
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The origin of isomerization of aniline revealed by high kinetic energy ion mobility spectrometry (HiKE-IMS)†
Cameron N.
Naylor
 *,
Christoph
Schaefer
,
Ansgar T.
Kirk
and
Stefan
Zimmermann
*,
Christoph
Schaefer
,
Ansgar T.
Kirk
and
Stefan
Zimmermann
Leibniz University Hannover, Institute of Electrical Engineering and Measurement Technology, Department of Sensors and Measurement Technology, 30167 Hannover, Germany. E-mail: naylor@geml.uni-hannover.de
First published on 14th December 2022
Abstract
Although aniline is a relatively simple small molecule, the origin of its two peaks observed in ion mobility spectrometry (IMS) has remained under debate for at least 30 years. First hypothesized as a difference in protonation site (amine vs. benzene ring), each ion mobility peak differs by one Dalton when coupled with mass spectrometry where the faster mobility peak is the molecular ion peak, and the slower mobility peak is protonated. To complicate the deconvolution of structures, some previous literature shows the peaks as unresolved and thus proposes these species exist in equilibrium. In this work, we show that when measured with high kinetic energy ion mobility spectrometry (HiKE-IMS), the two peaks observed in spectra of both aniline and all n-fluoroanilines are fully separated (chromatographic resolution from 2–7, Rp > 110) and therefore not in equilibrium. The HiKE-IMS is capable of changing ionization conditions independently of drift region conditions, and our results agree with previous literature showing that ionization source settings (including possible fragmentation at this stage) are the only influence determining the speciation of the two aniline peaks. Finally, when the drift and reactant gas are changed to nitrogen, a third peak appears at high E/N for 2-fluoroaniline and 4-fluoroaniline for the first time in reported literature. As observed by HiKE-IMS-MS, the new third peak is also protonated showing that the para-protonated aniline and resulting fragment ion, molecular ion aniline, can be fully separated in the mobility domain for the first time. The appearance of the third peak is only possible due to the increased separation of the other two peaks within the HiKE-IMS.
Introduction
Ion mobility spectrometry (IMS) is a gas-phase separation technique where ions traverse through a neutral-gas filled drift tube under the presence of an electric field.1–3 As the ions travel, they collide with the drift gas and are separated according to the size and shape of the ion. Larger ions experience more collisions and arrive later at the detector located at the end of the drift tube than smaller ions. Once the separated ion packet arrives at the detector, the ion current is recorded as a function of the time of arrival after the gate pulse, or the drift time. However, since analyte drift times change as a function of several different variables, ion mobilities are often reported in terms of an ion-neutral collisional cross-section value (CCS, Ω) or a reduced mobility value (K0) as defined below in eqn (1):3 | (1) |
However, analysis of small molecules using IMS is deceptively more complex than it would first appear. Aniline is a classic example of a small molecule with two distinctly different mobilities as first reported by Karpas et al.21 They observed two different mobilities for aniline in purified air and hypothesized that two different protonation sites (one at the amine, one at the benzene ring) caused the two distinct different conformers in the mobility domain. The protonation site theory was initially revisited a few years ago in studies that used traveling wave IMS and differential mobility spectrometry (DMS) to examine the different aniline conformers where a time of flight mass spectrometer (ToF-MS) was used to verify the m/z of each aniline conformer peak.22–24 The slower mobility peak (the N-protonated isomer in literature) has an associated m/z of only 94 Da, whereas the faster mobility peak (C-protonated isomer in literature) has a variety of m/z values ranging from 92 Da, 93 Da, or 94 Da all in different abundances that change with instrumental conditions, but most commonly the most abundant m/z of the fast peak is 93 Da.22 The difference in 1 Da between the fast and slow aniline peaks has largely been ignored in favor of the difference in mobility between peaks. However, when the difference in m/z arises, usually the explanation given is fragmentation of the C-protonated isomer but the discussion of when (i.e. pre- or post-mobility separation) fragmentation occurs rarely arises. Alternatively, Lalli et al. propose a rearrangement of molecular ion aniline into azepine, explaining the m/z 93 Da mass spectrum, which then can lose an additional neutral hydrogen atom to form the radical azatropylium ion (m/z 92 Da).22 These rearrangement and fragmentation pathways are largely absent from the ion mobility literature but appear more prominently in the electron impact mass spectrometry literature.25,26
Regardless of the actual chemical identity of each peak, the ion mobilities of these two protonated isomers, or protomers, are overlapping in most literature, which raises the question if these two species are in equilibrium or if these experiments simply lacked the resolving power to baseline separate the peaks with the IMS platforms used in these studies.21–23,27,28 The hypothesis that both peaks are in equilibrium is supported by the change in intensities of each peak that are easily modified as a function of ion source voltages. Recent literature demonstrates that both peaks are produced from atmospheric pressure chemical ionization (APCI), electrospray ionization (ESI), and helium plasma ionization (HePI), but the most abundant peak always shifts from the N-protonated isomer to the C-protonated isomer as the cone inlet voltage on the instrument increases.22,23,27,28 In contrast, Asbury and Hill report that aniline only has one peak with ambient pressure ESI even when changing the drift gas.29 However, it is entirely possible that their data acquisition was not fast enough to capture both peaks since their aniline spectra in three of the four drift gases have a significant shoulder that could easily be the missing peak.29
Since the aniline peaks seem to change abundance related to ionization source settings and a high resolving power is needed to separate the peaks, high kinetic energy ion mobility spectrometry (HiKE-IMS) offers a unique opportunity to examine the influence of both parameters on measurements of aniline. HiKE-IMS is a variant of a drift tube ion mobility spectrometer that operates with the same basic principles of drift tube IMS with a few key differences.30,31 Notably, the electric fields in the drift region and reaction region (similar to the desolvation region on an ESI-IMS) can be independently changed allowing for different reduced electric field strengths in both regions simultaneously. These differences in the electric field allow ionization conditions to be altered to favor different ionization mechanisms by changing the abundances of reactant ions which results in ionizing analytes, such as benzene, that other sources cannot.30,32–34 Additionally, the HiKE-IMS achieves high resolving powers (Rp > 140) despite being operated at reduced pressure, to allow for the reduced electric field strength to span over an order of magnitude (5–125 Td). Finally, HiKE-IMS has the capability of measuring first-principles ion mobilities, which allows for the calculation of accurate K0 values.35
Due to these advantages, aniline and its derivatives are interesting molecules to analyze with HiKE-IMS. First, the high resolving power capable with the HiKE-IMS and the ability to change the equilibrium of reactant ions within the reaction region can clarify whether the two aniline peaks are truly in equilibrium in the mobility experiment as a function of ionization source settings or if they are simply two peaks that can be separated with a sufficiently high resolving power. Additionally, the capability of the HiKE-IMS to give accurate first-principles derived reduced mobilities opens the possibility to observe changes in mobility when ionization conditions change confirming possible fragmentation based on the mass spectra when HiKE-IMS is coupled to a ToF-MS. This experiment can determine if the C-protonated isomer described in the literature is actually a protomer, or if it is a molecular ion radical formed from charge transfer. The final experimental parameter that can identify the aniline peak identities is changing the concentrations of the reactant ions by adding water to the drift gas or changing the drift gas from air to nitrogen to reduce oxygen radical ions. Both of these parameters change the corona ionization conditions and can favor either charge transfer ionization or proton transfer ionization conditions. Through these sets of experiments plus computational modeling, fully characterizing aniline and 3 fluorinated aniline isomers by changing ionization conditions can be accomplished. These combinations of experiments may also benefit applications such as drug development and discovery, where identification of the ion mobility peaks can be difficult, or cases where ionization or ion transfer optics settings may wish to be optimized to achieve a specific ion abundance.36
Material and methods
Sample preparation
The following aniline compounds were purchased from Sigma Aldrich (Taufkirchen, Germany): Aniline (Purity 99.5% ACS; Sigma number: 242284-100ML), 4-fluoroaniline (Purity 99%; Sigma number: F3800-25G), 2-fluoroaniline (Purity 99%; Sigma number: F3401-25G-A), and 3-fluoroaniline (Purity 99%; Sigma number: F3606-25G). The affiliated ionization properties with each aniline are listed in Table 1. Samples of aniline, 4-fluoroaniline, 3-fluoroaniline, and 2-fluoroaniline were prepared by transferring each sample into a 1 mL polyethylene transfer pipette, sealing it, and placing it in a permeation oven (Vici Dynacalibrator 150, Schenkon, Switzerland) set at 45.00 °C. The sample gas exiting the permeation oven containing the analyte in either purified air or N2 was then diluted with either purified air or N2 and varying water vapor concentrations using model F-200DV-ABD (Bronkhorst, Kamen Germany) mass flow controllers for each gas source. The total flow rate of this diluted mixture of sample gas was 2 L min−1. The weight of the pipette in the permeation oven was taken every few days to determine the following gas-phase concentration inserted into the drift tube for each analyte: aniline at 1.26 μg s−1, 4-fluoroaniline at 0.734 μg s−1, 3-fluoroaniline at 0.276 μg s−1, and 2-fluoroaniline at 2.06 μg s−1. The water vapor pressure was monitored with an Easidew EA2 (Michell Instruments GmbH, Friedrichsdorf, Germany) immediately before insertion into the drift tube to determine the actual water concentration entering the HiKE-IMS. For these experiments, the dew point varied between −78 °C to −30 °C, corresponding to a water vapor concentration of 0.74 ppmv to 375 ppmv immediately before insertion into the reaction region.| Proton affinities (kJ mol−1) | Gas-phase basicity ΔG (kJ mol−1) | Ionization energy (eV) | |
|---|---|---|---|
| Aniline | 882.5 | 850.6 | 7.72; 8.05 |
| 4-Fluoroaniline | 871.5 | 839.7 | 8.18 |
| 3-Fluoroaniline | 867.3 | 835.5 | 8.33 |
| 2-Fluoroaniline | 8.18 |
Instrumentation
The HiKE-IMS with a tri-state ion shutter and the HiKE-IMS-MS used in these experiments have been described in detail previously.31,32,38 In this work, three experiments were performed with regards to varying the reduced electric field strength in the drift region (EDR/N) and varying the reduced electric field strength in the reaction region (ERR/N) as detailed in Table 2. The HiKE-IMS is operated at a reduced pressure with an MVP-040 membrane pump controlled by an EVN-116 valve and monitored with a CMR-362 capacitance manometer (all from Pfeiffer Vacuum, Asslar Germany). Ions were generated using a corona ion source with an applied voltage of 1500–1600 V depending on the analyte. Ions are detected with a Faraday plate and a commercial DLPCA-200 transimpedance amplifier (Femto Messtechnik GmbH, Berlin Germany) attached. The analog signal from the amplifier is converted into a digital signal through a Keysight U1082A digitizer (Keysight, Santa Rosa CA USA), where each individual spectrum was averaged 50 times. An additional 300–500 of these averaged ion mobility spectra were taken in triplicate depending on the ionization efficiency of the sample, for a total of three replicates of 15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 to 25000 spectra (where each mobility spectrum requires 1 ms to 24 ms to collect, 15 s to 3 min total for one replicate). The HiKE-IMS-MS also operates under reduced pressure with the same parts for pressure regulation on the HiKE-IMS listed above and the specific instrument parameters listed in Table 2. Additionally, the lab-built mass spectrometer consists of an Einzel lens followed by an orthogonal time-of-flight with a spectrum acquisition rate of 50 Hz.32,38
000 to 25000 spectra (where each mobility spectrum requires 1 ms to 24 ms to collect, 15 s to 3 min total for one replicate). The HiKE-IMS-MS also operates under reduced pressure with the same parts for pressure regulation on the HiKE-IMS listed above and the specific instrument parameters listed in Table 2. Additionally, the lab-built mass spectrometer consists of an Einzel lens followed by an orthogonal time-of-flight with a spectrum acquisition rate of 50 Hz.32,38
| Experimental variable | HiKE-IMS value | HiKE-IMS-MS value |
|---|---|---|
| Reaction region length | 7.7 cm | 10.5 cm |
| Drift region length | 30.65 cm | 15.7 cm |
| Pressure | 14–15 mbar | 14–15 mbar |
| Temperature | 43–45 °C | 25–30 °C |
| Corona needle voltage | 1500–1600 V | 1400 V |
| Gate pulse width | 1 μs | 3 μs |
| Reaction region scan parameters | ||
| Reaction region reduced electric field strength | 6–120 Td | 40–120 Td |
| Reduced electric field strength step | 2 Td | 20 Td |
| Drift region reduced electric field strength | 60 Td or 120 Td | 60 Td or 120 Td |
| Drift region scan parameters | ||
| Reaction region reduced electric field strength | 38 Td | |
| Drift region reduced electric field strength | 6–120 Td | |
| Reduced electric field strength step | 1 Td | |
Data acquisition on both instruments was performed using custom software developed using LabView 2018 version 18.0f2 (National Instruments Corp., Austin, TX USA). Matlab R2020a version 9.8.0.1323502 for win64 (Mathworks Inc., Natick, MA USA) was used for data analysis (Fig. 1).
