
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFirst investigation of the morphological and luminescence properties of HfO2 nanoparticles synthesized by photochemical synthesis†
Irene
Villa‡
 *a,
Lenka Prouzová
Procházková
ab,
Eva
Mihóková
a,
Vladimir
Babin
a,
Robert
Král
a,
Petra
Zemenová
a,
Alexandra
Falvey
a,
Václav
Čuba
b,
Matteo
Salomoni
cd,
Fiammetta
Pagano
cd,
Roberto
Calà
cd,
Isabel
Frank
ce,
Etiennette
Auffray
c and
M.
Nikl
a
*a,
Lenka Prouzová
Procházková
ab,
Eva
Mihóková
a,
Vladimir
Babin
a,
Robert
Král
a,
Petra
Zemenová
a,
Alexandra
Falvey
a,
Václav
Čuba
b,
Matteo
Salomoni
cd,
Fiammetta
Pagano
cd,
Roberto
Calà
cd,
Isabel
Frank
ce,
Etiennette
Auffray
c and
M.
Nikl
a
aFZU Institute of Physics, Czech Academy of Sciences, Cukrovarnicka 10, 162 00 Prague, Czech Republic. E-mail: villa@fzu.cz
bFaculty of Nuclear Sciences and Physical Engineering CTU in Prague, Czech Republic
cEuropean Organization for Nuclear Research (CERN), Geneva, Switzerland
dDipartimento di Fisica “Giuseppe Occhialini”, Università degli Studi Milano-Bicocca, Piazza della Scienza 3, 20126 Milano, Italy
eLudwig Maximilian University of Munich, Geschwister-Scholl-Platz 1, Munich, Germany
First published on 30th June 2023
Abstract
For the first time, hafnia (HfO2) nanoparticles have been produced by photochemical synthesis. The photochemical route has been proven to be scalable, affordable, and straightforward to create monoclinic HfO2 nanoparticles with a size of tens of nanometers. The exploitation of this route offers a chance to create large amounts of dense nanoparticles with reduced costs and time of production for future creation of large area composite scintillators for fast timing techniques. Specific annealing treatments from 450 °C to 1000 °C have been targeted to tune the structural and morphological properties and optimize the luminescence of the nanoparticles. Hafnia nanoparticles annealed at low temperature display an amorphous structure. After thermal treatment at 1000 °C, HfO2 nanoparticles crystallize into a monoclinic phase, as evidenced by thermal analyses and X-ray diffraction. Radioluminescence and photoluminescence of HfO2 have been investigated at room temperature and 77 K. The emission band of hafnia covers a range from 300 to 600 nm and it can be attributed to defects within the matrix. In particular, the highest radioluminescence intensity appears in the crystalline sample annealed at 1000 °C, thanks to the higher crystallinity degree and the reduction of non-radiative channels and quenching defects. Photoluminescence and scintillation decay have evidenced the presence of the fast decay in the nanosecond time range. Thus, due to their size, density, and spectroscopic and timing features, monoclinic hafnia nanoparticles obtained by photochemical synthesis are attractive for potential creation of large area scintillating composites.
1. Introduction
Hafnium oxide (hafnia, HfO2) has come into the scientific world as an excellent and versatile material for electronic and industrial applications thanks to its dielectric constant, hardness, and chemical stability.1–4 Thanks to its wide band gap (Eg = 5.6 eV), optical transparency in the UV-visible (vis) range, high density, and high Hf atomic number (ρ (HfO2) = 9.6 g cm−3; Z (Hf)= 72), HfO2 has been proposed in lighting5 and scintillation applications.6,7 In the literature, most studies on the luminescence properties of hafnia under excitation by optical light and ionizing radiation have been explored in depth on thin films, and pure and doped nanoparticles (NPs).7–14 In contrast, the research of the spectroscopic properties of HfO2 single crystals appears only in a limited number of papers.15–17 This discrepancy is related to the complexity of the production of hafnia single crystals due to their high melting point temperature (above 2700 °C).4,18 The most used synthetic routes for the manufacture of ultra-small metal oxide NPs are wet chemical syntheses, like hydrothermal and Pechini-based sol–gel methods,19–21 together with microemulsion processes22 and precipitation routes.23 Lastly, the non-hydrolytic sol–gel method (NHSG) has been successfully used to obtain ultra-small metal oxide NPs.24,25 The exploitation of these syntheses allows the production of nanostructured crystalline HfO2 with controlled surface and morphology characteristics, size, and crystalline phase by using appropriate solvents and growth conditions and/or annealing treatments.26–28 The manipulation of the structural and morphological properties of hafnia NPs results in the tuning of their light output efficiency and colour of the emission, under both ionizing radiation and optical light. The chance to create hafnia NPs with tailored luminescence qualities has been more and more attracting the materials science community towards the use in cutting edge technologies and in advanced radio-oncological therapeutic protocols.29,30 Notably, thanks to the high stopping power and – hence – the ability to interact with ionizing radiation, luminescent hafnia NPs have been recently proposed as a key constituent of composite scintillators for the design of next generation scintillation detectors for applications in fast-timing techniques, such as advanced time of flight positron emission tomography (ToF-PET) and high energy physics experiments, or homeland security measures and border controls.31–34 These composite scintillators are generally made with a dense nanoscintillator, like HfO2, coupled to a dye featuring high quantum yield and fast lifetime and a polymeric host matrix.35,36 Next generation detectors designed on composite scintillators are expected to have superior timing and sensitivity performances with respect to those based on inorganic bulk and plastic scintillators, currently employed in fast timing technologies.36–38 However, the creation of such detectors implies the manufacture of reliable, affordable and large area plastic scintillators. Recent studies in the chemistry field have been focusing on establishment of new syntheses to scale up the production of dense metal oxide NPs with reduced costs and times. In this context, photo-induced synthesis presents a valid and solid route to produce highly efficient luminescent nanomaterials with respect to conventional approaches.39–44 The perspective of the up scaling of the method has already been shown.45,46 The advantage of exploiting the photochemical route for the production of large amounts of NPs comes from several evidence, among which, the softening of strict control of temperature (T) and pH parameters, required in the majority of chemical methodologies. Moreover, by choosing the stoichiometry of the precursors and reaction times, the chemical product of the synthesis can be easily doped, and its size can be scaled down to the nanometer size, a peculiarity that is mandatory for the creation of scintillating nanocomposites to avoid scattering and the subsequent reduction of transparency.32,36,47 However, so far, the photochemical synthesis has not been employed to produce HfO2 NPs.Encouraged by the increasing interest in plastic composite scintillators based on hafnia and by the known versatility of the photochemical routes to produce scalable quantities of nanomaterials, we have explored the reliability of photochemical synthesis for the synthesis of hafnia NPs. Hafnia NPs have been produced and annealed at different temperatures (from 450 °C to 1000 °C). Amorphous hafnia has been obtained at the lowest annealing temperature, while pure monoclinic hafnia with a dimension in the range of tens of nm has been achieved by the 1000 °C annealing treatment, as revealed by morphological and structural analyses (X-ray diffraction (XRD), transmission electron microscopy (TEM), and differential scanning calorimetry and thermogravimetry (DSC-TGA) coupled to mass spectrometry (MS)). The luminescence properties of HfO2 have been tested by the steady state and time resolved radioluminescence (RL) and photoluminescence (PL) measurements from RT down to 77 K. The amorphous and crystalline HfO2 prepared by photochemical synthesis present a broad luminescence band spanning from 300 to 600 nm under excitation by both UV and ionizing radiation. These findings agree with the spectroscopic properties of HfO2 NPs produced by different synthesis routes, as it is attested by a large number of publications in the literature. In particular, the luminescence of nanocrystalline hafnia is commonly attributed to oxygen vacancy related defects, colour centres,5,48–50 as well as to radiative decays of excitons.51–53
For the first time, this investigation reveals that the photochemical synthesis is suitable for a straightforward manufacture of monoclinic hafnia NPs with reduced costs and times. Interestingly, the visible emission band of hafnia NPs lies in the wavelength range matching the absorption spectrum of many common fast organic dyes, a condition that is required for the creation of composite scintillators.35 Moreover, time resolved (TR) PL and scintillation measurements have evidenced that the luminescence band of crystalline hafnia annealed at 1000 °C has fast decay in the order of a few ns. These qualities make the obtained monoclinic hafnia NPs suitable for the creation of large area composite scintillators for foreseen applications in medical imaging and high energy physics.
