
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Halogen-bonded liquid-crystalline complexes formed from 4-alkoxyphenylpyridines with iodine and with interhalogens†
Linda J.
McAllister
,
James
Taylor
 ,
Natalie E.
Pridmore
,
Alice J.
McEllin
,
Adrian C.
Whitwood
,
Peter B.
Karadakov
* and
Duncan W.
Bruce
*
,
Natalie E.
Pridmore
,
Alice J.
McEllin
,
Adrian C.
Whitwood
,
Peter B.
Karadakov
* and
Duncan W.
Bruce
*
Department of Chemistry, University of York, Heslington, York YO10 5DD, UK. E-mail: duncan.bruce@york.ac.uk; Tel: +44 (0)1904 324085
First published on 20th February 2023
Abstract
Strongly halogen-bonded complexes are formed between 4-alkoxyphenyl-4-pyridines and iodine as well as the interhalogen compounds ICl and IBr, and examples of each are characterised by single crystal X-ray crystallography. On heating, all but one of the complexes display a liquid-crystalline smectic A phase, although there is evidence of decomposition as the materials are heated through the mesophase. For such short molecules, the mesophases are rather stable and small-angle X-ray scattering shows that the complexes form a type of antiparallel, head-to-head dimeric arrangement in the mesophase. Quantum chemical calculations at the DFT (M06-2X) and MP2 levels of theory show the complexes to have very high dipole moments (between ≈ 9–12 D) and the mesophase stability of the complexes is rationalised in terms of antiparallel correlations induced by the strong molecular dipoles.
Introduction
Self-assembly using halogen bonding is now an exceptionally well-embedded strategy that has enriched supramolecular approaches to a very wide variety of material types, providing new design possibilities that can be exploited in their own right or in conjunction with other non-covalent interactions.1 The strength of a halogen bond, a non-covalent interaction between a Lewis base and an electrophilic halogen, is often evaluated by consideration of its length when compared to the sum of the van der Waals radii of the two atoms from which it is formed. Typically for a pyridine nitrogen and an iodine as in, for example, iodopentafluorobenzene, the value comes out at around 80% corresponding in this case to a distance of ca. 2.8 Å.2 Of course, this length depends on the donor ability of the Lewis acid and also on the nature of the halogen, and particularly strong halogen bonds are known to form when molecular iodine is employed as the halogen bond donor.3One of the applications of halogen bonding has been in the area of liquid crystals4–7 and there has been significant interest in this field since the first reports of the formation of a nematic (N) and a smectic A (SmA) phase from a halogen-bonded complex formed between 4-alkoxystilbazoles and iodopentafluorobenzene.8 Many of these liquid-crystal complexes exhibit lability of the halogen bond so that repeated thermal cycles can lead to decomposition,9,10 which in turn stimulates interest in systems that contain a more robust halogen bond.
Halogen-bonded complexes of molecular halogens and interhalogens formed the very origins of halogen bonding going back to reports from, for example Guthrie in the 1860s, of iodine adducts of ammonia. Following on from a review by Bent,11 much of this history is given in an excellent review by Pennington et al.,3 both of whom note the significant breakthrough made by Hassel and Hvoslef in reporting the solid-state structure determination of the 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 adduct between 1,4-dioxane and Br2.12 Gas-phase studies have been reviewed by Legon.13
:1 adduct between 1,4-dioxane and Br2.12 Gas-phase studies have been reviewed by Legon.13
Halogen bonds to molecular halogens and interhalogens have proved to be some of the strongest found and Pennington's review provides extensive tables that illustrate well the sorts of intermolecular distances involved.3 To that end, we reported liquid crystals formed between alkoxystilbazoles and molecular iodine (Fig. 1), in which the N⋯I distance was 2.353 Å, exceptionally short at 67% of the sum of the van der Waals radii. Indeed, the robustness of the system was shown by the observation of liquid crystal mesophases at temperatures in excess of 200 °C.14 A remarkable feature of these complexes was the fact that they retained a head-to-head dimeric arrangement in the mesophase, although it was not obvious how this orientation was held together at such elevated temperatures, not least as the I⋯I interactions ought to have been type I and thus repulsive.
 | ||
| Fig. 1 Liquid-crystalline materials formed by iodine complexes of alkoxystilbazoles. The diagram shows the relative disposition of pairs of complexes as found in the solid state. | ||
In attempting to prepare the analogous complexes with dibromine, we observed that the bromine initially added across the double bond of the stilbazole before eliminating HBr regiospecifically to leave a stilbazole with a bromine on a carbon of its double bond (Fig. 2).14
 | ||
| Fig. 2 Scheme showing the reaction between an alkoxystilbazole and bromine. | ||
Later, Chen et al. reported the preparation of halogen-bonded complexes of both iodine and bromine using azo pyridines (Fig. 3) with structures related to the stilbazoles in Fig. 1.15 By analogy with our stilbazole derivatives, these complexes also showed a SmA phase and this included the complexes with bromine, which in this case could be isolated as bromine cannot add across the N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N double bond. The DSC traces show that, with the exception of the homologue with n = 6, these complexes appear unstable at elevated temperatures as the clearing point is significantly lower on cooling compared to that on heating (e.g. around 20 °C for n = 8 when complexed with Br2).
N double bond. The DSC traces show that, with the exception of the homologue with n = 6, these complexes appear unstable at elevated temperatures as the clearing point is significantly lower on cooling compared to that on heating (e.g. around 20 °C for n = 8 when complexed with Br2).
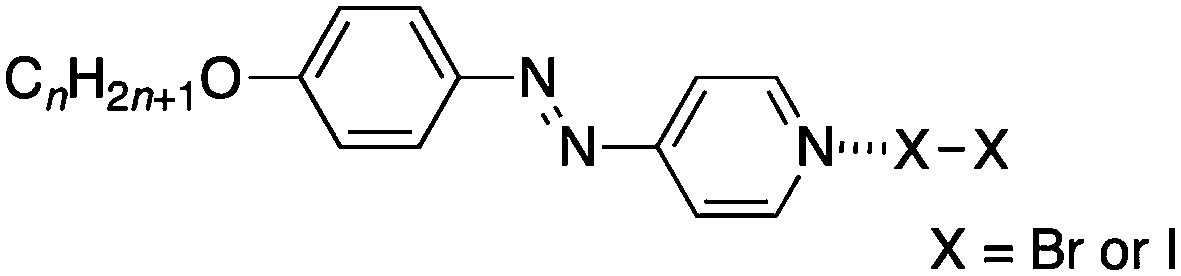 | ||
| Fig. 3 Molecular halogen complexes of azopyridines showing liquid crystal mesophases. | ||
While considering other approaches to liquid crystals halogen bonded to both dibromine and other reactive species such as interhalogens, we elected to remove the source of the reactivity in the stilbazole used previously (the CC double bond) by simply preparing 4-alkoxyphenyl-4-pyridines instead. These were readily obtained via a palladium-catalysed Suzuki–Miyaura coupling between 4-alkoxybromobenzene and pyridine-4-boronic acid as shown in Fig. 4. They were also found to form isolable complexes with iodine and interhalogen compounds and the results of these studies are now reported.
