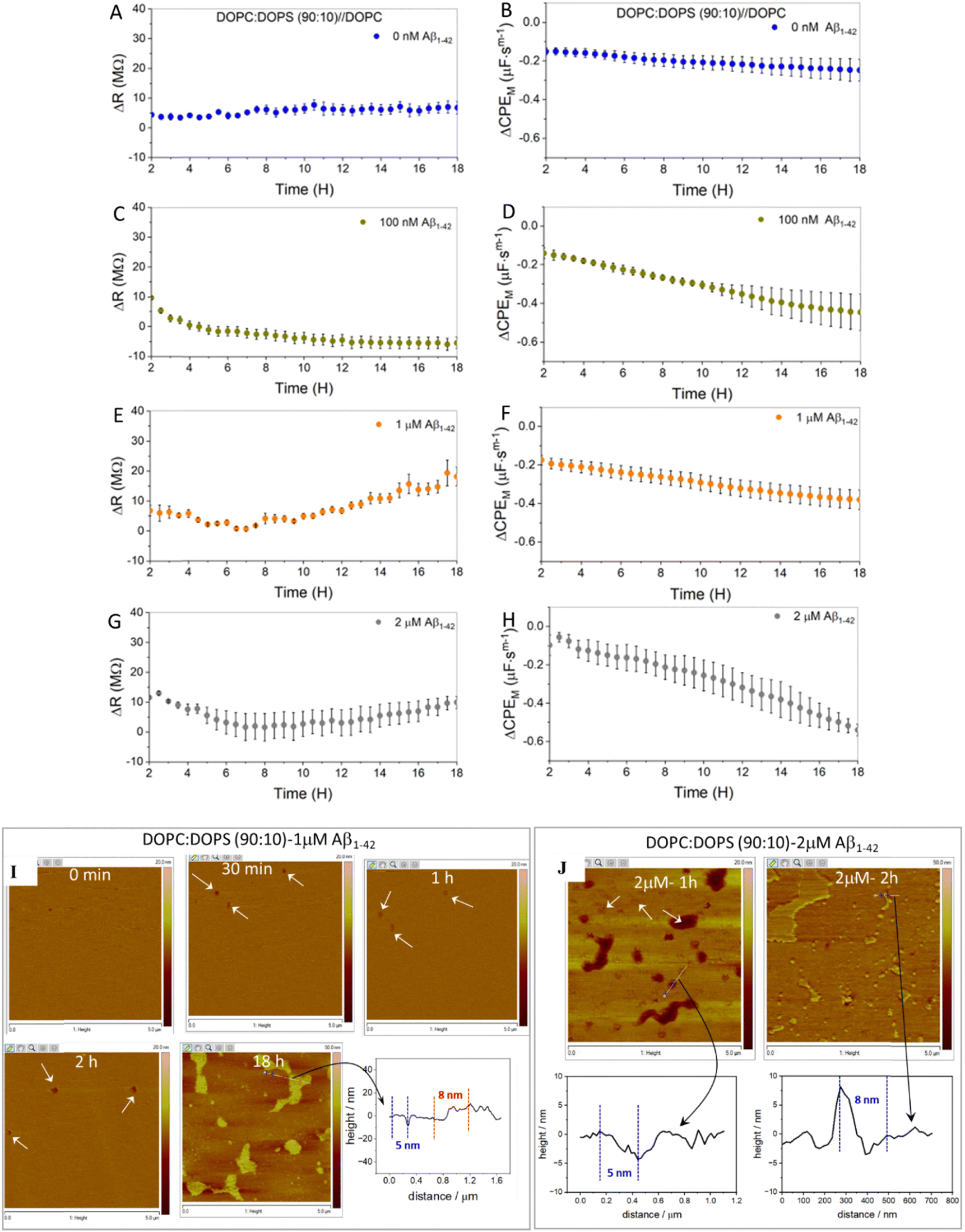 | ||
| Fig. 5 Temporal evolution of changes in relative resistance and capacitance of DOPC:DOPS (90:10)//DOPC asymmetric bilayer without and in presence of different concentrations of Aβ1–42. In the absence of Aβ1–42 the relative change in (A) resistance and (B) capacitance showing the stability of an asymmetric DOPC:DOPS (90:10) MSLB. In presence of 100 nM Aβ1–42 real time temporal evolution of (C) resistance and (D) capacitance changes are shown. (E and F) Illustrates the resistance and capacitance change respectively with incubation of 1 μM Aβ1–42. The corresponding resistance and capacitance change in presence of 2 μM Aβ1–42 are shown in panel (G) and (H) respectively. For EIS measurements, all samples were in contact with HEPES buffer at pH 7.4. In all the panels A–H, the experimental data is presented, ranging 2 to 18 hours. The error bars represent the standard error across the samples (N = 3). Temporal evolution of topographic AFM images of asymmetric DOPC:DOPS (90:10) bilayer deposited on mica before and after incubation with (I) 1 μM Aβ1–42 and (J) 2 μM Aβ1–42, as well as height profile analysis. In panel I and J, the image size was 5 μm × 5 μm. Aβ1–42 induced lipid disruption is indicated by the white arrow in panels I and J. The line profile analyses plots corresponding to the image are indicated by black arrow and was taken from a cross-section in the region of interest, marked in white solid line in the respective images. All images are acquired under liquid and the membrane was prepared via LB-VF method. | ||
For all three Aβ1–42 concentrations (100 nM, 1 μM and 2 μM), the resistance decreased considerably over the first several hours as shown in Fig. 5C, E and G. The magnitude of the change suggests membrane damage; loss of lipid or possibly pore formation, which is consistent with the carpet model.22–24,95 Such a large resistance drop was not observed at the pristine DOPC (without DOPS) membrane, implying that the anionic charge of the DOPC:DOPS facilitates the effect. This is consistent with previous observations that PS in the bilayer catalyses Aβ1–42 aggregation on the membrane.37,41 Notably for 100 nM Aβ1–42, the membrane resistance (Fig. 5C) continued to decrease and then equilibrated. The corresponding capacitance (Fig. 5D) also decreased, and the extent of decrease remained large when compared to the capacitance without Aβ1–42. A modest decrease in pristine membrane capacitance (0 nM Aβ1–42) may be due to the HEPES buffer as observed in our earlier study.61
Increasing amyloid concentrations over 100 nM resulted in resistance increase (recovery) after 7 hours of decreasing resistance in 1 μM Aβ1–42; where final membrane resistance eventually surpassed the initial membrane resistance (Fig. 5E). We speculate that this is due to adhesion of Aβ1–42 oligomers across the bilayer interface, which is supported by the AFM data below. The corresponding membrane capacitance (Fig. 5F) decreases, also indicating membrane thickening, but the extent of change and kinetics remain low when compared to 100 nM Aβ1–42. This is attributed to concomitant pore-formation followed by oligomer formation at higher Aβ1–42 concentrations. Notably, at 2 μM Aβ1–42, the increasing (recovering) resistance (at 18 h) does not surpass the initial membrane resistance (at 2 h time point); this is attributed to competing effects of layer formation and concomitant damage inflicted on the membrane. Although there was a clear decrease in capacitance overall, on initial addition of 2 μM Aβ1–42 it was observed to increase for the first 1–2 h before progressively declining over the following 18 h (cf.Fig. 5H). This initial increase in capacitance could be due to the removal of lipids as a result of the high concentration of Aβ1–42 (2 μM), possibly a detergent-like effect.