2. Experimental section
2.1. Synthesis and annealing treatments
The HfO2 NPs were prepared via photo-induced synthesis according to ref. 54, based on the reaction of dissolved salts with products of water photolysis followed by the photo-induced precipitation of the solid products (Fig. 1). The principle of the synthesis lies in the photochemical precipitation of amorphous/weakly crystalline solid precursors in a sequence of steps, as listed below:1. UV irradiation of aqueous solutions.
2. Formation of finely dispersed solid precursors in the amorphous or weakly crystalline phase.
3. Separation of solid precursors via microfiltration. Afterwards, products were washed with ethanol and deionized water and dried at laboratory temperature.
4. Annealing procedures at different temperatures to obtain final products in the form of white powders.
 | ||
| Fig. 1 Photo-induced synthesis; left – aqueous solution before irradiation, middle – precipitation of the solid phase during irradiation, right – gelatinous product after irradiation and sedimentation. | ||
In detail (steps 1–3), the irradiated solution contains 2.8 × 10−3 mol dm−3 Hf4+, 2 × 10−2 mol l−1 HCOONH4 (99.995%, Sigma Aldrich) and 0.5 mol dm−3 H2O2 (30%, penta). HfCl4 (99.9%, Sigma Aldrich) was used as a precursor for Hf4+; it exothermally hydrolysed to HfOCl2 and then it is dissolved in deionized water. Continuously stirred solution in a 2 l photoreactor was irradiated for 3 hours with low-pressure mercury lamps (254 nm) with a total nominal power of 100 W. The product in a gel-like consistency was then sedimented for 2–4 hours and separated via decantation and microfiltration using a Millipore HAWP 0.45 μm filter. The product was washed with deionized water and ethanol and dried at 40 °C for 24–48 h. The product was then treated at 450 °C and 1000 °C in air for 1 hour.
The formation of HfO2 solid precursors is not fully understood, and this topic deserves further investigation; however we can assume the occurrence of photoinduced reactions similar to the case of ZnO in the presence of H2O2.40,45 Chemical yields of UV-light reaction routes can be close to 80–95%; however the precipitation yield can vary with both the concentration of the salts in solutions and the irradiation time.45
The annealing treatment (step 4) of the samples was carried out in a vacuum furnace 0415 VAK (Clasic s. r. o.) with a regulator, enabling us to set up the temperature, temperature gradient, and dwell time at a given temperature. The treatment proceeded in air with a temperature gradient of 10 °C min−1 for 1 hour.
2.2. Characterization
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1). An empty alumina crucible was used as a reference. The DSC-TGA apparatus was calibrated in the temperature range of 25–1300 °C using the following standards (Sn, Ag2SO4, Al, Ag, and Au). The standard deviation of the performed calibrations is in the range of ±0.8 K. Each DSC-TGA measurement was preceded by a blank experiment with two empty alumina crucibles and recorded under the same conditions as the experiment itself.
:1). An empty alumina crucible was used as a reference. The DSC-TGA apparatus was calibrated in the temperature range of 25–1300 °C using the following standards (Sn, Ag2SO4, Al, Ag, and Au). The standard deviation of the performed calibrations is in the range of ±0.8 K. Each DSC-TGA measurement was preceded by a blank experiment with two empty alumina crucibles and recorded under the same conditions as the experiment itself.
Mass spectrometry (MS) was performed using a quadrupole mass spectrometer Pfeiffer OmniStar GSD 320 equipped with a controlled and heated anti-corrosive quartz capillary, a coupling element enabling a connection with a Themys 24 yttria coated iridium filament as a source of ionization, secondary electron multiplier (SEM) as a detector (detection limit <1 ppm of gases), and a turbo drag pumping system with an interstage turbo pump and diaphragm pump as a vacuum source. The measurements were conducted in a multiple ion detection regime (MID) with a gas flow of 1–2 cm3, 850–1450 V at the SEM detector, a mass detection dwell of 50 ms, a resolution of 50, in the m/z range of 1–50 amu, and a signal sensitivity below 1 ppm.
Thermomechanical analysis (TMA) of the samples was performed with a thermomechanical analyser Setaram Labsys 1400 in the temperature range of 25–700 °C and an inert N2 atmosphere, at a heating rate of 10 K min−1, and a load of 10 g. The powder samples were placed into an alumina crucible and covered with an alumina cylinder, which was pressed with the TMA sensor. The TMA analyser was calibrated with the standards (aluminium, silver, and corundum), and a blank run (baseline) was carried out before each TMA measurement of the sample.
The data from all performed analyses (DSC-TGA-MS, TMA) were processed by the Calisto Processing software.
| FWHM2 = b2 + bstrain2 + bsize2 |
| l = Kλ βsizecosθhkl, |
keV and a mean energy of 15 keV, excited with a pulse diode laser (PDL 800-B, PicoQuant), was used. The scintillation light was collected in the reflection mode using a hybrid photomultiplier tube (HPM 100-07, Becker & Hickel), operating in the TCSPC mode, and processed with an amplifier and timing discriminator (model 9237, ORTEC). This processed HPM output signal was used as the stop signal for the time-to-digital converter (TDC xTDC4, chronologic), while the start signal was given by the external trigger of PDL. An optical band-pass filter (450 nm with a FWHM of 40 nm) cuts the air luminescence excited by X-rays. The scintillation pulse was fitted by a convolution of the instrument response function (IRF) of the whole system with a full width at half maximum (FWHM) of 160 ps and the collected scintillation.56
3. Results and discussion
3.1. Structural and morphological qualities of NPs – evidence of the creation of monoclinic HfO2