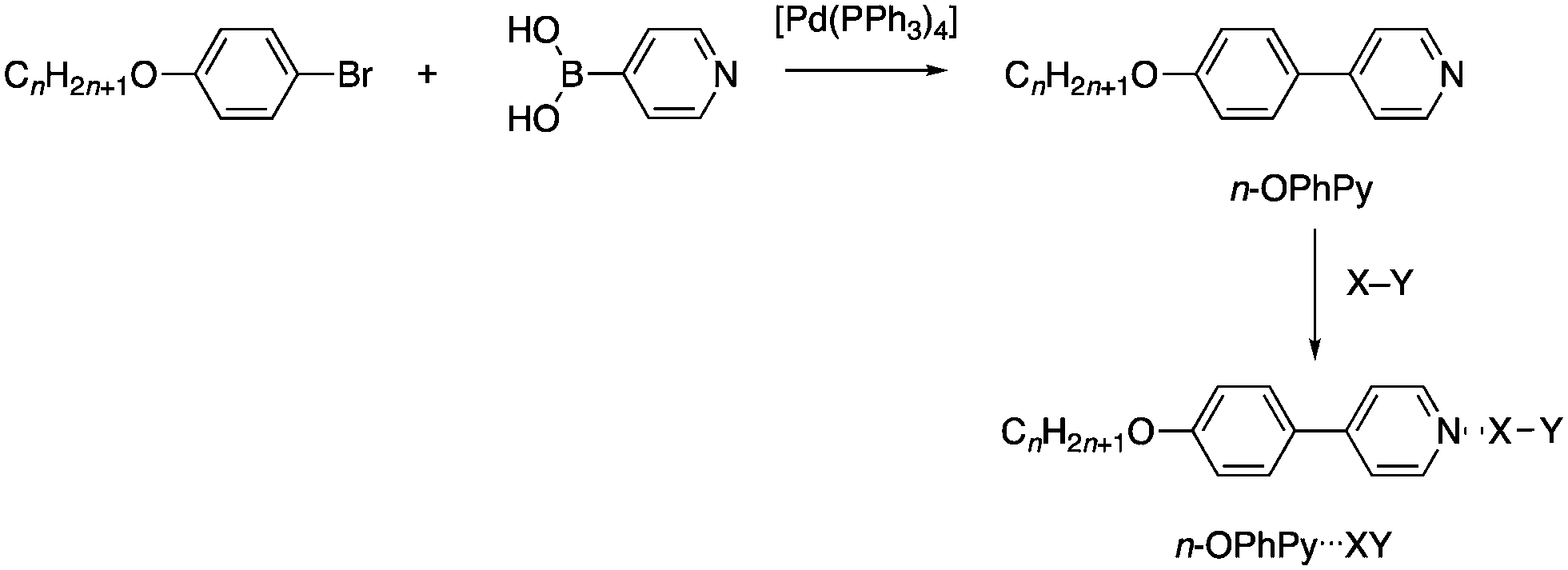 | ||
| Fig. 4 Preparation of the 4-alkoxyphenyl-4-pyridines (n-OPhPy) and their halogen-bonded complexes with I2 (X = Y = I), Br2 (X = Y = Br), ICl (X = I, Y = Cl) and IBr (X = I, Y = Br). | ||
Results
Synthesis
According the route shown in Fig. 4, five chain lengths of the alkoxyphenylpyridine were prepared – C4, C6, C8, C10 and C12 – and the materials were readily characterised by their 1H NMR spectra in which there were signals arising from two aromatic AA′XX′ spin systems (Fig. S1†); the resonances associated with the hydrogens ortho to the ring nitrogen being to the highest field at ca. δ = 8.6 ppm.The alkoxyphenylpyridines are not themselves mesomorphic, simply melting directly to an isotropic liquid. However, the analogous N-oxides are known to display SmA phases with increasing thermal stability as the alkyl chain length increases16 and it was postulated that the terminal, polar N+–O− unit promoted anti-parallel molecular correlations, so stabilising the mesophase by analogy with behaviour that is well known for cyanobiphenyl liquid crystals.17
Complexes of the alkoxyphenylpyridines with I2, ICl and IBr were then obtained by adding a hexane solution of the halogen/interhalogen to a chloroform solution of the appropriate alkoxyphenylpyridine, with the complexes being isolated in low to moderate yields. For each of these, it was possible to obtain an X-ray single crystal structure (the butoxy derivative was chosen) by crystallising the precipitate. Attempts were also made to obtain complexes with Br2 and these are also described.
Molecular structures of the complexes
X-Ray data are shown in Tables S1 and S2,† and some key distances are collected in Table 1. CCDC numbers for the complexes are 2123966–2123974 and the cif and cifcheck files are found as part of the ESI.†| I–Y | d(N⋯I)/Å | % vdW(N+I)a | d(I–Y)/Å | % I–Y(free)b |
|---|---|---|---|---|
| a N⋯I separation expressed as a % of the sum of the van der Waals radii of nitrogen and iodine. b Length of the covalent bond in the (inter)halogen in the complex as a % of its value in the free molecule. | ||||
| I2 | 2.398(3) | 68 | 2.8144(4) | 105 |
| ICl | 2.276(2) | 64 | 2.5174(7) | 108 |
| IBr | 2.289(3) | 65 | 2.6715(5) | 106 |
4-OPhPy⋯I2
The complex crystallised from dichloromethane in the P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) space group and the molecular structure is shown in Fig. 5a, while the packing is shown in Fig. 5b. The adduct crystallises showing a non-covalent N⋯I separation of 2.398(3) Å, which is 68% of the sum of the van der Waals radii of iodine and nitrogen, a percentage similar to that observed for the complex between iodine and octyloxystilbazole.8 The halogen bond is very close to linear (N⋯I–I angle = 176.92(7)°) as would be expected for a strong interaction. The length of the I–I bond is 2.8143(4) Å, which is 5% longer than that in isolated molecular iodine where the bond length is 2.68 Å.18 Apart from the inter-complex I⋯I interaction (below), there are no other short contacts and the complexes simply pack end-to-end as shown in Fig. 5b.
space group and the molecular structure is shown in Fig. 5a, while the packing is shown in Fig. 5b. The adduct crystallises showing a non-covalent N⋯I separation of 2.398(3) Å, which is 68% of the sum of the van der Waals radii of iodine and nitrogen, a percentage similar to that observed for the complex between iodine and octyloxystilbazole.8 The halogen bond is very close to linear (N⋯I–I angle = 176.92(7)°) as would be expected for a strong interaction. The length of the I–I bond is 2.8143(4) Å, which is 5% longer than that in isolated molecular iodine where the bond length is 2.68 Å.18 Apart from the inter-complex I⋯I interaction (below), there are no other short contacts and the complexes simply pack end-to-end as shown in Fig. 5b.
 | ||
| Fig. 5 Structure of the iodine complex 4-OPhPy⋯I2 showing: (a) the dimeric unit with the inter-complex I⋯I separation and (b) the packing viewed down the b-axis. | ||
There is a secondary I⋯I interaction between the unbound iodine atoms of adjacent complexes with an angle at that iodine of 175.13(1)° and a separation of 3.6543(5) Å, which is 92% of twice the van der Waals radius of iodine. This is significantly shorter than the intermolecular I⋯I interaction observed for the halogen-bonded complex between molecular iodine and octyloxystilbazole and can be considered as attractive. However, the almost linear arrangement of the iodine atoms is surprising and the I⋯I–I angle is larger than what would be expected for a typical type I interaction. Indeed, a search of the CSD shows that in fact there are only five other structures with such a motif (Fig. S2†).19–23 This interaction is surprising because if the electrostatic surface potential of iodine is considered, then the regions of positive potential would be directed towards one another and any interaction would be expected to be repulsive in nature. This is the subject of a computational investigation and subsequent discussion below.