 | ||
| Fig. 1 The HiKE-IMS has been described previously, but briefly, the HiKE-IMS is divided into two regions: the reaction region and the drift region.31 Both sections have an electric field that can be changed independently of each other which allows for examination of the speciation of each ion based on explicit operating parameters. | ||
Results
Mobility and separation of isomer peaks
The first point to address from the literature about the mobility of aniline isomers is whether the two observed peaks are in equilibrium in the drift tube or the instruments used in the literature are unable to resolve the stable peaks because their resolving power is too low. Using the model developed by Dodds et al.,39 a minimum resolving power of 38 would be needed to fully baseline-separate the two aniline isomers based on the literature K0 values for both peaks as reported from Karpas et al. (1.93 cm2 V−1 s−1 and 2.07 cm2 V−1 s−1 respectively, although these peaks are not fully baseline resolved in the spectra they show).21 This minimum resolving power can easily be obtained using HiKE-IMS. In Fig. 2, the example spectra for all aniline peaks are always baseline resolved even though the abundances of the peaks change as a function of E/N in the reaction region (ERR/N). The mass spectra affiliated for each peak can be used to aid in structure identification later. Unsurprisingly, the most abundant species of Peak 1 is the molecular ion peak (m/z 93 Da for aniline or 111 Da for fluoroanilines), and Peak 2 is the protonated peak (m/z 94 Da or 112 Da). Although the signal-to-noise ratio of the mass spectra is low due to a duty-cycle mismatch between the home-built ToF-MS and the IMS, these results agree with results prolifically covered in the literature.22,23,28 To be abundantly clear for the rest of this work until structures are associated with each peak, Peak 1 will refer to the molecular ion peak (m/z 93 or 111) and Peak 2 will refer to the protonated peak (m/z 94 or 112). | ||
| Fig. 2 Example IMS spectra of aniline in air at the same drift region E/N (EDR/N), but at low (orange) and higher (blue dotted) E/N in the reaction region (ERR/N). The reactant ions are present in expected quantities based on previous HiKE-IMS experiments.32,40 The intensities of the aniline peaks change correlating to the presence of the reactant ions, where Peak 1 increases in abundance with increased amounts of NO+ and O2+ radical reactant ions. The peaks are also baseline separated with a chromatographic resolution above 1.5 (Fig. 3). All pertinent experimental details are given in Table 2. | ||
While Fig. 2 shows that the resolution of the mobility peaks does not change as a function of the reduced reaction region field strength ERR/N, each peak changes mobilities as a function of the reduced drift region field strength EDR/N at different rates that may potentially cause overlap between the two peaks.31 Using the two aniline isomer peaks, the chromatographic resolution between these peaks can be measured as the electric field in the drift region is changed (EDR/N) (Fig. 3). Although the 2-fluoroaniline and the 3-fluoroaniline pairs have higher standard deviations due to a lower signal-to-noise ratio, they still show, on average, significantly higher resolution than is required for baseline separation of the peaks (R > 1.5). Interestingly, the resolution is not static between the peaks as the electric field changes in the drift region. Instead, there is a maximum resolution between the peaks between 60–70 Td depending on ion identity. One explanation for this effect is the voltage that gives the maximum resolving power is different for each peak (Fig. S1, ESI†) and has been previously discussed in the literature.31 Additionally, the resolving powers decrease at different rates as functions of EDR/N.
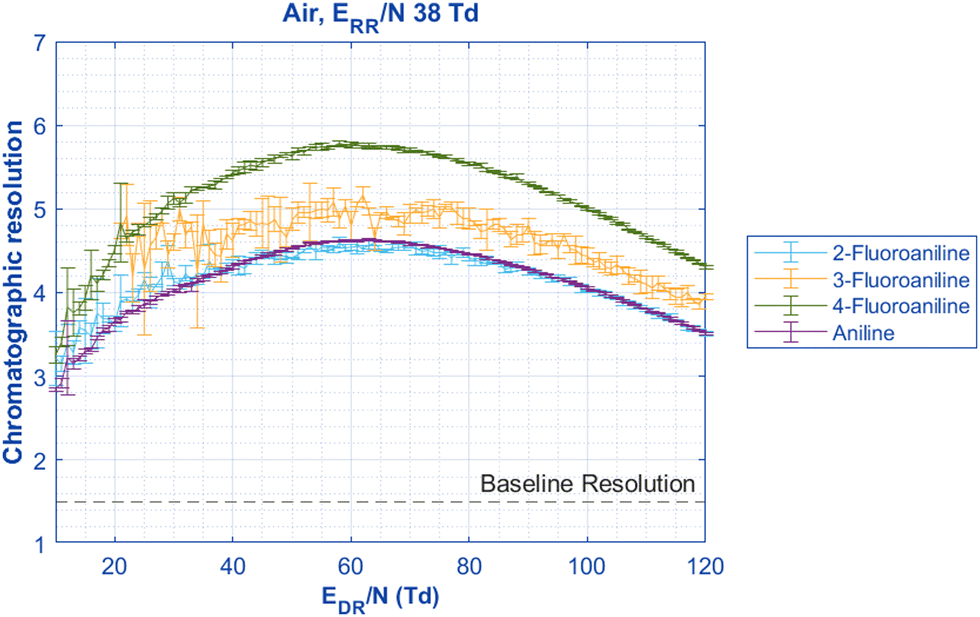 | ||
| Fig. 3 When the reduced drift region field strength (EDR/N) is changed to observe changes in mobility, the chromatographic resolution of the Peaks 1 and 2 can be compared. All pairs are significantly above the definition of baseline resolution of 1.5 between two peaks. Interestingly, the resolution is not static for all pairs of aniline isomers; instead, a maximum separation is reached between 60–70 Td for all species. This observed maximum resolution is at the mean EDR/N of the maximum resolving power occurring at different EDR/N for the two peaks (Fig. S1, ESI†). All pertinent experimental details are given in Table 2. | ||
Additional interesting observations about reduced mobilities as a function of EDR/N can be made from Fig. 4. Unsurprisingly, the mobilities of both aniline peaks are higher than those of the fluoroaniline isomers, and the mobilities of 2-fluoroaniline are higher than those of the other isomers suggesting a more compact molecular structure. Interestingly, Peak 2 of both 3-fluoroaniline and 4-fluoroaniline have the same mobility at most EDR/N values and Peak 1 for 3-fluoroaniline and 4-fluoroaniline have nearly the same mobilities at only very low EDR/N. At low EDR/N, the mobility values of aniline differ from the reported mobilities and abundance ratio from Karpas et al. One possible cause for the discrepancy between the mobilities here and those reported by Karpas et al.21 is from temperature and field heating effects. The system from Karpas et al. was operated at 250 °C and ours is between 43–45 °C, however, when the reduced electric field is increased in the HiKE-IMS, the mobilities for both peaks in air are within error of the values from Karpas et al. (Table 3).21 The resulting field heating effect on the mobilities measured in the HiKE-IMS agreeing with the mobilities from a heated DTIMS system has some interesting implications for correlating field heating with traditional heated systems, however, that discussion lies outside the scope of this work.41 Additionally, Borsdorf et al. report the mobilities of n-fluoroaniline in purified air under similar temperatures, and ionization source (50 °C temperature, corona discharge ionization source) as the HiKE-IMS used here and agree with our results at low E/N within less than 1% difference for all peaks.42 Using two-temperature theory and corrections to the Mason-Schamp equation as outlined by Siems et al., ion-neutral collision cross-section values can be calculated for aniline to compare to the last literature values.41,43–45 At relatively low EDR/N (6–15 Td), the CCS values of each aniline peak at low EDR/N in nitrogen agree within the 95% confidence interval of previously reported values from Kune et al. of each peak respectively.28 It should be stated that the corresponding reduced mobilities from Kune et al. are not given, and the cross-sections were obtained from a calibrated trapped ion mobility spectrometer using reference mobilities taken at room temperature, which may be one possible source of discrepancy.28,46
