
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Perchlorate-induced structural diversity in thiosemicarbazone copper(II) complexes provides insights to understand the reactivity in acidic and basic media†
Rubén
Gil-García
a,
Gotzon
Madariaga
 *bc,
Alondra
Jiménez-Pérez
a,
Ignacio
Herrán-Torres
a,
Adrián
Gago-González
a,
María
Ugalde
a,
Vaidas
Januskaitis
a,
Joaquín
Barrera-García
a,
Maite
Insausti
d,
María
S. Galletero
e,
Joaquín
Borrás
f,
José Vicente
Cuevas
a,
Rosa
Pedrido
g,
Patricia
Gómez-Saiz
a,
Luis
Lezama
d and
Javier
García-Tojal
*a
*bc,
Alondra
Jiménez-Pérez
a,
Ignacio
Herrán-Torres
a,
Adrián
Gago-González
a,
María
Ugalde
a,
Vaidas
Januskaitis
a,
Joaquín
Barrera-García
a,
Maite
Insausti
d,
María
S. Galletero
e,
Joaquín
Borrás
f,
José Vicente
Cuevas
a,
Rosa
Pedrido
g,
Patricia
Gómez-Saiz
a,
Luis
Lezama
d and
Javier
García-Tojal
*a
aDepartamento de Química, Universidad de Burgos, 09001 Burgos, Spain. E-mail: qipgatoj@ubu.es
bDepartamento de Física, Universidad del País Vasco, Aptdo. 644, 48080 Bilbao, Spain
cEHU Quantum Center, University of the Basque Country, UPV/EHU, E-48080 Bilbao, Spain
dDepartamento de Química Orgánica e Inorgánica, Universidad del País Vasco, Aptdo. 644, 48080 Bilbao, Spain
eServicio Central de Espectrometría de Masas, Universidad de Valencia, Av. Dr. Moliner 100, Burjassot, Valencia, Spain
fDepartamento de Química Inorgánica, Facultad de Farmacia, Universidad de Valencia, 46100 Burjassot, Valencia, Spain
gDepartamento de Química Inorgánica, Facultade de Química, Campus Vida, Universidade de Santiago de Compostela, 15782 Santiago de Compostela, Spain
First published on 6th March 2023
Abstract
The use of perchlorate ancillary ligands with relatively low coordination ability nicely illustrates a rich structural diversity in thiosemicarbazone copper(II) (TSC-Cu(II)) systems. Five compounds with the formulae {[CuL(OH2)][CuL(OClO3)]}n·nClO4·2nH2O (1), [{CuL(OClO3)}2] (2), [Cu(HL)(OClO3)2(OH2)] H2O (3), [Cu2L3](ClO4)·2H2O (4) and [Cu(HL)(NCS)](ClO4) (5) are studied, where HL = pyridine-2-carbaldehyde thiosemicarbazone. The crystal structures show the presence of polynuclear species in the compounds containing an anionic L− thiosemicarbazone ligand: 1D chains (1) and dinuclear (2 and 4) arrangements. Meanwhile, complexes with a neutral HL ligand give rise to mononuclear entities (3 and 5). The chemical and structural parallelism of these complexes with the analogous TSC–Cu(II)–nitrate system is discussed. The thiocyanate ligand in compound 5 comes from partial desulfurization of the thiosemicarbazone in acid medium and results in an incommensurate modulated structure. On the other hand, it is proposed that [Cu2L3]+ species present in 4 could play a possible role in desulfurization reactions. Experimental evidence and theoretical calculations support a proposed mechanism for the partial desulfurization of preformed TSC-Cu(II) entities in basic and physiological media to yield the [CuL(LCN)] (HLCN = pyridine-2-ylmethylene(hydrazine carbonitrile)) and [{CuL(SH)}2)] compounds.
Introduction
Thiosemicarbazones (TSCs) and their metal complexes have been proposed to exhibit a wide range of biological activities1–15 and tested as drugs against microbial diseases (malaria,16–18 tuberculosis,19,20 smallpox,21,22 influenza,23 leishmaniasis24,25), neurological pathologies (Alzheimer's,26–29 Parkinson's30) and cancer.31–35 Several biochemical targets have been identified,36–43 while the formation of TSC-Fe and TSC-Cu complexes seems to play an important role in their affectation to cells.44–62The promising results in cells and animals prompted the researchers to test these substances as antitumor therapeutics, being evaluated in phase I,63–69 phase II and even phase III clinical trials.70 To date, the results in humans have been poorer than those obtained in cells and animal models, except for combination therapy,71 and this decrease in the effectiveness has been associated with methemoglobin formation in patients, hypoxia, lower clearance times than those observed in mice, and toxicity effects.72,73 Glucuronide formation and an increase in sulfate contents in plasma have also been reported, which suggested possible transformations of TSCs in physiological medium.74 Taking into account that reactions with intracellular copper seem to be pivotal to understanding the biological activity of TSCs,75–77 desulfurization processes such as those described for PTSC-Cu(II) species in aqueous medium ranging from basic to physiological pH 7.4 values (HPTSC = HL = pyridine-2-carbaldehyde thiosemicarbazone, Scheme 1)78 deserve special attention. This reaction is not detected for complexes of HL with other metal ions such as Hg(II), Pb(II), Fe(II/III), Co(III), and Ni(II).79,80 Notwithstanding, desulfurization processes triggered by coordination to different metal ions have been described for other TSC systems.81–90 In fact, these kinds of transformations could be responsible, at least in part, for the biological behaviour and its control could be key to improving the use of these substances as antitumor agents.91
 | ||
| Scheme 1 HL ligand and the corresponding atom labels for the present work. | ||
In order to obtain insight into the coordination behaviour, chemical speciation and desulfurization processes in TSC-Cu(II) complexes, our starting hypothesis was to consider that ligands with low coordination ability, such as perchlorate anions, lead to an increase in the variety of entities in the isolated solids. Our experience with nitrate ions suggested this idea, but nitrate derivatives were often difficult to crystallize. Thus, we have synthesized and characterized a series of perchlorate derivatives of TSC: {[CuL(OH2)][CuL(OClO3)]}n·nClO4·2nH2O (1), [{CuL(OClO3)}2] (2), [Cu(HL)(OClO3)2(OH2)]H2O (3), [Cu2L3](ClO4)·2H2O (4) and [Cu(HL)(NCS)](ClO4) (5). Acidic media induce the loss of the sulfur thiosemicarbazone atoms released as a part of molecular fragments arising from a breakage reaction, such as the thiocyanate groups present in the aperiodically ordered compound 5. The crystal structures and spectroscopic properties are described and compared with those of analogous thiosemicarbazone systems, emphasizing the role that perchlorate anions play in the structural and chemical behaviour. In fact, the parallelism with the HL–Cu–nitrate system is analysed in depth and supported by an extensive characterization provided as ESI† so as not to hinder the discussion of the rest of the results. The relevance of the [Cu2L3]+ entities present in the crystal structure of 4, also detected in aqueous solutions at physiological pH 7.4, is discussed in relation with ESI+ measurements and different structural and spectroscopic evidence of thiosemicarbazone desulfurization processes in neutral and basic media. A mechanistic proposal is given, filling the gaps in desulfurization mechanisms for TSCs previously published. In addition, it rationalizes the relationships among different species independently isolated for decades.
Results and discussion
Synthesis
The chemical complexity of the reported perchlorate system is analogous to that found in other HL-Cu(II) compounds containing O-donor inorganic ancillary ligands derived from strong acids, such as sulfato92,93 and nitrato.84,94–96 This behaviour contrasts with the simple structural chemistry found in more coordination ligands, such as chloride (square pyramidal geometries for monomers and centrosymmetric dimers in acidic and basic media, respectively) and acetate derivatives (only centrosymmetric dimers, because anionic ligands derived from weak acids are not compatible with the neutral HL form).7 Concerning the nitrate ligands, there is a marked parallelism with the composition and structures reported here and those of previously published nitrate compounds, as discussed in the crystallographic section and the chapter entitled Summary about the parallel Cu2+/HL/NO3− system in the ESI.† In particular, there are important analogies in the chemical composition, spectroscopic properties and crystal structures of CuL(NO3) (actually, [CuL(ONO2)]n)96vs. compound 1, [Cu(HL)(ONO2)(OH2)](NO3)95vs. compound 3, Cu2(HL)L2(NO3)2(H2O)2vs. compound 4 (see the ESI†), and [Cu(HL)(NCS)](NO3)80vs. compound 5.Some of the crystals whose structures are reported here were serendipitously obtained when we were looking into the reaction of the HL-Cu(II) system with nucleobases and nucleotides.97,98 For that reason, we focused on the search for rational methods to attain them (see Experimental section). It is worth noting the nearly ubiquitous presence of compound 1, even as an impurity, in many of the assays carried out in spite of its high solubility in water. In the case of compound 3, the only successful synthetic method required an ethanolic acid medium because of the high solubility in water, long reaction times and also several filtrations to remove impurities of 1 and 5. Regarding 5, it was also attained in a highly acidic medium, which emphasizes the great affinity between HL and Cu(II) ions. Unfortunately, we failed in the reproducibility of the syntheses of compounds 2 and 4 (Fig. S1.17 and S1.18†). In spite of this, the physicochemical characterization suggests that the powder samples retained the main figures of the crystal structures: CuL(ClO4) motifs in an anhydrous lattice (2) and a compound with presumable [Cu2L3](ClO4)·3H2O formulae (4, see Experimental section).
Considering the latter results, a complete mass spectrometry study was performed in aqueous solution at pH 4.0, 7.4, 9.0, 11.0 and 13.0 with the aim of elucidating the experimental conditions that boost the formation of [Cu2L3]+ entities (see the ESI;† Fig. S3.1–S3.4). The work was carried out on aqueous solutions of the CuL(NO3) compound, a better understood complex whose behaviour in solution is expected to be quite parallel to that of its homologue 1 because of the low coordination abilities of the counterions that allow easy release of the nitrate and perchlorate groups. Peaks at m/z 662.8 and 664.7 attributed to [Cu2L3]+ and [Cu2(HL)2L]+ species, respectively, were detected in aqueous solutions only at physiological pH 7.4 (Fig. S3.5–S3.8†). Both peaks were also observed in the mass spectra of the solids isolated from experiments at different pH values. The formation of [Cu2L3]+ species in aqueous solutions at physiological pH is in good agreement not only with the measured pH 7.6 value in the medium that yielded the crystals of 4 but also with the studies reported for analogous [Cu2(TSC)3]+ species detected in the 3-aminopyridine-2-carbaldehyde thiosemicarbazone copper(II) system.99,100
Regarding the origin of the thiocyanate ligands in compound 5, they could only arise from a desulfurization process involving a breakage of the thiosemicarbazone ligand induced by the acid medium because no other sulfur-containing reactant was present in the reaction. A possible mechanism will be discussed later in the section dealing with desulfurization reactions.