4-OPhPy⋯ICl
Single crystals were obtained by slow evaporation from a solution of dichloromethane; the structure and aspects of the organisation are shown in Fig. 6.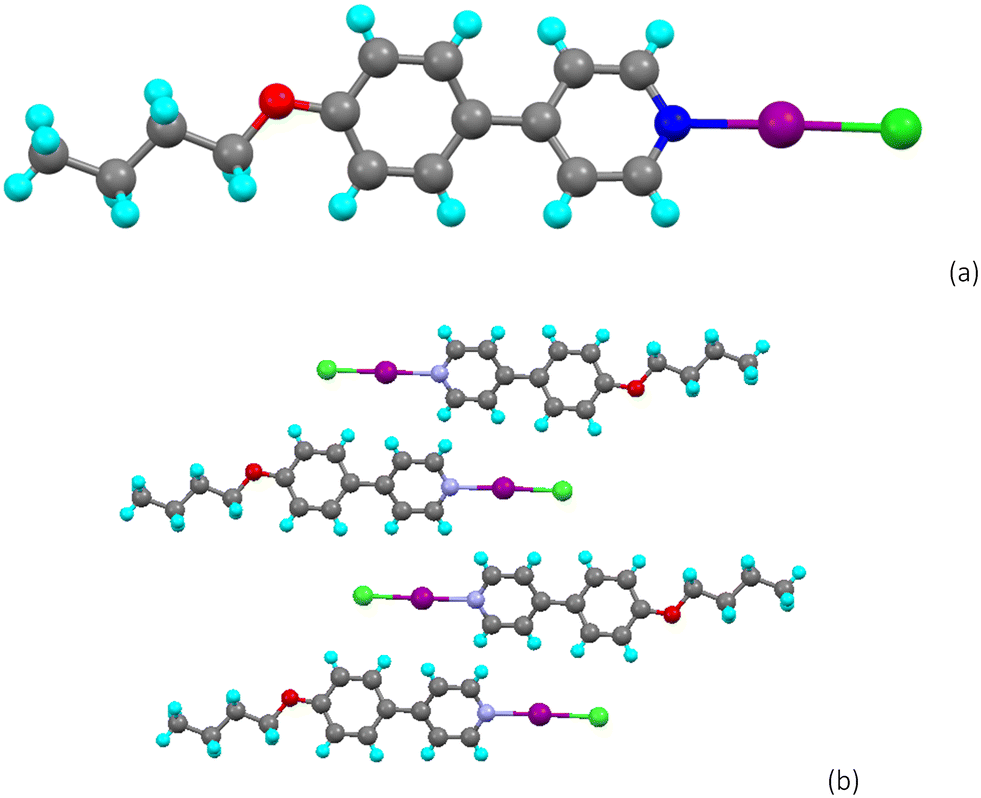 | ||
| Fig. 6 (a) Molecular structure of the halogen-bonded complex between iodine monochloride and butoxyphenylpyridine (4-OPhPy⋯ICl); (b) the interdigitated, packed structure. | ||
The complex crystallised in space group P with two complexes in the unit cell and the N⋯I separation is 2.276(2) Å, which at 64% of the sum of the van der Waals radii is remarkably short; the Cl–I⋯N angle is almost linear at 178.74(5)° indicating a strong interaction. This is consistent with Legon's observations that ICl is a particularly strong halogen-bond donor.24 The I–Cl bond length is 2.5174(7) Å, which is an elongation of 8% compared to isolated ICl, where the bond length is 2.323 Å.23
The N⋯I separation is shorter than those found in most complexes between ICl and nitrogen bases, and the only examples with shorter separations are complexes with the powerful Lewis base DMAP25 and trimethylsilyltrimethylphosphoranimine,26 which have I⋯N separations of 2.246(2) and 2.228(8) Å, respectively (Fig. S3†).
Unlike the structure between iodine and butoxyphenylpyridine, the chlorine atom in this structure does not participate in any inter-complex interactions. Fig. 6b shows the anti-parallel arrangement of the complexes, which is similar to the interdigitated arrangement observed in the liquid crystal phase of the N-oxides of alkoxyphenylpyridines.16
4-OPhPy⋯IBr
Single crystals of 4-OPhPy⋯IBr were obtained by slow evaporation from solution in dichloromethane and the molecular structure obtained from XRD is shown in Fig. 7. This complex also crystallised in the P space group and there were two complexes in the unit cell, the major difference being in the disposition of IBr with respect to the ring. Thus, in one of them (complex 1), the angle at nitrogen measured between the ipso 4-carbon of the pyridine ring, the ring nitrogen and iodine is close to linear at 177.0(2)°, while in the other (complex 2) it is significantly bent at 172.7(2)°. Consideration of the packing shows no particular driving force for this difference.
 | ||
| Fig. 7 (a) The molecular structure of the halogen bonded complex between iodine monobromide and butoxyphenylpyridine (4-OPhPy⋯IBr); (b) packing of 4-OPhPy⋯IBr showing both the antiparallel arrangement and the more bent nature of one of the halogen bonding interactions. | ||
The N⋯I separations (2.303(4) Å and 2.289(3) Å for complexes 1 and 2, respectively) are equivalent statistically and represent ca. 65% of the sum of the van der Waals radii of iodine and nitrogen. This separation is very slightly longer than that for the complex with iodine monochloride and shorter than that for the complex with iodine, which is consistent with the strength of iodine monobromide as a halogen bond donor being between I2 and ICl. The halogen bond is approximately linear at iodine with N⋯I–Br angles of 177.39(8)° and 178.59(8)° (complexes 1 and 2, respectively), consistent with a strong halogen bond. However, the I–Br bond distance is sensitive to the bend angle at nitrogen being shorter at 2.6464(5) Å in the more linear complex 1 (an increase of 5.0% compared with isolated IBr (2.521(4) Å)27 and a little longer (2.6715(5) Å) in complex 2 (an increase of 6.0% compared with isolated IBr (2.521(4) Å).27 Similar to the complex with iodine monochloride, the bromine atom does not participate in any additional interactions in the structure.
There are only five other examples of complexes between IBr and nitrogen bases. The complex with pyridine28 has an N⋯I separation of 2.26(4) Å – shorter than the present example, while a DABCO derivative29 has an N⋯I separation of 2.28(1) Å which is statistically the same. Other complexes with 2,2-bipyridine,30 tetra-2-pyridyl-pyrazine31 and a 2-(phenylselenopyridine)32 (Fig. S4†) have longer N⋯I separations of 2.4607(5), 2.405(3) Å and 2.411(3) Å, respectively.
As in the case with the ICl complex, there are no meaningful inter-complex interactions and once more, the complexes adopt an anti-parallel arrangement as shown in Fig. 7b, consistent with their dipolar nature.