In order to explore the significant effects of annealing on the morphology and structural properties of the HfO2 NPs, the untreated sample (i.e., the pristine product of the photochemical synthesis) and HfO2 NPs annealed at 1000 °C (HfO2-1000 °C, i.e., at the maximum annealing temperature) have been investigated by DSC-TGA-MS analyses. These measurements (under Ar + 20 mol% O2 atm) have demonstrated the decomposition of organic precursors (residuals) from the sample surface at temperatures starting above 50 °C in the pristine sample as shown in Fig. 2.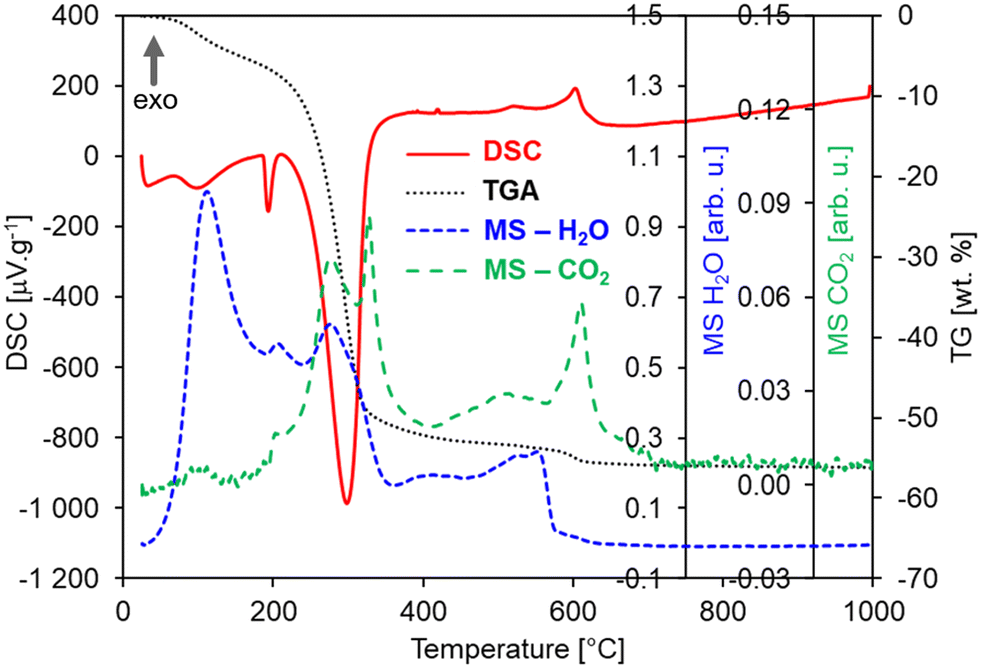 | ||
| Fig. 2 DSC-TGA-MS curves of the HfO2 untreated sample under an Ar + 20 mol% O2 atmosphere. | ||
Multiple endothermic and exothermic effects on DSC (red curve, Fig. 2) have been recorded in a wide temperature range from 50 to 700 °C mostly accompanied with a significant mass loss of ca. 55 wt% (TG, black curve, Fig. 2). The endo-peaks with onset temperatures of 82 °C, 187 °C, and 244 °C and a mass loss of ca. 42% are accompanied by the release of H2O and CO2 fragments, which can be mostly ascribed to products of such decomposition, combustion or evaporation of organic residuals, as shown in the MS curves (Fig. 2).57–60 The following three exo-peaks (528, 573, and 619 °C) with a small mass loss of ca. 4% can be ascribed to the crystallization of hafnium carbonate followed by its decomposition into the HfO2 crystalline monoclinic phase.57,58 This is also supported by the evolution of H2O and CO2 gases or their fragments, which are released from pores while the crystallization proceeds, or decomposition of residual carbonates as it was observed for ZnO NPs.61 In this case, the intensity of the MS peaks is lower than that of the endo-peaks pointing to a lower content of e.g. carbonates and formates. The origin of residuals is related to the specific procedures of the photochemical synthesis since when NPs are formed, their surfaces are covered by an organic layer coming from an excess of the ammonium formate ((HCOO)NH4) precursor. Generally, before getting the final metal oxide crystalline phase, any wet chemical method, such as photochemical synthesis, produces a gelatinous/solid precursor state of the metal oxide. This may contain chlorides, hydroxy groups, carbonates, organics, and a lot of loosely bound water. Fragments of all these compounds (e.g. CH3, OH, NH3, CO, O2, CO2, HCOO−, NO2) were recorded on the MS curves accompanying all DSC and TGA effects, see Table S1;† however, for simplicity, they are not displayed in Fig. 2(left). Most wet chemical methods for the synthesis of oxides requires annealing treatments to get rid of the water, hydroxy groups, and other volatile compounds. According to this evidence and the results obtained for untreated NPs, the DSC-TGA-MS curves of the NPs of hafnia annealed at 450 °C (HfO2-450 °C) are expected to be very similar to those of pristine HfO2, apart from the effects below 450 °C that have been irreversibly removed by annealing at 450 °C. In contrast, HfO2-1000 °C displays expectedly a different DSC-TGA record than that of the untreated HfO2, containing neither DSC peaks nor mass loss at all, see Fig. 2(right). In general, the influence of the Ar and Ar–O2 atmospheres on the DSC-TGA-MS of the untreated hafnia has not been observed, see Fig. S1 in the ESI.† The DSC curves under both atmospheres are almost identical and contained the same exo- and endo-peaks as described in Fig. 2(left). In contrast, the TGA curve under the Ar atmosphere has evidenced a smaller mass loss of ca. 45 wt% in total. This could be ascribed to the organic residuals experiencing only decomposition or evaporation and no oxidization.
Furthermore, the thermomechanical (TMA) properties of the hafnia samples have been investigated, as shown in Fig. S2(left),† where the TMA, DSC, and TGA curves of the HfO2 untreated sample are displayed for comparison. The TMA curves show two effects both matching the peaks in the DSC curve. The first sharp effect at 189 °C is related to the expansion of the sample for ca. 10%, which could suggest that the first order transition occurred and not the decomposition (or evaporation), as discussed above. By contrast, the MS curves prove opposite that a decomposition is occurring at this temperature as well. Thus, possibly both the transition and decomposition (or evaporation) take place simultaneously and they can be ascribed to organic residuals in the untreated NPs. This hypothesis is also corroborated by the fact that in HfO2-1000 °C (DSC, TGA, MS, TMA), these effects have not been evidenced, see Fig. 2(right) and S2(right).† On the other hand, oxidization can be excluded from the explanation as the TMA effect is occurring in both DSC curves measured under Ar and Ar–O2 atmospheres, see Fig. S1.† The second TMA effect in Fig. S2(left)† is more complex containing a slope change. First, there is a gradual decrease (at 196 °C), followed by very steep one (at 287 °C) for more than 60%, well matching the decomposition effects in DSC and TGA. Interestingly, HfO2-1000 °C is not expended as could be expected; its displacement remains constant up to 1000 °C and afterwards it slightly decreases for ca. 4% reaching a very low average coefficient of linear thermal expansion (CLTE) of 2.47 × 10–6 °C−1 at 50 °C, see Fig. S2(right) in the ESI,† which is in good agreement with the value for one of lattice parameters (a) reported in ref. 62. A possible explanation of this result can be attributed to powder densification and sintering during the thermal treatment. The massive impact of the temperature in removing the majority of organic residuals, and thus, in driving the structural change of NPs has been confirmed by the XRD patterns and TEM images obtained on the NPs annealed under diverse conditions. In the untreated NPs (Fig. S3†) and in the sample treated at low temperature (Fig. 3a), the XRD results have demonstrated the amorphous nature of the structure, where the atoms and ions are not in their thermodynamic equilibrium, while the TEM images of HfO2-450 °C have unveiled the presence of agglomerated NPs (Fig. 3b and c). As previously stated, crystallization requires heating at higher temperatures (above 500 °C) (Fig. 2). The XRD patterns have pointed out that HfO2-1000 °C features a pure monoclinic crystalline phase (space group P21/c) (Fig. 3a).
