
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Melting pseudosymmetry and thermal expansion in 3-benzoylpropionic acid†
Tomasz
Poręba
 *ab,
Marcin
Świątkowski
c and
Giorgia
Confalonieri
a
*ab,
Marcin
Świątkowski
c and
Giorgia
Confalonieri
a
aEuropean Synchrotron Radiation Facility, 71 Avenue des Martyrs, 38000 Grenoble, France. E-mail: tomasz.poreba@esrf.fr
bLaboratory of Quantum Magnetism, Institute of Physics, École Polytechnique Federale de Lausanne, Lausanne CH-1015, Switzerland
cInstitute of General and Ecological Chemistry, Lodz University of Technology, Żeromskiego 116, 90-924 Łódź, Poland
First published on 4th October 2023
Abstract
The thermal behavior of 3-benzoylpropionic acid (BPA) was investigated using single-crystal and synchrotron powder X-diffraction, as well as differential scanning calorimetry analysis. The study revealed the existence of two subtly distinct structures of BPA, with different numbers of molecules in the asymmetric unit. A detailed analysis of conformational changes in the molecules, supported by quantum mechanical calculations, allowed for an explanation of the origins of the two phases. The continuous transition upon cooling is linked to the emergence of pseudosymmetry. The transition mechanism was revealed by thermal expansion analysis. Furthermore, the research provided evidence to refute the previously reported premise of BPA's colossal thermal expansivity.
Introduction
Generally speaking, molecular crystalline solids are built-up of crystallographic asymmetric units that tessellate the space in a regular manner, through the combination of the three-dimensional space group symmetry operations and the translational symmetry of the unit cell. However, the molecules within the asymmetric unit are free to associate in the most energy-efficient way, without the symmetry restrictions. The same molecule presents different packing arrangements in different polymorphic phases, often in different molecular conformations. The very same molecule can also be combined with other species in the asymmetric unit, forming unique crystalline structures such as in co-crystals, solvates, clathrates, and inclusion compounds. The manifestation of polymorphs and multicomponent crystals has important practical implications in the pharmaceutical industry. Crystalline biologically-active compounds with different molecular arrangements not only differ in physicochemical properties (bioavailability, stability, dissolution rate, density, etc.) but also may be considered to have formally different drug identity profiles, which opens a door for a patent application.1,2 The existence of polymorphs and multicomponent crystals can be rationalized as a correlation (or competition) between the tendency to form close-packed structures and energetically-favorable intra- and intermolecular interactions at given conditions.3 Subtle interplay between conformational and crystal lattice energies determines which structure is favored, often already at the nucleation stage.4,5In most cases, there is only one chemically-identical molecule in the crystal asymmetric unit. Such structures can be described with the parameter Z′ = 1, which denotes the number of formula units in the given unit cell divided by the number of general positions. The range of Z′ values in the reported crystal structures varies from 1/96 to 32, and the structures with Z′ > 1 constitute about 9% of the total number of structures deposited in the Cambridge Structural Database (CSD).6,7 The presence of disorder or twinning can obscure as higher Z′ if inappropriately treated. On top of that, the fundamental issue of adequate space-group assignment may lead to apparently high Z′ values, as in relatively frequent cases of misassignment of P1 (Z′ = 2) instead of P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) (Z′ = 1) space group.8 Finally, some of the structures with Z′ > 1 can be more appropriately described by the modulated structure, consisting of a higher-symmetry subcell and modulation vector(s).9 For example, the superspace approach to a hydrate of sodium saccharinate allows us to adequately describe its complex structure (Z′ = 16) as a modulated eightfold substructure.10
(Z′ = 1) space group.8 Finally, some of the structures with Z′ > 1 can be more appropriately described by the modulated structure, consisting of a higher-symmetry subcell and modulation vector(s).9 For example, the superspace approach to a hydrate of sodium saccharinate allows us to adequately describe its complex structure (Z′ = 16) as a modulated eightfold substructure.10
There are a few factors that contribute to the formation of structures with more than one molecule in the asymmetric unit. The frustration between the closed-pack structure and optimal intermolecular interactions may result in the location of two different molecules in two different intermolecular environments.6,11 In this sense, such structures are similar co-crystals, where the stable packing arrangement is achieved by incorporation of the guest molecules. The strong tendency to form chains or rings in Z′ > 1 structures has been recognized in molecules which able to form strong intermolecular interactions.12 Monoalcohols and monoamines with large substituents can only achieve a good H⋯lone-pair overlap either by incorporation of the other symmetry-independent molecule in the asymmetric unit, or aggregation around the high-order (3,4 or 6) screw axis.13 As a result, the incidence of Z′ > 1 in such structures is very high (51% versus 9% in the whole CSD). Some dimeric supramolecular synthons were identified to have a higher propensity to occur in structures with Z′ > 1, which is linked to the presence of strong intermolecular interactions and geometrical considerations.12 For example, in the case of the P21 space-group, frequent for Z′ > 1 structures (19.4%), two symmetry-unrelated species are required to form asymmetric dimers, as it is otherwise difficult to a form strongly-bound pair of molecules (e.g. alcohols, dimeric carboxylic acids, and amides) over the screw axis.
The variety of intermolecular interactions found frequently in Z′ > 1 subset include π–π stacking, C–H⋯π, halogen–halogen, OH⋯alkyne or aryl, alkane-chain van der Waals interactions, to name a few.6,14 These common motifs show indeed that the strong interactions in strongly-bound symmetry-independent molecules, often related by pseudosymmetry, yield more efficient packing, in comparison to Z′ = 1 structures. The formation of (asymmetric) molecular dimers may occur already in solution, from which crystal forms, and some of the authors suggest that Z′ > 1 structures are thermodynamically-metastable crystallographic “fossils” that preserve connectivity patterns found in the solution or melt.15–17 A curious observation was made recently, that small and presumably rapidly grown crystals studied at synchrotrons show an elevated incidence of Z′ > 1 structures, compared to large, well-formed crystals used in neutron diffraction experiments.18 However, the presence of the strong intermolecular interactions is not mandatory, and high Z′ values are also observed in the structures bound only by weak interactions.
The delicate balance between conformational and lattice energy in crystals can be shifted by external stimuli. The influence of temperature on Z′ value is varied. In some of the structures, cooling causes an increase in Z′ through the transition from dynamic to ordered behavior, for example, the ordering of isopropyl groups in a Tröger's base derivative (transition from Z′ = 1 to 3 below 130 K).19 Such “frozen” disorder can be well-described using modulated structure and/or pseudosymmetry. A striking example of trimethyltin hydroxide, which forms polymeric strands, shows the opposite trend. The Z′ reduction from 32 to 1 upon cooling, shows that a high Z′ structure results in better packing, and is related to the composite motion of the individual strands in a high-temperature polymorph, and cooling below 100 K results in a commensurately modulated twofold P212121(00γ)00s superstructure.20,21 Later studies, however showed that room temperature Z′ value is 4, and the previously observed higher order reflections are in fact structured diffuse scattering signal.22 Similarly, already mentioned highly disordered sodium saccharinate showed concerted setting into a fully correlated disorder upon cooling.23 On the other hand, higher pressures increase the crystal energy without elevating the entropic term and therefore typically favour more symmetric, better-packed structures with lower Z′ with respect to ambient-pressure structures.24 High pressure may also activate competing intermolecular interactions, which are otherwise absent at ambient conditions. The frustration between NH⋯N and I⋯I interactions in 2,4,5-triiodo-(1H)-imidazole at high pressure was shown to optimize both the packing and intermolecular interactions, which is associated with the increase of Z′.25 Finally, pressure-induced structure densification can result in pseudosymmetry, as well as twinning, which can be mistaken for higher Z′ structure.26
In this study, we investigate crystals of 3-benzoylpropionic acid (BPA) using single-crystal and synchrotron powder X-diffraction (SC-XRD and PXRD, respectively) at variable temperatures. Its structure shows a combination of multiple intermolecular interactions typical for Z′ > 1 structures and a flexible conformation (Fig. 1). In fact, 25% of γ-ketoacids reported in CSD show Z′ > 1. Yet, two structural studies at 235 (refcode: VERMAG)27 and 295 K (refcode: VERMAG01),28 reported that it forms only a Z = 1 structure, but the room-temperature structure shows aberrant values of intramolecular bond lengths and angles. These unusual features may be related to the overlooked phase transition to a higher Z′ structure. Our interest in this compound was also piqued because of its apparent colossal volume expansion coefficient of 2333.5 MK−1, the highest positive among the known organic solids (168.8 MK−1 on average).29 Since the thermal expansivity was determined only from the two mentioned datasets, we endeavored to systematically study its structural and thermal behavior, to confirm or refute the claimed colossal thermal expansion.