Attempted formation of complexes with molecular bromine
While it was readily possible to obtain single crystals of complexes with ICl, IBr and I2, those containing molecular bromine proved much more elusive. For example, reaction of the hexyloxyphenylpyridine with elemental bromine in hexane led to the formation of a precipitate, whose composition by combustion analysis was consistent with formation of 6-OPhPy⋯Br2. Furthermore, on dissolution, its 1H NMR spectrum (Fig. S1†) was also consistent with the formation of the bromine complex as evidenced by the chemical shift of the hydrogen atoms ortho to the pyridine nitrogen. Thus, at δ = 8.55 ppm, it was consistent with those observed for the I2 (8.57 ppm), IBr (8.56 ppm) and ICl (8.56 ppm) complexes, and even the free ligand (8.61 ppm) but different from a protonated alkoxyphenylpyridine (e.g. δ = 8.73 ppm for [6-OPhPyH][Cl]). However, the isolated complex 6-OPhPy⋯Br2 was found not to be liquid-crystalline and it decomposed upon heating, releasing bromine. Given that all of the other complexes with (inter)halogens at this chain length did show liquid crystal properties, no other bromine complexes were studied in this regard.Crystallisation, however, did not lead to isolation of the halogen-bonded complex, rather one of three outcomes was observed. Thus, crystallisation of 10-OPhPy·Br2 from THF/cyclohexane in which the THF had not been dried led to the formation of the alkoxyphenylpyridinium cation with tribromide as the counter anion (Fig. 8a). There is a hydrogen bond between the pyridinium hydrogen and a terminal bromine of Br3− (dH⋯Br = 2.35(5) Å) and the structure propagates in the ab plane (Fig. 8b) with what is currently described as a tetrel bond33 to the terminal carbon of the alkoxy chain in a neighbouring cation with dC⋯Br = 3.543(5) Å. The Br–Br distances in the anion are 2.4381(6) and 2.6584(6) Å.
 | ||
| Fig. 8 (a) Molecular structure of the tribromide salt [10-OPhPyH][Br3] obtained from crystallisation from THF/cyclohexane and (b) its propagation in the ab plane. | ||
Crystallisation of 12-OPhPy·Br2 from THF/cyclohexane which had been dried then led to a different outcome, this time with protonation of only 50% of the alkoxyphenylpyridines leading to the formation of a hydrogen-bonded dimer between a neutral alkoxyphenylpyridine and a protonated alkoxyphenylpyridinium (Fig. 9a). Here we assume the presence of some adventitious water despite the precautions we took. The N⋯N separation is 2.652(4) Å and the covalent and non-covalent NH distances are 0.859 and 1.804 Å, respectively. Tribromide is once more the counter anion, but this arrangement precludes the hydrogen bonding arrangement described above and so that anion sits alongside the positively charged pyridinium ring as shown in Fig. 8a. The Br–Br distances are 2.5370(5) and 2.5595(5) Å and the Br⋯H separations are 3.029, 3.0246 and 3.0371 Å. That the Br3− anion associates with the pyridinium ring is consistent with observations made for the association of a triflate counter anion in an N-phenylpyridinium salt and what will be the positive electrostatic potential on this ring, a connection supported by calculation.34 However, the Br⋯H separations are on the very edge of the sum of the van der Waals radii suggesting a purely electrostatic interaction. The structure propagates in the bc planes as shown in Fig. 9b.
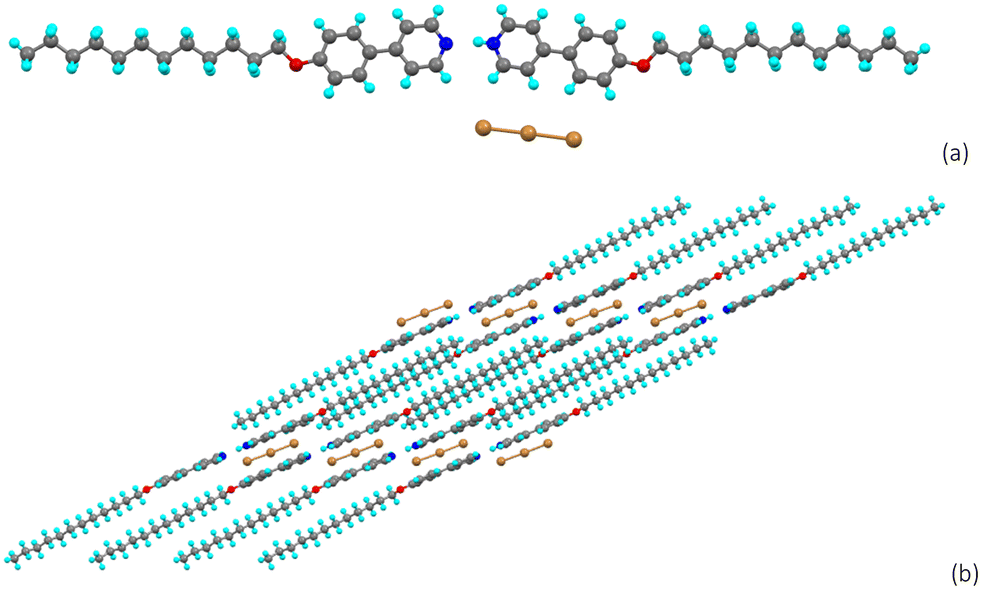 | ||
| Fig. 9 (a) Molecular structure of the hydrogen-bonded tribromide salt [(12-OPhPy)2H][Br3] obtained by crystallisation of 12-OPhPy·Br2 from dried THF/cyclohexane and (b) its propagation in the bc plane. | ||
Finally, crystallisation from THF gave a different outcome and for four chain lengths, the result was a simple alkoxyphenylpyridinium bromide, which organised into a hydrogen-bonded 2 + 2 dimer as shown in Fig. 10 for [6-OPhPyH]+ Br−. Analogous structures were found for [4-OPhPyH]+ Br−, [8-OPhPyH]+ Br− and [12-OPhPyH]+ Br− (Fig. S5–S7†).
 | ||
| Fig. 10 Hydrogen-bonded, dimeric arrangement for [6-OPhPyH]+Br−. | ||
Liquid crystal properties of complexes with alkoxyphenylpyridines
As noted already, following the observation that heating the bromine adduct 10-OPhPy⋯Br2 simply led to decomposition with release of the bromine, none of the other homologues was studied for liquid crystal behaviour. However, the remaining complexes were, with one exception, liquid crystalline. The behaviour of 8-OPhPy⋯ICl, typical of the others, is recorded here. Thus, 8-OPhPy⋯ICl was found to melt into the SmA phase at 130 °C, characterised by the streaky texture with very small focal conic areas as shown in Fig. 11a. Upon further heating, some decomposition of the complex was observed from approximately 165 °C, resulting in isotropic regions with the SmA phase as characterised by the appearance of bright filaments within an isotropic background (Fig. 11b). The partly decomposed sample cleared into the isotropic phase at 196 °C. The phase behaviour observed upon heating could not be replicated upon cooling, confirming that the sample had decomposed in common with the azo complexes reported by Chen et al.15 The other complexes behaved in a similar way and the melting points are found in Table 2, being plotted in Fig. 12. As the materials decomposed on heating through the SmA phase, it is not possible to quote a reliable clearing point. Noteworthy, however, is 4-OPhPy⋯I2 which was a little different. Thus, on heating the complex appeared to melt over several degrees and appeared initially to give an isotropic liquid, but on heating further a SmA phase was clearly visible and the sample had darkened considerably. This is similar to the behaviour reported for shorter-chain stilbazole complexes of I2, for which it was speculated that a 2:1 stilbazole:I2 complex formed on extrusion of I2.
 | ||
| Fig. 11 Optical micrographs of the textures observed for the halogen-bonded complex 12-OPhPy⋯ICl at a) 135 °C and b) 162 °C. | ||
 | ||
| Fig. 12 Melting points of the complexes of n-OPhPy as a function of alkoxy chain length. | ||
The one very surprising observation was that 4-OPhPy⋯ICl was not liquid crystalline and that it melted at such a low temperature compared to the other complexes. Naturally the preparation and observations were repeated, but on each occasion the complex simply melted to form an isotropic liquid, for which we are unable to offer an explanation.