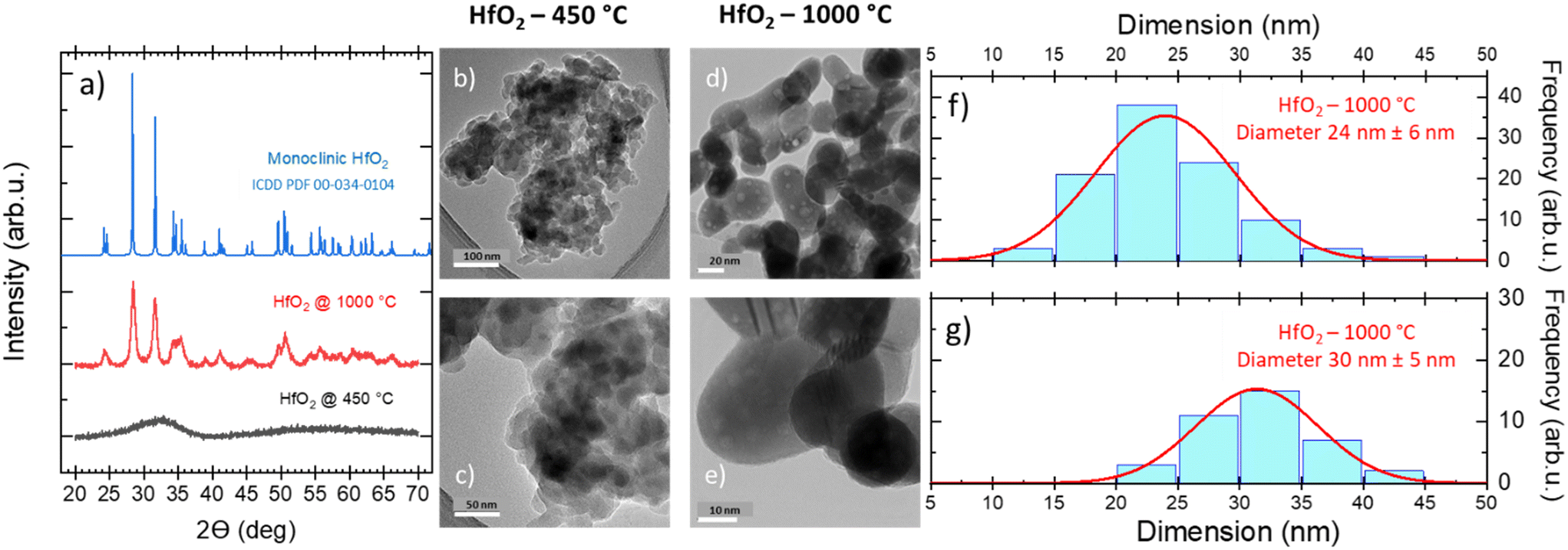 | ||
| Fig. 3 (a) Structural evolution of monoclinic hafnia NPs annealed at different temperatures. (a) XRD analysis performed on HfO2 suggests the amorphous nature of NPs; after annealing at 1000 °C, the XRD pattern reveals a crystalline domain growth (the pattern of monoclinic HfO2 [ICDD PDF: 00-034-0104] is reported for reference); (b–e) HR-TEM micrographs of HfO2 nanoparticles under two diverse annealing conditions. (f and g) Particle size distributions of elongated crystalline NPs annealed at 1000 °C, measured along the two axes. | ||
After thermal treatment at high T, the NPs present an elongated shape with a size averaged in two dimensions to 27 nm (Fig. 3d–g). The average crystallite size of 29 nm for HfO2-1000 °C calculated from the XRD patterns (values calculated from the XRD patterns by applying the Scherrer formula on the (111) plane diffraction located at 2θ = 31.62°) has been found to be in a good agreement with the one evaluated from the TEM images. The morphological changes, the improved crystallinity degree, and the removal of the organic residuals from the surface of the NPs induced by the annealing treatment and depicted above can modify the NP luminescence properties, as discussed in the next paragraph.
3.2. Luminescence features and timing properties of HfO2 NPs under light and soft X-ray irradiation
Considering the similarity of the amorphous structures of the pristine sample and the one annealed at 450 °C (XRD patterns in Fig. 3a and S3†), together with the DSC-TGA data evidencing that the crystallization of HfO2 occurs only after annealing at 1000 °C, we decided to explore in detail the luminescence properties only of the annealed samples under optical light and X-ray excitation and as a function of temperature. In this way, one can make sure to study the bands typical of HfO2 that are not perturbed by the presence of large amounts of residuals from the synthesis, as evidenced by thermogravimetric analyses.In Fig. 4, the PL//PLE spectra for the HfO2 NPs annealed at 450 °C and 1000 °C (Fig. 4a and c and b and d respectively) are displayed. The PLE spectra of both amorphous and crystalline samples have been acquired at 460 nm, where the RL maximum occurs. They reveal the presence of the excitation onset at wavelengths lower than 230 nm, the value close to the hafnia band gap. The emission of HfO2-450 °C can be excited solely at 250 nm, while the luminescence in HfO2-1000 °C can be excited at an additional band at around 310 nm. PL measurements for the HfO2 NPs annealed at 450 °C and 1000 °C have been performed by exciting the NPs under the 255 nm excitation. The PL spectrum of HfO2-450 °C shows a broad emission band peaking at about 420 nm. The luminescence band of HfO2-1000 °C is narrower, with a slight redshift of the peak towards 440 nm. The wider PL spectrum of the amorphous NPs with respect to those of crystalline hafnia can be attributed to various factors, such as the effect of inhomogeneous broadening, typical of amorphous materials, and the presence of defects specific to the amorphous state, as well as the luminescence contribution of the residual organic moieties remaining on the NP surface at low annealing temperature.63 PL under excitation in the UV excitation has also been investigated at 300 K and 77 K (Fig. 5c and d), to give a deeper insight into the observed emissions. The results show a general increase of the PL intensity at 77 K, mostly evident in the NPs annealed at 1000 °C. PL spectra of HfO2-1000 °C have been measured under diverse excitations (Fig. S5†). PL spectra are similar for all excitations, especially for those below the typical band gap of HfO2, suggesting that the PL emission is related to the defectiveness of the matrix.64 The radioluminescence of the annealed samples at 450 °C and at 1000 °C is presented in Fig. S4 in the ESI,† together with that of the pristine NPs. The radioluminescence of the annealed NPs is analogous to PL, characterized by an emission band peaking in the blue spectral region. Before annealing at high temperature, the RL intensity of the NPs is weak. Also, the RL measurements on hafnia NPs have been performed as a function of the temperature from room temperature to 77 K. The RL spectra at room temperature in HfO2 annealed at 450 °C and 1000 °C show the luminescence band peaking at about 440 nm (Fig. 5a and b). In addition, HfO2-1000 °C NPs present a shoulder at 330 nm, whose intensity increases with decreasing temperature. In agreement with the PL findings, HfO2-1000 °C features the highest RL intensity. In this context, in order to discover the occurrence of thermal quenching mechanisms and to assess the RL intensity temperature dependence, the RL spectra obtained by decreasing the temperature have been integrated within 420–600 nm. The RL intensity starts to drop at T >150 K, likely due to the thermal quenching effect as evidenced in Fig. S5(left) in the ESI.† A numerical fit of the RL intensity experimental data has been performed to determine the activation energies E of the thermal quenching using eqn (1) (Fig. S6(right) and Table S2, ESI†). The estimated E is 0.1 eV, comparable to the values of E reported in the literature for crystalline NPs of hafnia and other metal oxide nanosystems, produced by various chemical syntheses.64–66 This work also aims to address the timing properties of the emission of monoclinic crystalline hafnia NPs. The PL decay and the scintillation decay have been compared (Fig. 6a and b, respectively) by collecting the luminescence signals in the wavelength range of around 400–500 nm. Both the decays show the presence of a fast component with an average lifetime of a few nanoseconds (Exc 255 nm, τave = 3.0 ns; scintillation decay τave = 2.2 ns). In addition, the scintillation decay evidences a higher background than the PL decay, related to the occurrence of slower components that cannot be detected in the ns time window (cf.Fig. 6b). These slow components may be due to the presence of shallow charge traps acting in the transfer stage of the scintillation mechanism.