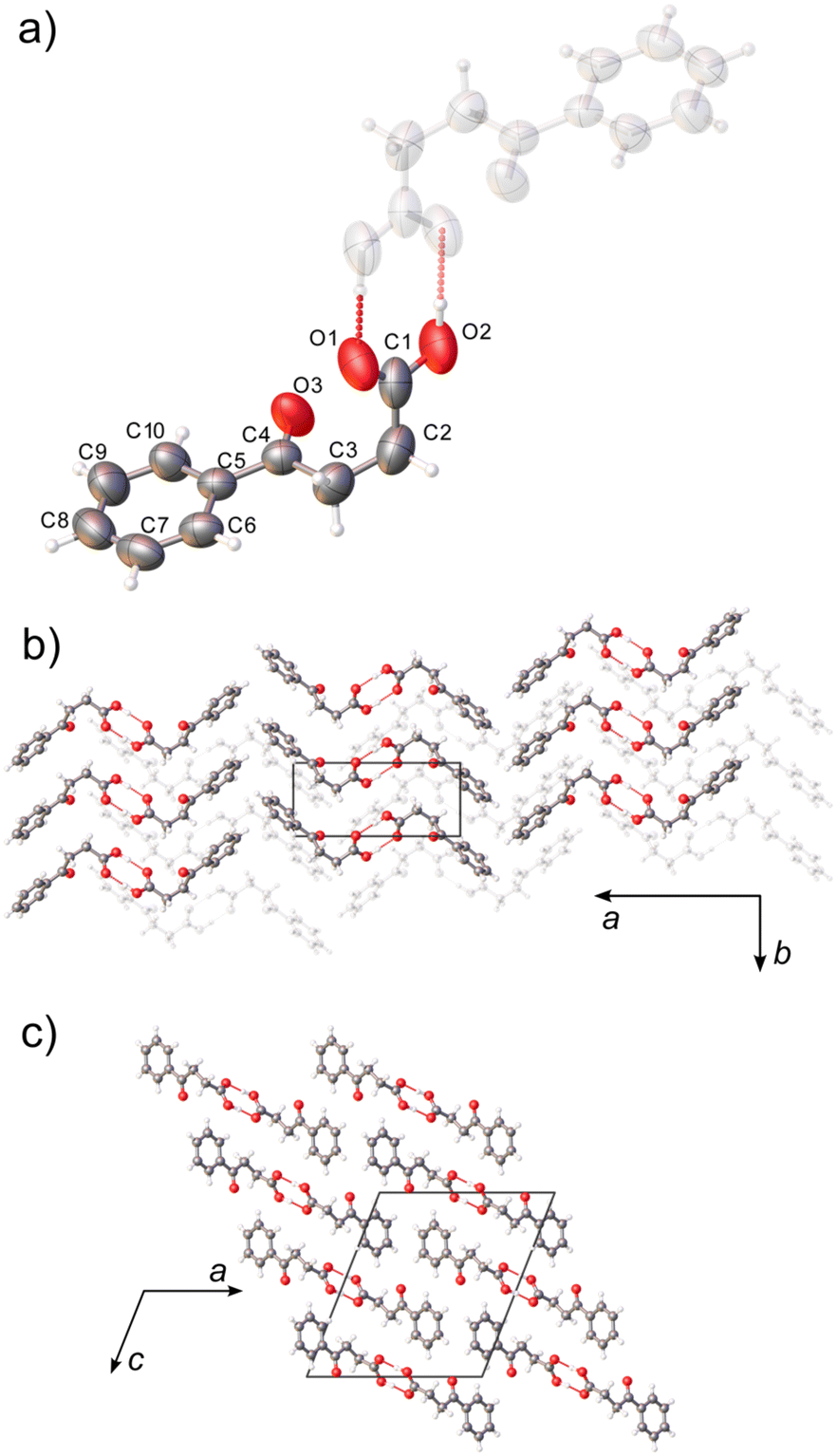 | ||
| Fig. 1 Molecular structure, numbering scheme (a), and packing diagrams (b and c) of BPA at 290 K. Hydrogen-bonded dimers (a) form extended zig-zag motif along approximately [502] (b and c). Thermal ellipsoids are plotted at a 50% probability level. | ||
Experimental
Synthesis and structural investigation
BPA was synthesized by Friedel–Crafts condensation, according to literature procedures.30 The material was subsequently recrystallized from water at 278 K. Transparent, plate-like crystals were grown over the course of two weeks.DSC measurements were performed with a NETZSCH DSC 200 F3 differential scanning calorimeter under a nitrogen flow of 20 cm3 min−1. The sample was placed in a sealed aluminum crucible. Data were collected during heating and cooling in the temperature range of 190–295 K with a heating/cooling rate of 5 K min−1.
Single-crystal X-ray diffraction data were collected on the XtaLAB Synergy Dualflex Pilatus 300 K diffractometer (CuKα = 1.54056 Å). The temperature was controlled by a Nitrogen Cryostream Cryostream (Oxford Cryosystems 800 series, ±0.1 K). The determination of the unit cell and preliminary data reduction were carried out in CrysAlis Pro software.31 The structures were solved with SHELXT, using the intrinsic phasic algorithm.32 All non-hydrogen atoms were refined anisotropically, using the full-matrix, least-squares method on F2 by the SHELXL software.33 Hydrogen atoms were located from the Fourier difference map, and their positions were refined freely. Structural visualizations were made in Mercury 2022.3.0 and Olex2 software.34,35 CCDC 2283189–2283199 contains the ESI† crystallographic data for this paper.
Synchrotron powder diffraction data were collected at ID22 high-resolution beamline at ESRF.36 Monochromatic wavelength was set at 0.35420 Å with a beam size of 1 × 1 mm. Diffraction intensities were recorded by an Eiger2 X CdTe 2M-W detector preceded by 13 Si(111) analyzer crystals. The finely-ground powder has been tightly packed in ø 1.00 mm borosilicate capillary together with 1% (w/w) NaCl (≥99.5%, Sigma-Aldrich) as an internal temperature calibrant. The data was collected in the transmission mode as a function of temperature using a Cryostream (Oxford Cryosystems Cryostream 7+ nitrogen blower). Diffraction data were fitted using the Le Bail method, as implemented in TOPAS7.37,38
Computational details
Calculations were performed with the Gaussian16 program package.39 Geometry optimizations and the constraint scan of the dihedral angle C3–C2–C1–O2 (Fig. 1) were carried out with the B3LYP/6-31+G(d) level of theory40–42 including Grimme's DFT-D3 dispersion correction43 and the SMD implicit solvation model using the dielectric constant of water.44 The initial BPA conformer was confirmed to be a minimum of the potential energy surface using frequency calculations, and the most stable conformer based on the Gibbs free energies calculated at 298 K.Lattice energy and interaction energies were calculated in CrystalExplorer software using CE-B3LYP model energies and were based on the experimental crystal structures.45,46 The cluster radius for the lattice energy calculation was set to 30 Å, to ensure their convergence. CrystalExplorer was also used to generate and plot Hirshfeld surfaces.