Irrespective of the concentration of Aβ1–42 used, our data suggests carpet model behaviour, especially within the incubation widow of 1–7 hours. However, the resistance increase (recovery) over longer length-scales, after reaching a minimum, deviates from the carpet model (Fig. 5E and F). Chang et al.95 noticed this ambiguity when analysing antimicrobial peptide (AMPs) interactions with model membrane, using EIS within a 25 minutes time span and termed it as carpet ‘’raft’’. The Aβ1–42 aggregate formation is a very slow process that differs from AMPs, and our data captures this longer-scale process. As pointed out earlier by Chang et al.95 because the carpet model does not precisely explain how peptide rafts destabilize the membrane, our EIS results (particularly in the case of 1 and 2 μM Aβ1–42) cannot definitively rule out or agree with the carpet model. Membrane instability and aggregation may occur in nanoscale regimes within the bilayer, that do not lead to full destruction, given we see detectable resistance during incubation. Resistance increase could be linked to network-like, possibly stacked Aβ1–42 layers that forms across the bilayer surface. To investigate this further we performed AFM studies to visualize any structures forming at the membrane directly.
Fig. 5I shows AFM images of the DOPC:DOPS SLB membrane on cleaved mica before, and at different time intervals following, incubation with 1 μM Aβ1–42. Before addition of Aβ1–42, the DOPC:DOPS membrane is quite homogeneous (Fig. 5I, 0 min). Within 30 minutes of incubation with 1 μM Aβ1–42, nanoscale depressions (see arrow in Fig. 5I) appear on the bilayer surface. The density and dimensions of the features grow over time until 4–6 h (for reference, the impact of Aβ1–42 at different time intervals such as 0.5, 1 and 2 h data are shown in Fig. 5I). This observation is strongly consistent with EIS data, which show a decrease in membrane resistance (Fig. 5E) over this window. The Aβ1–42 oligomer is stacked in patches and distributed heterogeneously over the membrane's surface when observed at 18 hours. The micron-scale patches, attributed to network-like layered aggregates of Aβ1–42 oligomers, protrude from the bilayer surface at a height of ∼8–10 nm, as shown in the bottom right panel of Fig. 5I. This is consistent with a report by D'Ursi et al. for AFM study on Aβ25–35 interaction with bilayer membrane.92 The Aβ1–42 oligomer network formation commences after 6 hours of incubation (data not shown) and continues to grow, which is again strongly consistent with our electrochemical data, in which membrane resistance increased and capacitance decreased over the same time period.
Within 1 hour of incubating 2 μM Aβ1–42 at the membrane, dramatic changes in the AFM of the DOPC:DOPS membrane were evident, with extensive pore formation and membrane damage (cf.Fig. 5J). At this concentration, Aβ1–42 seems to behave like a detergent, removing patches of membrane from the bilayer surface and creating pores with dimensions of (10–30 nm diameter). The regions where bilayer patches have been completely removed appear dark in the images (Fig. 5J, left). The line profile analyses (bottom left, in panel J) distinguish two regions: areas of lipid removal, with a height difference between mica and the top surface membrane of 5 nm, consistent with the known thickness of DOPC membrane, and circular pore like regimes with a depression of 1.6 nm from the bilayer surface. At the center of these circular features, globular Aβ1–42 aggregates with lateral dimensions of ∼4–6 nm can be distinguished, likely the oligomeric seed. Although this observation correlates with EIS data, considering the significant change in morphology, one might expect the extent of resistance drop and capacitance rise during the initial incubation time-window to be substantially higher. One plausible explanation for this variation relates to the experimental incubation conditions, where in AFM, the incubation was carried out a horizontally oriented substrate in a ∼200 μL liquid cell versus a vertically oriented substrate in a 5 mL volume for EIS measurement. Although absolute Aβ1–42 concentrations were the same, the effects of gravity and low volume in the AFM measurement cell, is likely to lead to higher local concentration of peptide incident at the membrane surface in the AFM experiment which may accelerate the process compared to the EIS set-up. Nevertheless, the image acquired at 18 hours of incubation, shows heterogeneous large scale Aβ1–42 aggregates had developed and covered up to ∼20% of the bilayer surface. Fig. 5J shows a representative image of the layered as well as globular aggregates covering the bilayer surface, that protrude at a height of ∼8 nm (right panel, Fig. 5J). These AFM results, which were highly reproducible across multiple replicates, explain the resistance increase (recovery) observed in EIS in terms of oligomeric growth and the associated drop in capacitance, which causes the formation of a peptide layer at the membrane interface, leading to membrane thickening. Overall, the data agree well with the hypothesized theory illustrated in Fig. 1 that the interaction of Aβ at the negatively charged membrane follows initial binding, pore formation, lipid dissolution, and growth in oligomer formation, and also provides additional insights towards the concentration dependence and the time scale of Aβ oligomer formation.
To further confirm pore formation by 1 μM Aβ1–42 on the DOPC:DOPS membrane and its influence on bilayer integrity, confocal fluorescence imaging was carried out. Fig. 6 shows the impact of 1 μM Aβ555 after 3 hours of incubation on an asymmetric DOPC:DOPS (90:10) membrane suspended over PDMS cavity array. Fig. 6A shows the confocal image obtained from DOPC:DOPS MSLB labeled with DOPE-Atto655. The association of Aβ555 (green emission) to the membrane is shown in Fig. 6B. As expected, pore-like features could not be distinguished due to the diffraction limit of optical microscopy. However, it is clear that after 3 hours of incubation with 1 μM Aβ555 there is extensive loss of the pyranine from the pores (Fig. 6C). This behaviour contrasts sharply with images collected under identical conditions in Fig. 3C at the DOPC-only membrane, where following Aβ555 incubation, pyranine remained trapped by the intact membrane, and brightly emissive from the cavity walls. From the overlayed images of DOPC (Fig. 3D) and DOPC:DOPS (Fig. 6D) after Aβ incubation it is clear that the pyranine escapes from the cavity attributed to leakage due to poration of the DOPC:DOPS membrane.