Crystal structures
The backbone of 1 contains two different thiosemicarbazone copper(II) units connected through a hydrazine nitrogen atom: [CuL(ClO4)] (entity 1) and [CuL(OH2)]+ (entity 2) that constitute {([CuL(OH2)][CuL(ClO4)])+}n chains (Fig. 1). These chains run parallel to the [1 1 0] and [−1 1 0] directions (Fig. 2). A perchlorate counterion and two water molecules complete the crystal network. The donor atoms around the metal centres adopt a distorted square pyramidal arrangement,101τ = 0.15 and 0.10 for Cu1 and Cu2, respectively. Thiosemicarbazone ions behave as tetradentate ligands by coordination to a Cu(II) ion through their N1, N2 and S atoms, while the N3 atom links to the neighbouring metal centre: [Cu1–N11, N12, S1, N23 and O21: 1.995(3), 1.975(3), 2.2339(14), 2.004(3) and 2.678(3) Å] and [Cu2–N21, N22, S2, N13 and O1W: 2.012(3), 1.972(3), 2.2428(15), 2.005(3) and 2.546(3) Å] (see Table S1.1†). The distance between two consecutive Cu(II) ions inside the chain is Cu1⋯Cu2: 4.857(1) Å and the angle of the NNS planes of their thiosemicarbazone ligands is 72.14°. The activation of the thiosemicarbazone N3 hydrazine donor atom to act as a single linker to neighbouring Cu(II) ions is not usual.94,102,103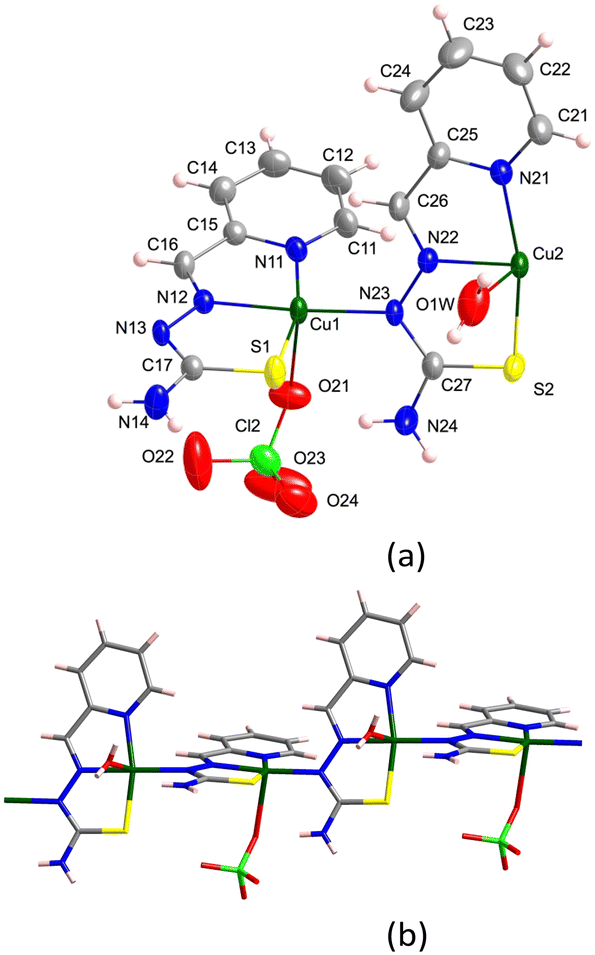 | ||
| Fig. 1 (a) Crystal structure of the asymmetric unit [{CuL(OClO3)}{CuL(OH2)}]+ in 1. Thermal ellipsoids are drawn at 30% probability level. (b) Detail of the monomer arrangement in the chain. | ||
 | ||
| Fig. 2 Detail of the chains parallel to the [−1 1 0] (red) and [1 1 0] (blue) directions in crystal 1. | ||
The crystal structure of 2 is formed by dimers (Fig. 3), where Cu(II) ions in distorted square pyramidal symmetries (τ = 0.25) are linked to oxygen atoms of two perchlorate ligands that act as bridges. The X-bridged dimer system formed in this way is structurally expected in the case of HL-Cu(II) compounds for coligands containing four or more non-hydrogen atoms, as the perchlorate ones, being lighter coligands, are prone to generate S-bridged systems through the sulfur thiosemicarbazone atoms.104 The NNS thiosemicarbazone donor atoms and an oxygen of a perchlorate anion constitute the basal plane [Cu–N1, N2, S and O12: 2.015(7), 1.954(6), 2.283(2) and 1.920(9) Å, respectively], while the apical position is occupied by an oxygen atom of the other perchlorate ion [Cu–O12i: 2.324(6) Å ((i) = −x + 1/2, −y + 1/2, −z)] (Table S1.1†). The distance between the metal centres inside the dimer is Cu⋯Cui: 3.322(2) Å and the minimum interdimeric distance is Cu⋯Cuii: 5.927(6) Å ((i) = −x + 1/2, −y + 1/2, −z; (ii) = −x, −y + 1, −z).
 | ||
| Fig. 3 Crystal structure of 2. Thermal ellipsoids are drawn at 30% probability level. | ||
The crystal lattice of 3 consists of [Cu(HL)(OClO3)2(OH2)] distorted octahedral monomers (Fig. 4) and one crystallization water molecule per metal complex formula. Perchlorate groups act as unidentate ligands, placed in trans position with respect to the [Cu(HL)(OH2)]2+ equatorial fragment, while the aqua ligand O1W atom sits 0.22 Å above the plane formed by the NNS chelating centres of the thiosemicarbazone ligand. The Cu–ligand bond lengths are Cu–N1, N2, S, O1W, O11 and O12′: 1.998(6), 1.950(4), 2.282(2), 1.933(5), 2.697(5) and 2.440(12) Å, respectively (Table S1.2†). The mononuclear species are essentially the same as those reported for the dihydrate [Cu(HL)(OClO3)2(OH2)]·2H2O derivative.105
 | ||
| Fig. 4 Crystal structure of the [Cu(HL)(OClO3)2(OH2)] unit in 3. Thermal ellipsoids are drawn at 30% probability level. | ||
[Cu2L3]+ units (Fig. 5), a perchlorate anion and two water molecules per formula make up the crystal lattice of 4. The dinuclear entity has two different Cu(II) ions. Cu1 is bonded to the NNS chelating centres of thiosemicarbazone 1 [Cu1–N11, N21 and S11: 2.043(3), 1.973(3) and 2.2591(14) Å] and to the sulfur atoms of the other two TSC ligands [Cu1–S12 and S13: 2.2848(14) and 2.8329(13) Å] in a distorted square pyramidal disposition (τ = 0.26). On the other hand, Cu2, which has a distorted octahedral geometry, is coordinated to NNS atoms of thiosemicarbazones 2 and 3 [Cu2–N12, N22, S12, N13, N23 and S13: 2.307(5), 2.046(3), 2.7361(13), 2.080(4), 1.973(3) and 2.3269(14) Å, respectively] (Table S1.3†). The intramolecular distance between metal centres is Cu1⋯Cu2: 3.562(1) Å and the minimum interdinuclear distance is Cu1⋯Cu2i: 6.305(1) Å ((i) = −x + 1, −y, −z + 1). It must be highlighted that the structure of this compound shows important resemblances to those reported for the 2-acetylpyridine N(4)-phenylthiosemicarbazone copper(II)106 and the 2-acetylpyridine 4,4-dimethyl-3-selenosemicarbazone copper(II) analogues.107 In both compounds, the Cu(ClO4)2 salt was used as starting material and the media were basified with NaOAc and Et3N, respectively, as in the case of compound 4.
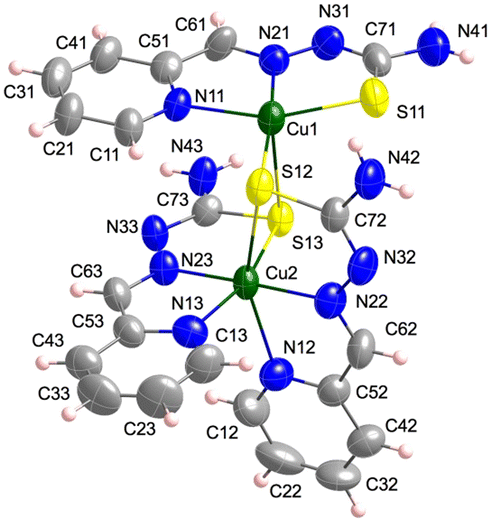 | ||
| Fig. 5 Crystal structure of the [Cu2L3]+ unit in 4. Thermal ellipsoids are drawn at 30% probability level. | ||
The structure of compound 5 is aperiodic and has been refined using a rigid body approximation; see details in the Experimental section and the ESI† (Fig. S1.19 and S1.20 and Table S1.10). It contains [Cu(HL)(NCS)]+ cationic monomers (Fig. 6a) and perchlorate counterions. Note that the thiocyanate ligands arise from the breakage of some of the thiosemicarbazone molecules by desulfurization. Bond lengths in the coordination polyhedron are Cu–N1, N2, S and N5: 2.013(7), 1.939(5), 2.290(2) and 1.888(6) Å, respectively (Table S1.2†). The value of Yang's parameter t4 is 0.13, very close to a square planar geometry.108 The [Cu(HL)(NCS)]+ entities stack, forming ladders along the [0 1 0] direction through Cu⋯S pseudo-contact distances of 3.3318(10) Å that relate the closest Cu⋯Cu ions at 5.7114(19) Å (Fig. 6b).
 | ||
| Fig. 6 (a) Crystal structure of the [Cu(HL)(NCS)]+ motif in 5. Thermal ellipsoids are drawn at 30% probability level. (b) Detail of the pseudo-chains in 5, depicting an approximation for the actual modulated structure of the perchlorate anions. | ||
The molecular structures of the five compounds described here mostly obey the structural features deduced for Cu(II) complexes derived from tridentate TSC ligands that allow a differentiation between the neutral (HL) and the anionic (L−) forms of the thiosemicarbazone ligand109(see Table S1.4 in the ESI†).
Noncovalent interactions stabilize the crystal building, such as hydrogen bonds (Fig. S1.1–S1.3 and Table S1.5†), π–π stacking (including chelate–π linkages, Fig. S1.4 and Table S1.6†), anion–π (Fig. S1.5 and Table S1.7†) and CH–π (only in 4, Fig. S1.6 and Table S1.8†) interactions.