Results and discussion
Crystal structure and pseudosymmetry melting
The crystal structure of BPA at room temperature corresponds to the structure reported by Lalancette et al. (VERMAG, collected at 235 K).27 BPA crystallizes in the monoclinic P21/n space group with one molecule in the asymmetric unit (Z = 4). The non-standard space-group setting was kept for the sake of consistency with the reported data, and to minimize the correlation between parameters with respect to the crystallographic a and c axes (monoclinic β angle in the standard oblique P21/c setting is equal to 129.4°). The molecule of BPA is V-shaped, where the 3-benzoylpropionic fragment lies essentially on one plane, and the carboxylic carbon forms 102.8(1)° to that plane (Fig. 1a). The ketone carbonyl group, however, is tilted out of the aromatic-ring plane by 3.8(2)° in the direction of the carboxyl group. It might be indicative of weak intramolecular carbonyl–carbonyl interaction, as the distance between the carbonyl oxygen and the carboxylic carbon atom is less than their sum of van der Waals radii: 2.962(2) Å. The molecules form hydrogen-bonded dimers (dO–O = 2.649(2) Å), related by the crystallographic inversion center, and the two molecules in the dimer are conformational enantiomers (Fig. S1†). The calculated interaction energy in the dimer of −79.8 kJ mol−1, is typical for aromatic carboxylic acids, and accounts for 65% of the total lattice energy (Elatt = −126.5 kJ mol−1).47The ketone carbonyl group is not involved in the hydrogen bonding, and parallel aromatic rings do not form strong π–π stacking, as the rings are shifted 3.803(3) Å in respect to each other. However, the two perpendicular aromatic rings from the neighboring molecules, at 87.06° and 5.11 Å centroid distance, were found to form a strong, structure-directing, π–π stacking, as evaluated by the CSD Aromatic Analyser neural network model.34 In the absence of other strong intermolecular interactions, the zig-zag shape of the hydrogen-bonded dimers interacting through perpendicular π–π interactions dictates the supramolecular architecture in BPA. The zig-zag motif in BPA extends in approximately [502] direction, forming layers of parallel molecules stacked along the crystallographic b-axis (Fig. 1b and c).
Upon cooling to 200 K, additional diffraction peaks can be seen to arise in the h1l plane (Fig. 2). The extra reflections from the doubled cell gradually disappear as the temperature raises. This process was tracked with variable-temperature SC-XRD (Table 1). At 200 K, only about 2/3 of the reflections can be indexed using the room temperature (RT) unit cell. When the temperature reaches 240 K, nearly all the reflections can be indexed in RT unit-cell, as almost all of the supercell reflections disappear. The process is not associated with any detectable thermal events in the DSC thermogram (Fig. S3†), ruling out the first-order phase transition. To reveal the mechanism of the transition, first, the possibility of twinning was examined. It has been reported that in some cases apparent superlattice reflections were in fact a superposition of the diffraction signals from different twin domains.48,49 In the case of BPA, no satisfactory twinning law, with [010] as a twin axis, was found. Additionally, the analysis of the cumulative intensity distribution points out a lack of twinning (Fig. S2†). We confirmed this observation, by performing high-resolution PXRD at 200 and 300 K (Fig. 3). Since the distribution of the crystallites is random during the measurement, any additional Bragg superlattice reflections must come from the genuine phase change and not simply from the oriented crystalline domains. Careful analysis of the diffractograms shows the presence of (100) superlattice reflection around 2θ angle of 1.333° (d = 15.2273 Å) at 200 K, which disappears at 300 K. At this temperature, the corresponding (101) reflection (vide infra) is observed at 2θ = 2.654° (d = 7.6476 Å, red curve Fig. 3), confirming the halving of this crystallographic axis upon heating. The other calculated reflection positions from the SCXRD model are in excellent agreement with the PXRD results, both in terms of peak 2θ position and relative intensities calculated from the experimental structure factors.
 | ||
| Fig. 2 Pseudosymmetry melting observed on the precession photographs of BPA at 200 and 290 K (left and middle panel) as a disappearance of the supercell reflections. This gradual process can be tracked with the overall number of observed reflections which can be indexed in a given unit cell. | ||
| CCDC number | 2283189 | 2283190 | 2283191 | 2283192 | 2283193 | 2283194 |
|---|---|---|---|---|---|---|
| Empirical formula | C10H10O3 | |||||
| Formula weight | 178.18 | |||||
| Temperature (K) | 200.0(1) | 210.0(1) | 220.0(1) | 230.0(1) | 235.0(1) | 240.0(1) |
| Radiation | Cu Kα (1.54184 Å) | |||||
| Space group | P21/c | P21/c | P21/c | P21/c | P21/c | P21/n |
| a (Å) | 15.3694(3) | 15.3811(3) | 15.3909(4) | 15.4013(4) | 15.4043(4) | 12.7470(4) |
| b (Å) | 5.2160(1) | 5.21845(12) | 5.22159(12) | 5.2236(1) | 5.2267(1) | 5.22886(14) |
| c (Å) | 22.3953(5) | 22.4123(5) | 22.4298(6) | 22.4510(6) | 22.4631(5) | 14.4507(4) |
| β (°) | 97.797(2) | 97.767(2) | 97.735(2) | 97.710(2) | 97.697(2) | 111.302(3) |
| Volume (Å3) | 1778.76(6) | 1782.43(7) | 1786.17(8) | 1789.86(8) | 1792.29(8) | 897.37(5) |
| Z | 8 | 8 | 8 | 8 | 8 | 4 |
| ρ calc (g cm−3) | 1.331 | 1.328 | 1.325 | 1.322 | 1.321 | 1.319 |
| μ (mm−1) | 0.817 | 0.816 | 0.814 | 0.812 | 0.811 | 0.81 |
| F (000) | 752 | 752 | 752 | 752 | 752 | 376 |
| θ range for data collection (°) | 3.985 to 79.926 | 3.981 to 79.730 | 3.978 to 79.567 | 3.961 to 79.267 | 3.972 to 79.433 | 3.969 to 79.378 |
| Index ranges | −19 ≤ h ≤ 18 | −19 ≤ h ≤ 19 | −19 ≤ h ≤ 19 | −19 ≤ h ≤ 19 | −19 ≤ h ≤ 19 | −16 ≤ h ≤ 14 |
| −6 ≤ k ≤ 5 | −6 ≤ k ≤ 5 | −6 ≤ k ≤ 5 | −5 ≤ k ≤ 6 | −6 ≤ k ≤ 5 | −5 ≤ k ≤ 6 | |
| −23 ≤ l ≤ 28 | −23 ≤ l ≤ 28 | −23 ≤ l ≤ 28 | −23 ≤ l ≤ 28 | −23 ≤ l ≤ 28 | −16 ≤ l ≤ 18 | |