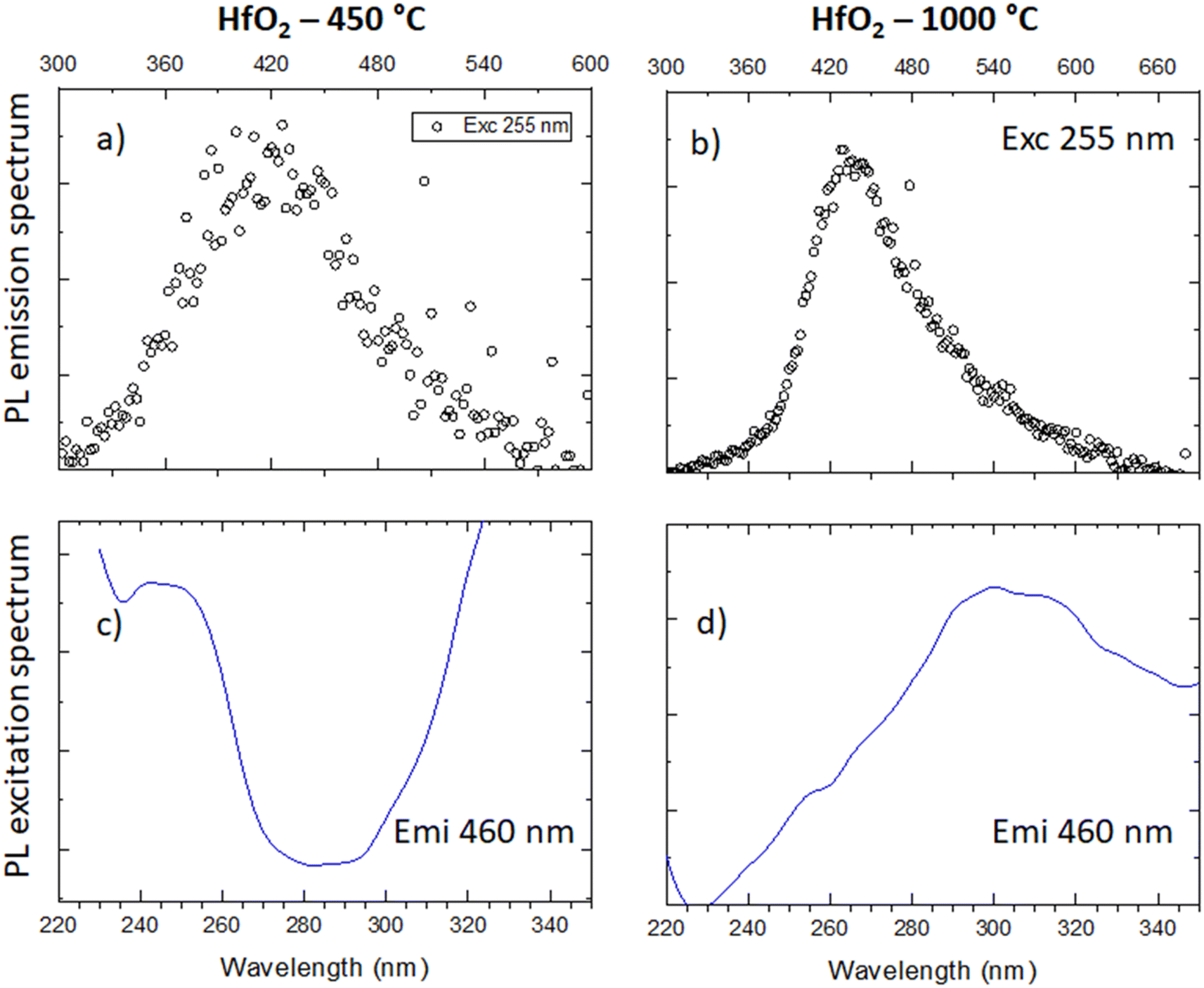 | ||
| Fig. 4 (a and b) PL emission spectra under 255 nm of HfO2 annealed at 450 °C and 1000 °C; (c and d) PLE spectra of the emission of the two samples monitored at 460 nm. | ||
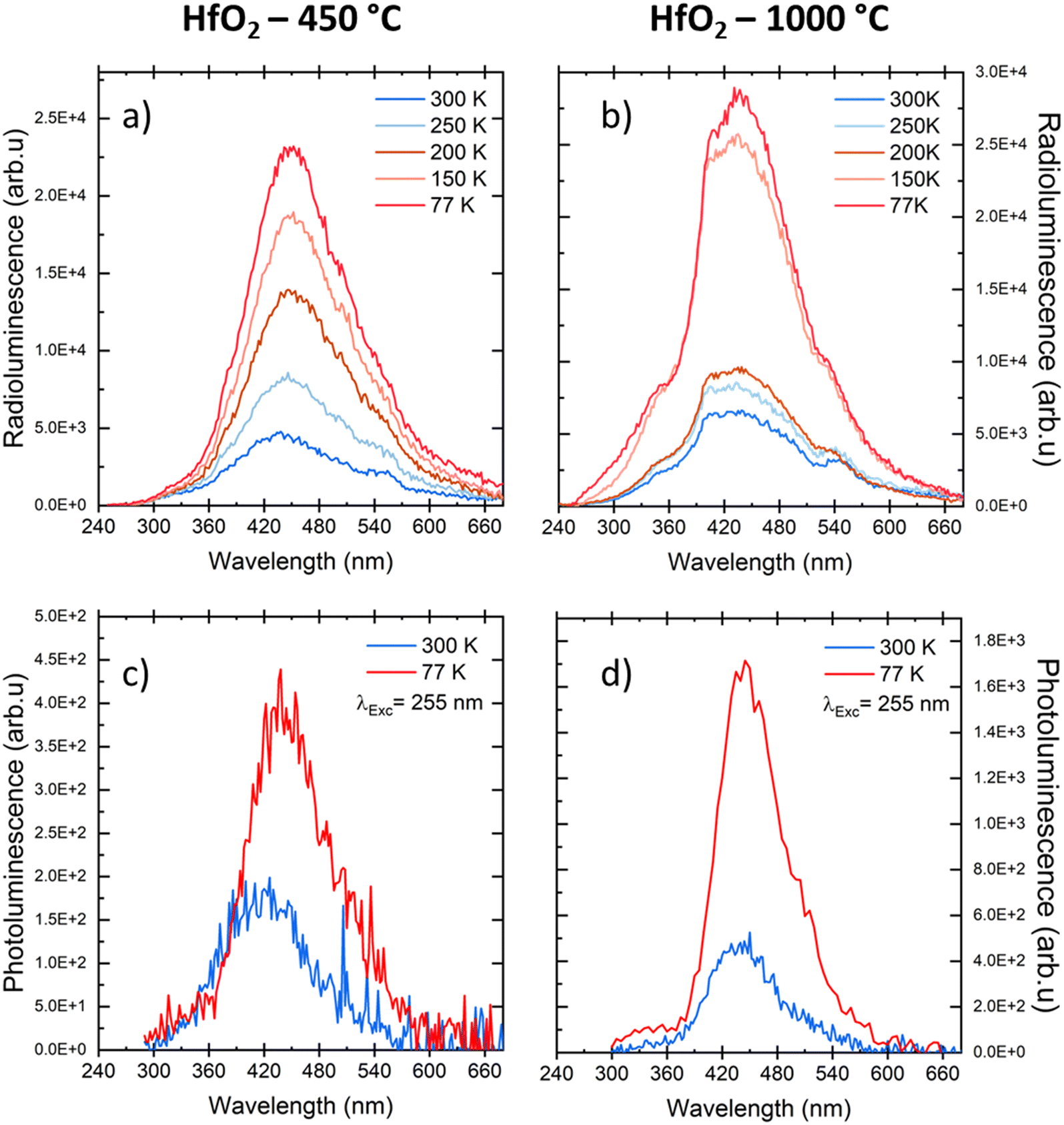 | ||
| Fig. 5 (a and b) RL spectra of HfO2 NPs annealed at 450 °C and 1000 °C measured at different temperatures; (c and d) PL spectra of the same samples under UV excitation at RT and at 77 K. | ||
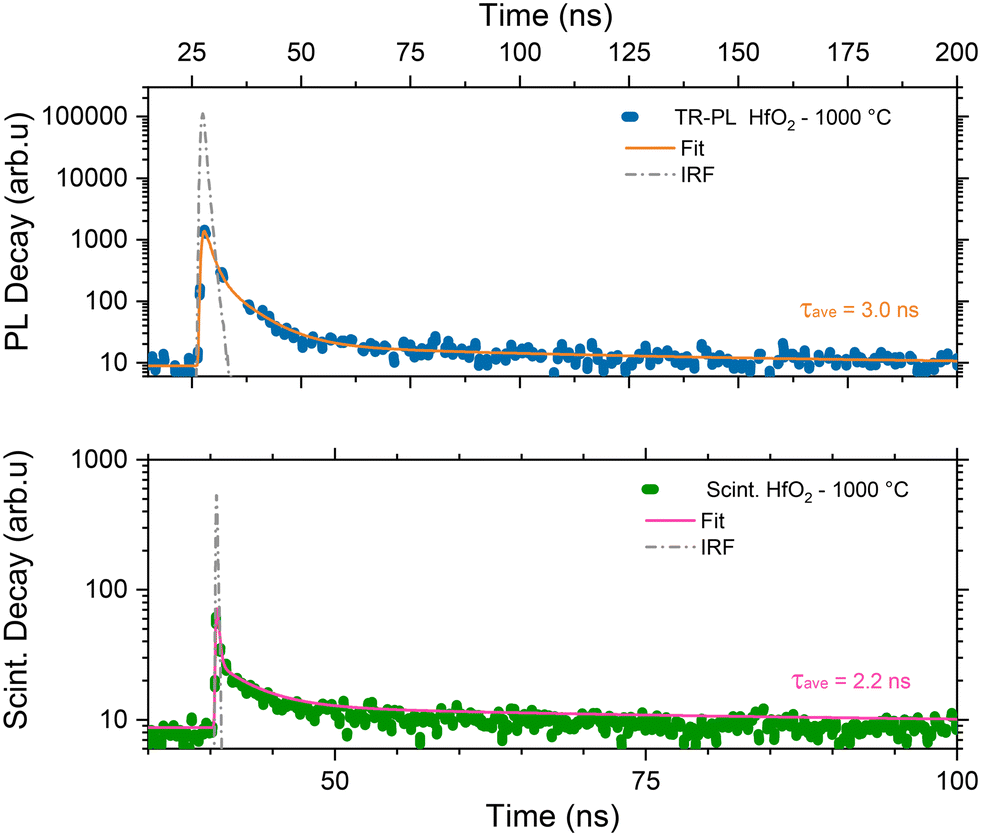 | ||
| Fig. 6 PL decay of monoclinic HfO2 annealed at 1000 °C under excitation at 255 nm. The luminescence has been collected through a low-pass filter cutting at 400 nm (top); scintillation decay spectra of the same NPs under X-ray excitation with a band pass filter peaked at 450 nm ± 40 nm (bottom). In the panels, the experimental data (blue and green dots) are presented together with the instrumental responses (dashed grey line) and the numerical fit of the decays (solid orange and pink line) together with the average calculated lifetime. The complete data set of the fit parameters is in Table S2 in the ESI.† | ||
3.3. Discussion
In this paragraph, the key points for the creation of pure monoclinic HfO2 from photochemical synthesis are presented together with the spectroscopic and timing features of the produced HfO2 NPs in order to highlight the most important indications helping to assess their suitability for scintillating composite materials.Photo-induced synthesis has enabled the quick production of the HfO2 NPs with the size close to 30 nm, just by choosing the appropriate mixture of starting solutions (see the details in the Experimental part) and the temperature conditions of the annealing treatments. In this work, the annealing strategies have been utilised as a tool to monitor the change and improvement of NP luminescence. The investigation of the PL and RL of the NPs as a function of annealing temperature has uncovered the relationship between the structural and morphological characteristics of the NPs and their luminescence. Our work has highlighted that only annealing at high temperatures allows the removal of organic residuals from the synthesis, the re-ordering of the crystal lattice matrix, the improvement of the crystallinity degree with amorphous hafnia NPs turning into a pure monoclinic phase, and the reduction of quenching surface defects that can alter and deteriorate the final luminescence of the NPs. Except for a slight broadening of the luminescence band at room temperature in the amorphous NPs, the amorphous and crystalline samples feature similar PL and RL spectra (Fig. 5). These can be explained by the fact that amorphous HfO2 can be described as a material with a short-range order and the similarity with the defectiveness of the monoclinic crystalline NPs, responsible for the luminescence emission, can be assumed. The luminescence of amorphous and crystalline hafnia produced by photochemical synthesis can be related to radiative transitions from excited states of the same recombination centres, as also validated by the similarity of the activation energies E of the emissions in the samples annealed at 450 °C and at 1000 °C (Table S2, ESI†).64
The reliability of exploiting the photochemical synthesis as a route for the creation of nanometric HfO2 NPs is supported by a plethora of publications on the morphological and spectroscopic characterization of HfO2, obtained by diverse chemical methodologies. Indeed, the NPs achieved by the photochemical route and by a subsequent annealing at high temperature feature the characteristic crystalline monoclinic structure of hafnia, and a visible fast emission peaked at around 440 nm, typically attributed to the defectiveness of the material, as it is largely proven in the literature for diverse nanometric metal oxide systems.5,16,67–73 