There is a clear chemical and structural parallelism between the perchlorate and the nitrate systems derived from pyridine-2-carbaldehyde thiosemicarbazone copper(II) entities. Note, in this regard, the analogies between 1 and the extended 2D framework of [CuL(ONO2)]n, where chains formed by [CuL]+ monomers linked through the hydrazine N3 atom are connected by m-O2 nitrate bridges to form sheets in the [010] direction.96 The structure is even closer to that of [CuL(ONO2)(DMSO)]n, where each of the Cu(II) ions completes its polyhedron by nitrate and dimethyl sulfoxide ligands, generating 1D chains.94 The formation of coligand-bridged centrosymmetric dimers (X-bridge systems) such as 2 has been reported for the quite similar [{Cu(PTSC*)(ONO2)}2] derivative (HPTSC* = pyridine-2-carbaldehyde 4N-methylthiosemicarbazone).110 The monomeric nature of the 3 derivative containing the neutral ligand is reflected by the analogous [Cu(HL)(OH2)(ONO2)](NO3) compound, notwithstanding the square-pyramidal geometry of the latter.95 Besides, there is a thiocyanate [Cu(HL)(NCS)](NO3) derivative very close to 4, obtained under similar acidic conditions, together with a compound which exhibits a 28% replacement of NCS− by NO3− ligands.80 Finally, in spite of the lack of [Cu2L3](NO3) crystal structures with the same ligand, a preliminary synthetic study points to the presence of analogous [Cu2L3]+ and [Cu2(HL)L2]2+ entities in some of the experiments performed with copper(II) nitrate (see the ESI†). The similarity between both nitrate and perchlorate systems opens up other possibilities for increasing the structural complexity of the HL-Cu-perchlorate derivatives, such as the quite distorted dinuclear [{Cu(PTSC*)}2(NO3)](OH) compound found in the closely related system.84 In this sense, a solid with the proposed (CuL)2(NO3)(OH) formula obtained at pH 9.0 is also described in the ESI† (chapter 3, Fig. S3.8 and S3.11).
Spectroscopic and magnetic characterization
Despite the complexity of the spectra that precludes a complete assignment of the bands, they clearly exhibit the usual infrared spectroscopy (IR) features in the pyridine-2-carbaldehyde thiosemicarbazone copper(II) system111 (see Fig. S1.7–S1.9, S2.12 and S2.13 in the ESI,† Experimental section and Table S1.9 for tentative assignments). In this regard, one of the most informative items is the ratio between the intensity of the bands around 1620–1590 cm−1 (region A in Fig. 7) and those in the 1480–1430 cm−1 range (region B). The bands at 1620–1590 cm−1 are assigned to ν(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N)azomethinic, ν(CN)pyridinic and thioamide I absorptions with high δ(NH2) contribution, and those absorptions observed about 1480–1430 cm−1 are attributed to thioamide II vibrations. The bands in region B are more intense than the bands in region A in compound 1 with anionic deprotonated ligands. On the other hand, the medium intensity band at 1646 cm−1 in 1 could be related to the imine C2N3 group involved in the linkage to neighbouring Cu(II) ions. This band is weaker (compound 2) or absent in the spectra of compounds without this kind of connectivity (see the ESI†). In different experiments carried out at variable pH values obtaining solid mixtures where 1 is one of the present phases, the intensity of this band decreases on increasing pH, with concomitant appearance of absorptions in the 2200–2000 cm−1 region, probably due to breakage of the TSC ligand (see the ESI†).
N)azomethinic, ν(CN)pyridinic and thioamide I absorptions with high δ(NH2) contribution, and those absorptions observed about 1480–1430 cm−1 are attributed to thioamide II vibrations. The bands in region B are more intense than the bands in region A in compound 1 with anionic deprotonated ligands. On the other hand, the medium intensity band at 1646 cm−1 in 1 could be related to the imine C2N3 group involved in the linkage to neighbouring Cu(II) ions. This band is weaker (compound 2) or absent in the spectra of compounds without this kind of connectivity (see the ESI†). In different experiments carried out at variable pH values obtaining solid mixtures where 1 is one of the present phases, the intensity of this band decreases on increasing pH, with concomitant appearance of absorptions in the 2200–2000 cm−1 region, probably due to breakage of the TSC ligand (see the ESI†).
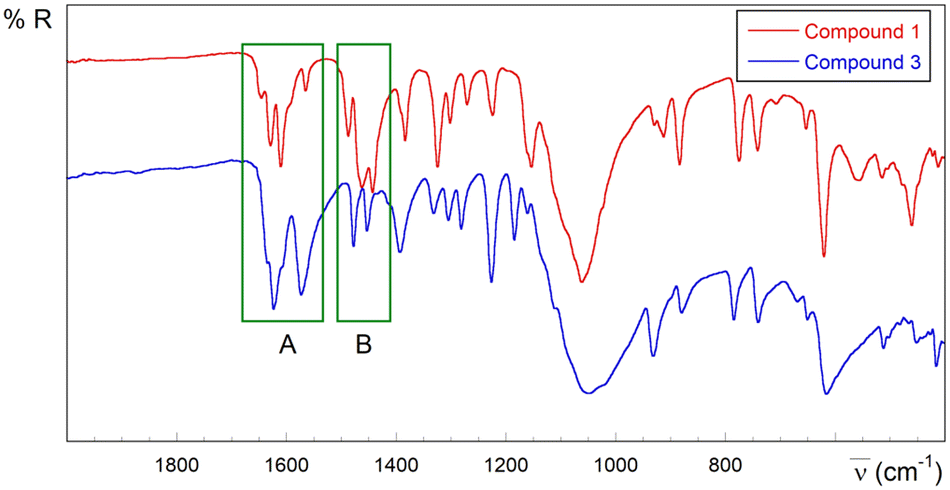 | ||
| Fig. 7 Selected IR region (2000–400 cm−1) for derivatives containing anionic L− (compound 1, red) and neutral HL ligands (compound 3, blue). | ||
Bands observed at 1080–1015 cm−1 (very strong and broad, vs-b) and 640–610 cm−1 (strong, s) are attributed to the ν3 and ν4 perchlorate group modes, respectively. In addition, the spectrum of 5 exhibits a strong band at 2136 cm−1 assigned to the thiocyanate ν(NCS) stretching vibration. Note that the energy of this band is higher than those observed in the [{CuL(NCS)}2] and [Cu(HL)(NCS)](NO3) compounds (2100/2072 and 2097 cm−1, respectively),80,95 perhaps due to an increase in the intensity of the Cu–NCS bond, in agreement with the bond distance of 1.888(6) Å for 5, 1.906–1.918 Å for [Cu(HL)(NCS)](NO3) and two values, 1.96(1) and 1.89(1) Å, for these bond lengths in the asymmetric [{CuL(NCS)}2] compound (see Fig. S1.10†).
The electron paramagnetic resonance (EPR) measurements performed for compound 1 reveal the presence of at least two signals (Fig. S1.15†). The most intense and broad absorption can be attributed to an antiferromagnetic phase. In addition, a weak and sharper line, with an approximate contribution of 1% to the whole intensity at RT, predominates at low temperatures. The latter could be ascribed to a paramagnetic or even ferromagnetic impurity. In fact, about 0.2% of impurities are deduced from the best fitting of the magnetic susceptibility data (Fig. S1.21†), while the predominant strong antiferromagnetic interactions (J/k = −100 K, −69.44 cm−1) can be attributed to exchange pathways in the chains through the –Cu–N3–N2–Cu′–N3′– moieties that involve the closest Cu(II) centres at 4.84 Å and run parallel to the lobes of the dx2−y2 orbitals of the metal ions. This behaviour resembles that observed in [CuL(ONO2)]n.96 Axial and orthorhombic EPR spectra are obtained for 3 and 5, respectively (Fig. S1.15 and S1.16†), which are characteristic of S = 1/2 Cu(II) ions with a dx2−y2 ground state, in good agreement with the tetragonally distorted octahedral and square-planar geometries elucidated in the crystallographic studies. The broad isotropic spectrum of the compound containing the same [Cu2L3]+ motif as that of 4 (see experimental details) suggests dominant dipolar interactions between Cu(II) ions, as expected in a dimer with short Cu–Cu distances but inefficient exchange pathway.
Desulfurization studies
As explained before, compound 5 exhibits a thiocyanate group bonded to the Cu(II) ion. A proposal for a plausible pathway to explain the origin of the thiocyanate ligand in compound 5 is depicted in Scheme 2. The coordination to Cu(II) ions would increase the electron-withdrawing properties of the thioamide carbon atom in the thiosemicarbazone neutral ligand, which would undergo nucleophilic attack of a water molecule. Then, an electron reorganization process could lead to the breakage of the thiosemicarbazone ligand with the release of 2-(hydrazinomethyl)pyridine (RNH2 in Scheme 2 and compound 5 in the Experimental section and the ESI†), and carbamothioic O-acid. The dehydration of the latter, probably facilitated by its coordination to metal ions, and further deprotonation would give rise to thiocyanate groups. Desulfurization reactions in thiosemicarbazone–metal aqueous solutions induced by pH and temperature have been described to yield [CuL(LCN)]78 [HLCN = pyridine-2-ylmethylene(hydrazine carbonitrile), Scheme 3], [CuLCl]2[Cu(pic)2]80 (Hpic = picolinic acid), sulfate, sulfide and thiocyanate derivatives.80,84 In fact, partial transformation of [CuL]+ entities seems to operate even at physiological pH 7.4. | ||
| Scheme 2 (a) Mechanism proposed for desulfurization process in acid medium leading to compound 5. (b) Structure of the R–NH2 product. | ||
 | ||
| Scheme 3 Nitrile LCN− derivative arising from desulfurization of HL in basic medium. | ||
The possibility of breakage of the thiosemicarbazone ligand with a concomitant increase in the acidity of the medium could influence the biological activity of these substances. In this sense, the reactivity against reduced glutathione seems to be notably enhanced with acidity in thiosemicarbazone copper(II) complexes112 and, therefore, the reactive oxygen species (ROS) production.
On the other hand, the processes related with transformations of the thiosemicarbazone ligand in physiological and basic aqueous media seem to be quite complex, as demonstrated by the experiments performed on the CuL(NO3) system under different basic conditions (see chapter 3 in the ESI,† entitled Studies in aqueous basic media). Thus, peaks at m/z 628.99 and 630.99 are detected in the mass spectra of solids filtered from solutions at pH ≥7.4 (Fig. S3.7 and S3.8†), which are attributed to metal cationic [Cu2L2(LCN)]+ and [Cu2(HL)2(LCN)]+ complexes derived from HLCN (see Scheme 3). It must be emphasized that the [Cu2(HL)2(LCN)]+ formula is merely nominal and does not necessarily imply the presence of Cu(I) ions in the species, because it is possible that both hydrogen atoms could be incorporated into the LCN– ligand by transforming the triple bond to a double bond without affecting the negative charge in the new ligand. The intensity of these peaks increases in solutions heated to 80 °C. In these hot solutions, the greater the pH (9.0 vs. 7.4) and the longer the time of treatment (3 h vs. 1 h), the more intense are the peaks at 628.99 and 630.99 (Fig. S3.9 and S3.10†). In addition, medium to intense bands at about 2116–2106 cm−1 appear in the IR spectra of samples obtained at pH 7.4 and 9.0 (Fig. S3.11 and S3.12†). These IR bands are characteristic of the (N–C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N) cyanamide/(NCN) carbothioamide moiety in the LCN– anionic ligands arising from desulfurization reactions of HL.78,80 Surprisingly, IR bands in the 2150–2050 cm−1 region (Fig. S3.12†) do not appear in compound CuL2, synthesized at pH 13 by addition of Cu(II) salts to basic aqueous solutions of HL with metal-to-ligand stoichiometry (1
N) cyanamide/(NCN) carbothioamide moiety in the LCN– anionic ligands arising from desulfurization reactions of HL.78,80 Surprisingly, IR bands in the 2150–2050 cm−1 region (Fig. S3.12†) do not appear in compound CuL2, synthesized at pH 13 by addition of Cu(II) salts to basic aqueous solutions of HL with metal-to-ligand stoichiometry (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2).78 It seems that the presence of preformed [CuL]+ entities in solution before the addition of base could be required for the desulfurization. Taking into account all these observations, it can be proposed that formation of dinuclear [Cu2L3]+ entities could be an intermediate stage in a desulfurization reaction whose sequence, inspired by an already published mechanism,78 is provided in Scheme 4.