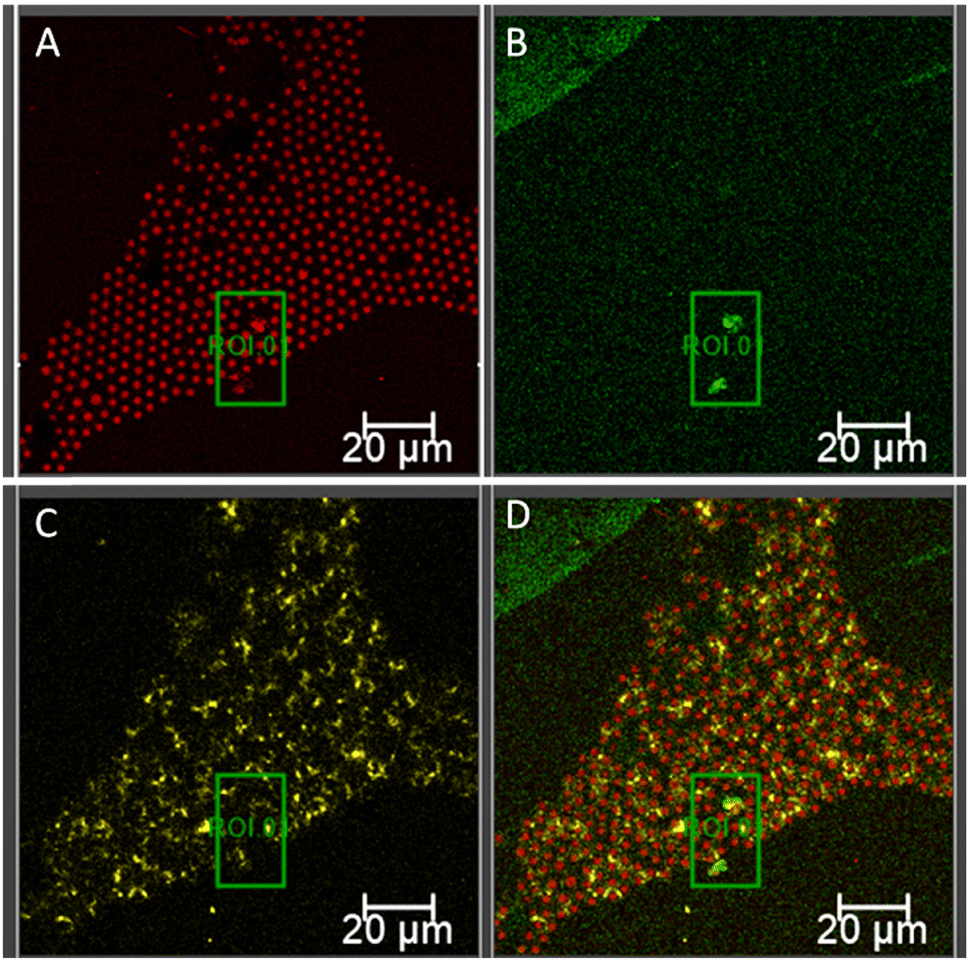 | ||
| Fig. 6 Confocal fluorescence microscopy images of an asymmetric DOPC:DOPS (90:10) bilayer spanning a pyranine (5 μM) filled PDMS microcavity array after 3 hours of incubation with 1 μM Aβ555. Confocal image of DOPC:DOPS MSLB displaying the fluorescence signal of fluorescently labelled (A) DOPE-Atto655 excited at 640 nm with detection window 650–780 nm; (B) Aβ555 excited at 561 nm with a detection window of 570–630 nm; and (C) pyranine fluorescence image obtained with an excitation wavelength of 405 nm and a detection window of 440–560 nm showed dramatic loss of pyranine emission signal. (D) Overlayed fluorescence images displaying the association of Aβ555 with the bilayer surface, resulting leaky pyranine signal and Aβ555 aggregates as indicated by region of interest, ROI 0. The scale bar in each panel measures 20 μm. | ||
Furthermore, according to the hypothesized mechanism described in Fig. 1, when Aβ self-assembly leads to larger scale oligomers/fibrils, they are released from the membrane into the contact solution.55,99,100 To explore this, the contacting electrolyte solution from the impedance experiments after 24 hours of Aβ incubation with the membrane were collected and studied via UV/Vis and fluorescence, following treatment with thioflavin T (Tht). Tht is a fluorescent probe is known to bind specifically to amyloid fibrils but not to amyloid monomers. In the absence of amyloid, the absorbance of Tht alone in HEPES buffer was 411.5 nm, and the emission was 480 nm. When Tht is added to the electrolyte, along with the emission peak at 480 nm, another peak at 510 nm appears (Fig. S5, ESI†) attributed to the Tht bound to the Aβ fibrils/aggregates,101,102 indicating that fibrils/oligomers are indeed released from the membrane into the contact solution after 24 hours incubation of 1 μM Aβ1–42 with asymmetric DOPC:DOPS (90:10)//DOPC membrane.
Effects of DOPS concentration within asymmetric DOPC:DOPS//DOPC membrane
Next we evaluated the impact of DOPS content on oligomerization, first by applying 1 μM Aβ1–42 to a MSLB containing 5 mol% DOPS in the distal leaflet (Fig. 7A and B, orange). The resistance decreased over time (Fig. 7A, orange), with no evidence of increase resistance (recovery) as observed earlier at 10 mol% DOPS concentration (vide infra, cf.Fig. 5E). Simultaneously, the membrane capacitance increased systematically with time (Fig. 7B, orange), indicating membrane thinning or pore formation. Consistent with the EIS observation, AFM images of 5 mol% DOPS containing membrane, as shown in Fig. S8 (ESI†), reveal pores but no aggregation or layers form, even after prolonged incubation with 1 μM Aβ1–42.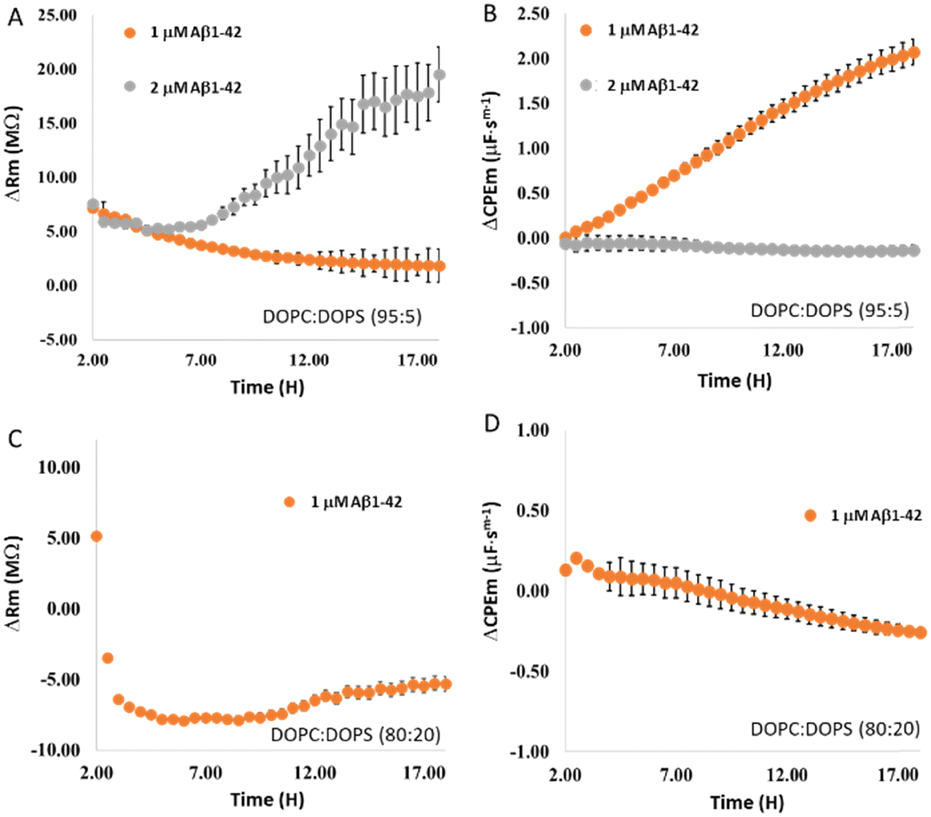 | ||
| Fig. 7 Temporal changes in relative (A) resistance and (B) capacitance of a DOPC:DOPS (95:5)//DOPC membrane following addition of 1 μM (orange) and 2 μM (grey) Aβ1–42. All EIS measurements were performed in HEPES buffer at pH 7.4. After 1.5 hours of membrane equilibration (cf. Fig. S6, ESI†), designated concentration of Aβ1–42 was added. Panels C and D show the relative temporal changes in resistance and capacitance of the DOPC:DOPS (80:20)//DOPC asymmetric bilayer membrane (orange) after the addition of 1 μM Aβ1–42. Prior to the addition of Aβ1–42, membrane's stability was established, and it was found that beyond 1.5 hours, the membrane resistance and capacitance remained unchanged, establishing the time point for Aβ1–42 incubation. All measurements are performed in triplicates (N = 3), and the error bars reflect standard error. | ||