| Reflections collected | 18![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 337 337 |
18362 |
18382 |
18393 |
18408 |
9279 |
| Independent reflections | 3710 | 3714 | 3721 | 3731 | 3734 | 1873 |
| Data/restraints/parameters | 3710/0/309 | 3714/0/311 | 3721/0/309 | 3731/0/309 | 3734/0/309 | 1873/0/158 |
| R int | 0.0366 | 0.0356 | 0.0415 | 0.0352 | 0.0334 | 0.0323 |
| Completeness (%) | 99.9 | 99.8 | 99.8 | 99.8 | 99.8 | 99.9 |
| Goodness-of-fit on F2 | 1.022 | 1.022 | 1.061 | 1.060 | 1.071 | 1.045 |
| Final R indexes [I > 2σ(I)] | R 1 = 0.0426 | R 1 = 0.0426 | R 1 = 0.0451 | R1 = 0.0482 | R 1 = 0.0493 | R 1 = 0.0404 |
| wR2 = 0.1139 | wR2 = 0.1139 | wR2 = 0.1346 | wR2 = 0.1485 | wR2 = 0.1505 | wR2 = 0.1106 | |
| Final R indexes (all data) | R 1 = 0.0496 | R 1 = 0.0496 | R 1 = 0.0562 | R 1 = 0.0652 | R 1 = 0.0696 | R 1 = 0.0465 |
| wR2 = 0.1191 | wR2 = 0.1191 | wR2 = 0.1434 | wR2 = 0.1620 | wR2 = 0.1650 | wR2 = 0.1152 | |
| Largest diff. peak and hole (e Å−3) | 0.167 and −0.183 | 0.167 and −0.183 | 0.208 and −0.179 | 0.211 and −0.185 | 0.207 and −0.168 | 0.135 and −0.170 |
| CCDC number | 2283195 | 2283196 | 2283197 | 2283198 | 2283199 | |
|---|---|---|---|---|---|---|
| Temperature (K) | 250.0(1) | 260.0(1) | 270.0(1) | 280.0(1) | 290.0(1) | |
| Radiation | Cu Kα (1.54184 Å) | |||||
| Space group | P21/n | P21/n | P21/n | P21/n | P21/n | |
| a (Å) | 12.7632(4) | 12.7762(4) | 12.7932(5) | 12.8072(4) | 12.8223(5) | |
| b (Å) | 5.23201(15) | 5.23515(15) | 5.23468(18) | 5.24245(15) | 5.2450(2) | |
| c (Å) | 14.4635(4) | 14.4736(5) | 14.4811(6) | 14.4951(4) | 14.5064(5) | |
| β (°) | 111.364(4) | 111.409(4) | 111.423(5) | 111.513(4) | 111.569(4) | |
| Volume (Å3) | 899.46(5) | 901.28(5) | 902.77(6) | 905.42(5) | 907.28(6) | |
| Z | 4 | 4 | 4 | 4 | 4 | |
| ρ calc (g cm−3) | 1.316 | 1.313 | 1.311 | 1.307 | 1.304 | |
| μ (mm−1) | 0.808 | 0.807 | 0.805 | 0.803 | 0.801 | |
| F (000) | 376 | 376 | 376 | 376 | 376 | |
| θ range for data collection (°) | 3.964 to 79.186 | 3.959 to 79.037 | 3.955 to 79.723 | 3.948 to 80.150 | 3.943 to 80.032 | |
| Index ranges | −16 ≤ h ≤ 14 | −14 ≤ h ≤ 16 | −15 ≤ h ≤ 15 | −16 ≤ h ≤ 14 | −16 ≤ h ≤ 14 | |
| −5 ≤ k ≤ 6 | −5 ≤ k ≤ 6 | −5 ≤ k ≤ 6 | −5 ≤ k ≤ 6 | −5 ≤ k ≤ 6 | ||
| −16 ≤ l ≤ 18 | −16 ≤ l ≤ 18 | −16 ≤ l ≤ 18 | −16 ≤ l ≤ 18 | −16 ≤ l ≤ 18 | ||
| Reflections collected | 9290 | 9326 | 9246 | 9383 | 9404 | |
| Independent reflections | 1875 | 1881 | 1785 | 1891 | 1893 | |
| Data/restraints/parameters | 1875/0/158 | 1881/0/158 | 1785/0/158 | 1891/0/158 | 1893/0/158 | |
| R int | 0.0354 | 0.0368 | 0.0401 | 0.0377 | 0.0275 | |
| Completeness (%) | 99.8 | 99.8 | 99.8 | 99.9 | 99.9 | |
| Goodness-of-fit on F2 | 1.069 | 1.059 | 1.040 | 1.061 | 1.080 | |
| Final R indexes [I > 2σ(I)] | R 1 = 0.0411 | R 1 = 0.0418 | R 1 = 0.0424 | R 1 = 0.0436 | R 1 = 0.0433 | |
| wR2 = 0.1148 | wR2 = 0.1196 | wR2 = 0.1159 | wR2 = 0.1207 | wR2 = 0.1203 | ||
| Final R indexes (all data) | R 1 = 0.0476 | R 1 = 0.0490 | R 1 = 0.0518 | R 1 = 0.0516 | R 1 = 0.0519 | |
| wR2 = 0.1196 | wR2 = 0.1241 | wR2 = 0.1232 | wR2 = 0.1273 | wR2 = 0.1268 | ||
| Largest diff. peak and hole (e Å−3) | 0.141 and −0.165 | 0.131 and −0.169 | 0.168 and −0.167 | 0.136 and −0.167 | 0.141 and −0.155 | |
 | ||
| Fig. 3 High-resolution powder diffractograms of BPA were collected for low (200 K, blue) and room temperature (300 K, red) polymorphs. The appearance of superlattice (100) upon cooling is depicted in insets. | ||
The low intensity of the superlattice reflections points out the small deformation of electron density distribution between room temperature (RT) and low temperature (LT) phases. The new LT unit cell can be indexed with the monoclinic supercell, with the doubled volume with respect to the RT cell. The unit cell of the LT phase has two molecules in the asymmetric unit (Z′ = 2, compared to Z′ = 1 in the RT phase). These two molecules are conformational enantiomers, initially related by an n-glide plane in the RT phase (Fig. 4). The symmetry-generated molecules belong to the parallel stacks of zig-zag chains along the crystallographic c direction (plotted as light gray in Fig. 1b). Upon cooling, the two molecules (and therefore stacks) become symmetry-independent, approximately related by a pseudoglide. It is not uncommon for tightly bound dimers, such as one observed in BPA, to present Z′ = 2 with approximate symmetry. It was shown that 83% percent of the asymmetric crystal structures show some kind of pseudosymmetry (tolerance threshold of 0.5 Å molecule−1), including 60% of pseudoglides.12 Although the packing in the two phases is essentially the same, the difference lies in the subtle variations of the BPA molecular conformation. Such supramolecular arrangement resembles the one found in aspirin IV polymorph (Z′ = 2), which consists of stacks of hydrogen-bonded symmetric dimers which are, in turn, symmetry-unrelated to one another.50 The continuous character of the phase transition, associated with a partial loss of translational symmetry upon cooling, ascribes it, according to Landau theory,51 to the klassengleiche (k type) phase transition. This is in line with DSC and SC-XRD results, as thermograms showed a lack of any thermal events, in the continuous temperature range 200–310 K, which can be associated with (discontinuous) first-order phase transition (Fig. S3†). Notably, no hysteresis was observed via SC-XRD experiments during heating–cooling cycles in 200–290 K range. A recent CSD data-mining study revealed several low-temperature structures which undergo k-transitions into a more symmetric phase upon heating, associated with the loss or change of pseudosymmetry.52 It seems that in multiple Z′ > 1 crystals, including BPA, they are indeed the only thermodynamically stable forms at low temperatures and not merely “fossil relics” of the nucleation process, as generally crystallizations are carried out, instead, around room temperature, and produce mostly Z ≤ 1 structures.
