
Theoretical insight into the relevance between the oxidation states of CeO2 supported Pt4+/2+/1+/0/2− and their HER performance†
Hongxian
Liu‡
ab,
Changling
Liu‡
c,
Chao
Ma
b,
Dunhua
Hong
a,
Zhenxing
Fang
 *a,
Haitao
Zhao
e,
Tongwei
Wu
*d and
Yanning
Zhang
*b
*a,
Haitao
Zhao
e,
Tongwei
Wu
*d and
Yanning
Zhang
*b
aSchool of Physics and Electronic Science, Zunyi Normal University, Zunyi 563000, Guizhou, China
bInstitute of Fundamental and Frontier Sciences, University of Electronic Science and Technology of China, Chengdu 610054, Sichuan, China. E-mail: yanningz@uestc.edu.cn
cCollege of Materials Science and Engineering, Jilin Institute of Chemical Technology, Jilin City 132022, Jilin Province, China
dState Key Laboratory of Electronic Thin Films and Integrated Devices, University of Electronic Science and Technology of China, Chengdu 610054, Sichuan, China
eMaterials Interfaces Center, Shenzhen Institutes of Advanced Technology, Chinese Academy of Sciences, Shenzhen 440305, China
First published on 17th November 2022
Abstract
The oxidation states (OSs) of Pt catalysts play a key role in the hydrogen evolution reaction (HER), which has become a focus in recent years. As for Pt/CeO2 catalysts, we have systematically explored the effect of Pt4+/2+/1+/0/2− OSs on the HER. H atoms could not adsorb on the Pt4+ and Pt4c2+ sites, and they prefer to locate on the adjacent O atoms (labeled as Pt4+–O and Pt4c2+–O). As for the other OSs, the adsorption ability of H atoms follows the order Pt0 > Pt3c2+ > Pt1+ > Pt2−. The electron-enriched Pt2− is the most suitable oxidation state for the HER, no matter the reaction mechanism, which is the Volmer–Heyrovsky or Volmer–Tafel mechanism. The Pt OSs would greatly affect the electronic structures of Pt catalysts, which will further impact their overall catalytic properties. Our study could shed light on the research of Pt-based catalysts for the HER.
1. Introduction
The development of modern society mainly depends on fossil fuels, which would cause fossil fuel exhaustion and environmental issues. Hydrogen has a very high energy density and the combustion product is only water, thus hydrogen is one of the most promising clean and sustainable energy sources. To date, platinum (Pt) is still the most effective electrocatalyst for the hydrogen evolution reaction (HER), though the scarcity of Pt has severely limited its large-scale applications.1–3 One of the most straightforward methods to address this issue is to improve the utilization efficiency of Pt atoms, reducing the size of Pt electrocatalysts from nanoparticles (NPs) to nanoclusters (NCs) or even to single atoms (SAs).4–6 A very recent study found that Pt single-atoms anchored to functional CeOx nanoglues could remain dispersed in both oxidizing and reducing environments at high temperatures.7 As is well known, the catalytic activity of Pt/CeO2 is affected by multiple factors, such as the size and loading of Pt, and the surface facets and morphology of CeO2.8–13 Remarkably, the importance of OSs present in CeO2 supported Pt catalysts has been gradually discovered in determining the catalytic activity for the HER.14The electrons of Pt catalysts are usually taken away by metal–support interactions because of their difference in electronegativity, resulting in electron-deficient Pt (Ptδ+, 0 < δ < 4).15–18 Many studies have shown that the HER activity of oxidized Ptδ+ can be obviously better than that of metal Pt0, and Pt–O bonds act as an active site superior to Pt0.19–21 Strikingly, Wei et al. found that the rich oxygen vacancies of porous TiO2 could reverse the electron transfer from TiO2 to Pt NCs, leading to the ultrahigh mass activity of Ptδ− NCs/TiO2.22 Yan et al. found that Ptδ− NPs supported on MgO nanosheets with rich oxygen vacancies could create a local acid-like environment in an alkaline medium, which brings about excellent HER performances.23 Yan et al. found that electron-enriched Pt (Ptδ−) NCs caused by the size-dependent electron transfer reversal are far more active than electron-deficient Pt single atoms for the HER.24 On the other hand, Shi et al. found that the decrease of single-atom Pt OSs could optimize the HER performance in an acidic environment.25 Similarly, Fang et al. also discovered that the near-free single-atom Pt resembling a free metallic atom is responsible for the superior HER activity.26
The Pt OSs depend on the kind of support and specific synthesis conditions, and various Pt OSs would lead to diverse reaction data.14 In this article, we have systematically explored the effect of CeO2 supported Pt4+/2+/1+/0/2− OSs on the HER by density functional theory (DFT) calculations.