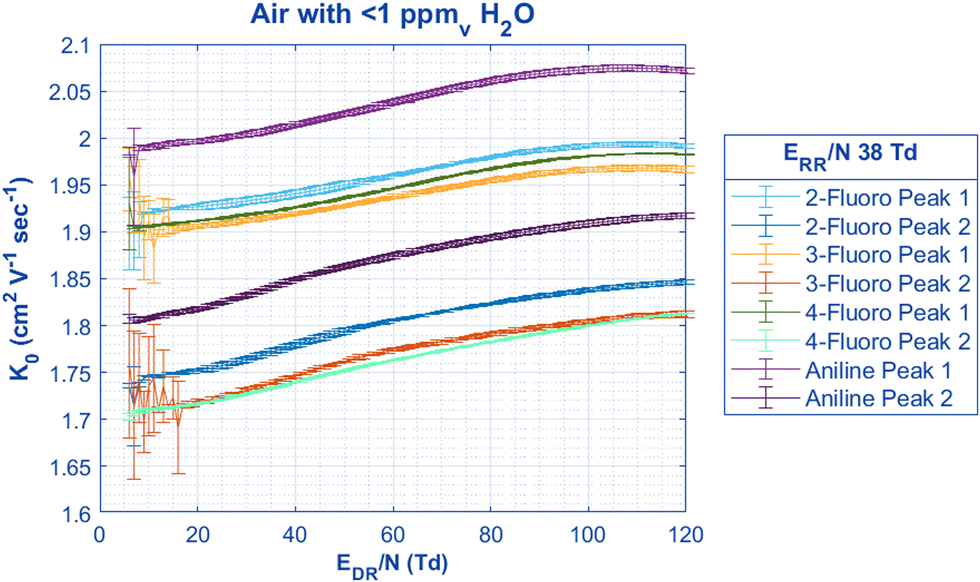 | ||
| Fig. 4 By keeping the ERR/N constant at a value where both aniline peaks are present in nearly equal quantity, the change in mobility in air can be found for both peaks when the EDR/N is scanned. Unsurprisingly, the mobility of both aniline peaks is significantly higher than the respective charge-located peaks for each of the n-fluoroaniline isomers. Interestingly, the mobilities for the Peak 2 of both 4-fluoroaniline and 3-fluoroaniline are the same except for the EDR/N region between 40 to 90 Td where 3-fluoroaniline has a greater increase with mobility than 4-fluoroaniline. All pertinent experimental details are given in Table 2. | ||
| Peak 1 | Peak 2 | Gas temperature | Drift gas | |
|---|---|---|---|---|
| HiKE-IMS (low field <15 Td) | 1.99 ± 0.02 cm2 V−1 s−1 | 1.801 ± 0.003 cm2 V−1 s−1 | 43.75 °C | Air |
| HiKE-IMS (high field >110 Td) | 2.073 ± 0.002 cm2 V−1 s−1 | 1.916 ± 0.002 cm2 V−1 s−1 | 43.69 °C | Air |
| Karpas et al.21 | 2.07 cm2 V−1 s−1 | 1.93 cm2 V−1 s−1 | 250 °C | Air |
| HiKE-IMS (low field) | 112.7 ± 4 Å2 | 124.8 ± 4 Å2 | 44.56 °C | N2 |
| Kune et al.28 | 111 ± 2 Å2 | 120 ± 2 Å2 | Not given | N2 |
With distinct mobilities that match literature values in their respective drift gases and temperatures and the chromatographic resolution presented in Fig. 2 and 3, it is safe to assume that both aniline species are the same as those observed in previous literature. Additionally, these two peaks are stable within the mobility experiment at all EDR/N, and there is not an equilibrium of the two species interconverting within the drift tube, especially since there is not a raised baseline between the peaks as has been observed in previous literature.21,23 Since the relative abundance of each aniline peak does not change with the drift region conditions, this means that in the HiKE-IMS the conditions in the reaction region and ionization parameters alone must determine aniline speciation. Lastly, it should be noted that the mobilities of all the peaks are significantly different from each other within error (one standard deviation of 3 replicates of 400 averaged spectra) above EDR/N = 20 Td, which demonstrates the HiKE-IMS's ability to not only separate the distinct aniline peaks but also to distinguish between the n-fluoroaniline isomers based on mobility. Since first-principles ion mobility measurements are possible with the HiKE-IMS (i.e., no calibration is necessary to obtain K0 values), these mobilities are accurate enough to be used to identify the structure of the ions for each peak.
Influence of reactant gas on speciation of isomer peaks
In order to discuss the effects of changing the gas and adding moisture on aniline speciation, HiKE-IMS ionization chemistry should be discussed first. Briefly, in HiKE-IMS the abundances of the three most common reactant ions (NO+, O2+, and H3O+) are directly determined by the electric field in the reaction region (Fig. 5). Each reactant ion can interact differently with the analyte where O2+ and NO+ primarily cause charge transfer, and H3O+ causes proton transfer. At low ERR/N, these three reactant ions are able to cluster with neutral water in the reaction region via a combination of low kinetic energy and longer reaction times. These clusters often contain more than one neutral water molecules at low ERR/N and begin to dissociate from increased kinetic energy as the ERR/N increases. Additionally at higher ERR/N, the conversion of O2+ and NO+ into hydronium via a chain reaction is inhibited by shorter reaction times and cluster dissociation, resulting in more bare O2+ and NO+ ions present as the reactant ions. Adding water vapor into the analyte gas can also directly influence the speciation of the reactant ions by increasing the abundance of hydronium ions and the size of hydronium-water clusters.32,47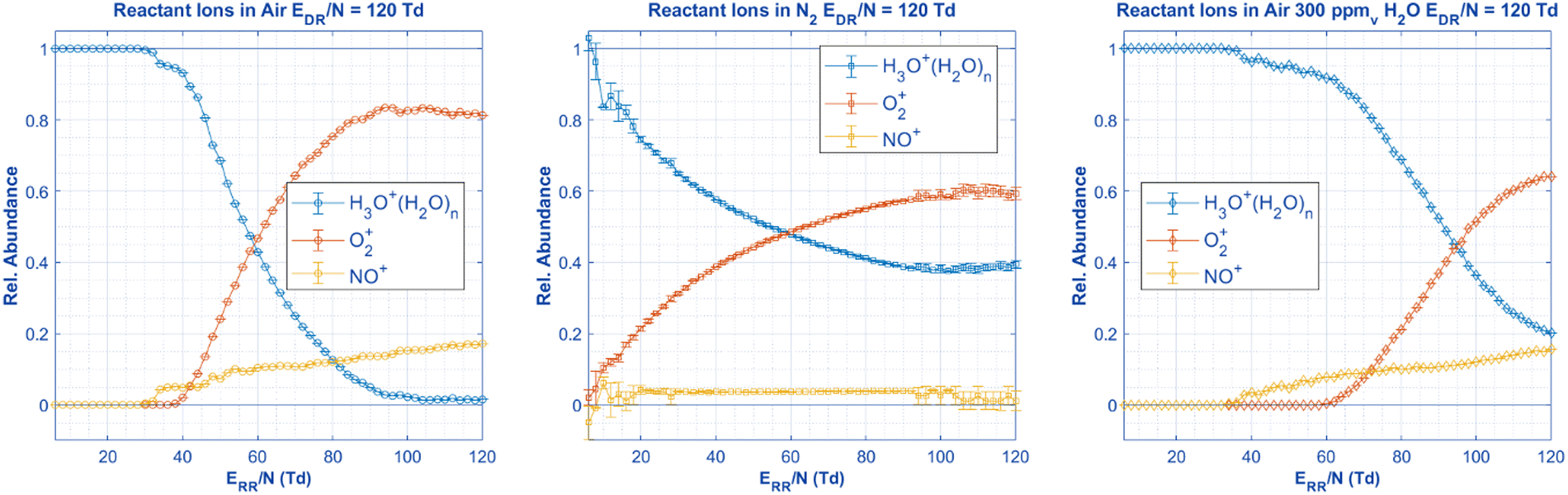 | ||
| Fig. 5 With no aniline present in the ionization region, the following reactant ions are measured in the HiKE-IMS under the same experimental settings as Fig. 6 and 7. Since HiKE-IMS has a corona source, these reactant ions are responsible for ionizing the analyte, and therefore, the speciation and identity of the three aniline peaks must originate from these reactant ion species alone. All relevant details are in Table 2. | ||
With the chemistry of HiKE-IMS reactant ions in mind and the two aniline isomers established as stable within the time of the IMS experiment (i.e., abundances do not change within the drift region), we can discuss the identity and generation of each isomer and the parameters that affect the relative abundances. Previous literature focuses on changing cone voltage settings and implementing CID to change aniline speciation,23,24 however, with the complicated reactant ion chemistry in the HiKE-IMS, parameters of the bath gas need to be addressed before examining the sole influence of ERR/N. The first experiment to influence possible ionization mechanisms is to change the water concentration in the drift tube. Increasing the water vapor concentration in the reaction region should increase the amount of hydronium and water clusters within the drift tube, and therefore increase the abundance of Peak 2, the structure assumed to be protonated at the amine group. Additionally, by assuming that the drift conditions in the drift region are the same as the ones in the reaction region (i.e., ERR/N = EDR/N), the mobilities measured by changing the EDR/N can be used to obtain the effective temperature of each ion at each reaction region reduced electric field strength (Fig. 6). For example, at 70 Td, aniline has a reduced mobility value of 2.0502 cm2 V−1 s−1 and 1.8846 cm2 V−1 s−1 for Peaks 1 and 2, respectively. Using the modified Wannier equation from Siems et al.,41,45 the mobility of each ion species can be converted into an effective temperature of 462 K and 489 K, respectively. For clarity, since the Wannier equation requires the m/z of the ion for calculating effective temperature, the observed masses in the mass spectrum (protonated for Peak 2 and molecular ion for Peak 1) were used for this calculation. It is crucial to keep in mind effective temperature is ion-specific,43 and in this case, even though there is only one analyte in the permeation oven at a time, there are two peaks with two distinct mobilities, two distinct mass to charge ratios, and two different structures. Therefore, the mobilities must be measured over the full range of electric fields in the drift region for each ion under the same conditions (i.e., water concentration) when performing the separate experiment of scanning the electric field in the reaction region. This is the only way to obtain a comparable effective temperature for the ions to correlate with reduced electric field strength.