:2).78 It seems that the presence of preformed [CuL]+ entities in solution before the addition of base could be required for the desulfurization. Taking into account all these observations, it can be proposed that formation of dinuclear [Cu2L3]+ entities could be an intermediate stage in a desulfurization reaction whose sequence, inspired by an already published mechanism,78 is provided in Scheme 4.
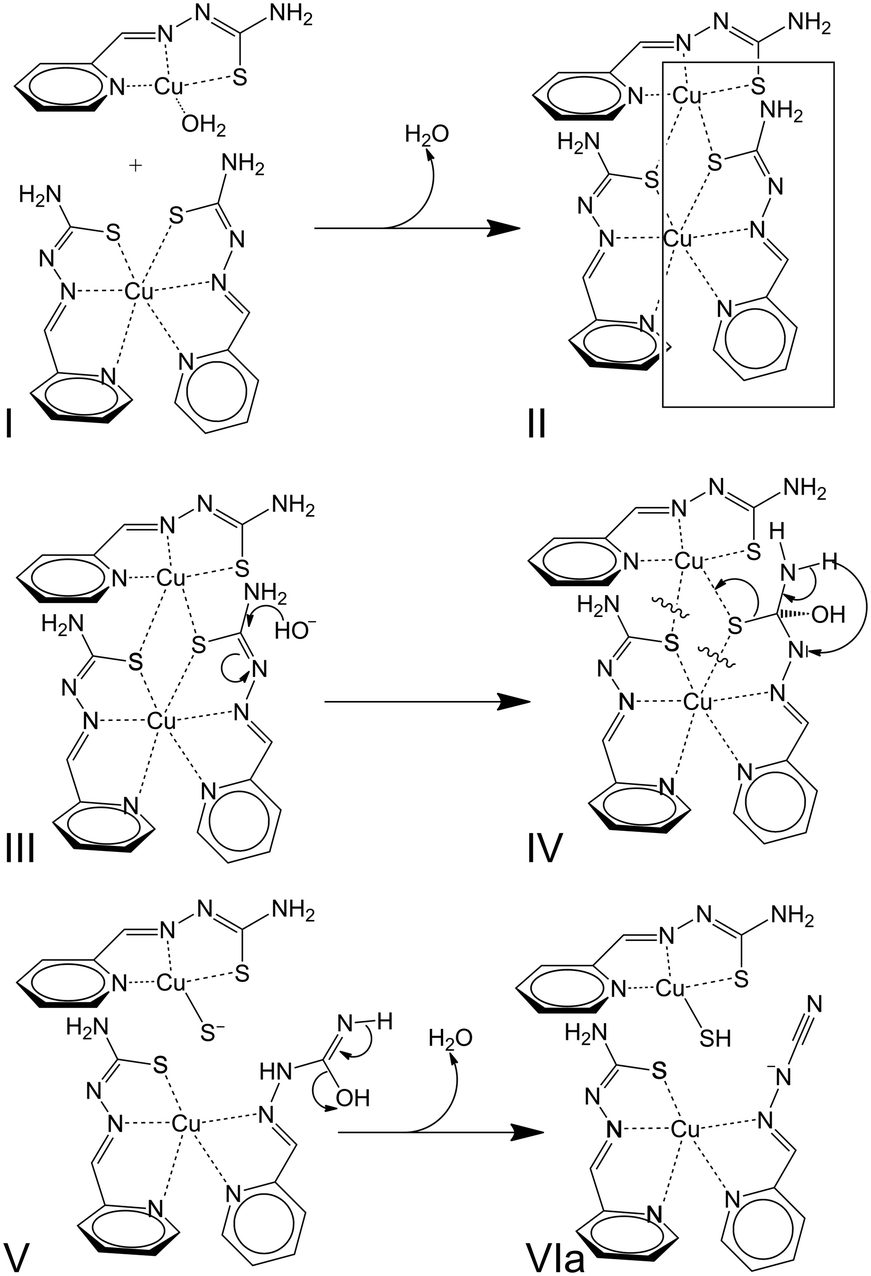 | ||
| Scheme 4 Mechanism proposed for the desulfurization process in basic medium involving [Cu2L3]+ intermediates. | ||
Briefly, the approximation of a [CuL(OH2)n]+ species to a [CuL2] neutral complex (I) leads to the formation of [Cu2L3]+ entities (II). Further nucleophilic attack of hydroxide OH− anions (III) triggers transformations inside one of the thiosemicarbazone ligands (IV + V) that lead to the formation of [CuL(LCN)] and [CuL(SH)] complexes (VIa). The desulfurization process can be summarized by reactions (1) and (2). The plausibility of this process is based not only on the results described in the present study but also on the existence of crystal structures for all the species mentioned in the reactions, i.e. [CuL(OH2)]+ entities in [{CuL(OH2)}2](SiF6)·4H2O,111 [CuL2],78 [CuL(LCN)],78 and [{CuL(SH)}2].80 Notwithstanding, the formation of [Cu2L3]+ could also be influenced by the effects of the concentration of [CuL]+ entities in the medium. Finally, the influence or concomitance of other processes in the desulfurization reaction cannot be ruled out as redox reactions involving the Cu(II) ions. Regarding this possibility, note that three sulfur atoms are placed around one of the Cu(II) ions in [Cu2L3]+, which represents a reducing environment in the coordination polyhedron.
| [CuL(OH2)]+ + [CuL2] → [Cu2L3]+ + H2O | (1) |
| [Cu2L3]+ + OH− → [CuL(LCN)] + [CuL(SH)] + H2O | (2) |
Computational studies
The viability of the proposed mechanism shown in Scheme 5 has been tested through quantum chemical calculations (see the Experimental section for details). The first step is the formation of a dinuclear intermediate II by the displacement of a water molecule from I. This step is calculated to be exergonic with ΔG = −57.9 kcal mol−1. The next step (represented in structure III of Scheme 4) is the nucleophilic attack of a hydroxide anion to the carbon bonded to a bridging sulfur atom to yield structure IV. This process is calculated to be exergonic with ΔG = −146.1 kcal mol−1. Then, it is proposed a prototropy from the –NH2 group next to the carbon atom that suffered the nucleophilic attack; while the C–S bond is breaking, a new Cu–S bond is forming and a single bond C–N is transforming in a double bond to reach structure V. The transformation of IV into V is found to be an exergonic process, ΔG = −15.6 kcal mol−1. The final step is the release of a new water molecule with the transformation of the newly formed double bond CN in the previous step into a triple CN bond, as shown in structure VIa. This step was found to be endergonic, ΔG = 37.4 kcal mol−1; nevertheless, the coordination of the leaving water molecule to the copper atom that had received the sulfur atom (structure VIb in Scheme 5) transforms this step into an exergonic step, ΔG = −11.3 kcal mol−1. Finally, the splitting of the units integrating VIb release a pentacoordinate copper complex (entity VII in Scheme 5) that undergoes a dimerization with a calculated free energy, ΔG = −47.1 kcal mol−1. Packing these [{CuL(OH2)(SH)}2] dimers (VIII) to form the crystal structure would lead to the loss of the water molecules compensated for by different intermolecular interactions. In this sense, it is well established that strong non-covalent interactions stabilize the lattice of the [{CuL(SH)}2] compound (see Supporting Information in ref. 80). Regarding hydrogen bonds, it is worth noting that the N4⋯N3 distance is as short as 3.05 Å. Notwithstanding, the π–π interactions are even more relevant in this structure. Three π–π interactions associate the pyridine fragment and two chelating rings in each of the TSC-Cu moieties with their analogues in a neighbouring dimer, with centroid-to-centroid distances of 3.46, 3.33 and 3.46 Å, respectively.
 | ||
| Scheme 5 Final steps of the desulfurization mechanism according to DFT calculations. | ||
The mechanism is of paramount relevance because the presence of [CU2L3]+ species has also been detected in experiments carried out with the antitumor drug Triapine® and Cu(II) ions,99,100 and it could be even more general. Besides, it has been proved that Cu(II) ions have an essential role in the biological activity of free TSCs inside the cell.91
Experimental
Materials and methods
Copper(II) perchlorate hexahydrate, copper(II) nitrate trihydrate, sodium hydroxide, pyridine-2-carbaldehyde and thiosemicarbazide, methanol, acetone, guanine (Hgua) and adenosine-5′-monophosphate monohydrate (H2AMP) were purchased from commercial sources and used as received. Published methods were used to synthesize HL,113,114 [CuL(ONO2)]n (ref. 92) and [CuL2].78 The preparation of complexes of the HL–Cu(II)–nitrate system is described in the ESI.† In particular, compounds with the following formulae are shown: [CuL2], Cu2(HL)L2(NO3)2(H2O)2 and Cu2L2(NO3)(OH) together with the [{CuLCl}2] complex for comparative spectroscopic studies. CAUTION: Perchlorate salts and their respective metal complexes can be explosive and the use of small amounts of reactants is strongly recommended. In fact, thermal measurements attempted on the products were not possible due to small explosions in the thermobalance.Synthetic procedures
![[small upsilon, Greek, macron]](https://www.rsc.org/images/entities/i_char_e0d5.gif) max/cm−1: 3401 m, 3309 s, 3207 m, 2173 w, 1637 m, 1626 m, 1613 s, 1570 m, 1488 m, 1461 s, 1448 s, 1382 m, 1326 m, 1305 m, 1272 m, 1225 m, 1157 m, 1051 vs (υ3-ClO4−), 971 w, 934 w, 914 w, 880 m, 786 w, 769 m, 739 m, 653 w, 619 s (υ4-ClO4−), 590 sh, 514 m, 473 m, 464 m, 451 m, 430 w, 415 m. None of the trials performed allowed us to establish a rational method to obtain compound 2.
max/cm−1: 3401 m, 3309 s, 3207 m, 2173 w, 1637 m, 1626 m, 1613 s, 1570 m, 1488 m, 1461 s, 1448 s, 1382 m, 1326 m, 1305 m, 1272 m, 1225 m, 1157 m, 1051 vs (υ3-ClO4−), 971 w, 934 w, 914 w, 880 m, 786 w, 769 m, 739 m, 653 w, 619 s (υ4-ClO4−), 590 sh, 514 m, 473 m, 464 m, 451 m, 430 w, 415 m. None of the trials performed allowed us to establish a rational method to obtain compound 2.