2. Computational methods
All DFT calculations were performed using the spin-polarized Kohn–Sham formalism as implemented in the Vienna ab initio simulation package (VASP).27 The exchange–correlation was described by the Perdew–Burke–Ernzerhof (PBE) functional of the generalized gradient approximation (GGA).28 The valence wave functions were expanded in plane-wave basis sets with a cutoff energy of 400 eV, and the Γ point was used for Brillouin zone integration. The convergence criteria of energy and force for structural optimization were set to 1 × 10−5 eV and 0.02 eV Å−1, respectively. The on-site Coulomb interaction via the Hubbard-like term U was adopted for the highly localized 4f electrons with an effective U of 5.0 eV. AIMD simulations were performed using the canonical (NVT) ensemble for 10 ps with a time step of 2 fs with the Nośe–Hoover thermostats at 300 K and 700 K.Pt incorporated in or loaded on ceria (CeO2) has been widely used in various catalytic reactions because of the well-defined structures.29,30 CeO2 (111) surface cells were adopted as the support to construct various Pt OSs, and have been always used in many experimental and theoretical studies.31–35 As for the combustion-synthesized Pt/CeO2 catalysts, Pt could be incorporated into the CeO2 lattice, mostly as Pt2+/Pt4+ and rarely as Pt0.36,37 Numerous studies have demonstrated that Pt SAs can be stabilized on CeO2 (111) steps through different preparation methods.31–33 The (4 × 4) CeO2 (111) model contains three O–Ce–O tri-layers separated by a 15 Å vacuum in the normal direction, and the bottom tri-layer was fixed during the geometry optimization.
The Gibbs free energy change (ΔGH*) in the hydrogen reduction was calculated according to the method developed by Nørskov et al.:38