Incubation of 2 μM Aβ1–42 with a membrane containing 5 mol% DOPS, on the other hand, shows resistance decreases over 3–4 h that subsequently recovers (grey, Fig. 7A). Simultaneously, membrane capacitance decreases modestly after 4 h (grey, Fig. 7B), indicating thickening of membrane/layer formation. The resistance and capacitance profiles are reminiscent of those for 1 μM Aβ1–42 at 10 mol% DOPS containing membrane (vide infra), indicating, that when DOPS content is reduced, more Aβ1–42 is required to induce oligomerization and form layered aggregates.
At membranes containing 20 mol% of DOPS, 1 μM Aβ1–42 elicited a dramatic decrease in resistance (Fig. 7C) within 1 h of incubation and remained steady over ∼9–10 h. Resistance began to recover modestly after this time point. Simultaneously, the membrane capacitance increased modestly within 1 h, then systematically decreased (Fig. 7D), suggesting the formation of layered aggregates. Overall, the data indicate that the rate and extent of oligomer formation are affected by the concentrations of both DOPS and Aβ1–42. When the membrane concentration of DOPS is decreased, a higher concentration of Aβ1–42 is required for the formation of oligomer. Combined, data across 18 hours indicate that at a fixed concentration of 1 μM Aβ1–42 and variable DOPS content, pore formation is only predominant when 5 mol% DOPS is present (as seen by a systematic rise in capacitance versus time, Fig. 7B (orange)), without concomitant fibril/oligomer formation; whereas when DOPS content is increased to 10 or 20 mol%, a modest capacitance rise is evident at within 1–2 h (Fig. 7D and 5F), but overall remains decreased beyond this time window.
Effects of Aβ1–42 on an asymmetric DOPC//DOPC:DOPS (90:10) membrane on optothermal damage
Varying the concentration of DOPS (20, 10 and 5 mol%) in the proximal leaflet in a bilayer with only DOPC present in the exterior leaflet, we then attempted to mimic discrete areas of PS translocation that might occur in lipid damage due to oxidative stress and apoptosis of neurons associated with aging of the brain. This was accomplished by optothermally damaging the membrane with a laser to initiate DOPS transversal diffusion as reported prevously.61 In the absence of photodamage, DOPC//DOPC:DOPS (90:10) bilayers were found to be stable for at least 10 hours, as shown previously by assessing transversal diffusion of DOPS via annexin binding.61 After initial equilibration, the MSLB was irradiated for 10 seconds with a 405 nm laser at 3.12 mW power. As reported previously, for all DOPS concentrations used in this study, irradiation led to a decrease in resistance confirming optothermal induced increase in membrane admittance associated with bilayer annealing and increased local fluidity.62 Correspondingly, the capacitance does not change significantly during this process, indicating that the membrane was not seriously damaged by the laser. When 20 mol% DOPS was present at the proximal leaflet, the resistance reduced considerably when 1 μM Aβ1–42 was added following photothermal damage. Although, the drop in resistance is significant (Fig. 8A), the extent of decrease in resistance and the associated kinetics remains low when compared to the case when 20 mol% DOPS was present on the distal leaflet (Fig. 7C, vide infra).