Further experiments were carried out in order to perform a rational method to obtain compound 3. Some of them involved the reaction between Cu(ClO4)2 and HL in perchloric acid media. In a representative assay, a solution of Cu(ClO4)2·6H2O (1.0 mmol, 0.370 g) in ethanol (15 ml) was acidified with 9 drops of HClO4(c). A few minutes later, a solution of HL (1.0 mmol, 0.180 g) in ethanol (50 ml) was added dropwise and the mixture was stirred for 3 h. A brown precipitate was immediately filtered out and identified as compound 1. The mother liquor was allowed to slowly evaporate for several days, yielding green crystals of compound 5, which were removed. Two months later, dark bluish green crystals of compound 3 were isolated (0.045 g, 9%). No successful elemental analysis was obtained presumably due to the explosive character of the compound. In fact, an attempt to carry out thermogravimetric studies gave rise to a small explosion inside the thermobalance. IR bands (ATR-FTIR) max/cm−1: 3480 b, sh, m, 3380 s, sh, 3302 s, 3210 sh, 3200 s, 3168 sh, 3053 sm, 2980 vw, 2947 w, 2897 vw, 2656 vw, 1635 sh, 1624 s, 1605 sh, 1574 s, 1478 m, 1453 m, 1394 ms, 1332 m, 1304 m, 1281 m, 1227 s, 1185 m, 1160 w, 1130, 1107, 1082, 1049 vs and vb (υ3-ClO4−), 931 m, 880 w, 784 m, 740 m, 666 w, 650 w, 616 s (υ4-ClO4−), 511 w, 448 vw, 450 vw, 415 w. ESI+ mass spectrometry (m/z): 241.97 [CuL]+, 528.94 [Cu2L2(HCO2)]+. X-band EPR signals at RT: g‖ = 2.226, g⊥ = 2.053.
728 charge transfer transition), 298 (50454, intraligand). IR bands (ATR-FTIR) max/cm−1: 3585 b, vw, 3440 m, sh, 3330 s, 3265 mw, 3168 sh, 3115 mb, 3020 vw, 2960 v w, 2705 vw, 1628 m, sh, 1602 s, 1586 s, 1558 m, 1511 m, 1480 m, 1430 vs, 1378 w, 1322 m, 1294 w, 1265 vw, 1253 vw, 1230 m, 1170 m, 1156 m, 1078, 1053, 1041 vs and vb (υ3-ClO4−), 1018 w, 945 vw, 929 vw, 913 w, 902 vw, 875 m, 825 mw, 770 m, 741 w, 722 w, 680 vw, 620 s (υ4-ClO4−), 548 vw, 515 w, 448 vw, 450 w, 415 m. ESI+ mass spectrometry (m/z): 181.05 [H2L]+, 241.97 [CuL]+, 422.02 [Cu(HL)L]+, 530.93 [Cu2L2(HCO2)]+, 664.98 [Cu2L3]+. X-band EPR signals at RT: g = 2.070. Analogous results were achieved by a third method, which required the use of the [CuL2] compound as starting material. However, this experiment yielded a greater amount of unidentified impurities. Briefly, reaction of Cu(ClO4)2·6H2O (1.0 mmol, 0.371 g) and HL (1.0 mmol, 0.180 g) in 20 ml of water was carried out for 1 h. Then, dilute NaOH was added to reach pH 3–4. Afterwards, solid [CuL2] (1.0 mmol, 0.422 g) was added and the reaction was kept with stirring for 3 h. A brown precipitate was obtained, washed with water and acetone and dried (0.683 g, 83%).
max/cm−1: 3525 m, 3339 m, 3275 m, 3165 s, 3066 w, 2934 w, 2890 w, 2136 s υ(NCS), 1614 s, 1568 s, 1475 w, 1454 w, 1390 w, 1330 w, 1297 vw, 1280 w, 1230 m, 1183 w, 1164 w, 1045 vs, broad, and 1017 s (υ3-ClO4−), 927 s, 877 mw, 818 vw, 777 w, 736 w, 671 vw, 698 w, 623 sh and 610 s (υ4-ClO4−), 515 w, 499 vw, 446 w, 409 m. ESI+ mass spectrometry (m/z): 121.05 [RNH2]+ (see Scheme 3), 241.97 [CuL]+, 467.1 [Cu(HL)(NCS)(ClO4)(H2O)2(MeOH)]+, 543.91 [Cu2L(NCS)(ClO4)(H2O)(MeOH)2]+. X-band EPR signals at RT: g1 = 2.196, g2 = 2.058 and g3 = 2.042; Q-band EPR signals at RT: g1 = 2.208, g2 = 2.065 and g3 = 2.045.
Physical measurements
The pH measurements were carried out using a Crison micro pH 2002 pH meter fitted with a combined glass electrode with 3 M KCl solution as a liquid junction. Conductivity measurements were acquired using a Crison 522 conductometer. Microanalyses were performed using a LECO CHNS-932 analyzer. FAB+ mass spectrometry data were obtained on a Micromass AutoSpec instrument. ESI+ measurements were carried out on DMSO solutions of the samples with a Bruker Esquire 3000 Plus LC-MS 6545 Q-TOF Agilent instrument, using as mobile phase a water–methanol (70:30, v/v) mixture acidified by formic acid (0.1%). XRD measurements were recorded at RT on powder samples on a PANalytical X'Pert Pro diffractometer using a copper fine focus X-ray tube, with Cu Kα1λ = 1.54059 Å with a secondary monochromator. Thermal analysis measurements were performed using Netzsch STA 449C and TA SDT 2960 instruments. Crucibles containing 20 mg samples were heated at 10 °C min−1 under a dry air atmosphere. UV-visible measurements were carried out using a Shimadzu UV-2450 spectrophotomer. IR spectra were obtained in the 4000–400 cm−1 region on a JASCO FT-IR 4200 spectrophotometer equipped with a single-reflection ATR PRO ONE device. The intensity of the reported IR bands is defined as vs = very strong, s = strong, m = medium and w = weak, while b stands for broad band. Room-temperature X-band EPR spectra of powder samples were recorded on a Bruker EMX spectrometer equipped with a Bruker ER 036TM NMR tesla meter and an Agilent 53150A microwave frequency counter to fit the magnetic field and the frequency inside the cavity. Q-band measurements were carried out with a Bruker ESP300 spectrometer equipped with a Bruker BNM 200 gaussmeter and a 5352B microwave frequency counter to fit the magnetic field and the frequency inside the cavity. Simulations of the EPR spectra were carried out using the SimFonia program.116 Magnetic measurements of powder samples were performed in the 5–300 K temperature range by using a Quantum Design MPMS-7 SQUID magnetometer with a magnetic field of 0.1 T. Diamagnetic corrections were calculated from Pascal tables.
Crystallography
The structure of 5 is aperiodic. The ESI† contains a succinct description of the modulated structure. Also shown are both the average C2/c structure where perchlorate anions are disordered and an ordered commensurate approximant of the incommensurate structure [q = −0.615a* + 0.345c*, superspace group C2/m(α0γ)] refined using the weak satellite reflections that appear in the diffraction diagram. Crystal data collection was performed on a STOE StadiVari Pilatus-100 K single-crystal diffractometer (multilayer monochromated Mo Kα radiation, λ = 0.71073 Å). Data frames were processed using the X-Area software package.117 Absorption corrections were made using Gaussian integration (compounds 1–3), spherical (4) and integration from the crystal shape (5). Direct methods (SIR97)118 were employed to solve the structures and then refined using the SHELXL-2018 computer program119 within WINGX120 and JANA2006 (ref. 121) (for the modulated structure of 5). The non-H atoms were refined anisotropically using weighted full-matrix least-squares on F2 and hydrogen atoms were placed in calculated positions using idealized geometries (riding model) and assigned fixed isotropic displacement parameters. Selected crystallographic and refinement details for the five crystal structures are shown in Table 1. CCDC 2217580–2217584 contain the supplementary crystallographic data for 1–5. The B-IncStrDB (https://www.cryst.ehu.eus/bincstrdb/search/) reference for the modulated structure of 5 is R50CsLsDyhg.| 1 | 2 | 3 | 4 | 5 | |
|---|---|---|---|---|---|
| Empirical formula | C14H18Cl2Cu2N8O10S2 | C14H14Cl2Cu2N8O8S2 | C14H22Cl4Cu2N8O19S2 | C21H25ClCu2N12O6S3 | C8H8ClCuN5O4S2 |
| Formula weight | 720.46 | 684.43 | 939.39 | 800.24 | 401.3 |
| Temperature (K) | 293(2) | 293(2) | 293(2) | 293(2) | 293(2) |
| Wavelength (Å) | 0.71073 | 0.71073 | 0.71073 | 0.71073 | 0.71073 |
| Crystal system | Monoclinic | Monoclinic | Monoclinic | Monoclinic | Monoclinic |
| Space group | C2/c | C2/c | C2/c | P21/n | C2/m |
| a (Å) | 15.172(4) | 14.812(14) | 14.429(6) | 10.963(3) | 14.950(5) |
| b (Å) | 11.583(5) | 9.144(4) | 18.170(13) | 10.1822(13) | 6.5697(17) |
| c (Å) | 29.439(5) | 17.494(9) | 13.688(5) | 18.387(3) | 15.010(5) |