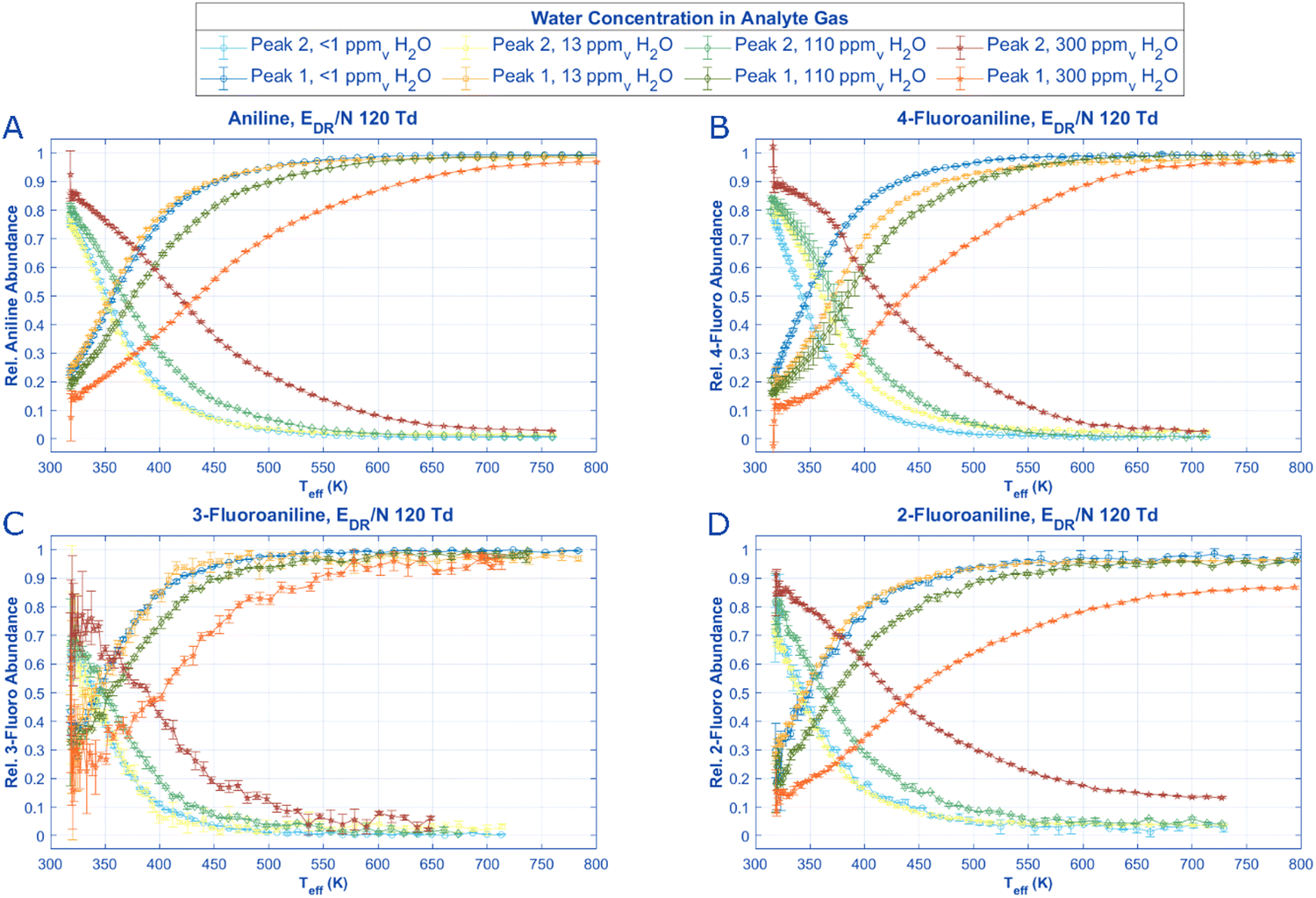 | ||
| Fig. 6 By scanning the reduced field strength in the drift region and establishing mobility data for each aniline and derivative isomer peak (Fig. 4), an effective temperature for each peak can be found for each ERR/N value. Assuming the conditions in the drift region from the previous experiments are the same as those in the reaction region at the same E/N settings, the relative peak intensities of each aniline isomer can be graphed against the Teff for each ion. Interestingly when the moisture content in the reaction region is high, 2-fluoroaniline never reaches a state at high temperature (Teff >500 K) or E/N where Peak 1 reaches an abundance of above 90%. All pertinent experimental details are given in Table 2. | ||
Once the effective temperatures for each ion at each concentration of water vapor in the drift tube are obtained, speciation diagrams can be constructed for the aniline protonated and molecular ion peaks as a function of effective temperature for each ion at the specific ERR/N in the reaction region (Fig. 6). For all species, Peak 2 has a slight increase in initial abundance (i.e. Teff <350 K) as water concentration in the reaction region increases, and a higher reduced electric field strength (i.e., a higher effective temperature) is required for Peak 1 to become the most abundant species. These change in abundances as water concentration increases are unsurprising if Peak 2 is formed via proton transfer or clustering reactions. Additionally, the change in inflection points between aniline and the n-fluoroanilines may offer insight into steric modifications and the role of water as a reactant ion. For example, 3-fluoroaniline has the lowest inflection points, requiring a maximum of 400 K at the highest water concentration tested. Compared with the other analytes, 3-fluoroaniline has a marginally lower proton affinity (Table 1), which may explain the less effective hydronium bonding.
The last experiment that can be performed to explore the role of reactant gas in ionization of each aniline species is changing the drift gas from air to N2. For both aniline and 3-fluoroaniline, changing the background gas has no effect on the speciation of both aniline peaks at ERR/N below 30 Td. Only at higher ERR/N is there a major difference in speciation of aniline and 3-fluoroaniline between air and N2; both of these analytes have nearly 100% abundance of Peak 1 when air is the background gas at higher E/N whereas <10% of Peak 2 remains at high E/N with N2 as the background gas. Similar observations can be made about 2-fluoroaniline and 4-fluoroaniline in the sense that initial abundances of each peak are similar between gases at very low E/N and trend to 100% abundance of Peak 1 in air at high E/N. However, for these two analytes when N2 is used as the bath gas, a third distinct peak (Peak 1.1) appears with a drift time between Peak 1 and 2 that is not present for any of the other analytes in either gas. The third peak increases in abundance as E/N increases, similar to the behavior of Peak 1 suggesting it may also originate from the specific reactant ion populations at higher E/N or result from the higher E/N itself. The appearance of this third peak, and the affiliated mass spectra, is the key to assigning the molecular structures for aniline and its isomers.
By measuring 4-fluoroaniline in nitrogen on the HiKE-IMS-MS, the mass to charge ratio of the ions under each of the 3 peaks can be obtained. Unsurprisingly, Peaks 1 is the molecular ion peak and Peak 2 is a protonated peak for 4-fluoroaniline, the same results as in Fig. 2 for aniline. Peak 1.1, however, is also protonated. In the mobility spectra for Fig. 8 and in Fig. 7B and D, at high E/N Peaks 1 and 1.1 have nearly the same abundance, however, at lower ERR/N in Fig. 7, Peak 1 has a much higher abundance than Peak 1.1. With two protonated peaks and a molecular ion peak clearly visible for 2-fluoroaniline and 4-fluoroaniline, how are these peaks formed and why are there only 2 peaks for aniline and 3-fluoroaniline?