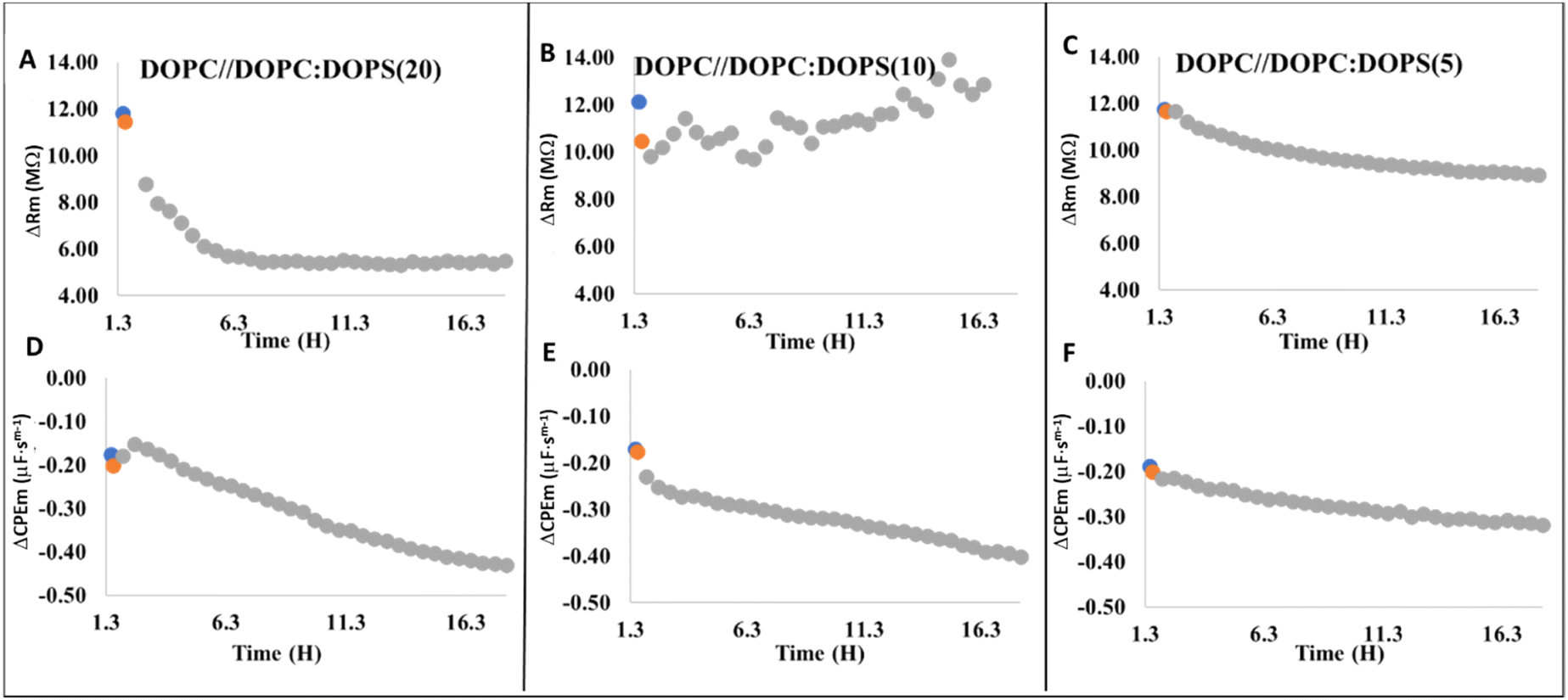 | ||
| Fig. 8 Representative relative resistance (A–C) and relative capacitance (D–F) changes as a result of 1 μM Aβ1–42 binding at different DOPS containing asymmetric DOPC//DOPC:DOPS MSLBs: (A and D) DOPC//DOPC:DOPS (80:20); (B and E) DOPC//DOPC:DOPS (90:10); and (C and F) DOPC//DOPC:DOPS (95:5). After photodamaging the MSLBs for 10 s with a 405 nm laser, Aβ1–42 was introduced into each type of membrane. In each panel, blue circles represent the electrochemically equilibrated membrane's resistance and capacitance, stability time point, whereas orange circle represent the corresponding data points following photodamage and defines the time point when Aβ1–42 was introduced to the cell. Each panel's grey circles represent the temporal evolution of membrane resistance and capacitance after addition of 1 μM Aβ1–42. All data points are the mean of N = 3, and error bars have been eliminated for clarity. | ||
Within 1–2 hours of Aβ1–42 incubation following laser damage, the capacitance increases, but then systematically decreases over 18 hours. The overall magnitude of decrease in capacitance remains modest when compared to when 20 mol% DOPS is included at distal leaflet. Because optothermally induced DOPS transverse migration from the lower to upper leaflet results in much lower DOPS concentration at the exterior leaflet, Aβ1–42 binding is expected to be significantly lower. When 10 mol% DOPS is present at the proximal leaflet, laser damage induced a modest increase in overall resistance, although the resistance change fluctuated over this incubation window and this response was reproducible across multiple (N = 3) independently prepared substrates (Fig. 8B). This is surprising as we did not detect this behavior in any other type of membrane or at different concentrations of Aβ1–42. However, the trend of decreasing capacitance persisted (Fig. 8E). We speculate that transient Aβ1–42 interactions with the DOPS exposed at the outer leaflet may explain this dynamic interaction and tentatively attributed the observed electrochemical changes to competing effects of membrane damage/pore formation and rate of Aβ1–42 aggregation. In contrast to 20 and 10 mol%, when 5 mol% DOPS was present at the proximal leaflet of the membrane after photothermally initiation, a slow decrease of both membrane resistance (Fig. 8C) and capacitance (Fig. 8F) was observed. Overall, the data revealed that during optothermal damage, there is a pool of local DOPS content at the area of laser focus due to fast flip-flop of DOPS, which may potentially seed and plays a catalytic role in amyloid oligomers formation causing membrane damage. Under identical conditions, when no laser irradiation was performed, DOPS remain at the inner leaflet and impedance changes were comparable to that of DOPC only membrane (data not shown). It is worth noting that none of the optothermally treated DOPC//DOPC:DOPS membranes showed evidence of any large scale oligomerization, as no recovery in resistance is evident.
Conclusions
Microcavity, and mica supported lipid bilayers comprised of DOPC-only and, asymmetric bilayers composed of (external//interior leaflet) DOPC:DOPS//DOPC (with varying DOPS concentrations on the exterior leaflet), and DOPC//DOPC:DOPS (with varying DOPS concentrations on the interior leaflet) were evaluated for Aβ1–42 binding and aggregation using electrochemical impedance spectroscopy, AFM and confocal fluorescence microscopy.At pristine DOPC membrane, Aβ1–42 appears to adsorb at the zwitterionic membrane interface, without aggregation even at 1 μM of peptide. Using a membrane integrity test with pyranine, we observed that the DOPC membranes remained intact and impermeable to the probe in the presence of Aβ1–42.
The presence of anionic DOPS in the membrane dramatically alters the way Aβ1–42 interacts with the membrane. Oligomerization of Aβ1–42 monomer was observed by EIS and AFM, and confocal microscopy, when DOPS was exposed to the outer membrane leaflet. Over time, there was evidence of both pore formation (at short time frames) and oligomerization-with extensive membrane-oligomer multilayer binding and Aβ1–42 fibril formation (3 to 18 hours). The extent of oligomerization and membrane damage depended on the concentrations of DOPS in the membrane and Aβ1–42 in the contacting solution. The peptide aggregation was accelerated at the highest Aβ1–42 and DOPS concentrations, as expected, and the extent of membrane damage increased with Aβ1–42 concentration. The data suggested that all of the hypothesized membrane Aβ1–42 interactions (pore formation, dissolution and carpet effect) occur, but the outcome is dependent on the concentrations of both Aβ1–42 and DOPS.
In a simple model of DOPS translocation that occurs in damaged neuronal membranes associated with e.g., oxidative stress and apoptosis, asymmetric membranes with DOPS localized to the lower leaflet were prepared, and DOPS lipid transversal migration was instigated optothermally to create local regions of high concentration of DOPS. Our data indicated that higher concentrations of DOPS (>10 mol%) at the lower leaflet migration of anionic lipids can seed Aβ1–42 aggregation where DOPS appears to have a catalytic effect on Aβ oligomerization.