| β (°) | 95.35(2) | 104.95(9) | 118.29(4) | 91.860(14) | 106.96(2) |
| Volume (Å3) | 5151(3) | 2289(3) | 3160(3) | 3174.1(9) | 1410.1(7) |
| Z | 8 | 4 | 4 | 4 | 4 |
| ρ (mg m−3) | 1.858 | 1.986 | 1.975 | 1.675 | 1.890 |
| μ (mm−1) | 2.086 | 2.336 | 1.909 | 1.679 | 2.056 |
| Reflns meas. | 11721 |
8551 | 12025 |
34357 |
5377 |
| Reflns ref. (Rint) | 4472 (0.052) | 2226 (0.170) | 2879 (0.114) | 9647 (0.090) | 1762 (0.0864) |
| Reflns [I > 2σ(I)] | 3352 | 1099 | 1453 | 3516 | 801 |
| R all/[I > 2σ(I)] | 0.0532/0.0361 | 0.1265/0.0516 | 0.1104/0.0453 | 0.1870/0.0516 | 0.1265/0.0497 |
| wR all/[I > 2σ(I)] | 0.095/0.091 | 0.133/0.113 | 0.103/0.088 | 0.123/0.093 | 0.115/0.093 |
Computational details
Quantum chemical calculations were performed using the B3LYP exchange–correlation functional122,123 implemented in the Gaussian09 package.124 Cu and S centers were described with the Stuttgart RECPs and associated basis sets.125 6-31G** basis sets were used for all other atoms.126,127 See Chapter 4 in the ESI† dealing with computational studies for more details.Conclusions
Five compounds containing thiosemicarbazone copper(II) [CuL]+/[Cu(HL)]2+ entities and perchlorate anions have been crystallographically studied. The crystal structures show the presence of polynuclear species, 1D or dinuclear entities, in the compounds with anionic thiosemicarbazone L− ligands, while the linkage to neutral HL ligand leads to the formation of mononuclear complexes. This great structural diversity is ascribed to the low coordination ability exhibited by the perchlorate anion, its non-hydrolyzable character and the high solubility of its complexes. All these features provoke (i) the activation of the hydrazine N3 as a donor atom in 1, (ii) the building of X-bridged dimers in 2, and (iii) the coexistence in the same complex of perchlorate and the neutral HL form in 3 and 5. The structural richness is shared by other complexes with O-donor ligands derived from strong acids, such as sulfate and nitrate. In fact, a clear parallelism with the chemical, magnetic and structural behaviour in the analogous Cu(II)–HL–nitrate system can be established. Desulfurization processes operate in acidic media and give rise to the formation of thiocyanate ligands in compound 5. On the other hand, our studies point to a possible role that [Cu2L3]+ entities, such as those present in compound 4, could play in intermediate stages of desulfurization reactions developed in basic medium or even at physiological pH values. These and/or analogous species, at low concentrations and with short lifetimes, could be involved in different chemical processes triggered by thiosemicarbazone copper(II) complexes in biological medium, strongly affecting their potential therapeutic uses.Author contributions
Conceptualization, G. M., R. P. and J. G.-T.; methodology, R. G.-G., G. M., J. B., J. V. C., P. G.-S., L. L. and J. G.-T.; investigation, synthesis and characterization: R. G.-G., A. J.-P., I. H.-T., A. G.-G., M. U., V. J., J. B.-G., P. G.-S. and J. G.-T.; investigation, MS: M. S. G. and J. B.; investigation, TG: M. I.; investigation, XRD: G. M.; investigation, DFT calculations: J. V. C.; investigation, EPR & magnetism: L. L. and J. G.-T; resources, G. M., M. I., J. B., L. L. and J. G.-T.; writing—original draft preparation, R. G.-G., G. M., A. J.-P., J. V. C. and J. G.-T.; writing—review and editing, G. M., A. J.-P., J. V. C., R. P., L. L. and J. G.-T.; supervision, G. M., A. J.-P., J. V. C., R. P., L. L. and J. G.-T.; funding acquisition, G. M. and J. G.-T.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Ms. Eva García Ugalde for the drawing in the Graphical Abstract, Dr. J. J. Delgado, Dr. Pilar Castroviejo, Marta Mansilla Dr. Ana Erica Páramo and Ms. Zaida Cabello (PCT, Universidad de Burgos, Spain) for the elemental analysis and mass spectra. This work was supported by the European Union H2020-LC-SC3-2020-NZE-RES-CC, NMBP-16-2020-GA 953152 and DT-NMBP-04-2020 Projects, together with Ministerio de Ciencia, Innovación, for the project PID2021-127531NB-I00 (AEI/10.13039/501100011033/FEDER, UE), PDC2022-133955-I00 and Ministerio de Ciencia, Innovación y Universidades CTQ(QMC) RED2018-102471-T MultiMetDrugs Network (Spain), Consejería de Educación of Junta de Castilla y León and FEDER BU049P20, Fundacion Bancaria Caixa D. Estalvis i Pensions de Barcelona 001 and Ministerio de Ciencia e Innovación PID2019-106644GB-I00 and Gobierno Vasco IT1458-22. R. G.-G. and A. J.-P. wish to thank the Junta de Castilla y León for their Doctoral Fellowships.References
- A. I. Matesanz and P. Souza, Mini-Rev. Med. Chem., 2009, 9, 1389–1396 CrossRef CAS PubMed
.
- T. S. Lobana, R. Sharma, G. Bawa and S. Khanna, Coord. Chem. Rev., 2009, 253, 977–1055 CrossRef CAS
- S. Arora, S. Agarwal and S. Singhal, Int. J. Pharm. Pharm. Sci., 2014, 6, 34–41 CAS
- P. J. Jansson, D. S. Kalinowski, D. J. R. Lane, Z. Kovacevic, N. A. Seebacher, L. Fouani, S. Sahni, A. M. Merlot and D. R. Richardson, Pharmacol. Res., 2015, 100, 255–260 CrossRef CAS PubMed
- P. F. Salas, C. Herrmann and C. Orvig, Chem. Rev., 2013, 113, 3450–3492 CrossRef CAS PubMed
- D. Krajčiová, M. Melník, E. Havránek, A. Forgácsová and P. Mikuš, J. Coord. Chem., 2014, 67, 1493–1519 CrossRef
- J. García-Tojal, R. Gil-García, P. Gómez-Saiz and M. Ugalde, Curr. Inorg. Chem., 2011, 1, 189–210 CrossRef
- S. Priyarega, J. Haribabu and R. Karvembu, Inorg. Chim. Acta, 2022, 532, 120742 CrossRef CAS
- Z. X. He, J. L. Huo, Y. P. Gong, Q. An, X. Zhang, H. Qiao, F. F. Yang, X. H. Zhang, L. M. Jiao, H. M. Liu, L. Y. Ma and W. Zhao, Eur. J. Med. Chem., 2020, 210, 112970 CrossRef PubMed
- L. R. P. de Siqueira, P. A. T. de Moraes Gomes, L. P. de Lima Ferreira, M. J. B. de Melo Rêgo and A. C. L. Leite, Eur. J. Med. Chem., 2019, 170, 237–260 CrossRef CAS PubMed
- V. Jevtovic, H. Hamoud, S. Al-zahrani, K. Alenezi, S. Latif, T. Alanazi, F. Abdyulaziz and D. Dimic, Molecules, 2022, 27, 4809 CrossRef CAS PubMed
- N. C. Kasuga, K. Sekino, M. Ishikawa, A. Honda, M. Yokoyama, S. Nakano, N. Shimada, C. Koumo and K. Nomiya, J. Inorg. Biochem., 2003, 96, 298–310 CrossRef CAS PubMed
- H. Beraldo and D. Gambino, Mini-Rev. Med. Chem., 2004, 4, 31–39 CrossRef CAS PubMed
- E. Pahontu, F. Julea, T. Rosu, V. Purcarea, Y. Chumakov, P. Petrenco and A. Gulea, J. Cell. Mol. Med., 2015, 19, 865–878 CrossRef CAS PubMed
- R. Salehi, S. Abyar, F. Ramazani, A. A. Khandar, S. A. Hosseini-Yazdi, J. M. White, M. Edalati, H. Kahroba and M. Talebi, Sci. Rep., 2022, 12, 8316 CrossRef CAS PubMed
- P. F. Salas, C. Herrmann and C. Orvig, Chem. Rev., 2013, 113, 3450–3492 CrossRef CAS PubMed
- S. Savir, J. W. K. Liew, I. Vythilingam, Y. A. L. Lim, C. H. Tan, K. S. Sim, V. S. Lee, M. J. Maah and K. W. Tan, J. Mol. Struct., 2021, 1242, 130815 CrossRef CAS
- M. Adams, C. De Kock, P. J. Smith, K. M. Land, N. Liu, M. Hopper, A. Hsiao, A. R. Burgoyne, T. Stringer, M. Meyer, L. Wiesner, K. Chibale and G. S. Smith, Dalton
Trans., 2015, 44, 2456–2468 RSC
- Z. X. He, J. L. Huo, Y. P. Gong, Q. An, X. Zhang, H. Qiao, F. F. Yang, X. H. Zhang, L. M. Jiao, H. M. Liu, L. Y. Ma and W. Zhao, Eur. J. Med. Chem., 2020, 210, 112970 CrossRef PubMed
- R. Arancibia, C. Quintana, C. Biot, M. E. Medina, S. Carrère-Kremer, L. Kremer and A. H. Klahn, Inorg. Chem. Commun., 2015, 55, 139–142 CrossRef CAS
- D. L. Sun, S. Poddar, R. D. Pan, E. W. Rosser, E. R. Abt, J. Van Valkenburgh, T. M. Le, V. Lok, S. P. Hernandez, J. Song, J. Li, A. Turlik, X. Chen, C.-A. Cheng, W. Chen, C. E. Mona, A. D. Stuparu, L. Vergnes, K. Reue, R. Damoiseaux, J. I. Zink, J. Czernin, T. R. Donahue, K. N. Houk, M. E. Jung and C. G. Radu, RSC Med. Chem., 2020, 11, 392–410 RSC