The luminescence light output of the achieved NPs has been tested by comparing their RL intensities with the light output of a standard scintillator Bi4Ge3O12 (BGO) (Fig. S4†). From this comparison, it is evident that crystalline hafnia displays the most promising luminescence features in terms of light output, a crucial criterion to evaluate the scintillator performance.74 The exploitation of the photochemical route allows a large number of dense oxide NPs to be created with low cost and reduced production time. In particular, in this context it has allowed us to produce, for the first time, crystalline nanometric HfO2 with spectroscopic and timing features suitable for the creation of hafnia-based composite scintillators towards applications in fast-timing techniques.
4. Conclusions
Thanks to its density and high Z-number, luminescent hafnia has been proposed as an essential element for the design of novel composite scintillators, traditionally made with dense scintillators coupled to fast organic dyes and embedded into a plastic matrix. Composite scintillators are of primary importance for the creation of next generation, affordable, reliable, and large area detectors in fast timing techniques. This work has validated for the first time the exploitation of the photochemical synthesis as a scalable, affordable, and straightforward route for the production of HfO2 NPs. Structural and morphological analyses coupled to spectroscopic luminescence measurements have revealed that crystalline monoclinic NPs can be obtained after annealing treatment at 1000 °C. The luminescence and timing properties of monoclinic crystalline hafnia NPs have been investigated as a function of temperature, from RT to 77 K. The NP luminescence band under both light and ionizing radiation excitations has been found to peak at around 440 nm and is associated with intrinsic defects, with the decay time of 2–3 ns. Although a further optimization of the luminescence light output can be foreseen, this work has allowed improvements in the optimisation of the photochemical synthesis route as low cost but valid synthesis method to manufacture scalable quantities of metal oxide nanoparticles. In addition, it has highlighted that the use of photochemical synthesis followed by annealing enables the production of monoclinic hafnia NPs with spectroscopic and timing features appropriate for the next coupling with fast organic dyes, opening the way for the creation of large hafnia based composite scintillators.Author contributions
I. V. designed the investigation, performed the photoluminescence and radioluminescence studies, and wrote and edited the manuscript. L. P. produced the nanoparticles, analysed the structural and morphological properties, and cooperated in planning the investigation. P. Z. and A. F. performed the TGA/DSC analyses, mass spectroscopy, and TMA and discussed the obtained data. R. K. contributed to the measurements of TGA, DSC, MS, and TMA analyses and performed interpretation and discussion of the obtained data. V. B., F. P., M. S., I. F., and R. C. performed the scintillation experiments. M. N., E. M., V. C., and E. A. supervised the synthesis steps of the nanoparticles and the luminescence/scintillation measurements and discussed the obtained data. All the authors cooperated in the draft corrections.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was carried out in the frame of the Crystal Clear Collaboration. This work was supported by the CERN Budget for Knowledge Transfer to Medical Applications and the Marie Skłodowska-Curie Actions Widening Fellowships (MSCAWF) Grant No. 101003405 – HANSOME. Financial support from the Czech Science Foundation under Grant No. 23-05615S is gratefully acknowledged.References
- V. A. Gritsenko, T. V. Perevalov and D. Islamov, Phys. Rep., 2015, 613, 1–20 CrossRef.