Author contributions
T. E. K. designed and directed the research. J. R. and N. K. S. performed the experiment and analysed the data. J. R, N. K. S. and T. E. K. wrote the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This material is based upon work supported by Science Foundation Ireland under Grant No. [14/IA/2488], [19/FFP/6428] and [12/RC/2276_P2]. The National Biophotonics and Imaging Platform, Ireland, funded by the Irish Government's Programme for Research in Third Level Institutions, Cycle 4 and 5, Ireland's EU Structural Funds Programmes 2007–2013 and to Irish research council, are gratefully acknowledged for funding under project GOIPG/2020/589.Notes and references
- C. A. Ross and M. A. Poirier, Nat. Med., 2004, 10, S10–S17 CrossRef PubMed.
- J. Näslund, A. Schierhorn, U. Hellman, L. Lannfelt, A. D. Roses, L. O. Tjernberg, J. Silberring, S. E. Gandy, B. Winblad and P. Greengard, Proc. Natl. Acad. Sci. U. S. A., 1994, 91, 8378–8382 CrossRef.
- C.-C. Chang, E. Edwald, S. Veatch, D. G. Steel and A. Gafni, Biochim. Biophys. Acta, Biomembr., 2018, 1860, 1616–1624 CrossRef CAS PubMed.
- K. A. Bruggink, W. Jongbloed, E. A. L. M. Biemans, R. Veerhuis, J. A. H. R. Claassen, H. B. Kuiperij and M. M. Verbeek, Anal. Biochem., 2013, 433, 112–120 CrossRef CAS PubMed.
- K. H. Ashe, Alzheimer's Dement., 2020, 16, 1561–1567 CrossRef PubMed.
- C. G. Glabe, J. Biol. Chem., 2008, 283, 29639–29643 CrossRef CAS.
- K. L. Viola and W. L. Klein, Acta Neuropathol., 2015, 129, 183–206 CrossRef CAS.
- E. N. Cline, M. A. Bicca, K. L. Viola and W. L. Klein, J. Alzheimer's Dis., 2018, 64, S567–S610 CAS.
- N. P. Cook and A. A. Martí, ACS Chem. Neurosci., 2012, 3, 896–899 CrossRef CAS.
- E. Cerasoli, M. G. Ryadnov and B. M. Austen, Front. Chem., 2015, 3, 17 Search PubMed.
- C. G. Glabe, Neurobiol. Aging, 2006, 27, 570–575 CrossRef CAS PubMed.
- E. Aoraha, J. Candreva and J. R. Kim, Mol. BioSyst., 2015, 11, 2281–2289 RSC.
- M. Mapstone, A. K. Cheema, M. S. Fiandaca, X. Zhong, T. R. Mhyre, L. H. MacArthur, W. J. Hall, S. G. Fisher, D. R. Peterson, J. M. Haley, M. D. Nazar, S. A. Rich, D. J. Berlau, C. B. Peltz, M. T. Tan, C. H. Kawas and H. J. Federoff, Nat. Med., 2014, 20, 415–418 CrossRef CAS.
- B. H. Monien, L. G. Apostolova and G. Bitan, Expert Rev. Neurother., 2006, 6, 1293–1306 CrossRef.
- J. V. Rushworth and N. M. Hooper, J. Alzheimer's Dis., 2011, 2011, 1–14 CrossRef.
- E. Drolle, A. Negoda, K. Hammond, E. Pavlov and Z. Leonenko, PLoS One, 2017, 12, e0182194 CrossRef.
- M. Ahmed, J. Davis, D. Aucoin, T. Sato, S. Ahuja, S. Aimoto, J. I. Elliott, W. E. Van Nostrand and S. O. Smith, Nat. Struct. Mol. Biol., 2010, 17, 561–567 CrossRef CAS.
- D. Mrdenovic, I. S. Pieta, R. Nowakowski, W. Kutner, J. Lipkowski and P. Pieta, Int. J. Biol. Macromol., 2022, 200, 520–531 CrossRef CAS.
- H. H. Jarosz-Griffiths, E. Noble, J. V. Rushworth and N. M. Hooper, J. Biol. Chem., 2016, 291, 3174–3183 CrossRef CAS.
- D. J. Lindberg, E. Wesén, J. Björkeroth, S. Rocha and E. K. Esbjörner, Biochim. Biophys. Acta, Biomembr., 2017, 1859, 1921–1929 CrossRef CAS.
- L. Puglielli, R. E. Tanzi and D. M. Kovacs, Nat. Neurosci., 2003, 6, 345–351 CrossRef CAS.
- D. C. Bode, M. Freeley, J. Nield, M. Palma and J. H. Viles, J. Biol. Chem., 2019, 294, 7566–7572 CrossRef CAS.
- J. W. Um, H. B. Nygaard, J. K. Heiss, M. A. Kostylev, M. Stagi, A. Vortmeyer, T. Wisniewski, E. C. Gunther and S. M. Strittmatter, Nat. Neurosci., 2012, 15, 1227–1235 CrossRef CAS PubMed.
- G. D’Errico, G. Vitiello, O. Ortona, A. Tedeschi, A. Ramunno and A. M. D’Ursi, Biochim. Biophys. Acta, Biomembr., 2008, 1778, 2710–2716 CrossRef.
- X. Yu, Q. Wang, Q. Pan, F. Zhou and J. Zheng, Phys. Chem. Chem. Phys., 2013, 15, 8878 RSC.
- A. Khondker, R. Alsop and M. Rheinstädter, Membranes, 2017, 7, 49 CrossRef PubMed.
- C. Haass and D. J. Selkoe, Nat. Rev. Mol. Cell Biol., 2007, 8, 101–112 CrossRef CAS.
- A. Aguzzi and T. O’Connor, Nat. Rev. Drug Discovery, 2010, 9, 237–248 CrossRef CAS PubMed.
- S. Micelli, D. Meleleo, V. Picciarelli and E. Gallucci, Biophys. J., 2004, 86, 2231–2237 CrossRef CAS.
- C. Emre, K. V. Do, B. Jun, E. Hjorth, S. G. Alcalde, M.-A. I. Kautzmann, W. C. Gordon, P. Nilsson, N. G. Bazan and M. Schultzberg, Acta Neuropathol. Commun., 2021, 9, 116 CrossRef CAS.
- V. E. Kagan, B. Gleiss, Y. Y. Tyurina, V. A. Tyurin, C. Elenström-Magnusson, S.-X. Liu, F. B. Serinkan, A. Arroyo, J. Chandra, S. Orrenius and B. Fadeel, J. Immunol., 2002, 169, 487–499 CrossRef CAS.
- M. L. Bader Lange, G. Cenini, M. Piroddi, H. Mohmmad Abdul, R. Sultana, F. Galli, M. Memo and D. A. Butterfield, Neurobiol. Dis., 2008, 29, 456–464 CrossRef CAS PubMed.