| ΔG = ΔE + ΔEzpe − TΔS | (1) |
The d-band center (εd) was calculated according to the formula:39,40
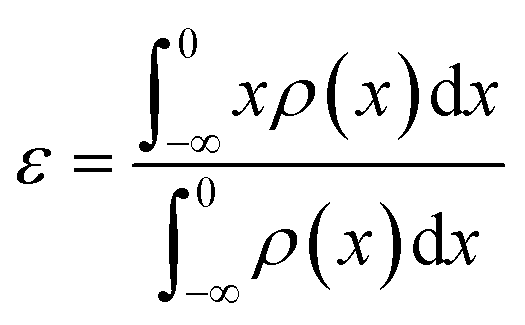 | (2) |
3. Results and discussion
3.1. Construction of Pt4+/2+/1+/0/2− OSs
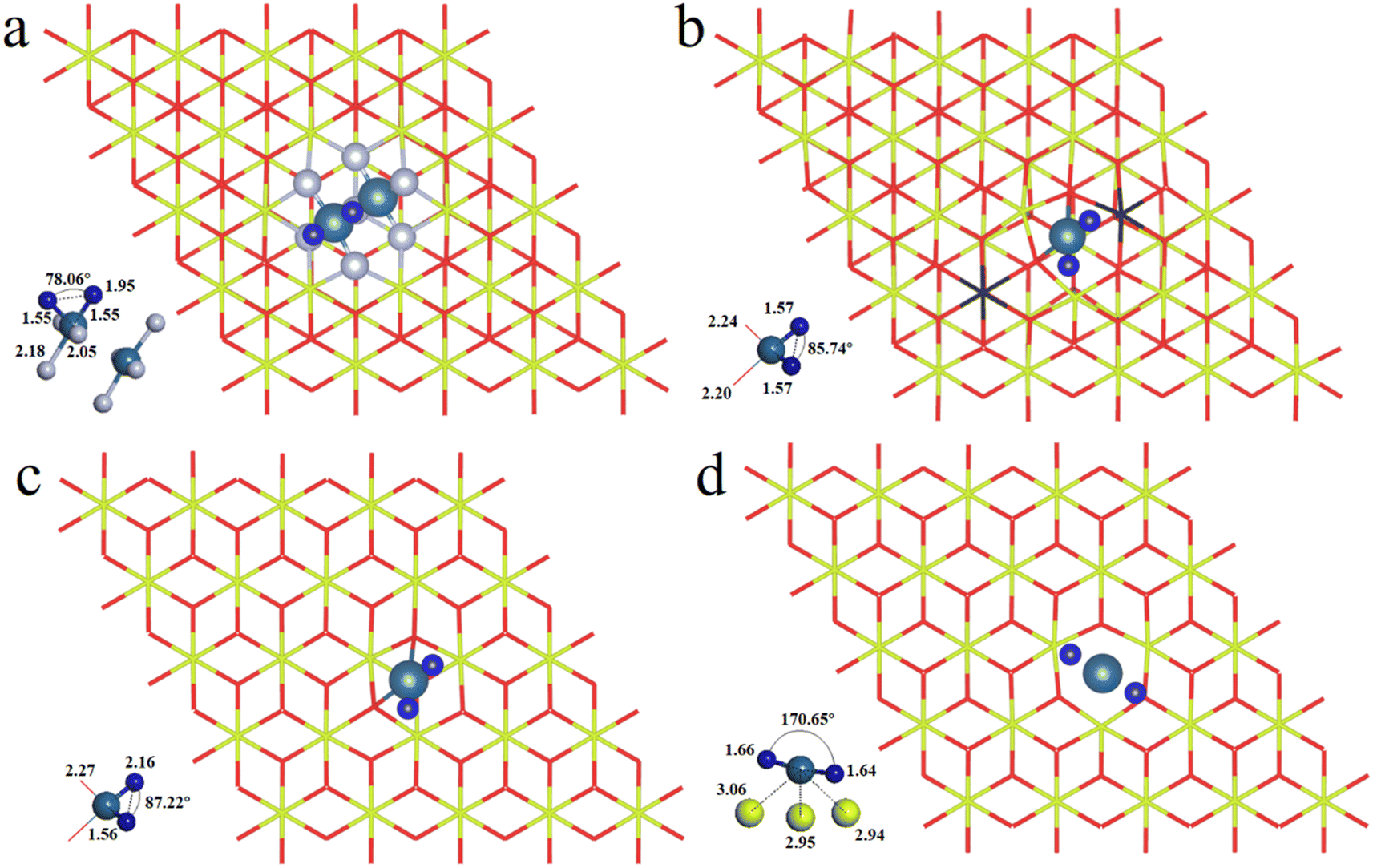
We have first explored the situation of Pt atoms incorporated into the CeO2 lattice, where one surface Ce cation is substituted by a single Pt atom (Fig. 1a) or two Pt atoms (Fig. 1b).34,44 There are three possible structures for the latter system, namely Pt2c + Pt4c, Pt3c + Pt3c, and Pt3c + Pt4c (Fig. S1†), and the Pt3c + Pt4c structure is the most energetically stable, where the subscripts represent the coordination number of the Pt atoms. Although identifying OSs is a challenge,45 the magnetic moment of CeO2 could well describe the Pt OSs of Pt/CeO2 single-atom catalysts by counting the number of Ce3+ species.46 Ce4+ presents no net magnetic moment, and the magnetic moment of Ce3+ is about ∼1μB on the 4f orbitals because of electron ejection.17 The magnetic moment of CeO2 has almost no significant changes when the surface Ce cation is substituted by one or two Pt atoms, suggesting that the OSs of the Pt single-atom and Pt3c + Pt4c should be similar to that of the replaced Ce4+. As for the situation of Ce4+ substituted by Pt3c + Pt4c, the Bader charge of Pt3c and Pt4c is 9.28 and 9.26 e respectively, which means that the valence states of Pt3c and Pt4c should be roughly equal, namely Pt3c2+ and Pt4c2+. The coordination number of Ce4+ is seven, and the number of nearest neighbor O atoms coordinated with Pt4+ is six. The Pt4+–O bonds of the upper three O atoms are about 2.16 Å, and the values for the lower three O atoms are about 2.10 Å. As for Pt4c2+–O and Pt3c2+–O, their average Pt2+–O bonds are all above 2.00 Å, which are shorter than the Pt4+–O bonds.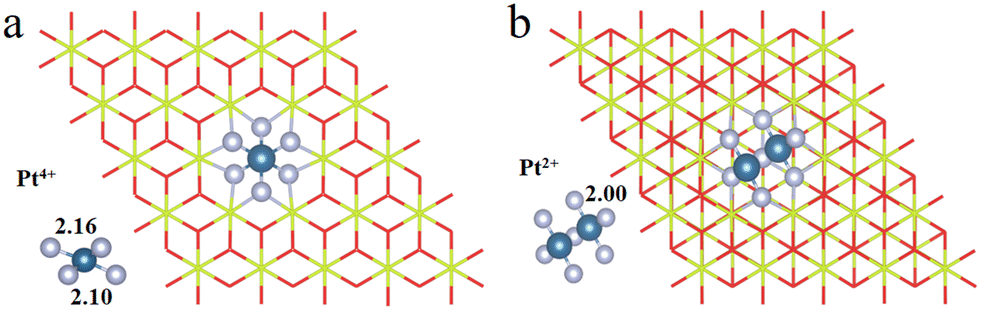 | ||