Unlike the complexes of iodine with alkoxystilbazoles, which formed smectic C (SmC) phases,14 these complexes of I2, ICl and IBr with alkoxyphenylpyridines only form SmA phases, although it is noticeable that the temperature range over which the phases are observed is broadly similar to those of the stilbazole-iodine complexes. The observation of a SmA phase is not atypical of smaller dipolar mesogens,35–37 although one might also expect to see a nematic phase, which is absent in these cases.
To investigate the nature of the SmA phase further, small-angle X-ray scattering data were acquired for 12-OPhPy⋯I2, 12-OPhPy⋯ICl and 12-OPhPy⋯IBr at 110, 135 and 120 °C, respectively (i.e. not so far above the melting point in each case, preventing the onset of any decomposition). Both complexes show a simple (001) reflection at low angle corresponding to the observed smectic periodicity and these were found at 2θ = 1.88° (ICl) and 1.81° (IBr), corresponding to spacings of 46.94 and 48.75 Å, respectively (data for 12-OPhPy⋯IBr in Fig. 13, while those for 12-OPhPy⋯ICl are in Fig. S8†). Curiously, the diffraction pattern for 12-OPhPy⋯I2 showed two reflections at low angle: 2θ = 1.77 and 1.95° corresponding to d = 49.85 and 45.25 Å (Fig. S9†). Optical microscopy did not show anything in the texture that would suggest other than a simple SmA phase and as such, the feature is interpreted as having its origin in incommensurate spacings arising from the periodicity of multiple heavy atoms (iodine).
 | ||
| Fig. 13 SAXS data for 12-OPhPy⋯IBr in the SmA phase at 120 °C. | ||
Given lengths of 29.01 Å, 28.59 Å and 28.76 Å, respectively for the three isolated complexes (calculated using data from the structures contained in this paper), then quite evidently these are not simple monolayer SmA phases. Thus, like the SmC phase observed for the iodine complexes of alkoxystilbazoles, there must be some sort of bilayer structure, which will be discussed below. Note also that the data for 12-OPhPy⋯IBr show a mid-angle reflection (2θ = 10.6°) corresponding to a spacing of 8.3 Å. This would appear not to be related to d(001) and, given its broad nature, is attributed to long-range periodicities in the mesophase associated with halogen–halogen interactions.
Quantum chemical calculations of halogen-bonded complexes between dihalogens and alkoxyphenylpyridines
The solid-state structure of 4-OPhPy⋯I2 shows an effectively in-line iodine⋯iodine interaction, yet it might be expected that the unbound, terminal iodine may show a σ-hole38 in which case the interaction may be expected to be repulsive. Therefore, in order to understand better the charges involved, the electrostatic potential (ESP) was calculated at the M06-2X/aug-cc-pVDZ optimised geometry for the halogen-bonded complex of the different halogens and interhalogens using pyridine as a model Lewis base. The resulting ESPs mapped onto electron density isosurfaces are shown in Fig. 14. For iodine, the plot shows that, as might have been anticipated, the ESP around the unbound iodine atom is slightly negative and that it features a well-defined σ-hole (Fig. 14a) with an ESP of +0.097 at the point on the isosurface most distant from pyridine. The terminal bromine is similarly polarised with a terminal ESP of +0.080, which drops more significantly for IBr (+0.046) and ICl (+0.018), not least on account of the electronegativity of Cl and Br. In relation to the iodine complex, the direct approach of two iodine atoms end-on with positive electrostatic potential as observed in the crystal structure remains surprising. However, the well-known type I interaction (e.g.Fig. 1) is similarly repulsive and so it is postulated that the unusual linear geometry observed for 4-OPhPy⋯I2 originates from crystal packing.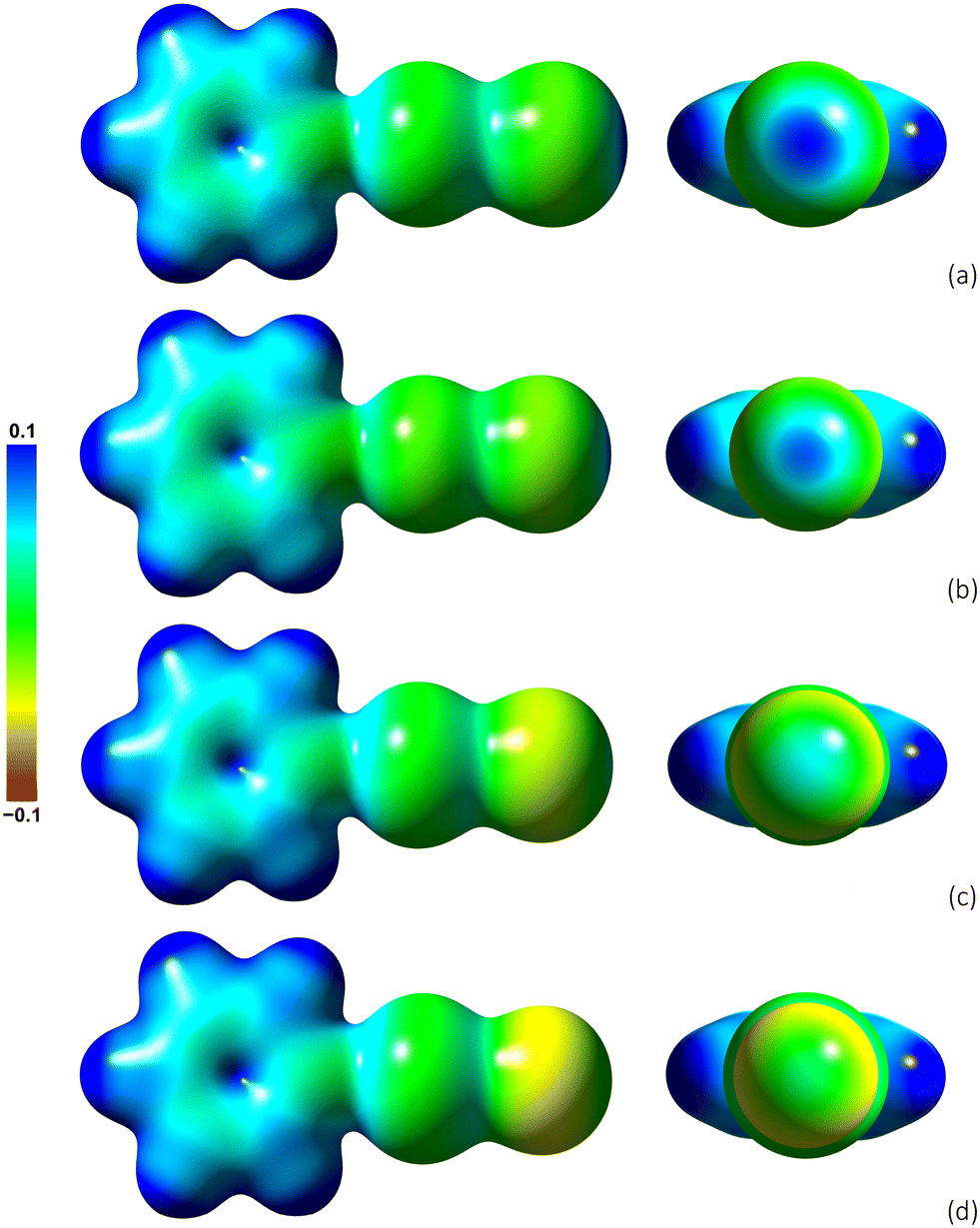 | ||
| Fig. 14 Views from above (left) and end-on (right) of the electrostatic potentials of the complexes: (a) Py⋯I2, (b) Py⋯Br2, (c) Py⋯IBr and (d) Py⋯ICl mapped onto the respective 0.01 e Bohr−3 electronic density isosurfaces. | ||
For calculations of other properties, geometries were optimised at the MP2 level using methoxyphenylpyridine (1-OPhPy) as the halogen bond acceptor, and at the M06-2X level using two halogen bond acceptors, 1-OPhPy and 4-OPhPy. Thus, Table 3 shows the N⋯X separations, dihalogen bond lengths, binding energies and dipole moments of these complexes. The M06-2X and MP2 results for the 1-OPhPy complexes are in reasonable agreement, which would suggest that the M06-2X functional is an appropriate choice for modelling these complexes using DFT. Comparing the N⋯I separations and I–X bond lengths for the complexes with I2, IBr and ICl to those in the crystal structures reveals that although absolute values are not reproduced, the trends are modelled correctly by the calculations.