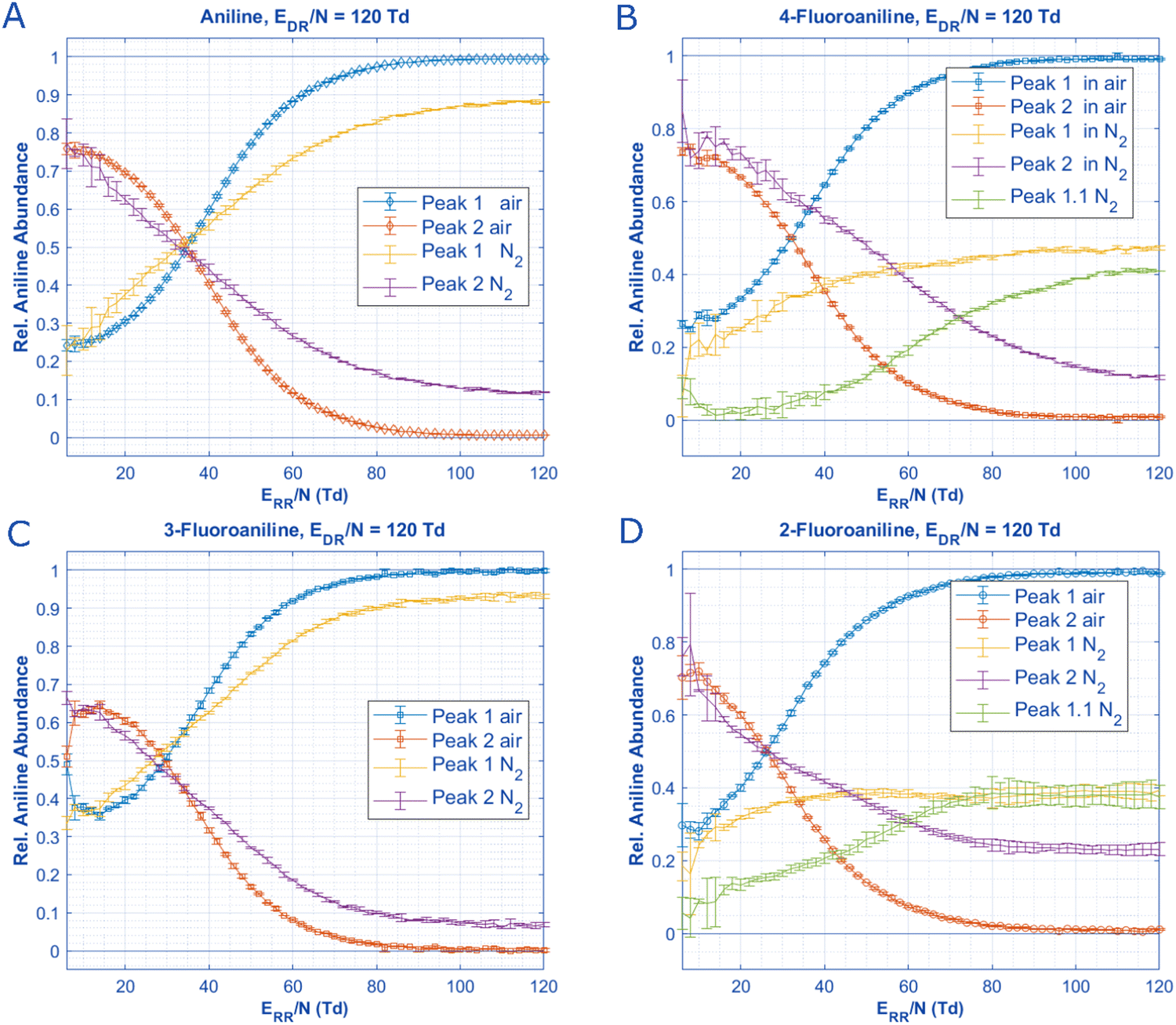 | ||
| Fig. 7 When no water is introduced into the reaction region and if the drift gas is changed from air to nitrogen, the speciation of aniline and n-fluoroaniline can be plotted as a function of ERR/N in both drift gases. For all analytes, the initial species present at low ERR/N are constant in concentration. Notably, 2-fluoroaniline and 4-fluoroaniline have an additional peak that is slightly slower than the molecular ion peak when N2 is used as the drift gas (Fig. S3 and S4, ESI†). This third peak (Peak 1.1) appears as a shoulder for 2-fluoroaniline and is baseline separated for 4-fluoroaniline, but this additional peak only reaches significant abundance at ERR/N above 60 Td. Additionally, the place where each peak is equal in abundance does not change as a function of the drift gas for aniline and 3-fluoroaniline. Arguably, this could be said for 2-fluoroaniline as well if the sum of Peak 1 and Peak 1.1 are treated as the same species. All pertinent experimental details are given in Table 2. | ||
Ionization and identity of three isomer peaks
With the presence of the three reactant ions in mind at different reaction region conditions from Fig. 5, Scheme 1 illustrates 4 possible reaction pathways in HiKE-IMS for the ionization, clustering, and fragmentation of aniline. The origin of Peak 2 is relatively straightforward to explain as this species is protonated and the abundance of this peak is most influenced by increased water vapor in the reaction region (Fig. 7). Therefore, Peak 2 most likely forms from proton transfer from hydrogen bonding between the hydronium to the amino group and may undergo clustering with water present in the IMS. These results for Peak 2, unsurprisingly, agree with previous literature including the original hypothesis from Karpas et al.21,23,28,48 and shortly afterwards by Smith et al. who confirm the amine protonation site with an HDX study.48 This proton-transfer mechanism can occur in the liquid or gas phase, which explains the presence of this peak with both ESI and APCI as ion sources.23,28,42 | ||
| Scheme 1 The two (or three for 4-fluoro and 2-fluoro) aniline peaks can originate from four possible gas-phase reaction pathways as described by the literature. The proton transfer and subsequent clustering pathway is the most likely origin of Peak 2. The fragmentation pathway describes the ionization of para-H aniline and subsequent fragmentation into molecular ion (MI) aniline. Lastly is the charge transfer/rearrangement pathway where the molecular ion aniline is directly formed from collisions with O2+ or NO+ radicals in the reaction region and subsequent rearrangement. Additionally, all named compounds in dotted boxes were modeled in IMoS (Table 4) to obtain reduced mobilities to later identify Peaks 1, 2, and 1.1 from Fig. 2 and 8. | ||
Peaks 1 and 1.1, however, are more interesting for discussion. Historically, Peak 1 has been referred to as the C-protonated, or para-protonated, isomer starting with Karpas et al., and continuing through recent literature.21,23,24,28,42,48 However, Peak 1 and 2 have never been fully baseline separated in the mobility domain, and the mass spectra affiliated with Peak 1 has always shown the molecular ion as the dominant species.21–23 Because Peaks 1 and 2 have never been fully separated, there is no chance Peak 1.1 would have appeared in previous literature. Previous literature hypothesized that para-substituted protonated aniline fragments into the molecular ion species of aniline, however, since the mobility peaks were not fully separated or in sufficient abundance, it was impossible to tell if the protonated m/z species underneath Peak 1 was truly from Peak 1 or if it was from the tail of Peak 2. Where the fragmentation occurs relative to the mobility separation also varies in the literature since Attygalle et al. fragment the aniline prior to the mobility separation, yet Walker et al. use CID after the mobility separation yet the same Peaks 1 and 2 are observed.23,24
Two possible origins for Peak 1 and Peak 1.1 in the literature rely on the presence of water vapor either in the IMS cell itself or in the ionization source. The first possible mechanism is suggested by Kune et al. where a high concentration of water vapor is present with aniline ionized via ESI, aniline may undergo a substituted nucleophilic reaction to form phenol via the para-substituted protonated form.28 However, this mechanism relies on fragmentation into a phenyl cation or a transition state with both an amine and hydroxyl group attached to the phenyl cation.28 Their mechanism is supported by the C-protonated isomer appearing with an affiliated mass spectrum peak of 95 Da, and the N-protonated isomer has an affiliated mass spectrum of 94 Da; neither of these peaks are baseline separated in only the mobility domain. Interestingly, even when we add high water concentration to our analyte gas used in APCI, we do not see a mobility shift in either aniline peak (Fig. S7, ESI†) compared to no added water vapor in Fig. 4 nor do we observe a shoulder on the aniline Peak 1 that corresponds to phenol's Peak 1 (Fig. S8 and S9, ESI†). The largest piece of evidence is we do not see a peak at 95 Da in the mass spectrum for aniline (Fig. 2) nor 113 Da for any of the n-fluoroanilines (Fig. 8), whereas the mass spectra in Kune et al. clearly show the 95 Da species.28 The lack of mobility shift and m + 2 Da peak in the mass spectra suggest that the substituted nucleophilic reaction to phenol does not happen under the APCI conditions in our HiKE-IMS.