| Fig. 1 The optimized structures of (a) Pt4+/CeO2 and (b) Pt2+/CeO2. The Ce, O and Pt atoms are denoted by yellow, red and light blue balls, respectively. The silver balls are the nearest O atoms to the Pt atoms. | ||
The possible OSs of Pt atoms on the Pt/CeO2 (111) interface with/without an oxygen vacancy were also explored (Fig. 2).35,47 For the adsorption of Pt on the stoichiometric CeO2 (111), the OS of the Pt site is zero (Pt0) as shown in Fig. 2a.35 The types of oxygen vacancies can be divided into (top) surface and subsurface oxygen vacancies, denoted as SV and SSV, respectively.48,49 The extraction of an oxygen vacancy would cause the reduction of two Ce4+ species to Ce3+, and the spin moments localized mainly on these two Ce3+ ions.50 As for the Pt/CeO2 containing SV/SSV (denoted as Pt + SV and Pt + SSV, respectively), the increase or decrease in the number of Ce3+ means the electron transfer between the Pt and CeO2 caused by the oxygen vacancies (Fig. 2b and c). The number of Ce3+ ions on Pt + SSV increases from 2 to 3 which means that one electron transfers from Pt to CeO2; hence the OS of Pt is +1. As for Pt1+/CeO2, there are five local spin minima (Fig. S2†), and the lowest energy structure is chosen for further study (Fig. 2b).51,52 There are no Ce3+ ions on Pt + SV suggesting that the CeO2 donates two electrons to the Pt site; hence the OS of Pt on Pt + SV is −2 (Fig. 2c). The Pt0–O bond is 2.14 Å, and that for Pt1+ is 1.98 Å. The distance between Pt2− and Ce4+ is 2.88 Å, which is larger than that of Ptδ+–O.