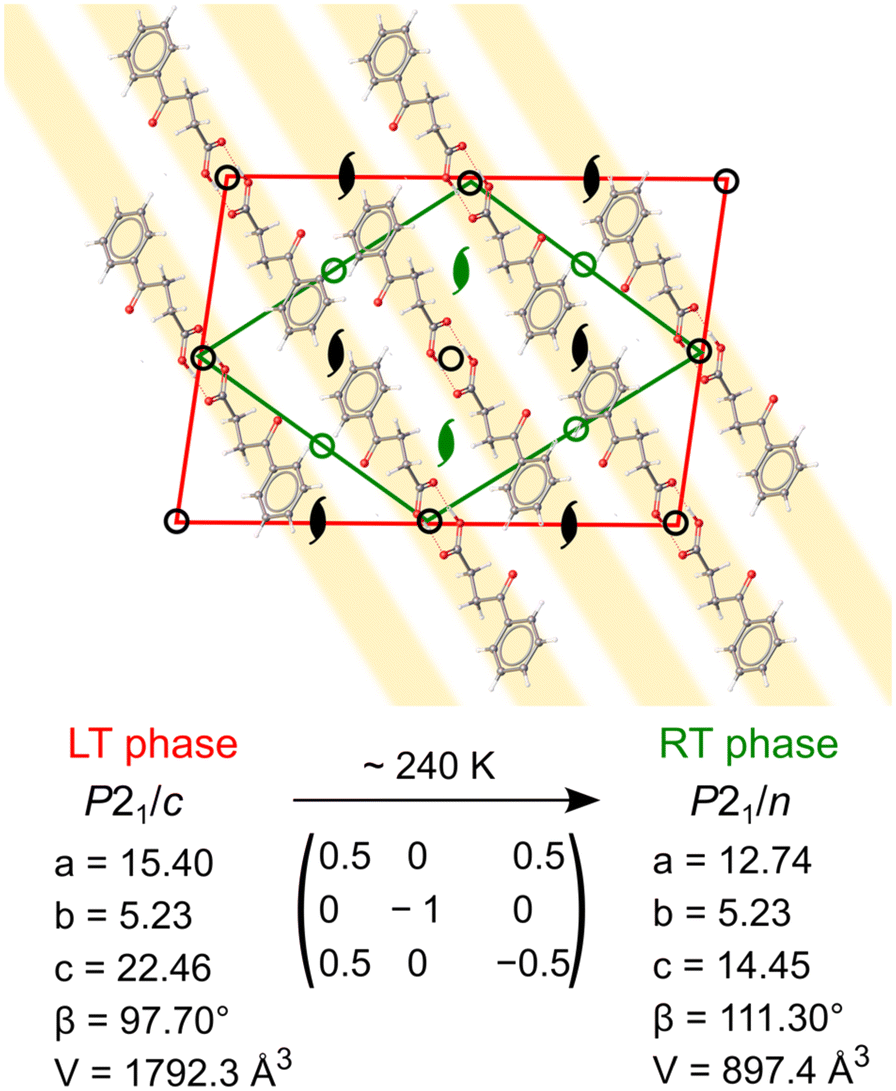 | ||
| Fig. 4 Geometrical relations between LT (red) and RT (green) phases of BPA, viewed along [010]. Upon phase transition, from RT to LT phase, certain symmetry elements are suppressed (in green), which results in symmetry inequivalence of pseudosymmetric parallel stacks of BPA molecules (highlighted in yellow). | ||
One of the parameters which can pinpoint the structural similarity between the molecules is the torsional angle (τ) formed through a carboxylic carbon and the propyl chain (C1–C2–C3–C4). At 200 K, the two molecules have significantly different τ of 68.9(1) and 67.7(1)°, respectively. As the temperature increases, τ values in two molecules converge towards ∼68.5° at 235 K, and become statistically indiscernible, leading to a lowering of Z′ from 2 to 1 (Fig. 5). This situation is coupled with the disappearance of the superlattice reflections, as described above. We note that the literature-reported structure (VERMAG) was coincidentally collected at 235 K, the point at which the structure transforms into the smaller cell. Further heating up to 290 K leads to the continuous increase of τ angle value to 69.0(2)°. Temperature-evolution of τ angle illustrates the lack of the intramolecular carbonyl–carbonyl interaction in BPA. It becomes more acute upon cooling, so the distance between the carboxylic oxygen (O1, vide Fig. 1) and ketone carbonyl (C4) shortens. Yet, the possible transformation from sp2 to sp3 hybridization, followed by pyramidalization around the carbonyl C4 atom, is not observed. The optimized structure of BPA shows indeed that all the atoms in the 3-benzoylpropionic fragment are co-planar, with the corresponding τ of 65.55°.
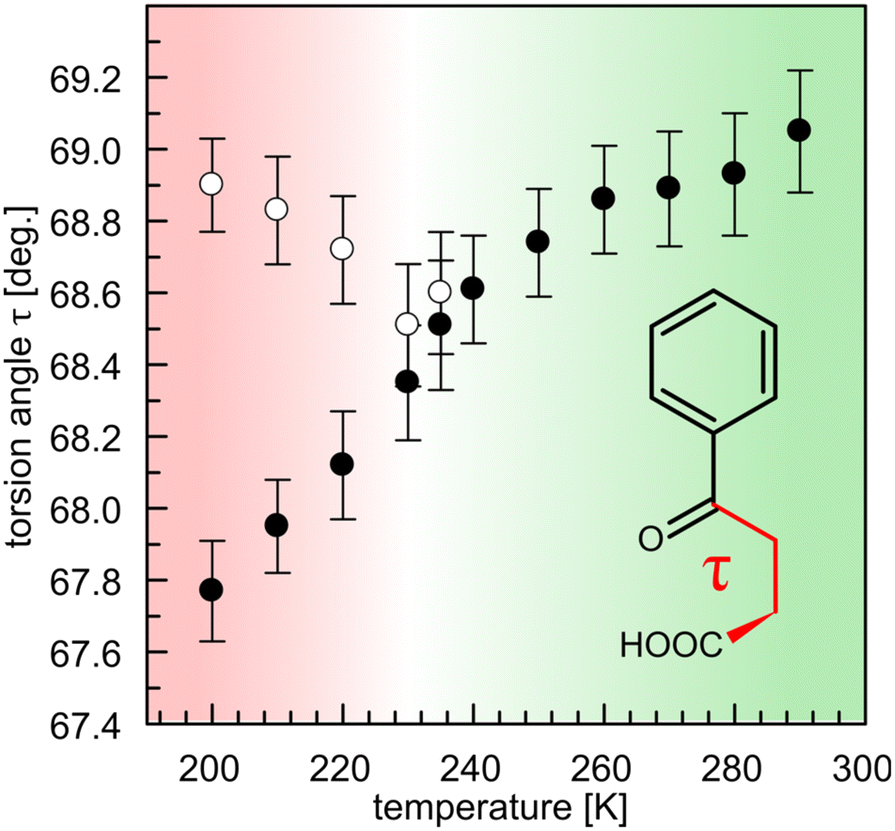 | ||
| Fig. 5 Evolution of the torsional angle τ (C1–C2–C3–C4) upon temperature change. The transition from supercell with two molecules in the asymmetric unit (red) into a symmetric half-volume cell (green) is accompanied by the geometry equilibration of the two molecules. | ||
Simulated rotation of the carboxyl group around the C1–C2 bond ±180° with respect to the C2–C3 bond allows to evaluate the influence of the deformation of the dimer, through the hydrogen bond, on the geometry and internal energy of BPA molecule (Fig. 6). Conformation of BPA found in crystal is only ∼1.5 kJ mol−1 higher in the potential energy than the optimized structure, and presents very similar geometry. Notably, the carboxylic group does not appreciably rotate in the studied temperature range (from 163.5(2) to 164.5(2)° upon heating from 200 to 290 K) from the optimal dihedral angle of 159.4°. Just like in the crystal, the lower molecular volume of the isolated dimer supports better stability. Therefore, small angular rotations of the carboxylic group and low τ are preferred, as they minimize the space occupied by the molecules. Similar conformational adjustments have been noted in 4-biphenylcarboxylic acid structure, in which they result in dimerization of the molecular stacks in two-fold superstructure upon cooling.53
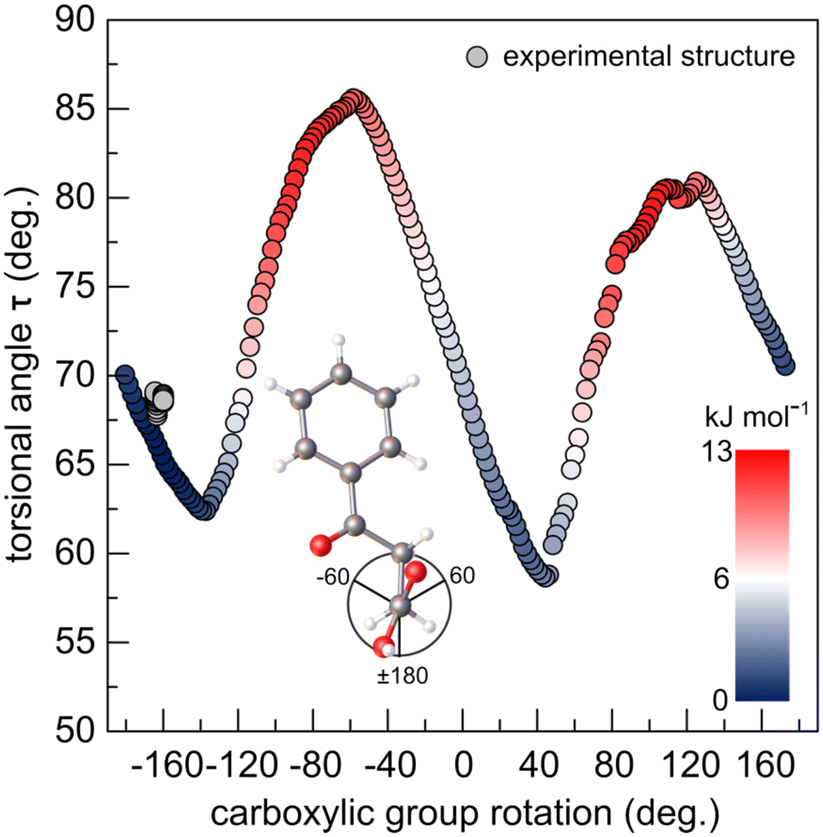 | ||
| Fig. 6 Potential energy (inset: colour scale) and torsional angle τ as a function of carboxylic group rotation (vide Fig. S4†) scan of the optimized BPA molecule. The experimental points, from SC-XRD collected in this study are depicted in grey. | ||