- N. Kumar, B. P. A. George, H. Abrahamse, V. Parashar, S. S. Ray and J. C. Ngila, Sci. Rep., 2017, 7, 9351 CrossRef PubMed.
- S. M. Woodley, S. Hamad, J. A. Mejías and C. R. A. Catlow, J. Mater. Chem., 2006, 16, 1927–1933 RSC.
- J. Wang, H. P. Li and R. Stevens, J. Mater. Sci., 1992, 27, 5397–5430 CrossRef CAS.
- I. Villa, A. Vedda, M. Fasoli, R. Lorenzi, N. Kränzlin, F. Rechberger, G. Ilari, D. Primc, B. Hattendorf, F. J. Heiligtag, M. Niederberger and A. Lauria, Chem. Mater., 2016, 28, 3245–3253 CrossRef CAS.
- M. Kirm, J. Aarik, M. Jürgens and I. Sildos, Nucl. Instrum. Methods Phys. Res., Sect. A, 2005, 537, 251–255 CrossRef CAS.
- C. LeLuyer, M. Villanueva-Ibañez, A. Pillonnet and C. Dujardin, J. Phys. Chem. A, 2008, 112, 10152–10155 CrossRef CAS PubMed.
- S. Lange, V. Kiisk, V. Reedo, M. Kirm, J. Aarik and I. Sildos, Opt. Mater., 2006, 28, 1238–1242 CrossRef CAS.
- R. Kumar, A. Vij and M. Singh, Optik, 2021, 248, 168121 CrossRef CAS.
- X. H. Guichard, F. Bernasconi and A. Lauria, Crystals, 2021, 11(9), 1042 CrossRef CAS.
- A. A. Rastorguev, V. I. Belyi, T. P. Smirnova, L. V. Yakovkina, M. V. Zamoryanskaya, V. A. Gritsenko and H. Wong, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 76, 235315 CrossRef.
- B. Cojocaru, D. Avram, R. Negrea, C. Ghica, V. G. Kessler, G. A. Seisenbaeva, V. I. Parvulescu and C. Tiseanu, ACS Omega, 2019, 4, 8881–8891 CrossRef CAS PubMed.
- Y. Sebti, T. Chauveau, M. Chalal, Y. Lalatonne, C. Lefebvre and L. Motte, Inorg. Chem., 2022, 61, 6508–6518 CrossRef CAS PubMed.
- I. Villa, F. Moretti, M. Fasoli, A. Rossi, B. Hattendorf, C. Dujardin, M. Niederberger, A. Vedda and A. Lauria, Adv. Opt. Mater., 2020, 8, 1901348 CrossRef CAS.
- M. Mattarelli, M. Montagna, F. Rossi, C. Tosello, N. D. Afify, M. Bettinelli, A. Speghini, C. Armellini, Y. Jestin, F. Rocca and S. Gialanella, Opt. Mater., 2009, 31, 1362–1365 CrossRef CAS.
- V. A. Gritsenko, D. R. Islamov, T. V. Perevalov, V. S. Aliev, A. P. Yelisseyev, E. E. Lomonova, V. A. Pustovarov and A. Chin, J. Phys. Chem. C, 2016, 120, 19980–19986 CrossRef CAS.
- S. Kurosawa, Y. Futami, V. Kochurikhin, B. Mikhail, Y. Yokota, T. Yanagida and A. Yoshikawa, Key Eng. Mater., 2012, 508, 81–86 CAS.
- R. Ruh and P. W. R. Corfield, J. Am. Ceram. Soc., 1970, 53, 126–129 CrossRef CAS.
- Y. Wan and X. Zhou, RSC Adv., 2017, 7, 7763–7773 RSC.
- B. L. Cushing, V. L. Kolesnichenko and C. J. O'Connor, Chem. Rev., 2004, 104, 3893–3946 CrossRef CAS PubMed.
- T. Taniguchi, N. Sakamoto, T. Watanabe, N. Matsushita and M. Yoshimura, J. Phys. Chem. C, 2008, 112, 4884–4891 CrossRef CAS.
- J. S. Quintero-García, B. A. Puente-Urbina, L. A. García-Cerda, O. S. Rodríguez-Fernández and E. Mendoza-Mendoza, Mater. Lett., 2015, 159, 520–524 CrossRef.
- A. Ramadoss, K. Krishnamoorthy and S. J. Kim, Mater. Res. Bull., 2012, 47, 2680–2684 CrossRef CAS.
- N. Pinna, G. Garnweitner, M. Antonietti and M. Niederberger, Adv. Mater., 2004, 16, 2196–2200 CrossRef CAS.
- A. D. Furasova, A. F. Fakhardo, V. A. Milichko, E. Tervoort, M. Niederberger and V. V. Vinogradov, Colloids Surf., B, 2017, 154, 21–26 CrossRef CAS PubMed.
- E. Rauwel, A. Galeckas and P. Rauwel, Mater. Res. Express, 2014, 1, 015035 CrossRef CAS.
- C.-H. Lu, J. M. Raitano, S. Khalid, L. Zhang and S.-W. Chan, J. Appl. Phys., 2008, 103, 124303 CrossRef.
- J. Zhao, Y. Zhang, H. Gong, Y. Zhang, X. Wang, X. Guo and Y. Zhao, Ceram. Int., 2015, 41(4), 5232–5238 CrossRef CAS.
- H. Fukushima, D. Nakauchi, T. Kato, N. Kawaguchi and T. Yanagida, Jpn. J. Appl. Phys., 2023, 62, 010506 CrossRef.
- S. Ding, L. Chen, J. Liao, Q. Huo, Q. Wang, G. Tian and W. Yin, Small, 2023, 2300341 CrossRef PubMed.
- C. Liu, T. J. Hajagos, D. Kishpaugh, Y. Jin, W. Hu, Q. Chen and Q. Pei, Adv. Funct. Mater., 2015, 25, 4607–4616 CrossRef CAS.
- C. Liu, Z. Li, T. J. Hajagos, D. Kishpaugh, D. Y. Chen and Q. Pei, ACS Nano, 2017, 11, 6422–6430 CrossRef CAS PubMed.
- M. Gandini, I. Villa, M. Beretta, C. Gotti, M. Imran, F. Carulli, E. Fantuzzi, M. Sassi, M. Zaffalon, C. Brofferio, L. Manna, L. Beverina, A. Vedda, M. Fasoli, L. Gironi and S. Brovelli, Nat. Nanotechnol., 2020, 15, 462–468 CrossRef CAS PubMed.
- K. Děcká, F. Pagano, I. Frank, N. Kratochwil, E. Mihóková, E. Auffray and V. Čuba, J. Mater. Chem. C, 2022, 10, 12836–12843 RSC.
- I. Villa, B. Santiago Gonzalez, M. Orfano, F. Cova, V. Secchi, C. Colombo, J. Páterek, R. Kučerková, V. Babin, M. Mauri, M. Nikl and A. Monguzzi, Nanomaterials, 2021, 11, 3387 CrossRef CAS PubMed.
- H. Burešová, L. Procházková, R. Martinez Turtos, V. Jary, E. Mihóková, A. Beitlerová, R. Pjatkan, S. Gundacker, E. Auffray, P. Lecoq, M. Nikl and V. Čuba, Opt. Express, 2016, 24, 15289 CrossRef.