- G. D. K. Kumar, G. E. Chavarria, A. K. Charlton-Sevcik, G. K. Yoo, J. Song, T. E. Strecker, B. G. Siim, D. J. Chaplin, M. L. Trawick and K. G. Pinney, Bioorg. Med. Chem. Lett., 2010, 20, 6610–6615 CrossRef CAS PubMed
- D. Rogolino, A. Bacchi, L. De Luca, G. Rispoli, M. Sechi, A. Stevaert, L. Naesens and M. Carcelli, J. Biol. Inorg. Chem., 2015, 20, 1109–1121 CrossRef CAS PubMed
- A. S. Nagle, S. Khare, A. B. Kumar, F. Supek, A. Buchynskyy, C. J. N. Mathison, N. K. Chennamaneni, N. Pendem, F. S. Buckner, M. H. Gelb and V. Molteni, Chem. Rev., 2014, 114, 11305–11347 CrossRef CAS PubMed
- Y. C. Ong, S. Roy, P. C. Andrews and G. Gasser, Chem. Rev., 2019, 119, 730–796 CrossRef CAS PubMed
- D. Palanimuthu, Z. Wu, P. J. Jansson, N. Braidy, P. V. Bernhardt, D. R. Richardson and D. S. Kalinowski, Dalton Trans., 2018, 47, 7190–7205 RSC
- C. Lambert, H. Beraldo, N. Lievre, A. Garnier-Suillerot, P. Dorlet and M. Salerno, J. Biol. Inorg. Chem., 2013, 18, 59–69 CrossRef CAS PubMed
- J. L. Hickey, P. J. Crouch, S. Mey, A. Caragounis, J. M. White, A. R. White and P. S. Donnelly, Dalton Trans., 2011, 40, 1338–1347 RSC
- M. Varma, B. Shravage, S. Tayade, A. Kumbhar, R. Butcher, V. Jani, U. Sonavane, R. Joshi and P. P. Kulkarni, J. Mol. Struct., 2021, 1235, 130265 CrossRef CAS
- J. M. Gardiner and J. Iqbal, Med. Chem. Commun., 2017, 8, 452–464 RSC
- L. Al-Akra, D. H. Bae, S. Sahni, M. L. H. Huang, K. C. Park, D. J. R. Lane, P. J. Jansson and D. R. Richardson, J. Biol. Chem., 2018, 293, 3562–3587 CrossRef CAS PubMed
- A. Steinbrueck, A. C. Sedgwick, J. T. Brewster, K.-C. Yan, Y. Shang, D. M. Knoll, G. I. Vargas-Zúñiga, X.-P. He, H. Tian and J. L. Sessler, Chem. Soc. Rev., 2020, 49, 3726–3747 RSC
- B. Englinger, C. Pirker, P. Heffeter, A. Terenzi, C. R. Kowol, B. K. Keppler and W. Berger, Chem. Rev., 2019, 119, 1519–1624 CrossRef CAS PubMed
- A. Mrozek-Wilczkiewicz, M. Serda, R. Musiol, G. Malecki, A. Szurko, A. Muchowicz, J. Golab, A. Ratuszna and J. Polanski, ACS Med. Chem. Lett., 2014, 5, 336–339 CrossRef CAS PubMed
- X.-G. Bai, Y. Zheng and J. Qi, Front. Pharmacol., 2022, 13, 1–15 Search PubMed
- Y. Yu, D. S. Kalinowski, Z. Kovacevic, A. R. Siafakas, P. J. Jansson, C. Stefani, D. B. Lovejoy, P. C. Sharpe, P. V. Bernhardt and D. R. Richardson, J. Med. Chem., 2009, 52, 5271–5294 CrossRef CAS PubMed
- E. M. Gutierrez, N. A. Seebacher, L. Arzuman, Z. Kovacevic, D. J. R. Lane, V. Richardson, A. M. Merlot, H. Lok, D. S. Kalinowski, S. Sahni, P. J. Jansson and D. R. Richardson, Biochim. Biophys. Acta, Mol. Cell Res., 2016, 1863, 1665–1681 CrossRef CAS PubMed
- A. M. Merlot, N. H. Shafie, Y. Yu, V. Richardson, P. J. Jansson, S. Sahni, D. J. R. Lane, Z. Kovacevic, D. S. Kalinowski and D. R. Richardson, Biochem. Pharmacol., 2016, 109, 27–47 CrossRef CAS PubMed
- C. Bailly, Chem. Rev., 2012, 112, 3611–3640 CrossRef CAS PubMed
- W. Hu, X. S. Huang, J. F. Wu, L. Yang, Y. T. Zheng, Y. M. Shen, Z. Y. Li and X. Li, J. Med. Chem., 2018, 61, 8947–8980 CrossRef CAS PubMed
- K. Haldys and R. Latajka, MedChemComm, 2019, 10, 378–389 RSC
- M. B. Mannargudi and S. Deb, J. Cancer Res. Clin. Oncol., 2017, 143, 1499–1529 CrossRef CAS PubMed
- V. Pósa, B. Hajdu, G. Tóth, O. Dömötör, C. R. Kowol, B. K. Keppler, G. Spengler, B. Gyurcsik and É. A. Enyedy, J. Inorg. Biochem., 2022, 231, 111786 CrossRef PubMed
- K. C. Park, L. Fouani, P. J. Jansson, D. Wooi, S. Sahni, D. J. R. Lane, D. Palanimuthu, H. C. Lok, Z. Kovačević, M. L. H. Huang, D. S. Kalinowski and D. Richardson, Metallomics, 2016, 874–886 CrossRef CAS PubMed
- D. B. Lovejoy, P. J. Jansson, U. T. Brunk, J. Wong, P. Ponka and D. R. Richardson, Cancer Res., 2011, 71, 5871–5880 CrossRef CAS PubMed
- M. Sobiesiak, T. Muzioł, M. Rozalski, U. Krajewska and E. Budzisz, New J. Chem., 2014, 38, 5349–5361 RSC
- C. Santini, M. Pellei, V. Gandin, M. Porchia, F. Tisato and C. Marzano, Chem. Rev., 2014, 114, 815–862 CrossRef CAS PubMed
- A. Mrozek-Wilczkiewicz, K. Malarz, M. Rams-Baron, M. Serda, D. Bauer, F. P. Montforts, A. Ratuszna, T. Burley, J. Polanski and R. Musiol, J. Cancer, 2017, 8, 1979–1987 CrossRef PubMed
- A. N. Nikam, A. Pandey, G. Fernandes, S. Kulkarni, S. P. Mutalik, B. S. Padya, S. D. George and S. Mutalik, Coord. Chem. Rev., 2020, 419, 213356 CrossRef CAS
- P. Heffeter, V. F. S. Pape, É. A. Enyedy, B. K. Keppler, G. Szakacs and C. R. Kowol, Antioxid. Redox Signaling, 2018, 30, 1062–1082 CrossRef PubMed
- M. Wehbe, A. W. Y. Leung, M. J. Abrams, C. Orvig and M. B. Bally, Dalton Trans., 2017, 46, 10758–10773 RSC
- R. A. C. Souza, W. R. P. Costa, E. de Franca Faria, M. A. de S. Bessa, R. de P. Menezes, C. H. G. Martins, P. I. S. Maia, V. M. Deflon and C. G. Oliveira, J. Inorg. Biochem., 2021, 223, 111543 CrossRef CAS PubMed
- D. J. Lane, T. M. Mills, N. H. Shafie, A. M. Merlot, R. Saleh Moussa, D. S. Kalinowski, Z. Kovacevic and D. R. Richardson, Biochim. Biophys. Acta, Rev. Cancer, 2014, 1845, 166–181 CrossRef CAS PubMed
- F. N. Akladios, S. D. Andrew and C. J. Parkinson, J. Biol. Inorg. Chem., 2016, 21, 931–944 CrossRef CAS PubMed
- M. Carcelli, M. Tegoni, J. Bartoli, C. Marzano, G. Pelosi, M. Salvalaio, D. Rogolino and V. Gandin, Eur. J. Med. Chem., 2020, 194, 112266 CrossRef CAS PubMed
- J. Shao, Z.-Y. Ma, A. Li, Y.-H. Liu, C.-Z. Xie, Z.-Y. Qiang and J.-Y. Xu, J. Inorg. Biochem., 2014, 136, 13–23 CrossRef CAS PubMed
- J. Deng, P. Yu, Z. Zhang, J. Wang, J. Cai, N. Wu, H. Sun, H. Liang and F. Yang, Eur. J. Med. Chem., 2018, 158, 442–452 CrossRef CAS PubMed
- E. Palma, P. Raposinho, M. P. C. Campello, D. Belo, J. F. Guerreiro, V. Alves, A. Fonseca, A. J. Abrunhosa, A. Paulo and F. Mendes, Eur. J. Inorg. Chem., 2021, 1337–1348 CrossRef CAS
- N. K. Singh, A. A. Kumbhar, Y. R. Pokharel and P. N. Yadav, J. Inorg. Biochem., 2020, 210, 111134 CrossRef CAS PubMed
- I. Stepanenko, M. V. Babak, G. Spengler, M. Hammerstad, A. P. Bijelic, S. Shova, G. Büchel, D. Darvasiova, P. Rapta and V. Arion, Biomolecules, 2021, 11, 862 CrossRef CAS PubMed
- M. Belicchi-Ferrari, F. Bisceglie, A. Buschini, S. Franzoni, G. Pelosi, S. Pinelli, P. Tarasconi and M. Tavonie, J. Inorg. Biochem., 2010, 104, 199–206 CrossRef CAS PubMed
- J. H. Bormio Nunes, S. Hager, M. Mathuber, V. Pósa, A. Roller, É. A. Enyedy, A. Stefanelli, W. Berger, B. K. Keppler, P. Heffeter and C. R. Kowol, J. Med. Chem., 2020, 63, 13719–13732 CrossRef CAS PubMed
- C. A. Kunos and S. P. Ivy, Front. Oncol., 2018, 8, 149 CrossRef PubMed
- J. Kolesar, R. C. Brundage, M. Pomplun, D. Alberti, K. Holen, A. Traynor, P. Ivy and G. Wilding, Cancer Chemother. Pharmacol., 2011, 67, 393–400 CrossRef CAS PubMed
- R. C. DeConti, B. R. Toftness, K. C. Agrawal, R. Tomchick, J. A. R. Mead, J. R. Joseph, B. Bertino, A. C. Sartorelli and W. A. Creasey, Cancer Res., 1972, 32, 1455–1462 CAS
- A. B. Miah, K. J. Harringtom and C. M. Nutting, Eur. J. Clin. Med. Oncol., 2010, 2, 87–92 Search PubMed
- Z.-L. Guo, D. R. Richardson, D. S. Kalinowski, Z. Kovacevic, K. C. Tan-Un and G. C.-F. Chan, J. Hematol. Oncol., 2016, 9, 98 CrossRef PubMed
- https://clinicaltrials.gov/ct2/show/NCT02688101?term=DpC&draw=2&rank=1, (access 06.10.2022).
- https://clinicaltrials.gov/ct2/show/NCT02433626?term=thiose-micarbazone&draw=2&rank=9, (access 06.10.2022).
- M. Serda, D. S. Kalinowski, N. Rasko, E. Potučková, A. Mrozek-Wilczkiewicz, R. Musiol, J. G. Małecki, M. Sajewicz, A. Ratuszna, A. Muchowicz, J. Gołąb, T. Šimunek, D. R. Richardson and J. Polanski, PLoS One, 2014, 9, e110291 CrossRef PubMed
- https://clinicaltrials.gov/ct2/show/NCT02466971?term=thiosemicarbazone&draw=2&rank=12, (access 06.10.2022).