| Level of theory | r(X⋯N)/Å | ΣvdW radii/% | ΔE/kJ mol−1 | μ/D | r(X–Y)/Å | |||
|---|---|---|---|---|---|---|---|---|
| (Inter) halogen | Complex | % Difference | ||||||
| a X = Y for I2 and Br2. | ||||||||
| 1-OPhPy⋯Br2 | MP2 | 2.410 | 70.9 | 45.28 | 9.42 | 2.324 | 2.422 | 4.22 |
| M06-2X | 2.460 | 72.3 | 38.63 | 9.13 | 2.300 | 2.375 | 3.26 | |
| 4-OPhPy⋯Br2 | M06-2X | 2.455 | 72.2 | 38.95 | 9.76 | 2.300 | 2.376 | 3.30 |
| 1-OPhPy⋯I2 | MP2 | 2.580 | 73.1 | 51.58 | 10.17 | 2.720 | 2.803 | 3.05 |
| M06-2X | 2.596 | 73.5 | 46.17 | 9.99 | 2.686 | 2.755 | 2.57 | |
| 4-OPhPy⋯I2 | M06-2X | 2.593 | 74.4 | 46.56 | 10.64 | 2.686 | 2.756 | 2.61 |
| 1-OPhPy⋯IBr | MP2 | 2.507 | 71.0 | 64.98 | 11.55 | 2.517 | 2.610 | 3.69 |
| M06-2X | 2.497 | 70.7 | 61.84 | 11.53 | 2.492 | 2.582 | 3.61 | |
| 4-OPhPy⋯IBr | M06-2X | 2.495 | 70.6 | 62.29 | 12.18 | 2.492 | 2.583 | 3.65 |
| 1-OPhPy⋯ICl | MP2 | 2.471 | 70.0 | 73.24 | 12.05 | 2.369 | 2.464 | 4.01 |
| M06-2X | 2.472 | 70.0 | 68.02 | 11.72 | 2.351 | 2.439 | 3.74 | |
| 4-OPhPy⋯ICl | M06-2X | 2.470 | 69.9 | 68.51 | 12.36 | 2.351 | 2.439 | 3.74 |
The computational data show that the complex with bromine has the smallest N⋯X (X = halogen) binding energy. This is followed by the complexes with iodine, then iodine monobromide with the complex with iodine monochloride showing the greatest binding energy. This is consistent with the Lewis acid strengths of the dihalogens which follows the trend Br2 < I2 < IBr < ICl.3 One surprising result is the increase in the N⋯X separation as a percentage of the sum of the van der Waals radii for 1-OPhPy⋯I2 and 4-OPhPy⋯I2 compared to 1-OPhPy⋯Br2 and 4-OPhPy⋯Br2, respectively. However, this could be due to the potential energy surfaces for these complexes being relatively flat, which can make the optimised geometries quite sensitive to the level of theory. The dipole moments of the complexes increase as the binding energy of the complex increases.
The longer alkyl chain in the 4-OPhPy complexes increases their dipole moments by about 0.6 D, but these slightly higher dipole moments continue to follow the trend observed for the 1-OPhPy complexes.
NBO analyses were then carried out on the M06-2X/aug-cc-pVDZ-optimised geometries of the 1-OPhPy complexes and the data are shown in Table 4. These results corroborate the observation of elongation of the X–Y bond in the crystal structures so that as the percentage elongation increases, the occupancy of the lone pair on the nitrogen decreases and the occupancy of the X–X anti-bonding orbital and the stabilisation energy increase.
| Dihalogen | Occupancy LP(N) | Occupancy σ*(X–X) | E ij/kJ mol−1 |
|---|---|---|---|
| Br2 | 1.797 | 0.149 | 138.04 |
| I2 | 1.812 | 0.134 | 130.00 |
| IBr | 1.784 | 0.166 | 184.26 |
| ICl | 1.781 | 0.167 | 191.84 |
Discussion
Speculating on the precise way in which decomposition in these complexes takes place is not something we have undertaken, accounting for the lack of speculation with regard to the fate of the complexes of ICl and IBr. That said, it was evident that Br2 was extruded from the complex studied. The proposal with respect to the iodine complex, however, is not without some foundation. Thus, the complex first melts to give an apparently isotropic liquid which, on further heating, reveals a SmA phase. Thermodynamically, the two phases cannot be from the same species and so a new species must have formed. As the uncomplexed ligand is not LC, then extrusion of iodine and the formation of a 2:1 complex would account for the observations from an LC point of view. The parallel with the stilbazole complexes14 is striking and in this earlier study the proposal was corroborated by TGA. As such, we believe that there are good grounds to anticipate analogous behaviour here.
While factors stabilising liquid crystal mesophases can be rather subtle, the common underlying structural feature is anisotropy, which gives rise to additional dispersion forces that stabilise a mesophase. Anisotropy is most effectively conferred by more rigid units such as a 1,4-phenylene or trans-1,4-cyclohexylene link with the high lattice energy this implies reduced by the use of flexible alkyl chains.35,36 However, where the anisotropy is insufficient, then the inclusion of a strong dipole moment is often beneficial. Thus, for example, 4-alkoxy-1,1′-biphenyl, 4-alkoxyphenylpyridine and 4-alkoxystilbazole are not liquid crystals yet by addition of a dipolar group (–CN {1}, –O− {2}, −M(Cl)(CO)2 – M = Rh, Ir {3}, respectively – Fig. 15), mesomorphism is induced in each.39 Such small, dipolar mesogens have a strong tendency to form nematic and SmA phases35–37 and in all of these new complexes, the stability of the mesophase increases with increasing terminal chain length. Complexes of series 1 and 3 show both N (shorter chains) and SmA phases, while those of series 2 show only a SmA phase.