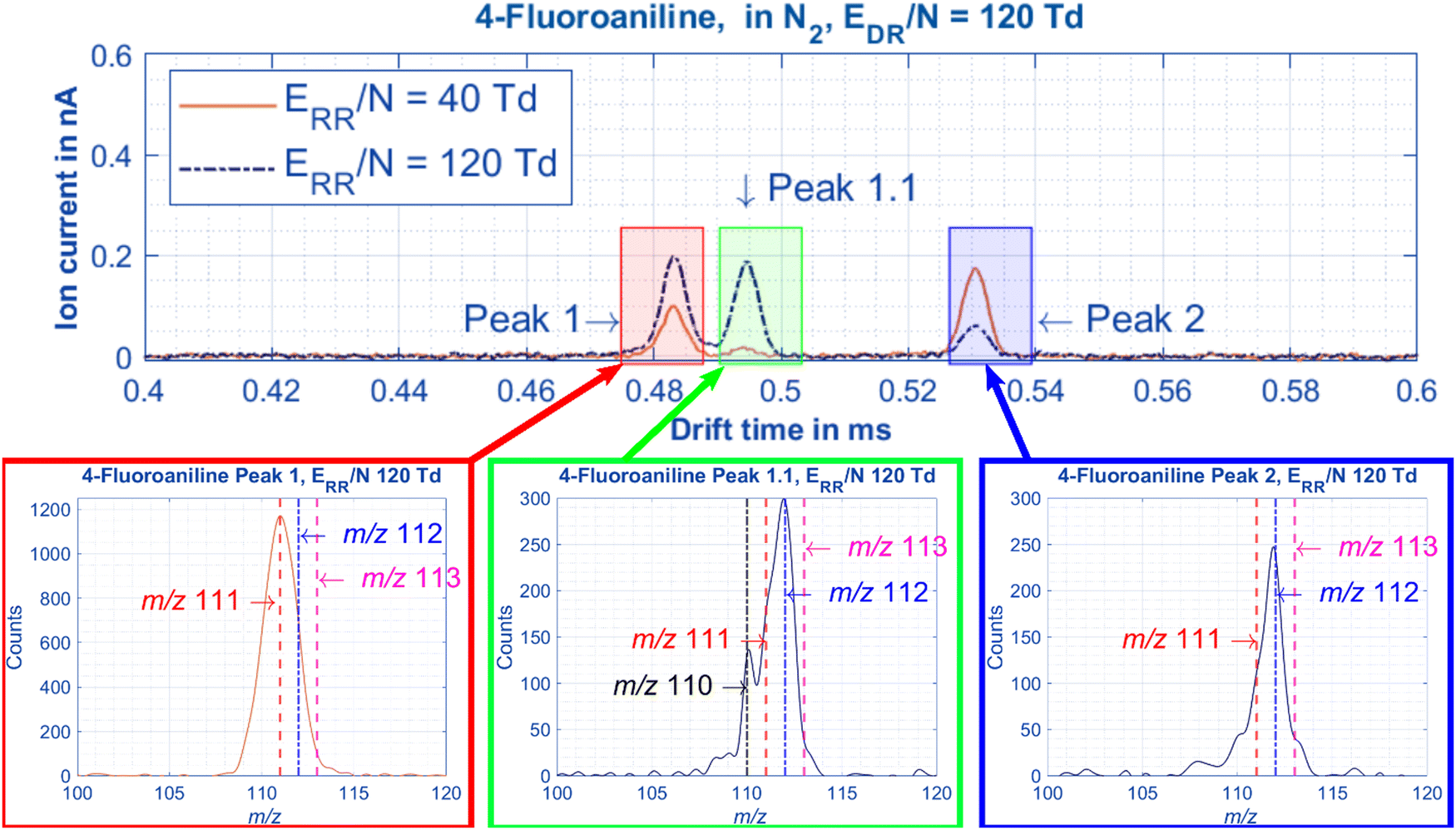 | ||
| Fig. 8 When the HiKE-IMS-MS is used to measure 4-fluoroaniline in nitrogen the following mobilities and mass spectra are recorded. The chromatographic resolution between Peak 1 and Peak 1.1 is 1.2 and the reduced mobilities of each peak are: Peak 1 = 1.971 cm2 V−1 s−1, Peaks 1.1 = 1.927 cm2 V−1 s−1, and Peak 2 = 1.794 cm2 V−1 s−1. The center of the Gaussian mass spectrum underneath each peak is as follows: Peak 1 = 111 Da (molecular ion peak), Peak 1.1 = 112 Da (para-protonated peak), and Peak 2 = 112 Da (amine-protonated peak). All pertinent experimental details are given in Table 2. | ||
The second mechanism for the appearance of Peak 1.1 is the label proton theory, where one proton systemically moves from the para-protonated position to the amine protonated position or vice versa. The original version of this idea comes from Karpas et al.21 where the hydrogen ‘walks’ around the benzene-ring to the amine-nitrogen before fragmenting with one of the amine's electrons to give the molecular-ion peak.23,49 An updated version of the mobile proton was first presented by Campbell et al. where they examined 4-aminobenzoic acid in the presence of water and methanol to examine the abundances of species with different protonation sites (at the amine or the carbonyl group) with DMS.50 They showed with HDX that when clusters are present, protons from the amine group can rapidly transfer to the carboxyl group via a solvent bridge across the molecule in the form of a water or methanol cluster in a Grotthuss-like mechanism. Xia and Attygalle perform a similar study a year later using benzocaine with a traveling wave IMS. They added water to their ion source to drive the speciation of the N-protomer and O-protomer, again suggesting speciation of the O-protomer arises from a solvent bridge.51
While this mechanism might be possible, the solvent mediated proton transfer likely is not happening in this work for two reasons. First, the studies by Campbell et al. and Xia and Attygalle examined molecules similar to aniline, but those molecules had 2 functional groups capable of hydrogen bonding to the clusters;50,51 aniline only has the amine group and hydrogen bonding to the benzene ring seems unlikely. Second, this mechanism relies on sizable water clusters capable of forming a bridge over the analyte (n > 5), and the corona ionization in our HiKE-IMS is operated at reduced pressure under the presence of strong electric fields, which discourages large cluster formation. If the solvent bridge was causing the appearance of the para-protonated aniline (Peak 1.1), we would see this peak appear in the presence of high water concentrations at low ERR/N (i.e. appear in Fig. 6). Instead, Peak 1.1 is only visible present at high ERR/N in pure N2 (Fig. 7 and 8). The appearance of Peak 1.1 under these conditions and these conditions only indicate that desolvated hydronium ions can protonate aniline at the para-position.
The last possible mechanism for Peak 1 to discuss is charge transfer from the O2+ and NO+ radicals in the reaction region of the HiKE-IMS to molecular ion aniline. With this mechanism, aniline loses an electron to produce neutral O2 or NO and produces an aniline radical explaining the molecular ion peak in the mass spectrometer. With aniline now a radical, it is possible rearrangement and possible subsequent fragmentation can occur (Scheme 1) as first suggested by Lalli et al.22 More details about the exact mechanism of the rearrangement are in the electron impact mass spectrometry literature, where the aniline radical can form into a more compact ring shape via azacaradiene then azepine.25,52,53 Since the ionization energy of aniline is lower than that of the reactant ions (8.05 eV) and activation energy for rearrangement is relatively low (ΔH azacaradiene = 27.7 kJ mol−1 or ΔH azepine = 88.8 kJ mol−1), the rearrangement pathway in the HiKE-IMS, especially at high reduced electric field strengths in the reaction region cannot be ruled out as a possiblility.22,25,37 Whether or not a rearrangement occurs, since the abundance of O2+ and NO+ are so high in the HiKE-IMS at high E/N, Peak 1 is almost certainly an aniline radical originated from these reactant ions.
Finally, the para-protonated species in Fig. 7 and 8 may be explained with observations from Langejuergen et al. where benzene was measured in the HiKE-IMS under similar conditions to the ones used in this work, and they observed protonated benzene that appears only at high ERR/N.34 Schaefer et al. also observed a protonated species of toluene, chlorobenzene, and fluorobenzene but the protonated species appears at a different E/N for each functional group.47 Langejuergen et al. explain the appearance of this protonated benzene is due to the decreased size in water clusters allowing for a greater chance of proton transfer from hydronium to benzene rather than hydronium to another larger water cluster.34 However, another thing that happens at high E/N is the extremely short drift times of the ion in HiKE-IMS. It is possible, both for benzene and the anilines used in this work, that the drift times of the ions are short enough that the protonated species do not have time to dissociate into the molecular ion via hydrogen atom abstraction.
With possible origins of these three peaks identified, it is worth emphasizing these aniline species are ionized via APCI. In the literature, similar abundances of the two aniline peaks were observed using instruments with ESI and helium plasma ionization (HePI).21–23,28
While HePI may operate similar to APCI and generate similar ion abundances, ESI is a softer ionization source that ionizes analytes with a very different mechanism. However, for many of the studies using ESI, source conditions were changed such as the cone inlet voltage immediately following the ESI source before transfer into the low pressure stage of the instrument. For instance, Attygalle et al. report at a sample cone voltage of 70 V, the aniline peaks are in equal abundance,23 which happens at an effective temperature of 350 K (or ERR/N = 38 Td with no water added) in our HiKE-IMS data in Fig. 6. By changing the cone voltage, fragmentation occurs before the mobility separation, which means in the measurements where ESI is the ionization source that the molecular ion radical has the same mobility as the para-protonated aniline. If these mobilities are the same, yet, we see three peaks for 2-fluoraniline and 4-fluoroaniline, do the instruments used in literature lack the resolving power to separate these species, or does Peak 1 actually undergo a rearrangement into azepine in the HiKE-IMS?
With accurate mobility spectra obtained over the entire reduced electric field strengths in the HiKE-IMS in both nitrogen and air that agree with literature mobility values, computationally modeled mobilities may aid in assigning structure to the two (or three) experimentally observed peaks. When comparing average experimental reduced mobilities of each peak to those modeled with IMoS (Table 4), helpful information can be obtained about the potential identity of each peak. Six possible prerequisite structures for each analyte (Boxed analytes in Scheme 1) were generated in Avogadro v1.2.0 before undergoing energy optimization using the B3LYP density functional theory with the 611(d) basis set with Gaussian 16.54,55 Those optimized structures were inserted into the ion mobility modeling software, IMoS v1.10 (https://www.imospedia.com/imos/) and CCS values and mobilities were calculated using the trajectory method accounting for the quadrupole moment in nitrogen with Lennard-Jones parameters.56,57 As a suitability test, methyl phosphonic acid and dimethyl methyl phosphonate were modeled using the workflow mentioned above, and the generated mobilities and cross-sections were the same as those reported by Kwantwi-Barrima et al. within 1% error of their theoretical CCS values using their experimental parameters to generate mobility models.9 It should also be noted that although the structures from Gaussian are optimized, the found minimum energy may not necessarily be the global minimum energy. Additionally, it should be pointed out that in the HiKE-IMS, the global energetic minimum structure may not be the most abundant species observed, especially at higher reduced electric field strengths.30,34,47
| Prerequisite Structure Modelled K0 (cm2 V−1 s−1) in N2 | Experimental Low EDR/N K0 (cm2 V−1 s−1) in N2 | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Azepine | MI Aniline | Azacaradiene | Para-H+ | NH3+ | Aniline-H3O+ | Aniline(H3O+), H2O | Aniline, (H3O+) H2O | Peak 1 | Peak 1.1 | Peak 2 | |
| Ion type | M+ | M + | M + | M + H | M + H | M + H3O | M + H2OH3O | M + H2OH3O | |||
| (m/z aniline) | 93 | 93 | 93 | 94 | 94 | 112 | 130 | 130 | |||
| (m/z n-fluoro) | 111 | 111 | 111 | 112 | 112 | 130 | 148 | 148 | |||
| Aniline | 2.0750 | 2.0772 | 2.0573 | 2.0792 | 1.9920 | 1.8335 | 1.7280 | 1.7506 | 1.956 ± 0.03 | 1.795 ± 0.03 | |
| 2-Fluoroaniline | 1.9638 | 1.9660 | 1.9283 | 1.9541 | 1.9120 | 1.7892 | 1.6927 | 1.6958 | 1.921 ± 0.02 | 1.749 ± 0.02 | |
| 3-Fluoroaniline | 1.9535 | 1.9230 | 1.9468 | 1.9212 | 1.8447 | 1.7140 | 1.6267 | 1.6450 | 1.898 ± 0.03 | 1.700 ± 0.03 | |
| 4-Fluoroaniline | 1.9297 | 1.9315 | 1.9277 | 1.9063 | 1.8377 | 1.7003 | 1.6193 | 1.6378 | 1.915 ± 0.03 | 1.848 ± 0.02 | 1.713 ± 0.03 |
Regardless, both the low-field experimental mobilities and modeled reduced mobilities from these calculations are listed in Table 4. Peak 1.1 for 2-fluoroaniline is not listed because it is not separated from Peak 1 at this reduced electric field strength. For Peak 2 in nitrogen, the closest modeled mobilities are that for the N-protonated isomer (aniline NH3+) and the aniline clustered with one hydronium ion (Aniline-H3O+). At low E/N it is possible that Peak 2 is a cluster in equilibrium between these two structures and the additional water molecule dissociates upon transfer to the mass spectrometer. Peak 1.1 is unmistakably the para-protonated structure, although it should be noted that this peak has the same mobility as Peak 1 at low E/N for both 2-fluoroaniline and 4-fluoroaniline. The experimental mobilities for both Peak 1 and Peak 1.1 are systematically lower than the modeled mobilities for all species, so direct comparison is impossible without adding an offset value to the modeled mobilities. Instead, since Peak 1 and Peak 1.1 are the same at low mobility for all species except 4-fluoroaniline, we can compare which modeled molecular ion structure is closest in mobility to the para-protonated structure. For all 4-fluoroaniline, the para-protonated structure has a mobility of 1.9063 cm2 V−1 s−1 and the molecular ion structure with the closest mobility is azacaradiene at 1.9277 cm2 V−1 s−1, but the mobilities of all molecular ion species differ by <0.005 cm2 V−1 s−1 which is too small of a difference accounting for experimental error to accurately assign structure. The other aniline and n-fluoroaniline isomers, however, the molecular ion structure has the closest mobilities to the para-protonated structure. For all species, even though there is an offset, the mobility for the para-protonated species and the molecular ion species in nitrogen is nearly the same for 3-fluoroaniline and aniline, and different enough for 2-fluoroaniline and 4-fluoroaniline that these two species can be separated in the HiKE-IMS. It is also possible that the mobility of these species are too similar in air to be resolved. Therefore, the appearance of the 3 peaks depends on the ability to gain a resolving power high enough to separate the molecular ion peak from the para-protonated peak.