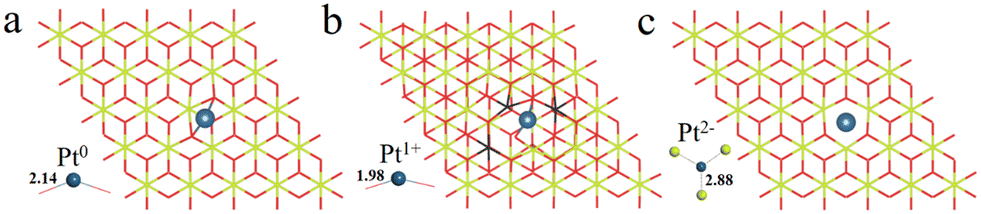 | ||
| Fig. 2 The optimized structures of (a) Pt0/CeO2, (b) Pt1+/CeO2 and (c) Pt2−/CeO2. The Ce, O and Pt atoms are denoted by yellow, red and light blue balls, respectively. The black color indicates the reduced Ce3+ atoms. | ||
3.2. The electronic structures of Pt4+/2+/1+/0/2− and their HER mechanisms
As is well known, the hydrogen adsorption free energy (ΔGH*) has been widely used as a useful descriptor for the evaluation of HER activity, and Pt exhibits the optimal level for the Volmer step with a near-zero ΔGH*, owing to its suitable d-band position.53,54 The first step of the HER is the Volmer step, in which electron transfer to the electrode surface is coupled to proton adsorption (H+ + e− → H*).55H atoms cannot be adsorbed on Pt4+ and Pt4c2+ sites because of the saturated coordination, and they prefer to adsorb on the O atoms adjacent to Pt4+ and Pt4c2+ (Fig. S3†), in which the ΔGH* values for Pt4+–O and Pt4c2+–O are −0.91 and −0.51 eV, respectively. The adsorption structures of H atoms on Pt3c2+, Pt1+, Pt0 and Pt2− sites are displayed in Fig. 3, and their ΔGH* values are −0.44 eV, −0.23 eV, −0.47 eV and −0.03 eV, respectively. To sum up, the ΔGH* values are in the following order: (Pt4+–O) > (Pt4c2+–O) > Pt0 > Pt3c2+ > Pt1+ > Pt2−. Too negative ΔGH* values mean the strong adsorption of H*, which will be disadvantageous to the subsequent desorption process. Meanwhile, too positive ΔGH* values mean the weak adsorption of H*, unfavorable for the subsequent reaction. The Pt1+ is more suitable for the H* adsorption than Pt4+/2+, and the H* adsorption on Pt0 is too strong. All in all, the electron-enriched Pt2− is the most favorable valence state for the Volmer step.
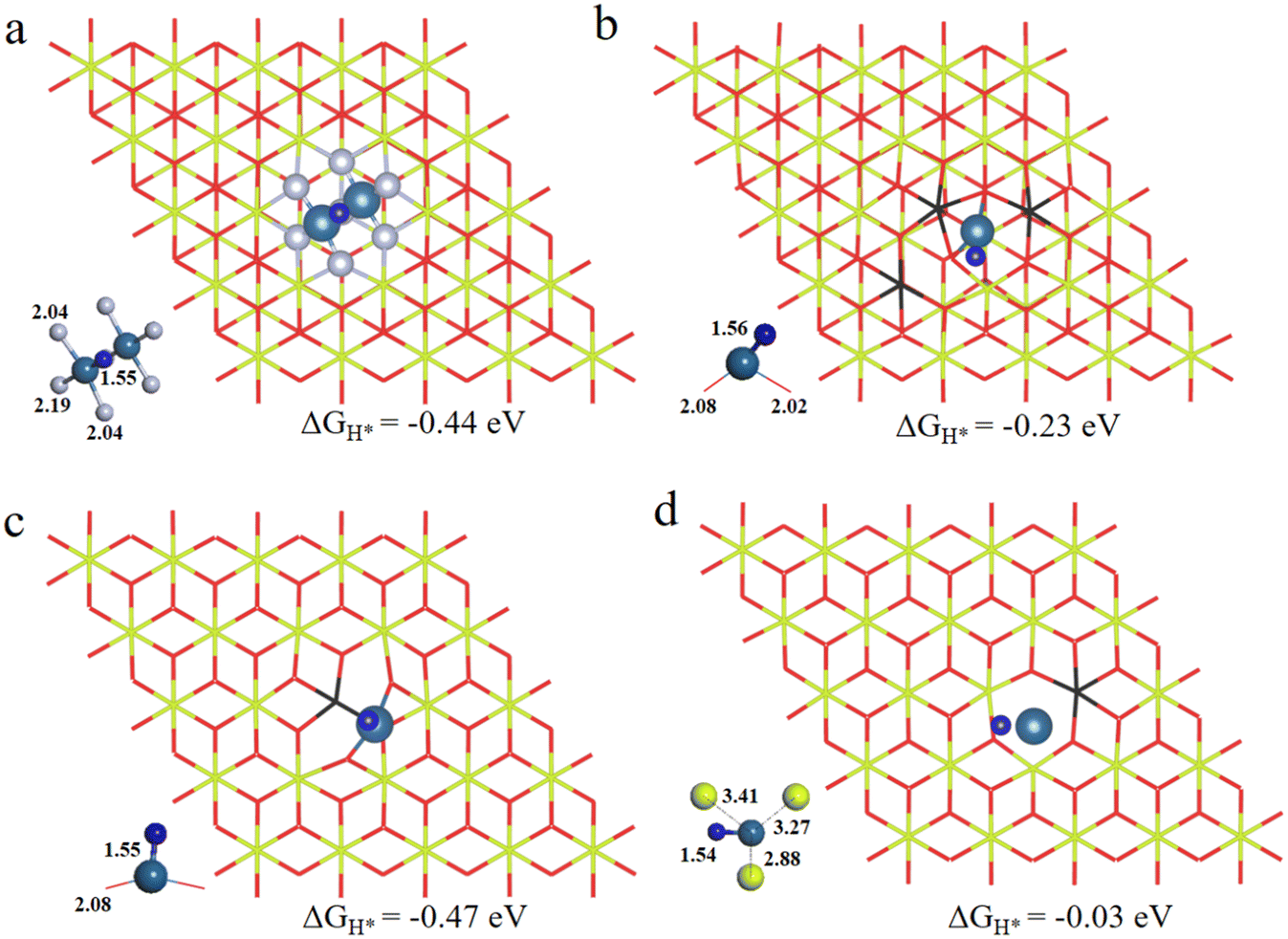 | ||
| Fig. 3 The optimized structures of (a) Pt3c2+/CeO2, (b) Pt1+/CeO2, (c) Pt0/CeO2 and (d) Pt2−/CeO2 with H* adsorption. The Ce, O, Pt and H atoms are denoted by yellow, red, light blue and dark blue balls, respectively. The black color indicates the reduced Ce3+ atoms. | ||
As for Pt4+/2+/1+, the magnetic moment of CeO2 would not change when the H* adsorption occurs, which leads to the formation of (Pt–H)4+/2+/1+ states. The CeO2 support of Pt0/CeO2 would accept one electron resulting in Ce3+ because of the H* adsorption; thus the adsorption state of Pt0 will turn into (Pt–H)1+. As for Pt2−/CeO2, the number of electrons donated from CeO2 to Pt2− would decrease from two to one during the Volmer step, forming a (Pt–H)1− state.