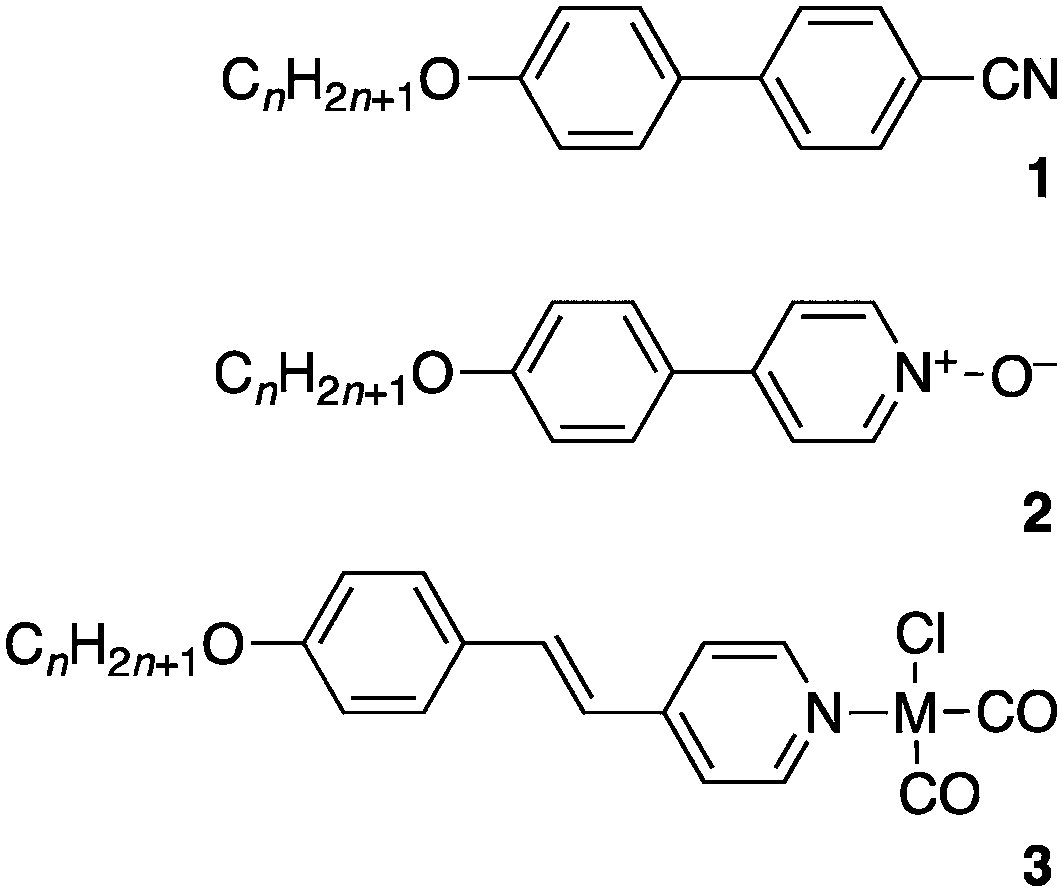 | ||
| Fig. 15 Structures of some simple dipolar mesogens (M = Rh or Ir). | ||
Compound 1 is of course exceptionally well-studied and, despite it being rather short in length, it is liquid crystalline thanks to the anti-parallel dipolar correlations induced by the cyano group, which increases the effective length of the mesogenic species. Similar arguments were advanced to account for the observation of the SmA phase in 2. Thus, the dipole moments for the three series of materials are 4.5 D (1), 6.78 D (2) and 6.9 D (3, M = Rh) or 7.4 D (3, M = Ir). The value for 2 is calculated at the MP2 level of theory using the aug-cc-pVDZ basis set (M06-2X gives 5.95 D).
However, these values are somewhat eclipsed by those found for the present materials where the smallest (MP2) value is 9.42 D (1-OPhPy⋯Br2) and the largest 12.05 D (1-OPhPy⋯ICl). Thus, it is entirely consistent to propose that liquid crystallinity is indeed induced owing to the strong longitudinal dipole that the materials possess. Then, in common at least with the cyanobiphenyl materials, the SAXS data show that the SmA phase has some sort of bilayer arrangement, given a calculated molecular length of ca. 29 Å and a lamellar periodicity of ca. 48 Å. Such a bilayer arrangement driven by antiparallel dipolar correlations is expected. Based on distances from X-ray single crystal structures in this study and assuming little chain folding/interdigitation as a first approximation, then Fig. 16 shows a proposal for the maximum extent of overlap of pairs of molecules consistent with the lamellar periodicity measured. Inevitably however, the terminal chains will not be in an extended, all-trans arrangement and so the extent of overlap will therefore be modulated.
 | ||
| Fig. 16 Sketch to show maximum extent of likely overlap of the halogen-bonded liquid crystals, with distances based on the structures of 4-OPhPy⋯IBr and of [12-OPhPyH]+ Br−. | ||
However, what is very clear is that the mesophase stability of these new halogen-bonded complexes is significantly higher than compounds 1–3 chosen for comparison, which it is proposed arises from their very high dipole moment acting to preserve the arrangement proposed in Fig. 16.
Conclusions
4-Alkoxyphenyl-4-pyridines form halogen-bonded complexes (n-OPhPy⋯XY) on addition of molecular iodine, molecular bromine or the interhalogen compounds IBr and ICl. Exemplar complexes of I2, IBr and ICl were characterised by X-ray crystallography and showed evidence of strong halogen bonding through the observation of short N⋯I distances (between 2.398(3) Å for n-OPhPy⋯I2 and 2.276(2) Å for n-OPhPy⋯ICl), with concomitant elongation of the X–Y bond by about 5–8%. These are very short and strong halogen bonds, typical of those seen previously with (inter)halogen complexes. While the complexes with IBr and ICl show an anti-parallel arrangement in the solid state, the complex with I2 shows an almost linear and direct inter-complex I⋯I interaction with an iodine⋯iodine separation of about 3.65 Å – some 92% of the sum of two iodine van der Waals radii. Calculation of the electrostatic potential shows a pronounced σ-hole at the extremity of each complexed iodine, suggesting that crystal packing effects overcome what is otherwise a repulsive interaction.While the alkoxyphenylpyridines do not show any liquid-crystalline behaviour, the halogen-bonded complexes do and all but one melt to form a SmA phase at temperatures between ca. 100–180 °C, showing the significant thermal stability of the halogen bond in these materials. However, at temperatures above the melting point there is evidence that the complexes are unstable thermally as evidenced both by darkening and the inability to thermally cycle the materials reproducibly. Furthermore, for the iodine complexes there is evidence of the thermal extrusion of iodine to generate a 2:1 alkoxyphenylpyridine:iodine complex, mirroring the behaviour observed in an earlier study of analogous complexes using alkoxystilbazoles as the nitrogen base.14
Investigation of the SmA phase of the materials using small-angle X-ray scattering shows that the lamellar spacing at ca. 1.7 times the length of the isolated complex. Calculations show that these complexes possess very high dipole moments of ca. 9.5–11.5 D and so by analogy with other simple, dipolar liquid crystals such as the cyanobiphenyls, the observed layer spacing will arise from the overlapping, antiparallel correlation of the complexes to, in effect, cancel out the dipoles.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank the University of York (LJM, NEP and AJM) for support.References
- G. Cavallo, P. Metrangolo, R. Milani, T. Pilati, A. Priimagi, G. Resnati and G. Terraneo, Chem. Rev., 2016, 116, 2478–2601 CrossRef CAS PubMed.