Steric hindrance between the carboxyl oxygens and carbonyl oxygen destabilizes the structure and forces τ to increase, in order to counteract the steric crowding. For this reason, LT phase with overall lower values of τ results in higher stability. Even though the energy gain from the τ-folding is small (given the rotation about the single C–C bond is worth 4–13 kJ mol−1),4 the pseudosymmetric LT phase is the only stable phase below 235 K. As the temperature of the system is increased from 200 to 290 K, thermal displacement parameters of the alkyl carbon atoms double (from ∼0.05 to 0.10 Å2). The thermal energy gained by the atoms at 240 K becomes large enough (RT ∼ 2.0 kJ mol−1) to activate the symmetrization of the supramolecular interaction pockets of two (initially) symmetry-independent molecules. Indeed, the calculated difference in Gibbs energy between the structures optimized at 0 and 298 K is only 3.0 kJ mol−1 – a fraction of the energy which is required to break a hydrogen bond. Symmetric dimer is preserved, while only the molecules in the parallel stacks differentiate due to the short intermolecular interactions between the stacks. To better understand the evolution of the short directional interactions in BPA, we have systematically studied its anisotropic thermal expansivity by high-resolution PXRD.
Thermal expansion
The discontinuities of the phase-change mechanism, which might be responsible for the abnormal thermal expansion in BPA, have been ruled out based on DSC and SC-XRD results, as described above. Subsequently, the thermal expansion has been investigated by multitemperature high-resolution synchrotron PXRD. The temperature of the sample has been calibrated by fitting diffraction data from NaCl internal calibrant, using low-temperature expansivity data from Meincke et al. (Table S2 and Fig. S5†).54The refined unit-cell parameters of BPA have shown an excellent agreement with the literature data for VERMAG01 (reported space group P21/n, V = 889 Å3 collected at 235 K) but not for VERMAG (reported space group P21/c, V = 1014 Å3 collected at 295 K). After the transformation of the P21/n structure into the standard setting, one can immediately see that the monoclinic β angle in VERMAG is essentially the same as in VERMAG01, while all the unit-cell axes are persistently overestimated by ∼4% (Table S1†). These erroneous values are reflected in the distorted molecular structure, where some of the C–C bonds are as long as 1.59–1.60 Å. The data in the study were collected using the Weissenberg camera, and one of the possible reasons for the discussed overestimation may arise simply from the incorrectly noted/determined effective camera radius. Although the crystals used in the two past studies were obtained from two different recrystallization solvents, methanol and benzene, respectively for VERMAG01 and VERMAG, they exhibit identical packing with a substantial difference in density, as additionally reported (in both cases) from a standard flotation technique at room temperature. Similarly, to the study of VERMAG01, we recrystallized BPA from methanol (both cold and from the boiling solution), to check the possibility of further polymorphism or chemical reaction with the solvent. Both recrystallizations resulted in the RT phase as reported in this study, and no formation of esters was observed.
The volumetric thermal expansion of BPA determined from high-resolution PXRD in the range 200–310 K is αv = 217(3) MK−1, which is a typical value for organic crystals. The lack of abnormal high thermal expansion, in contrary to what could be inferred from the two past studies, stems from the inaccurate data in one of them (VERMAG01).