- P. Quach, E. Gutierrez, M. T. Basha, D. S. Kalinowski, P. C. Sharpe, D. B. Lovejoy, P. V. Bernhardt, P. J. Jansson and D. R. Richardson, Mol. Pharmacol., 2012, 82, 105–114 CrossRef CAS PubMed
- K. Pelivan, L. M. Frensemeier, U. Karst, G. Koellensperger, P. Heffeter, B. K. Keppler and C. R. Kowol, Anal. Bioanal. Chem., 2018, 410, 2343–2361 CrossRef CAS PubMed
- W. A. Creasey, K. C. Agrawal, R. L. Capizzi and K. K. Stinson, Cancer Res., 1972, 32, 565–572 CAS
- D. B. Lovejoy, P. J. Jansson, U. T. Brunk, J. Wong, P. Ponka and D. R. Richardson, Cancer Res., 2011, 71, 5871–5880 CrossRef CAS PubMed
- M. Haeili, C. Moore, C. J. C. Davis, J. B. Cochran, S. Shah, T. B. Shrestha, Y. Zhang, S. H. Bossmann, W. H. Benjamin, O. Kutsch and F. Wolschendorf, Antimicrob. Agents Chemother., 2014, 58, 3727–3736 CrossRef PubMed
- A. E. Stacy, D. Palanimuthu, P. V. Bernhardt, D. S. Kalinowski, P. J. Jansson and D. R. Richardson, J. Med. Chem., 2016, 59, 4965–4984 CrossRef CAS PubMed
- P. Gómez-Saiz, R. Gil-García, M. A. Maestro, J. L. Pizarro, M. I. Arriortua, L. Lezama, T. Rojo and J. García-Tojal, Eur. J. Inorg. Chem., 2005, 3409–3413 CrossRef
- E. W. Ainscough, A. M. Brodie, W. A. Denny, G. J. Finlay and J. D. Ranford, J. Inorg. Biochem., 1998, 70, 175–185 CrossRef CAS PubMed
- R. Gil-García, R. Fraile, B. Donnadieu, G. Madariaga, V. Januskaitis, J. Rovira, L. González, J. Borrás, F. J. Arnáiz and J. García-Tojal, New J. Chem., 2013, 37, 3568–3580 RSC
- M. B. Ferrari, F. Bisceglie, G. Pelosi, P. Tarasconi, R. Albertini, A. Bonati, P. Lunghi and S. Pinelli, J. Inorg. Biochem., 2001, 83, 169–179 CrossRef CAS PubMed
- A. I. Matesanz, I. Cuadrado, C. Pastor and P. Souza, Z. Anorg. Allg. Chem., 2005, 631, 780–784 CrossRef CAS
- A. Castiñeiras and I. Garcia-Santos, Z. Anorg. Allg. Chem., 2008, 634, 2907–2916 CrossRef
- P. Gómez-Saiz, J. García-Tojal, V. Diez-Gómez, R. Gil-García, J. L. Pizarro, M. I. Arriortua and T. Rojo, Inorg. Chem. Commun., 2005, 8, 259–262 CrossRef
- R. Pedrido, M. J. Romero, M. R. Bermejo, M. Martínez-Calvo, A. M. González-Noya and G. Zaragoza, Dalton Trans., 2009, 8329–8340 RSC
- A. Castiñeiras and R. Pedrido, Dalton Trans., 2010, 39, 3572–3584 RSC
- P. I. D. S. Maia, H. H. Nguyen, D. Ponader, A. Hagenbach, S. Bergemann, R. Gust, V. M. Deflon and U. Abram, Inorg. Chem., 2012, 51, 1604–1613 CrossRef CAS PubMed
- P. Zhang, B. Shi, Y. Zhang, Q. Lin, H. Yao, X. You and T. Wei, Tetrahedron, 2013, 69, 10292–10298 CrossRef CAS
- S. Fernández-Fariña, L. M. González-Barcia, M. J. Romero, J. García-Tojal, M. Maneiro, J. M. Seco, G. Zaragoza, M. Martínez-Calvo, A. M. González-Noya and R. Pedrido, Inorg. Chem. Front., 2022, 531–536 RSC
- G. Pelosi, S. Pinelli and F. Bisceglie, Compounds, 2022, 2, 144–162 CrossRef
- K. Ishiguro, Z. P. Lin, P. G. Penketh, K. Shyam, R. Zhu, R. P. Baumann, Y. L. Zhu, A. C. Sartorelli, T. J. Rutherford and E. S. Ratner, Biochem. Pharmacol., 2014, 91, 312–322 CrossRef CAS PubMed
- A. G. Bingham, H. Bögge, A. Müller, E. W. Ainscough and A. M. Brodie, J. Chem. Soc., Dalton Trans., 1987, 493–499 RSC
- R. Gil-García, P. Gómez-Saiz, V. Díez-Gómez, B. Donnadieu, M. Insausti, L. Lezama and J. García-Tojal, Polyhedron, 2013, 54, 243–251 CrossRef
- Y. M. Chumakov, V. I. Tsapkov, E. Jeanneau, N. N. Bairac, G. Bocelli, D. Poirier, J. Roy and A. P. Gulea, Crystallogr. Rep., 2008, 53, 786–792 CrossRef CAS
- J. García-Tojal, L. Lezama, J. L. Pizarro, M. Insausti, M. I. Arriortua and T. Rojo, Polyhedron, 1999, 18, 3703–3711 CrossRef
- J. García-Tojal, R. Gil-García, V. I. Fouz, G. Madariaga, L. Lezama, M. S. Galletero, J. Borrás, F. I. Nollmann, C. García-Girón, R. Alcaraz, M. Cavia-Saiz, P. Muñiz, Ò. Palacios, K. G. Samper and T. Rojo, J. Inorg. Biochem., 2018, 180, 69–79 CrossRef PubMed
- B. García, J. Garcia-Tojal, R. Ruiz, R. Gil-García, S. Ibeas, B. Donnadieu and J. M. Leal, J. Inorg. Biochem., 2008, 102, 1892–1900 CrossRef PubMed
- R. Gil-García, M. Ugalde, N. Busto, H. J. Lozano, J. M. Leal, B. Pérez, G. Madariaga, M. Insausti, L. Lezama, R. Sanz, B. García and J. García-Tojal, Dalton Trans., 2016, 45, 18704–18718 RSC
- É. A. Enyedy, N. V. Nagy, É. Zsigó, C. R. Kowol, V. B. Arion, B. K. Keppler and T. Kiss, Eur. J. Inorg. Chem., 2010, 1717–1728 CrossRef
- O. Dömötör, N. V. May, K. Pelivan, T. Kiss, B. K. Keppler, C. R. Kowol and É. A. Enyedy, Inorg. Chim. Acta, 2018, 472, 264–275 CrossRef
- A. W. Addison, T. N. Rao, J. Reedijk, J. van Rijn and G. C. Verschoor, J. Chem. Soc., Dalton Trans., 1984, 1349–1356 RSC
- T. S. Lobana, P. Kumari, R. J. Butcher, J. P. Jasinski and J. A. Golen, Z. Anorg. Allg. Chem., 2012, 638, 1861–1867 CrossRef CAS
- M. T. Huseynova, M. N. Aliyeva, A. A. Medjidov, O. Şahin and B. Yalçın, J. Mol. Struct., 2019, 1176, 895–900 CrossRef CAS
- P. Gómez-Saiz, J. García-Tojal, A. Mendia, B. Donnadieu, L. Lezama, J. L. Pizarro, M. I. Arriortua and T. Rojo, Eur. J. Inorg. Chem., 2003, 518–527 CrossRef
- E. W. Ainscough, A. M. Brodie, J. D. Ranford and J. M. Waters, J. Chem. Soc., Dalton Trans., 1991, 2125–2131 RSC
- Y. Wang, Y. Fang, M. Zhao, M. Li, Y. Ji and Q. Han, Med. Chem. Commun., 2017, 8, 2125–2132 RSC
- Z. Al-Eisawi, C. Stefani, P. J. Jansson, A. Arvind, P. C. Sharpe, M. T. Basha, G. M. Iskander, N. Kumar, Z. Kovacevic, D. J. R. Lane, S. Sahni, P. V. Bernhardt, D. R. Richardson and D. S. Kalinowski, J. Med. Chem., 2016, 59, 294–312 CrossRef CAS PubMed
- L. Yang, D. R. Powell and R. P. Houser, Dalton Trans., 2007, 955–964 RSC
- R. Gil-García, R. Zichner, V. Díez-Gómez, B. Donnadieu, G. Madariaga, M. Insausti, L. Lezama, P. Vitoria, M. R. Pedrosa and J. García-Tojal, Eur. J. Inorg. Chem., 2010, 4513–4525 CrossRef
- P. Gómez-Saiz, J. García-Tojal, M. A. Maestro, F. J. Arnaiz and T. Rojo, Inorg. Chem., 2002, 41, 1345–1347 CrossRef PubMed
- R. Gil-García, P. Gómez-Saiz, V. Díez-Gómez, G. Madariaga, M. Insausti, L. Lezama, J. V. Cuevas and J. García-Tojal, Polyhedron, 2014, 81, 675–686 CrossRef
- E. Falcone, A. G. Ritacca, S. Hager, H. Schueffl, B. Vileno, Y. El Khoury, P. Hellwig, C. R. Kowol, P. Heffeter, E. Sicilia and P. Faller, J. Am. Chem. Soc., 2022, 144, 14758–14768 CrossRef CAS PubMed
- F. E. Anderson, C. J. Duca and J. V. Scudi, J. Am. Chem. Soc., 1951, 73, 4967–4968 CrossRef CAS
- D. J. Leggett and W. A. E. McBryde, Talanta, 1974, 21, 1005–1011 CrossRef CAS PubMed
- J. García-Tojal, M. K. Urtiaga, R. Cortés, L. Lezama, M. I. Arriortua and T. Rojo, J. Chem. Soc., Dalton Trans., 1994, 2233–2238 RSC
-
WINEPR SimFonia, version 1.25; Bruker Analytische Messtecnik GmβH, Rheinstetten, Germany, 1996 Search PubMed
-
X-Area, Software Manual, STOE & Cie Gmbh, Darmstadt, Germany, 2015
- A. Altomare, M. C. Burla, M. Camalli, G. L. Cascarano, C. Giacovazzo, A. Guagliardi, A. G. G. Moliterni, G. Polidori and R. Spagna, J. Appl. Crystallogr., 1999, 32, 115–119 CrossRef CAS
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed
- L. J. Farrugia, WINGX, J. Appl. Crystallogr., 2012, 45, 849–854 CrossRef CAS
- V. Petricek, M. Dusek and L. Palatinus, Crystallographic Computing System JANA2006: General features, Z. Kristallogr., 2014, 229, 345–352 CAS
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS
- C. T. Lee, W. T. Yang and R. G. Parr, Phys. Rev. B, 1988, 37, 785–789 CrossRef CAS PubMed
-
M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision D.01, Gaussian Inc., Wallingford, CT, 2009
- D. Andrae, U. Häußermann, M. Dolg, H. Stoll, H. Preuß, U. Wedig, H. Stoll and H. Preuss, J. Chem. Phys., 1987, 77, 123–141 Search PubMed
- P. C. Hariharan and J. A. Pople, Theor. Chim. Acta, 1973, 28, 213–222 CrossRef CAS
- W. J. Hehre, R. Ditchfield and J. A. Pople, J. Chem. Phys., 1972, 56, 2257–2261 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available: Details on the title compounds of the Cu2+/HL/ClO4− system, summary about the parallel Cu2+/HL /NO3− system, studies in aqueous basic media and computational studies. CCDC 2217580–2217584. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3ce00119a |
| This journal is © The Royal Society of Chemistry 2023 |