- See e.g., C. Präsang, A. C. Whitwood and D. W. Bruce, Cryst. Growth Des., 2009, 9, 5319–5326 CrossRef CAS; A. Wasilewska, M. Gdaniec and T. Pozonski, CrystEngComm, 2007, 9, 203–206 RSC.
- W. T. Pennington, T. W. Hanks and H. D. Arman, in Struct. Bond., Halogen Bonding, ed. P. Metrangolo and G. Resnati, 2008, vol. 126, pp. 65–104 Search PubMed.
- D. W. Bruce, in Struct. Bond., Halogen Bonding, ed. P. Metrangolo and G. Resnati, 2008, vol. 126, pp. 161–180 Search PubMed.
- D. W. Bruce, in Supramolecular Chemistry: From Molecules to Nanomaterials, ed. J. W. Steed and P. A. Gale, Wiley, Chichester, 2012, pp. 3493–3514 Search PubMed.
- D. Devadiga and T. N. Ahipa, J. Mol. Liq., 2021, 333, 115961 CrossRef CAS.
- C. Präsang and D. W. Bruce, Helv. Chem. Acta, 2023 DOI:10.1002/hlca.202300008.
- H. L. Nguyen, P. N. Horton, M. B. Hursthouse, A. C. Legon and D. W. Bruce, J. Am. Chem. Soc., 2004, 126, 16–17 CrossRef CAS PubMed.
- D. W. Bruce, P. Metrangolo, F. Meyer, C. Präsang, G. Resnati and A. C. Whitwood, New J. Chem., 2008, 32, 477–482 RSC.
- C. Präsang, A. C. Whitwood and D. W. Bruce, Chem. Commun., 2008, 2137–2139 RSC.
- H. A. Bent, Chem. Rev., 1968, 68, 587–648 CrossRef CAS.
- O. Hassel and J. Hvoslef, Acta Chem. Scand., 1954, 8, 873 CrossRef CAS.
- A. C. Legon, in Struct. Bond., Halogen Bonding, ed. P. Metrangolo and G. Resnati, Springer-Verlag, 2008, vol. 126, pp. 17–64 Search PubMed.
- L. J. McAllister, C. Präsang, J. P.-W. Wong, R. J. Thatcher, A. C. Whitwood, B. Donnio, P. O'Brien, P. B. Karadakov and D. W. Bruce, Chem. Commun., 2013, 49, 3946–3948 RSC.
- Y. Chen, H. Yu, L. Zhang, H. Yang and Y. Lu, Chem. Commun., 2014, 50, 9647–9649 RSC.
- D. J. Byron, D. Lacey and R. C. Wilson, Mol. Cryst. Liq. Cryst., 1981, 76, 253–260 CrossRef CAS.
- P. E. Cladis, Phys. Rev. Lett., 1975, 35, 48–51 CrossRef CAS.
- P. H. Svensson and L. Kloo, Chem. Rev., 2003, 103, 1649–1684 CrossRef CAS PubMed.
- P. Ravat, S. SeethaLekshmi, S. N. Biswas, P. Nandy and S. Varughese, Cryst. Growth Des., 2015, 15, 2389–2401 CrossRef CAS.
- C. B. Aakeröy, J. Desper, B. A. Helfrich, P. Metrangolo, T. Pilati, G. Resnati and A. Stevenazzi, Chem. Commun., 2007, 4236–4238 RSC.
- R. B. Walsh, C. W. Padgett, P. Metrangolo, G. Resnati, T. W. Hanks and W. T. Pennington, Cryst. Growth Des., 2001, 1, 165–175 CrossRef CAS.
- A. Peuronen, A. Valkonen, M. Kortelainen, K. Rissanen and M. Lahtinen, Cryst. Growth Des., 2012, 12, 4157–4169 CrossRef CAS.
- R. D. Bailey, G. W. Drake, M. Grabarczyk, T. W. Hanks, L. L. Hook and W. T. Pennington, J. Chem. Soc., Perkin Trans. 2, 1997, 2773–2780 RSC.
- J. B. Davey, A. C. Legon and E. R. Waclawik, Chem. Phys. Lett., 1999, 306, 133–144 CrossRef CAS.
- A. S. Batsanov, J. A. K. Howard, A. P. Lightfoot, S. J. R. Twiddle and A. Whiting, Eur. J. Org. Chem., 2005, 2005, 1876–1883 CrossRef.
- J. Grebe, K. Harms, F. Weller and K. Dehnicke, Z. Anorg. Allg. Chem., 1995, 621, 1489–1495 CrossRef CAS.
- L. N. Swink and G. B. Carpenter, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1968, 24, 429–433 CrossRef CAS.
- T. Dahl, O. Hassel and K. Sky, Acta Chem. Scand., 1967, 21, 592–593 CrossRef CAS.
- S. d'Agostino, D. Braga, F. Grepioni and P. Taddei, Cryst. Growth Des., 2014, 14, 821–829 CrossRef.
- S. Soled and G. B. Carpenter, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1974, 30, 910–914 CrossRef.
- M. C. Aragoni, M. Arca, F. A. Devillanova, M. B. Hursthouse, S. L. Huth, F. Isaia, V. Lippolis, A. Mancini, H. R. Ogilvie and G. Verani, J. Organomet. Chem., 2005, 690, 1923–1934 CrossRef CAS.
- R. Montis, M. Arca, M. C. Aragoni, A. J. Blake, C. Castellano, F. Demartin, F. Isaia, V. Lippolis, A. Pintus, E. J. Lenardão, G. Perin, A. E. O'Connor and S. Thurow, New J. Chem., 2018, 42, 10592–10602 RSC.
- A. Daolio, P. Scilabra, G. Terraneo and G. Resnati, Coord. Chem. Rev., 2020, 413, 213265 CrossRef CAS.
- J. D. Herod, M. A. Bates, A. C. Whitwood and D. W. Bruce, Soft Matter, 2019, 15, 4432–4436 RSC.
- G. W. Gray, Philos. Trans. R. Soc., A, 1983, 309, 77–92 CrossRef CAS.
- G. W. Gray, Philos. Trans. R. Soc., A, 1990, 330, 73–94 CAS.
- G. W. Gray, K. J. Harrison and J. A. Nash, Electron. Lett., 1973, 9, 130–131 CrossRef CAS.
- P. Politzer and J. S. Murray, ChemPhysChem, 2020, 21, 579–588 CrossRef CAS PubMed.
- D. W. Bruce, D. A. Dunmur, M. A. Esteruelas, S. E. Hunt, R. Le Lagadec, P. M. Maitlis, J. R. Marsden, E. Sola and J. M. Stacey, J. Mater. Chem., 1991, 1, 251–254 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2123966–2123974. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2ce01555b |
| This journal is © The Royal Society of Chemistry 2023 |