The linear thermal expansion coefficients were determined for the orthogonalized cell (Table 2 and Fig. 7a). The most compressible, crystallographic a axis coincides with the direction of the molecular zig-zag chains. The compression mechanism in this direction lies in accordion-like motion, during which the rigid hydrogen-bonded sections can tilt with respect to one another through perpendicular π–π interactions. The crystallographic b axis, perpendicular to the zig-zag stacks, is less compressible, as the BPA zig-zag tightly fits into another in this direction, stabilized by weak π–π stacking. The [103] direction is the least compressible and coincides with the normal to the main zig-zag chain axis. This direction is dominated by short intermolecular interactions between the atoms from adjacent stacks, some of them shorter than the sum of van der Waals radii already at ambient temperature. This property can be visualized with the de parameter (distance from the surface to the nearest nucleus outside the surface) mapped onto the Hirshfeld surface of BPA.55 As long as BPA remains in its Z′ = 2 form, the expansivity of ε2 and ε3 are nearly identical and start to deviate from one another above 240 K (Fig. 7a). The coincidence of the onset of this deviation with the phase transition to the Z′ = 1 structure, prompts us to relate the thermal expansion to the presence of pseudosymmetry. However, the polynomial character of the ε3 thermal expansion trend stems from the stronger repulsive character of multiple short intermolecular interactions in this direction (Fig. 7b), rather than from symmetry considerations. These repulsive forces appear to cause deformation of the parallel BPA molecules in the adjacent stacks which, have to adapt to the shorter collision distance. As a result, they assume two different most energetically-efficient conformations (Z′ = 2) in the crystal. Notably, the other two directions, ε1 and ε2, lack such short interactions, which is reflected in their linear character over the whole studied temperature range. The continuous character of phase change from Z′ = 1 to Z′ = 2 structure upon cooling, relies on the shortening of the repulsive interactions perpendicular to BPA stacks, as evidenced by the thermal expansion studies. Remarkably, no hysteresis was observed in heating–cooling cycles, both in thermal expansivity, and the onset of superlattice reflections (Table S3†).
| Axes | α [MK−1] | Approximate crystallographic axis |
|---|---|---|
| ε 1 | 112(2) | [100] |
| ε 2 | 69(3) | [010] |
| ε 3 | 33(4) | [103] |
| V | 217(3) |
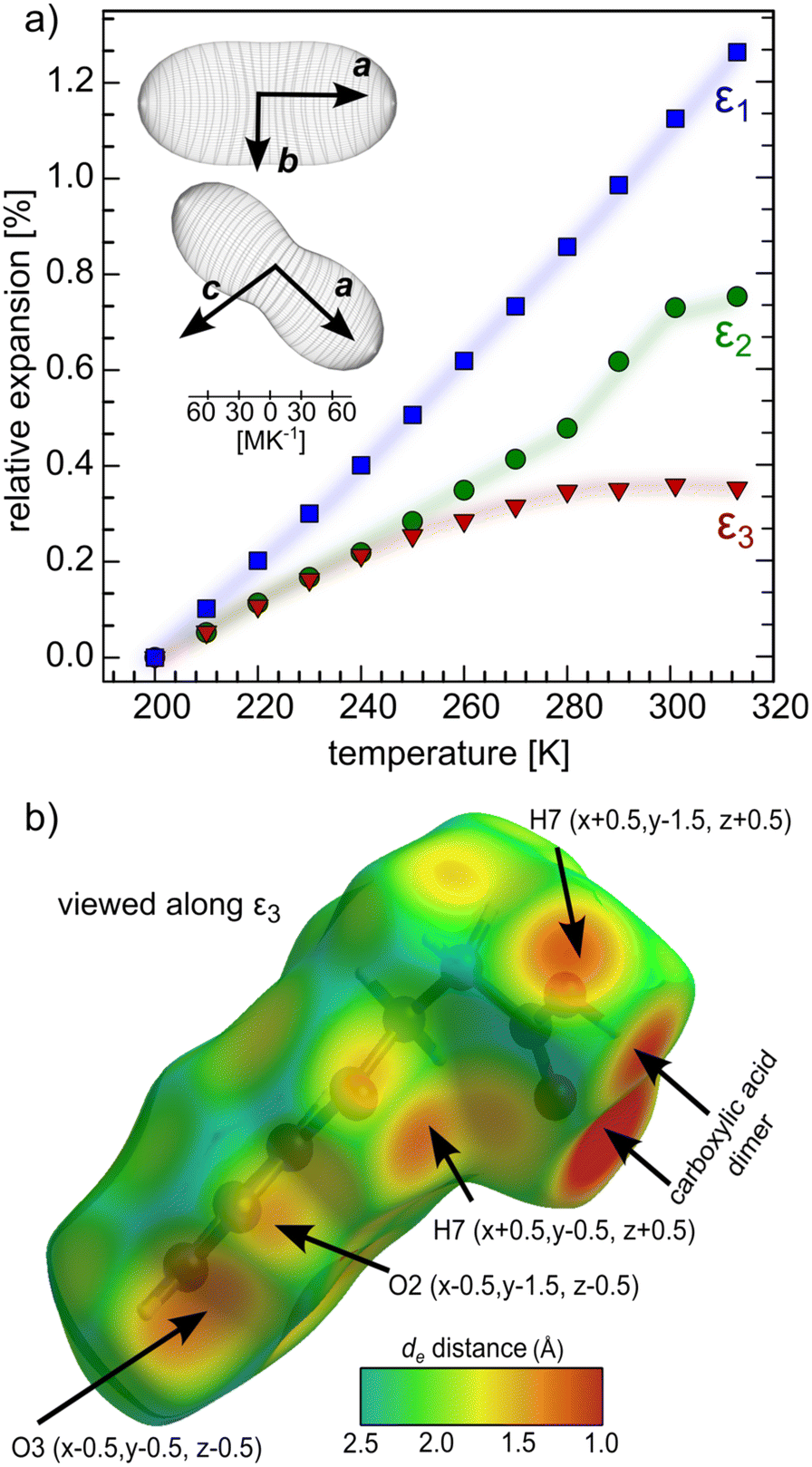 | ||
| Fig. 7 Anisotropic thermal expansion in BPA (a). Inset: Orientation of thermal-expansion indicatrices with respect to the unit cell of BPA in the RT polymorph (P21/n space group). Shortest intermolecular contacts to BPA molecule plotted as de parameter on the Hirshfeld surface view along the least expandable direction (b). Intermolecular contacts shorter than the sum of van der Waals radii are depicted with arrows. | ||
Conclusions
The current study revealed the existence of two polymorphs of BPA, the transition of which occurs at about 240 K. Transition is continuous and resembles Landau k-type transition into a less symmetric structure on cooling. The molecular packing in both of them is essentially the same, however, the competition between RT (Z′ = 1) and LT (Z′ = 2) structures results from subtle variations of the BPA molecular conformation, which can be expressed by the different torsional angles between carboxylic carbon and the propyl chain. The values of this angle are significantly different in two independent molecules of LT structure, but they converge becoming statistically indiscernible in RT structure. The thermal energy of the atoms at 240 K becomes large enough to activate the symmetrization of the supramolecular interactions, due to the small difference between the energies of LT and RT structures (∼2.0 kJ mol−1).The investigation of the thermal expansivity of BPA supported by DSC measurements, rules out the abnormal thermal expansion in BPA. The thermal expansion along [100] and [010] exhibits linear character, whereas the rather polynomial character of [103] thermal expansion trend, the direction normal to the molecular stacks, is a consequence of the presence of the short repulsive intermolecular interactions in this direction. These repulsive interactions are likely to cause deformation of the BPA molecules in the LT phase, which results in the increase of Z′ value from 1 to 2, due to the subtle sterically-induced differentiation of BPA conformations.
Author contributions
T. P. – conceptualization, data curation, formal analysis, investigation, methodology, visualisation, writing – original draft, writing – review & editing. M. S. and G. C. – investigation, writing – original draft, writing – review & editing. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge the European Synchrotron Radiation Facility (ESRF) for provision of synchrotron radiation facilities (ID22 beamline). T. P. thanks Dr. Stefano Racioppi for his help with carrying out quantum calculations.Notes and references
- R. Tandon, N. Tandon and R. K. Thapar, Pharm. Pat. Anal., 2018, 7, 59–63 CrossRef CAS PubMed.