Conclusions
HiKE-IMS offers unique opportunities to examine ion chemistry through the independent, variable electric fields in the reaction and drift regions. Here, changing the E/N separately in those regions allows for a closer examination of the ionization and separation of aniline as independent processes. Previous literature in different IMS platforms including traveling wave, TIMS, and drift tube IMS have never been able to fully baseline resolve the aniline peaks in the mobility domain alone, i.e. the tails of both peaks always overlapped to some degree.21–24,28 When observed in HiKE-IMS and HiKE-IMS-MS, both peaks were always baseline separated, reaching a maximum resolving power of 118, and each peak possessed the same m/z as reported in previous literature. Additionally, changing the drift region reduced electric field strength had no change on the speciation of each peak whereas increasing the reduced electric field strength in the reaction region changed the speciation from the protonated peak as the most abundant to the molecular ion peak as the most abundant. Greater electric field strengths were required to change the speciation when water was added as a reactant gas or when the buffer gas was changed from air to nitrogen. These observations when changing the ionization conditions and the parameters of the drift region separately determine that aniline speciation is dependent on ionization conditions only in this study.An additional finding when the buffer gas was changed to nitrogen was the appearance of a third peak for both 2-fluoroaniline and 4-fluoroaniline, where the third peak for 4-fluoroaniline was baseline-resolved from the other two peaks for the first time in the literature. This third peak was a protonated ion in the mass spectra suggesting showing for the first time that the C-protonated isomer can be separated in the mobility domain, without requiring a mass spectrometer, from the molecular ion aniline and also illustrates for the first time changing the drift gas from air to nitrogen can separate structural isomers. Additionally, the separation of para-protonated and molecular ion peaks may not be exclusive to aniline compounds and should be explored using other benzene derivatives such as toluene or phenol, or by changing the substituents at the para and ortho positions. As more ion mobility spectrometry instruments develop with higher resolving powers, more complex ionization mechanisms, thermodynamics, and the separation of distinctly different ion structures can be accomplished.
Author contributions:
Contributions: CNN performed all HiKE-IMS experiments, data analysis, and computational modeling. CS performed all HiKE-IMS-MS experiments. CS and ATK provided scientific advice. SZ supervised the research project, and gave scientific and conceptual advice. All authors contributed to discussions and the manuscript.Conflicts of interest
The authors have no conflicts of interest to declare.Acknowledgements
Funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) – 318063177 and 390583968.References
- G. E. Spangler and C. I. Collins, Peak shape analysis and plate theory for plasma chromatography, Anal. Chem., 1975, 47, 403–407 CrossRef CAS.
- H. E. Revercomb and E. A. Mason, Theory of Plasma Chromatography/Gaseous Electrophoresis- A Review, Anal. Chem., 1975, 47, 970–983 CrossRef CAS.
- G. A. Eiceman, Z. Karpas and H. H. J. Hill, Ion Mobility Spectrometry, Taylor & Francis Group, Boca Raton, FL, Third edn, 2014 Search PubMed.
- K. Jeanne Dit Fouque, A. Garabedian, F. Leng, Y.-C. Tse-Dinh, M. E. Ridgeway, M. A. Park and F. Fernandez-Lima, Trapped Ion Mobility Spectrometry of Native Macromolecular Assemblies, Anal. Chem., 2021, 93, 2933–2941 CrossRef CAS PubMed.
- M. F. Bush, Z. Hall, K. Giles, J. Hoyes, C. V. Robinson and B. T. Ruotolo, Collision cross sections of proteins and their complexes: a calibration framework and database for gas-phase structural biology, Anal. Chem., 2010, 82, 9557–9565 CrossRef CAS PubMed.
- A. Garabedian, D. Butcher, J. L. Lippens, J. Miksovska, P. P. Chapagain, D. Fabris, M. E. Ridgeway, M. A. Park and F. Fernandez-Lima, Structures of the kinetically trapped i-motif DNA intermediates, Phys. Chem. Chem. Phys., 2016, 18, 26691–26702 RSC.
- G. Nagy, K. Kedia, I. K. Attah, S. V. B. Garimella, Y. M. Ibrahim, V. A. Petyuk and R. D. Smith, Separation of β-Amyloid Tryptic Peptide Species with Isomerized and Racemized l-Aspartic Residues with Ion Mobility in Structures for Lossless Ion Manipulations, Anal. Chem., 2019, 91, 4374–4380 CrossRef CAS PubMed.
- M. A. Mäkinen, O. A. Anttalainen and M. E. T. Sillanpää, Ion mobility spectrometry and its applications in detection of chemical warfare agents, Anal. Chem., 2010, 82, 9594–9600 CrossRef PubMed.
- P. Kwantwi-Barima, H. Ouyang, C. J. Hogan Jr and B. H. Clowers, Tuning Mobility Separation Factors of Chemical Warfare Agent Degradation Products via Selective Ion-Neutral Clustering, Anal. Chem., 2017, 89, 12416–12424 CrossRef CAS PubMed.
- R. G. Ewing, D. A. Atkinson, G. A. Eiceman and G. J. Ewing, A critical review of ion mobility spectrometry for the detection of explosives and explosive related compounds, Talanta, 2001, 54, 515–529 CrossRef CAS PubMed.
- J. S. Babis, R. P. Sperline, A. K. Knight, D. A. Jones, C. A. Gresham and M. B. Denton, Performance evaluation of a miniature ion mobility spectrometer drift cell for application in hand-held explosives detection ion mobility spectrometers, Anal. Bioanal. Chem., 2009, 395, 411–419 CrossRef CAS PubMed.
- U. Chiluwal, G. Lee, M. Y. Rajapakse, T. Willy, S. Lukow, H. Schmidt and G. A. Eiceman, Tandem ion mobility spectrometry at ambient pressure and field decomposition of mobility selected ions of explosives and interferences, Analyst, 2019, 144, 2052–2061 RSC.
- J. R. Verkouteren, J. Lawrence, R. Michael Verkouteren and E. Sisco, Method for evaluating ion mobility spectrometers for trace detection of fentanyl and fentanyl-related substances, Anal. Methods, 2019, 11, 6043–6052 RSC.
- Z. Du, T. Sun, J. Zhao, D. Wang, Z. Zhang and W. Yu, Development of a plug-type IMS-MS instrument and its applications in resolving problems existing in in-situ detection of illicit drugs and explosives by IMS, Talanta, 2018, 184, 65–72 CrossRef CAS PubMed.
- T. P. Forbes and M. Najarro, Ion mobility spectrometry nuisance alarm threshold analysis for illicit narcotics based on environmental background and a ROC-curve approach, Analyst, 2016, 141, 4438–4446 RSC.
- K. M. Roscioli, M. R. Lamabadusuriya, C. S. Harden, A. J. Midey, C. Wu, W. F. Siems and H. H. Hill, Structure selective ion molecule interactions (SSIMI) in ion mobility spectrometry, Int. J. Ion Mobil. Spectrom., 2014, 17, 43–53 CrossRef CAS.
- B. Ungethüm, A. Walte, W. Münchmeyer and G. Matz, Comparative measurements of toxic industrial compounds with a differential mobility spectrometer and a time of flight ion mobility spectrometer, Int. J. Ion Mobil. Spectrom., 2009, 12, 131–137 CrossRef.
- R. F. LeBouf and C. C. Coffey, Effect of interferents on the performance of direct-reading organic vapor monitors, J. Air Waste Manag. Assoc., 2015, 65, 261–269 CrossRef CAS PubMed.
- K. Tuovinen, H. Paakkanen and O. Hänninen, Detection of pesticides from liquid matrices by ion mobility spectrometry, Anal. Chim. Acta, 2000, 404, 7–17 CrossRef CAS.
- M. T. Jafari and M. Azimi, Analysis of Sevin, Amitraz, and Metalaxyl Pesticides Using Ion Mobility Spectrometry, Anal. Lett., 2006, 39, 2061–2071 CrossRef.
- Z. Karpas, Z. Berant and R. M. Stimac, An ion mobility spectrometry/mass spectrometry (IMS/MS) study of the site of protonation in anilines, Struct. Chem., 1990, 1, 201–204 CrossRef CAS.