- J. Perego, I. Villa, A. Pedrini, E. Padovani, R. Crapanzano, A. Vedda, C. Dujardin, C. X. Bezuidenhout, S. Bracco and P. Sozzani, Nat. Photonics, 2021, 15, 393–400 CrossRef CAS.
- Y. S. Zhao, Z. Yu, A. Douraghy, A. F. Chatziioannou, Y. Mo and Q. Pei, Chem. Commun., 2008, 45, 6008–6010 RSC.
- J. Bárta, V. Čuba, M. Pospíšil, V. Jarý and M. Nikl, J. Mater. Chem., 2012, 22, 16590–16597 RSC.
- L. Procházková, T. Gbur, V. Čuba, V. Jarý and M. Nikl, Opt. Mater., 2015, 47, 67–71 CrossRef.
- V. Čuba, T. Pavelková, J. Bárta, V. Jarý, M. Nikl and I. Jakubec, J. Nanopart. Res., 2012, 14, 794 CrossRef.
- T. Gbur, V. Čuba, V. Múčka, M. Nikl, K. Knížek, M. Pospíšil and I. Jakubec, J. Nanopart. Res., 2011, 13, 4529–4537 CrossRef CAS.
- G. Cabello, L. Lillo, C. Caro, G. E. Buono-Core, B. Chornik, M. Flores, C. Carrasco and C. A. Rodriguez, Ceram. Int., 2014, 40, 7761–7768 CrossRef CAS.
- N. Jara, N. S. Milán, A. Rahman, L. Mouheb, D. C. Boffito, C. Jeffryes and S. A. Dahoumane, Molecules, 2021, 26, 4585 CrossRef CAS PubMed.
- J. Bárta, L. Prouzová Procházková, M. Škodová, K. Děcká, K. Popovich, T. Janoušková Pavelková, P. Beck and V. Čuba, React. Chem. Eng., 2022, 7, 968–977 RSC.
- A. Ross, M. Muñoz, B. H. Rotstein, E. J. Suuronen and E. I. Alarcon, Sci. Rep., 2021, 11, 5420 CrossRef CAS PubMed.
- T. J. Hajagos, C. Liu, N. J. Cherepy and Q. Pei, Adv. Mater., 2018, 30, 1706956 CrossRef PubMed.
- E. Aleksanyan, M. Kirm, E. Feldbach and V. Harutyunyan, Radiat. Meas., 2016, 90, 84–89 CrossRef CAS.
- Q. Li, K. M. Koo, W. M. Lau, P. F. Lee, J. Y. Dai, Z. F. Hou and X. G. Gong, Appl. Phys. Lett., 2006, 88, 182903 CrossRef.
- J. X. Zheng, G. Ceder, T. Maxisch, W. K. Chim and W. K. Choi, Phys. Rev. B, 2007, 75, 104112 CrossRef.
- A. Shilov, S. Savchenko, A. Vokhmintsev, V. Gritsenko and I. Weinstein, Temperature dependence of self-trapped exciton luminescence in nanostructured hafnia powder, 2021 Search PubMed.
- D. Muñoz Ramo, P. V. Sushko and A. L. Shluger, Phys. Rev. B, 2012, 85, 024120 CrossRef.
- V. A. Pustovarov, T. P. Smirnova, M. S. Lebedev, V. A. Gritsenko and M. Kirm, J. Lumin., 2016, 170, 161–167 CrossRef CAS.
- V. Čuba, L. Procházková, J. Bárta, A. Vondrášková, T. Pavelková, E. Mihóková, V. Jarý and M. Nikl, J. Nanopart. Res., 2014, 16, 2686 CrossRef.
- R. M. Turtos, S. Gundacker, S. Omelkov, E. Auffray and P. Lecoq, J. Lumin., 2019, 215, 116613 CrossRef CAS.
- F. Pagano, N. Kratochwil, I. Frank, S. Gundacker, M. Paganoni, M. Pizzichemi, M. Salomoni and E. Auffray, Front. Phys., 2022, 1085 Search PubMed.
- G. Štefanić, K. Molčanov and S. Musić, Mater. Chem. Phys., 2005, 90, 344–352 CrossRef.
- J. Wang, D. Zhou, W. Dong, Y. Yao, N. Sun, F. Ali, X. Hou and F. Liu, Adv. Electron. Mater., 2021, 7, 2000585 CrossRef CAS.
- C. Wang, S. Yang and Y. Chen, R. Soc. Open Sci., 2019, 6, 181193 CrossRef CAS PubMed.
- C.-H. Wang, S.-H. Yang, Y.-M. Chen, Y.-Z. Wu, J. He and C.-B. Tang, Trans. Nonferrous Met. Soc. China, 2017, 27, 694–700 CrossRef CAS.
- L. Procházková, J. Bárta, V. Čuba, C. Ekberg, S. Tietze and I. Jakubec, Radiat. Phys. Chem., 2016, 124, 68–74 CrossRef.
- R. P. Haggerty, P. Sarin, Z. D. Apostolov, P. E. Driemeyer and W. M. Kriven, J. Am. Ceram. Soc., 2014, 97, 2213–2222 CrossRef CAS.
- L. Skuja, J. Non-Cryst. Solids, 1998, 239, 16–48 CrossRef CAS.
- I. Villa, A. Lauria, F. Moretti, M. Fasoli, C. Dujardin, M. Niederberger and A. Vedda, Phys. Chem. Chem. Phys., 2018, 20, 15907–15915 RSC.
- V. Kiisk, L. Puust, K. Utt, A. Maaroos, H. Mändar, E. Viviani, F. Piccinelli, R. Saar, U. Joost and I. Sildos, J. Lumin., 2016, 174, 49–55 CrossRef CAS.
- A. V. Emeline and N. Serpone, Chem. Phys. Lett., 2001, 345, 105–110 CrossRef CAS.
- D. M. Ramo, J. Gavartin, A. Shluger and G. Bersuker, Phys. Rev. B, 2007, 75, 205336 CrossRef.
- D. Z. Gao, J. Strand, M. S. Munde and A. L. Shluger, Front. Phys., 2019, 7, 43 CrossRef.
- S. Papernov, M. D. Brunsman, J. B. Oliver, B. N. Hoffman, A. A. Kozlov, S. G. Demos, A. Shvydky, F. H. M. Cavalcante, L. Yang, C. S. Menoni, B. Roshanzadeh, S. T. P. Boyd, L. A. Emmert and W. Rudolph, Opt. Express, 2018, 26, 17608–17623 CrossRef CAS PubMed.
- A. Popov, E. Kotomin and J. Maier, Nucl. Instrum. Methods Phys. Res., Sect. B, 2010, 268, 3084–3089 CrossRef CAS.
- E. Cockayne, Phys. Rev. B, 2007, 75, 094103 CrossRef.
- V. Babin, V. Laguta, A. Maaroos, A. Makhov, M. Nikl and S. Zazubovich, Phys. Status Solidi B, 2011, 248, 239–242 CrossRef CAS.
- M. D'Arienzo, S. Mostoni, R. Crapanzano, C. Cepek, B. Di Credico, M. Fasoli, S. Polizzi, A. Vedda, I. Villa and R. Scotti, J. Phys. Chem. C, 2019, 123, 21651–21661 CrossRef.
- S. E. Derenzo, W. Moses, J. Cahoon, R. Perera and J. Litton, IEEE Trans. Nucl. Sci., 1990, 37, 203–208 CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: DSC-TGA-TMA data, XRD and RL results of untreated HfO2, table of thermal activation parameters, and TR-PL results. See DOI: https://doi.org/10.1039/d3ce00320e |
| ‡ Present address: Dipartimento di Scienza Dei Materiali, Università Degli Studi Milano-Bicocca, 20125 Milano, Italy. E-mail: E-mail: irene.villa@unimib.it |
| This journal is © The Royal Society of Chemistry 2023 |