- M.-A. Sani, J. D. Gehman and F. Separovic, FEBS Lett., 2011, 585, 749–754 CrossRef CAS PubMed.
- K. Matsuzaki, Biochim. Biophys. Acta, Biomembr., 2007, 1768, 1935–1942 CrossRef CAS.
- M. del Mar Martinez-Senac, J. Villalain and J. C. Gomez-Fernandez, Eur. J. Biochem., 1999, 265, 744–753 CrossRef CAS.
- E. Maltseva and G. Brezesinski, ChemPhysChem, 2004, 5, 1185–1190 CrossRef CAS PubMed.
- H. Ahyayauch, M. Raab, J. V. Busto, N. Andraka, J.-L. R. Arrondo, M. Masserini, I. Tvaroska and F. M. Goñi, Biophys. J., 2012, 103, 453–463 CrossRef CAS.
- E. Terzi, G. Hölzemann and J. Seelig, J. Mol. Biol., 1995, 252, 633–642 CrossRef CAS PubMed.
- C. Wu, M. M. Murray, S. L. Bernstein, M. M. Condron, G. Bitan, J.-E. Shea and M. T. Bowers, J. Mol. Biol., 2009, 387, 492–501 CrossRef CAS.
- L. Svennerholm, J. Lipid Res., 1968, 9, 570–579 CrossRef CAS.
- B. Bonev, A. Watts, M. Bokvist and G. Gröbner, Phys. Chem. Chem. Phys., 2001, 3, 2904–2910 RSC.
- J. J. Kremer and R. M. Murphy, J. Biochem. Biophys. Methods, 2003, 57, 159–169 CrossRef CAS.
- S. Dante, T. Hauss and N. A. Dencher, Biophys. J., 2002, 83, 2610–2616 CrossRef CAS PubMed.
- O. Simakova and N. J. Arispe, Biophys. J., 2012, 102, 657a CrossRef.
- C. H. Davis and M. L. Berkowitz, Biophys. J., 2009, 96, 785–797 CrossRef CAS PubMed.
- B. R. Sahoo, T. Genjo, S. J. Cox, A. K. Stoddard, G. M. Anantharamaiah, C. Fierke and A. Ramamoorthy, J. Mol. Biol., 2018, 430, 4230–4244 CrossRef CAS.
- J. Pilch, C. M. Franzin, L. M. Knowles, F. J. Ferrer, F. M. Marassi and E. Ruoslahti, J. Mol. Biol., 2006, 356, 876–885 CrossRef CAS.
- G. Lee, H. B. Pollard and N. Arispe, Peptides, 2002, 23, 1249–1263 CrossRef CAS.
- V. Martín, N. Fabelo, G. Santpere, B. Puig, R. Marín, I. Ferrer and M. Díaz, J. Alzheimer's Dis., 2010, 19, 489–502 Search PubMed.
- S. Banerjee, M. Hashemi, K. Zagorski and Y. L. Lyubchenko, ACS Chem. Neurosci., 2021, 12, 506–516 CrossRef CAS.
- L. Qiu, A. Lewis, J. Como, M. W. Vaughn, J. Huang, P. Somerharju, J. Virtanen and K. H. Cheng, Biophys. J., 2009, 96, 4299–4307 CrossRef CAS.
- D. Frankel, M. Davies, B. Bhushan, Y. Kulaberoglu, P. Urriola-Munoz, J. Bertrand-Michel, M. R. Pergande, A. A. Smith, S. Preet, T. J. Park, M. Vendruscolo, K. S. Rankin, S. M. Cologna, J. R. Kumita, N. Cenac and E. St John Smith, Aging, 2020, 12, 22266–22290 CrossRef CAS PubMed.
- W. Wood, F. Schroeder, U. Igbavboa, N. Avdulov and S. Chochina, Neurobiol. Aging, 2002, 23, 685–694 CrossRef CAS.
- C. E. Heo, C. R. Park and H. I. Kim, Chem. Phys. Lipids, 2021, 236, 105073 CrossRef CAS.
- V. Dubois, D. Serrano and S. Seeger, ACS Chem. Neurosci., 2019, 10, 4776–4786 CrossRef CAS PubMed.
- J. Lee, Y. H. Kim, F. T. Arce, A. L. Gillman, H. Jang, B. L. Kagan, R. Nussinov, J. Yang and R. Lal, ACS Chem. Neurosci., 2017, 8, 1348–1357 CrossRef CAS.
- S. Meker, H. Chin, T. N. Sut and N.-J. Cho, Langmuir, 2018, 34, 9548–9560 CAS.
- D. Mrdenovic, M. Majewska, I. S. Pieta, P. Bernatowicz, R. Nowakowski, W. Kutner, J. Lipkowski and P. Pieta, Langmuir, 2019, 35, 11940–11949 CrossRef CAS.
- Y. Tian, R. Liang, A. Kumar, P. Szwedziak and J. H. Viles, Chem. Sci., 2021, 12, 6896–6907 RSC.
- Y.-H. M. Chan and S. G. Boxer, Curr. Opin. Chem. Biol., 2007, 11, 581–587 CrossRef CAS.
- J. Robinson, G. B. Berselli, M. G. Ryadnov and T. E. Keyes, Langmuir, 2020, 36, 5454–5465 CrossRef CAS.
- G. B. Berselli, A. V. Gimenez, A. O’Connor and T. E. Keyes, ACS Appl. Mater. Interfaces, 2021, 13, 29158–29169 CrossRef CAS.
- G. B. Berselli, N. K. Sarangi, S. Ramadurai, P. V. Murphy and T. E. Keyes, ACS Appl. Bio Mater., 2019, 2, 3404–3417 CrossRef CAS.
- N. K. Sarangi, A. Prabhakaran and T. E. Keyes, Electroanalysis, 2020, 32, 2936–2945 CrossRef CAS.
- H. Basit, V. Gaul, S. Maher, R. J. Forster and T. E. Keyes, Analyst, 2015, 140, 3012–3018 RSC.
- S. Maher, H. Basit, R. J. Forster and T. E. Keyes, Bioelectrochemistry, 2016, 112, 16–23 CrossRef CAS PubMed.
- N. K. Sarangi, A. Stalcup and T. E. Keyes, ChemElectroChem, 2020, 7, 4535–4542 CrossRef CAS.
- N. K. Sarangi, A. Prabhakaran and T. E. Keyes, Langmuir, 2022, 38, 6411–6424 CrossRef CAS PubMed.
- K. Broersen, W. Jonckheere, J. Rozenski, A. Vandersteen, K. Pauwels, A. Pastore, F. Rousseau and J. Schymkowitz, Protein Eng., Des. Sel., 2011, 24, 743–750 CrossRef CAS.
- D. Mrdenovic, P. Zarzycki, M. Majewska, I. S. Pieta, R. Nowakowski, W. Kutner, J. Lipkowski and P. Pieta, ACS Chem. Neurosci., 2021, 12, 531–541 CrossRef CAS PubMed.