- P. M. Lalli, B. A. Iglesias, H. E. Toma, G. F. de Sa, R. J. Daroda, J. C. Silva Filho, J. E. Szulejko, K. Araki and M. N. Eberlin, Protomers: formation, separation and characterization via travelling wave ion mobility mass spectrometry, J. Mass Spectrom., 2012, 47, 712–719 CrossRef CAS PubMed.
- A. B. Attygalle, H. Xia and J. Pavlov, Influence of Ionization Source Conditions on the Gas-Phase Protomer Distribution of Anilinium and Related Cations, J. Am. Soc. Mass Spectrom., 2017, 28, 1575–1586 CrossRef CAS PubMed.
- S. W. C. Walker, A. Mark, B. Verbuyst, B. Bogdanov, J. L. Campbell and W. S. Hopkins, Characterizing the Tautomers of Protonated Aniline Using Differential Mobility Spectrometry and Mass Spectrometry, J. Phys. Chem. A, 2018, 122, 3858–3865 CrossRef CAS PubMed.
- M. Letzel, H.-F. Grützmacher, D. Stein and H. Grützmacher, Trends in the reactions of gaseous phenyl pnictogen radical cations C6H5EH2˙ + (E = N, P, As), Dalton Trans., J. Inorg. Chem., 2008, 0, 3282–3291 RSC.
- C. Lifshitz, Tropylium Ion Formation from Toluene: Solution of an Old Problem in Organic Mass Spectrometry, Acc. Chem. Res., 1994, 27, 138–144 CrossRef CAS.
- P. M. Lalli, Y. E. Corilo, M. Fasciotti, M. F. Riccio, G. F. de Sa, R. J. Daroda, G. H. M. F. Souza, M. McCullagh, M. D. Bartberger, M. N. Eberlin and I. D. G. Campuzano, Baseline resolution of isomers by traveling wave ion mobility mass spectrometry: investigating the effects of polarizable drift gases and ionic charge distribution, J. Mass Spectrom., 2013, 48, 989–997 CrossRef CAS PubMed.
- C. Kune, C. Delvaux, J. R. N. Haler, L. Quinton, G. Eppe, E. De Pauw and J. Far, A Mechanistic Study of Protonated Aniline to Protonated Phenol Substitution Considering Tautomerization by Ion Mobility Mass Spectrometry and Tandem Mass Spectrometry, J. Am. Soc. Mass Spectrom., 2019, 30, 2238–2249 CrossRef CAS PubMed.
- G. R. Asbury and H. H. Hill, Using Different Drift Gases To Change Separation Factors (α) in Ion Mobility Spectrometry, Anal. Chem., 2000, 72, 580–584 CrossRef CAS PubMed.
- J. Langejuergen, M. Allers, J. Oermann, A. Kirk and S. Zimmermann, High kinetic energy ion mobility spectrometer: Quantitative analysis of gas mixtures with ion mobility spectrometry, Anal. Chem., 2014, 86, 7023–7032 CrossRef CAS PubMed.
- A. T. Kirk, D. Grube, T. Kobelt, C. Wendt and S. Zimmermann, High-resolution high kinetic energy ion mobility spectrometer based on a low-discrimination tristate ion shutter, Anal. Chem., 2018, 90, 5603–5611 CrossRef CAS PubMed.
- M. Allers, A. T. Kirk, N. von Roßbitzky, D. Erdogdu, R. Hillen, W. Wissdorf, T. Benter and S. Zimmermann, Analyzing Positive Reactant Ions in High Kinetic Energy Ion Mobility Spectrometry (HiKE-IMS) by HiKE-IMS–MS, J. Am. Soc. Mass Spectrom., 2020, 31, 812–821 CrossRef CAS PubMed.
- M. Allers, A. T. Kirk, C. Schaefer, D. Erdogdu, W. Wissdorf, T. Benter and S. Zimmermann, Field-Dependent Reduced Ion Mobilities of Positive and Negative Ions in Air and Nitrogen in High Kinetic Energy Ion Mobility Spectrometry (HiKE-IMS), J. Am. Soc. Mass Spectrom., 2020, 31, 2191–2201 CrossRef CAS PubMed.
- J. Langejuergen, M. Allers, J. Oermann, A. Kirk and S. Zimmermann, Quantitative detection of benzene in toluene- and xylene-rich atmospheres using high-kinetic-energy ion mobility spectrometry (IMS), Anal. Chem., 2014, 86, 11841–11846 CrossRef CAS PubMed.
- B. C. Hauck, W. F. Siems, C. S. Harden, V. M. McHugh and H. H. Hill, E/N effects on K0 values revealed by high precision measurements under low field conditions, Rev. Sci. Instrum., 2016, 87, 075104 CrossRef PubMed.
- S. I. Merenbloom, T. G. Flick and E. R. Williams, How hot are your ions in TWAVE ion mobility spectrometry?, J. Am. Soc. Mass Spectrom., 2012, 23, 553–562 CrossRef CAS PubMed.
- E. John and R. Rumble, CRC Handbook of Chemistry and Physics (Internet Version 2018), CRC Press/Taylor & Francis, Boca Raton, FL, 99th Edition, 2018 Search PubMed.
- C. Schaefer, F. Schlottmann, A. T. Kirk and S. Zimmermann, Influence of Sample Gas Humidity on Product Ion Formation in High Kinetic Energy Ion Mobility Spectrometry (HiKE-IMS), J. Am. Soc. Mass Spectrom., 2022, 33, 1048–1060 CrossRef CAS PubMed.
- J. N. Dodds, J. C. May and J. A. McLean, Investigation of the Complete Suite of the Leucine and Isoleucine Isomers: Toward Prediction of Ion Mobility Separation Capabilities, Anal. Chem., 2017, 89, 952–959 CrossRef CAS PubMed.
- M. Allers, A. T. Kirk, M. Eckermann, C. Schaefer, D. Erdogdu, W. Wissdorf, T. Benter and S. Zimmermann, Positive Reactant Ion Formation in High Kinetic Energy Ion Mobility Spectrometry (HiKE-IMS), J. Am. Soc. Mass Spectrom., 2020, 31, 1291–1301 CrossRef CAS PubMed.
- W. F. Siems, L. A. Viehland and H. H. Hill, Correcting the fundamental ion mobility equation for field effects, Analyst, 2016, 141, 6396–6407 RSC.
- H. Borsdorf, K. Neitsch, G. A. Eiceman and J. A. Stone, A comparison of the ion chemistry for mono-substituted toluenes and anilines by three methods of atmospheric pressure ionization with ion mobility spectrometry, Talanta, 2009, 78, 1464–1475 CrossRef CAS PubMed.
- E. A. Mason and E. W. McDaniel, Transport Properties of Ions in Gases, Wiley-VCH Verlag GmbH, Weinheim, Federal Republic of Germany, 1988 Search PubMed.
- L. A. Viehland, Gaseous Ion Mobility, Diffusion, and Reaction, Springer International Publishing, 2019, vol. 3 Search PubMed.
- W. F. Siems, L. A. Viehland and H. H. Hill Jr, Improved momentum-transfer theory for ion mobility. 1. Derivation of the fundamental equation, Anal. Chem., 2012, 84, 9782–9791 CrossRef CAS PubMed.
- C. N. Naylor, T. Reinecke, M. E. Ridgeway, M. A. Park and B. H. Clowers, J. Am. Soc. Mass Spectrom., 2019, 30, 2152–2162 CrossRef CAS PubMed.
- C. Schaefer, M. Allers, A. T. Kirk, F. Schlottmann and S. Zimmermann, Influence of Reduced Field Strength on Product Ion Formation in High Kinetic Energy Ion Mobility Spectrometry (HiKE-IMS), J. Am. Soc. Mass Spectrom., 2021, 32, 1810–1820 CrossRef CAS PubMed.
- R. L. Smith, L. J. Chyall, B. J. Beasley and H. I. Kenttamaa, J. Am. Chem. Soc., 1995, 117, 7971–7973 CrossRef CAS.
- R. Flammang, N. Dechamps, L. Pascal, Y. Haverbeke, P. Gerbaux, P.-C. Nam and M. Nguyen, Ring Versus Nitrogen Protonation of Anilines, Lett. Org. Chem., 2004, 1, 23–30 CrossRef CAS.
- J. L. Campbell, A. M.-C. Yang, L. R. Melo and W. S. Hopkins, Studying Gas-Phase Interconversion of Tautomers Using Differential Mobility Spectrometry, J. Am. Soc. Mass Spectrom., 2016, 27, 1277–1284 CrossRef CAS PubMed.
- H. Xia and A. B. Attygalle, Untrapping kinetically trapped ions: The role of water vapor and ion-source activation conditions on the gas-phase protomer ratio of benzocaine revealed by ion-mobility mass spectrometry, J. Am. Soc. Mass Spectrom., 2017, 28, 2580–2587 CrossRef CAS PubMed.
- J. C. Choe, Does the Gaseous Aniline Cation Isomerize to Methylpyridine Cations Before Dissociation?, Bull. Korean Chem. Soc., 2013, 34, 3249–3252 CrossRef CAS.
- J. C. Choe, N. R. Cheong and S. M. Park, Unimolecular dissociation of aniline molecular ion: A theoretical study, Int. J. Mass Spectrom., 2009, 279, 25–31 CrossRef CAS.
- M. D. Hanwell, D. E. Curtis, D. C. Lonie, T. Vandermeersch, E. Zurek and G. R. Hutchison, Avogadro: an advanced semantic chemical editor, visualization, and analysis platform, J. Cheminform., 2012, 4, 17 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson and H. Nakatsujiet al., 2016.
- C. Larriba and C. J. Hogan Jr, Ion mobilities in diatomic gases: measurement versus prediction with non-specular scattering models, J. Phys. Chem. A, 2013, 117, 3887–3901 CrossRef CAS PubMed.
- C. Larriba-Andaluz and C. J. Hogan Jr, Collision cross section calculations for polyatomic ions considering rotating diatomic/linear gas molecules, J. Chem. Phys., 2014, 141, 194107 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Additional figures containing resolving power, K0 values of all species in nitrogen, and spreadsheets containing speciation measurements are contained as supplemental information. See DOI: https://doi.org/10.1039/d2cp01994a |
| This journal is © the Owner Societies 2023 |