- R. Censi and P. Di Martino, Molecules, 2015, 20, 18759–18776 CrossRef CAS PubMed.
- A. J. Cruz-Cabeza and J. Bernstein, Chem. Rev., 2014, 114, 2170–2191 CrossRef CAS PubMed.
- A. Nangia, Acc. Chem. Res., 2008, 41, 595–604 CrossRef CAS PubMed.
- H.-J. Zhang, S.-M. Peng, X.-S. Zhou and X. Ju, EPL, 2014, 107, 46002 CrossRef.
- K. M. Steed and J. W. Steed, Chem. Rev., 2015, 115, 2895–2933 CrossRef CAS PubMed.
- C. R. Groom, I. J. Bruno, M. P. Lightfoot and S. C. Ward, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2016, 72, 171–179 CrossRef CAS PubMed.
- R. E. Marsh, Acta Crystallogr., Sect. B: Struct. Sci., 1999, 55, 931–936 CrossRef PubMed.
- T. Rekis, A. Schönleber, L. Noohinejad, M. Tolkiehn, C. Paulmann and S. van Smaalen, Cryst. Growth Des., 2021, 21, 2324–2331 CrossRef CAS.
- T. Rekis, A. Schönleber and S. van Smaalen, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2020, 76, 18–27 CrossRef CAS PubMed.
- J. W. Steed, CrystEngComm, 2003, 5, 169–179 RSC.
- A. Gavezzotti, CrystEngComm, 2008, 10, 389–398 CAS.
- C. P. Brock and L. L. Duncan, Chem. Mater., 1994, 6, 1307–1312 CrossRef CAS.
- R. Taylor, J. C. Cole and C. R. Groom, Cryst. Growth Des., 2016, 16, 2988–3001 CrossRef CAS.
- L. N. Kuleshova, M. Y. Antipin and I. V. Komkov, J. Mol. Struct., 2003, 647, 41–51 CrossRef CAS.
- B. Sarma, S. Roy and A. Nangia, Chem. Commun., 2006, 4918 RSC.
- G. S. McGrady, M. Odlyha, P. D. Prince and J. W. Steed, CrystEngComm, 2002, 4, 271–276 RSC.
- G. S. Nichol and W. Clegg, CrystEngComm, 2007, 9, 959 RSC.
- C. M. L. Vande Velde, B. Tylleman, M. Zeller and S. Sergeyev, Acta Crystallogr., Sect. B: Struct. Sci., 2010, 66, 472–481 CrossRef CAS PubMed.
- K. M. Anderson, S. E. Tallentire, M. R. Probert, A. E. Goeta, B. G. Mendis and J. W. Steed, Cryst. Growth Des., 2011, 11, 820–826 CrossRef CAS.
- S. Dey, A. Schönleber, S. Mondal and S. van Smaalen, Z. Kristallogr. - Cryst. Mater., 2016, 231, 427–434 CrossRef CAS.
- S. Dey, A. Schönleber, S. Mondal, S. I. Ali and S. van Smaalen, Cryst. Growth Des., 2018, 18, 1394–1400 CrossRef CAS.
- T. Rekis, A. M. Schaller, S. R. Kotla, A. Schönleber, L. Noohinejad, M. Tolkiehn, C. Paulmann and S. van Smaalen, IUCrJ, 2021, 8, 139–147 CrossRef CAS PubMed.
- R. Lee, J. A. K. Howard, M. R. Probert and J. W. Steed, Chem. Soc. Rev., 2014, 43, 4300–4311 RSC.
- K. W. Rajewski, M. Andrzejewski and A. Katrusiak, Cryst. Growth Des., 2016, 16, 3869–3874 CrossRef CAS.
- K. Friese and A. Grzechnik, Z. Kristallogr. - Cryst. Mater., 2014, 229(2), 158–168 CAS.
- H. W. Thompson, P. A. Vanderhoff and R. A. Lalancette, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1991, 47, 1443–1445 CrossRef.
- S. Selladurai, M. S. Kumar and K. Subramanian, Proc. Indiana Acad. Sci., 1990, 102, 39–43 CrossRef CAS.
- A. van der Lee and D. G. Dumitrescu, Chem. Sci., 2021, 12, 8537–8547 RSC.
- C. F. H. Allen and L. F. Somerville, Org. Synth., 1933, 13, 12 CrossRef.
- Rigaku Oxford Diffraction, 2019 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8 CrossRef PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- C. F. Macrae, I. Sovago, S. J. Cottrell, P. T. A. Galek, P. McCabe, E. Pidcock, M. Platings, G. P. Shields, J. S. Stevens, M. Towler and P. A. Wood, J. Appl. Crystallogr., 2020, 53, 226–235 CrossRef CAS PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- A. Fitch, C. Dejoie, E. Covacci, G. Confalonieri, O. Grendal, L. Claustre, P. Guillou, J. Kieffer, W. de Nolf, S. Petitdemange, M. Ruat and Y. Watier, J. Synchrotron Radiat., 2023, 30, 1003–1012 CrossRef CAS PubMed.
- A. A. Coelho, J. Appl. Crystallogr., 2018, 51, 210–218 CrossRef CAS.
- A. Le Bail, Powder Diffr., 2005, 20, 316–326 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, R. B. 0. Gaussian 16, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B, 1988, 37, 785–789 CrossRef CAS PubMed.
- S. H. Vosko, L. Wilk and M. Nusair, Can. J. Phys., 1980, 58, 1200–1211 CrossRef CAS.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- A. V. Marenich, R. M. Olson, C. P. Kelly, C. J. Cramer and D. G. Truhlar, J. Chem. Theory Comput., 2007, 3, 2011–2033 CrossRef CAS PubMed.
- S. P. Thomas, P. R. Spackman, D. Jayatilaka and M. A. Spackman, J. Chem. Theory Comput., 2018, 14, 1614–1623 CrossRef CAS PubMed.
- C. F. Mackenzie, P. R. Spackman, D. Jayatilaka and M. A. Spackman, IUCrJ, 2017, 4, 575–587 CrossRef CAS PubMed.
- A. Gavezzotti, Acta Crystallogr., Sect. B: Struct. Sci., 2008, 64, 401–403 CrossRef CAS PubMed.
- A.-K. Larsson and A. G. Christy, Am. Mineral., 2008, 93, 103–113 CrossRef CAS.
- W. Sun, Y.-X. Huang, Y. Pan and J.-X. Mi, J. Solid State Chem., 2012, 187, 89–96 CrossRef CAS.
- A. G. Shtukenberg, C. T. Hu, Q. Zhu, M. U. Schmidt, W. Xu, M. Tan and B. Kahr, Cryst. Growth Des., 2017, 17, 3562–3566 CrossRef CAS.
- L. D. Landau, Zh. Eksp. Teor. Fiz., 1937, 7, 19–32 CAS.
- R. Strothmann, S. van Terwingen, I. Kalf and U. Englert, CrystEngComm, 2021, 23, 841–849 RSC.
- S. Dey, S. Sasmal, S. Mondal, S. Kumar, R. Chowdhury, D. Sarkar, C. Malla Reddy, L. Peters, G. Roth and D. Haldar, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2023, 79, 148–156 CrossRef CAS PubMed.
- P. P. M. Meincke and G. M. Graham, Can. J. Phys., 1965, 43, 1853–1866 CrossRef CAS.
- M. A. Spackman and D. Jayatilaka, CrystEngComm, 2009, 11, 19–32 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: DSC data and selected crystallographic parameters. CCDC 2283189–2283199. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3ce00940h |
| This journal is © The Royal Society of Chemistry 2023 |