- Y. Sokolov, J. A. Kozak, R. Kayed, A. Chanturiya, C. Glabe and J. E. Hall, J. Gen. Physiol., 2006, 128, 637–647 CrossRef CAS.
- W. B. Stine, K. N. Dahlgren, G. A. Krafft and M. J. LaDu, J. Biol. Chem., 2003, 278, 11612–11622 CrossRef CAS PubMed.
- S. Banerjee, M. Hashemi, K. Zagorski and Y. L. Lyubchenko, Int. J. Mol. Sci., 2020, 21, 1129 CrossRef CAS PubMed.
- A. Tiiman, J. Krishtal, P. Palumaa and V. Tõugu, AIP Adv., 2015, 5, 092401 CrossRef.
- S. Ramadurai, A. Kohut, N. K. Sarangi, O. Zholobko, V. A. Baulin, A. Voronov and T. E. Keyes, J. Colloid Interface Sci., 2019, 542, 483–494 CrossRef CAS.
- S. Ramadurai, N. K. Sarangi, S. Maher, N. MacConnell, A. M. Bond, D. McDaid, D. Flynn and T. E. Keyes, Langmuir, 2019, 35, 8095–8109 CrossRef CAS.
- S. Ramadurai, M. Werner, N. K. H. Slater, A. Martin, V. A. Baulin and T. E. Keyes, Soft Matter, 2017, 13, 3690–3700 RSC.
- A. Bouter, C. Gounou, R. Bérat, S. Tan, B. Gallois, T. Granier, B. L. D’Estaintot, E. Pöschl, B. Brachvogel and A. R. Brisson, Nat. Commun., 2011, 2, 270 CrossRef.
- M. A. Johnson, S. Seifert, H. I. Petrache and A. C. Kimble-Hill, Langmuir, 2014, 30, 9880–9885 CrossRef CAS.
- M. E. Haque, T. J. McIntosh and B. R. Lentz, Biochemistry, 2001, 40, 4340–4348 CrossRef CAS.
- V. Gerke and S. E. Moss, Physiol. Rev., 2002, 82, 331–371 CrossRef CAS PubMed.
- F. Oling, J. S. O. Santos, N. Govorukhina, C. Mazères-Dubut, W. Bergsma-Schutter, G. Oostergetel, W. Keegstra, O. Lambert, A. Lewit-Bentley and A. Brisson, J. Mol. Biol., 2000, 304, 561–573 CrossRef CAS PubMed.
- C. Pigault, A. Follenius-Wund, M. Schmutz, J.-M. Freyssinet and A. Brisson, J. Mol. Biol., 1994, 236, 199–208 CrossRef CAS PubMed.
- F. Oling, W. Bergsma-Schutter and A. Brisson, J. Struct. Biol., 2001, 133, 55–63 CrossRef CAS.
- H.-Y. Kim, B. X. Huang and A. A. Spector, Prog. Lipid Res., 2014, 56, 1–18 CrossRef CAS PubMed.
- L.-F. Lue, Y.-M. Kuo, A. E. Roher, L. Brachova, Y. Shen, L. Sue, T. Beach, J. H. Kurth, R. E. Rydel and J. Rogers, Am. J. Pathol., 1999, 155, 853–862 CrossRef CAS.
- K. Rajasekhar, M. Chakrabarti and T. Govindaraju, Chem. Commun., 2015, 51, 13434–13450 RSC.
- J. R. Cirrito, P. C. May, M. A. O’Dell, J. W. Taylor, M. Parsadanian, J. W. Cramer, J. E. Audia, J. S. Nissen, K. R. Bales, S. M. Paul, R. B. DeMattos and D. M. Holtzman, J. Neurosci., 2003, 23, 8844–8853 CrossRef CAS PubMed.
- D. Puzzo, L. Privitera, M. Fa, A. Staniszewski, G. Hashimoto, F. Aziz, M. Sakurai, E. M. Ribe, C. M. Troy, M. Mercken, S. S. Jung, A. Palmeri and O. Arancio, Ann. Neurol., 2011, 69, 819–830 CrossRef CAS.
- G. M. J. A. Klug, D. Losic, S. Subasinghe, M.-I. Aguilar, L. L. Martin and D. H. Small, Eur. J. Biochem., 2003, 270, 4282–4293 CrossRef CAS PubMed.
- K. Garai, R. Sureka and S. Maiti, Biophys. J., 2007, 92, L55–L57 CrossRef CAS.
- M. Sublimi Saponetti, M. Grimaldi, M. Scrima, C. Albonetti, S. L. Nori, A. Cucolo, F. Bobba and A. M. D’Ursi, PLoS One, 2014, 9, e115780 CrossRef PubMed.
- B. Soreghan, J. Kosmoski and C. Glabe, J. Biol. Chem., 1994, 269, 28551–28554 CrossRef CAS.
- M. Zhu, P. O. Souillac, C. Ionescu-Zanetti, S. A. Carter and A. L. Fink, J. Biol. Chem., 2002, 277, 50914–50922 CrossRef CAS.
- W. K. Chang, W. C. Wimley, P. C. Searson, K. Hristova and M. Merzlyakov, Biochim. Biophys. Acta, Biomembr., 2008, 1778, 2430–2436 CrossRef CAS PubMed.
- P. Zimmermann, K. Schlenstedt, S. Schwarz, D. Vehlow, M. Blanke, A. Fery and J. Nagel, ACS Appl. Polym. Mater., 2022, 4, 5189–5198 CrossRef CAS.
- Z. Chen, Y. Wang, Y. Shang, A. Umar, P. Xie, Q. Qi and G. Zhou, Sci. Rep., 2017, 7, 2713 CrossRef PubMed.
- N. Tarutani, Y. Tokudome, K. Nakanishi and M. Takahashi, RSC Adv., 2014, 4, 16075–16080 RSC.
- K. Sasahara, K. Morigaki and K. Shinya, Phys. Chem. Chem. Phys., 2013, 15, 8929 RSC.
- N. P. Reynolds, A. Soragni, M. Rabe, D. Verdes, E. Liverani, S. Handschin, R. Riek and S. Seeger, J. Am. Chem. Soc., 2011, 133, 19366–19375 CrossRef CAS.
- S. Sugimoto, K. Arita-Morioka, Y. Mizunoe, K. Yamanaka and T. Ogura, Nucleic Acids Res., 2015, 43, e92–e92 CrossRef.
- C. Xue, T. Y. Lin, D. Chang and Z. Guo, R. Soc. Open Sci., 2017, 4, 160696 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Detailed fabrication methods, bilayer stability studies, confocal fluorescence and additional AFM data are provided. See DOI: https://doi.org/10.1039/d2cp03344e |
| ‡ These authors contributed equally to this work. |
| This journal is © the Owner Societies 2023 |