The projected density of states (PDOS) was calculated to study the effect of the Pt OSs on electronic structures (Fig. 4 and S4†). Pt4+, Pt2+ and Pt0 exhibit semiconductor behavior, and the band gaps for Pt4+, Pt4c2+, Pt3c2+ and Pt0 are 1.22 eV, 0.58 eV, 0.50 eV and 0.50 eV, respectively. It is noteworthy that Pt1+ shows metallic properties, and the PDOS of Pt2− is highly localized. The d-band center has been widely used in revealing the relationship between electronic structures and hydrogen adsorption. Surprisingly, the εd is not relevant to the ΔGH* values (Fig. 5a), in which the εd values of Pt4+ and Pt4c2+ are not considered because H atoms could not adsorb on these metal sites and thus the Pt2+ in Fig. 5 refers to Pt3c2+. As is well known, the valence electronic structures of Pt2+/1+/0/2− vary largely in the electron number, whereas this difference is not considered in the calculation of εd. Furthermore, the s–d orbital interaction of Pt2+/1+/0/2− would also change with the various valence electronic structures.56 Considering these facts, the d-band center has been revised as:
| εd′ = (−εd + εs) × [(10 − OSs)/10] | (3) |
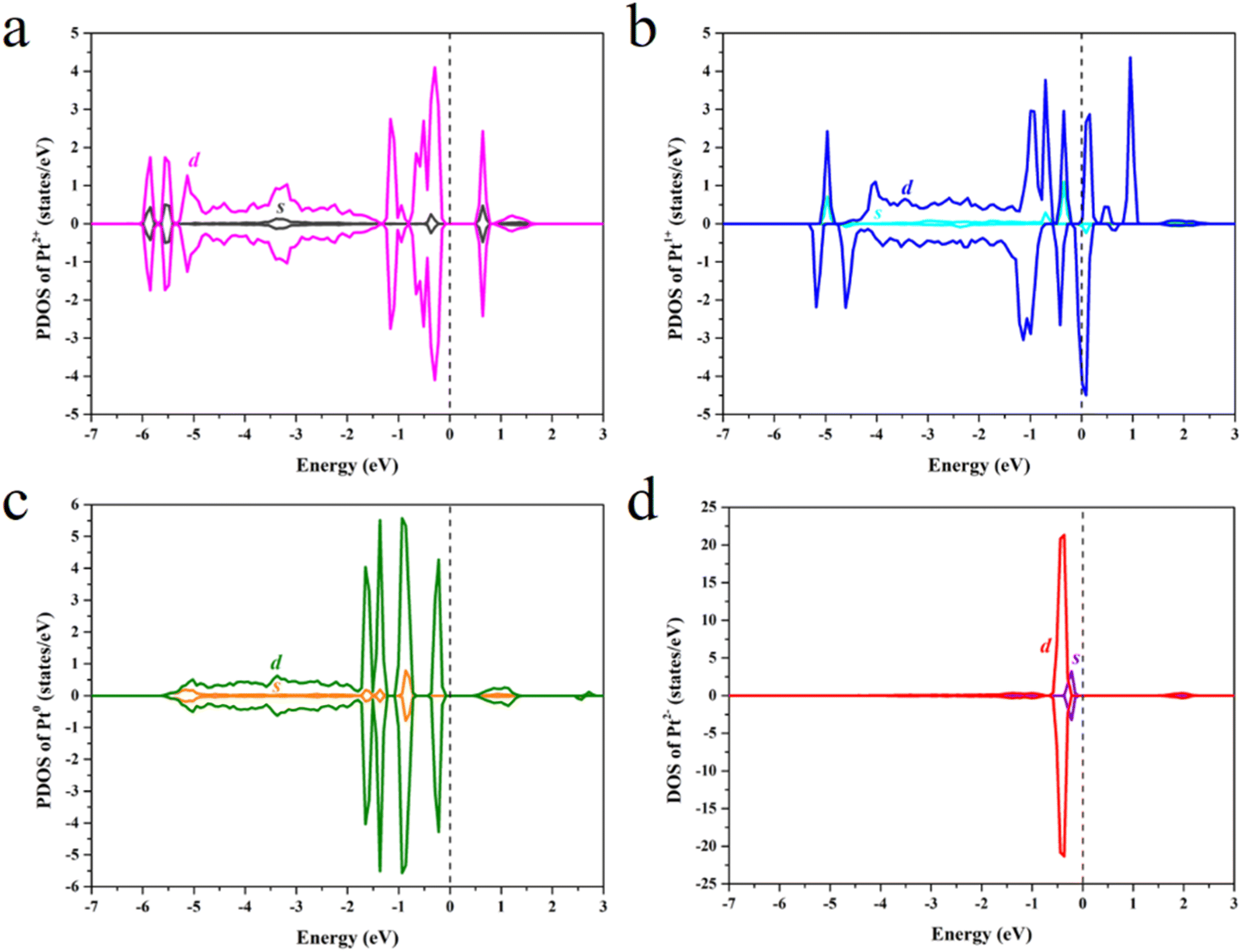 | ||
| Fig. 4 The PDOS of (a) Pt3c2+, (b) Pt1+, (c) Pt0 and (d) Pt2−. | ||
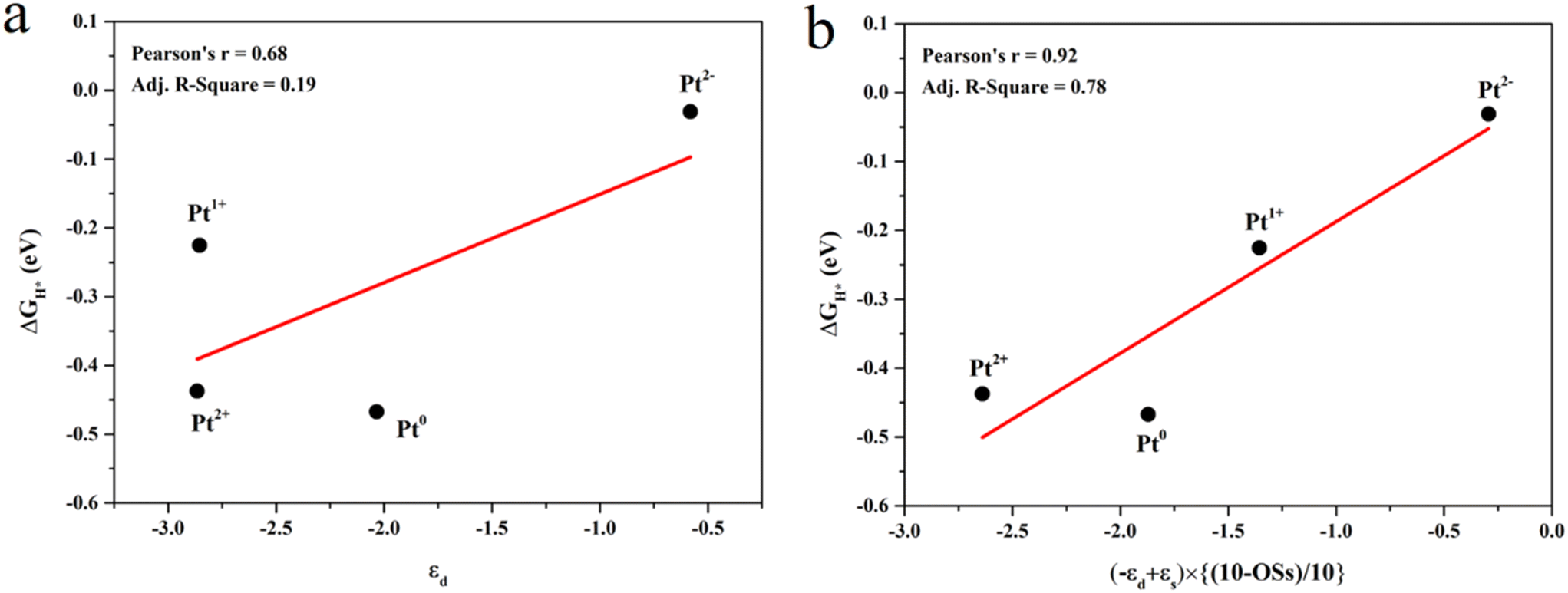 | ||
| Fig. 5 The relationship between d-band centers and ΔGH* values: (a) εd–ΔGH* and (b) εd′–ΔGH*. | ||
The second step of the HER is the process of H2 formation, which may occur via two different pathways: the Heyrovsky and Tafel reactions. In the Heyrovsky reaction, the transfer of a second electron to the H* is coupled with another proton adsorption.55 The active sites for the Volmer reaction will retain catalytic activity for the Heyrovsky reaction when the overpotential is zero, and their ΔGH* values have an opposite relationship.42,60 Through the above analysis, we can clearly see that the Pt4+ and Pt4c2+ sites are not suitable for the Volmer step; thus these two metal sites are no longer involved in the following research, in which Pt2+ below refers to Pt3c2+ for the sake of argument. As can be seen from Fig. 6, Pt2− exhibits the best performance for the Volmer–Heyrovsky reaction, followed by Pt1+, Pt2+ and Pt0.
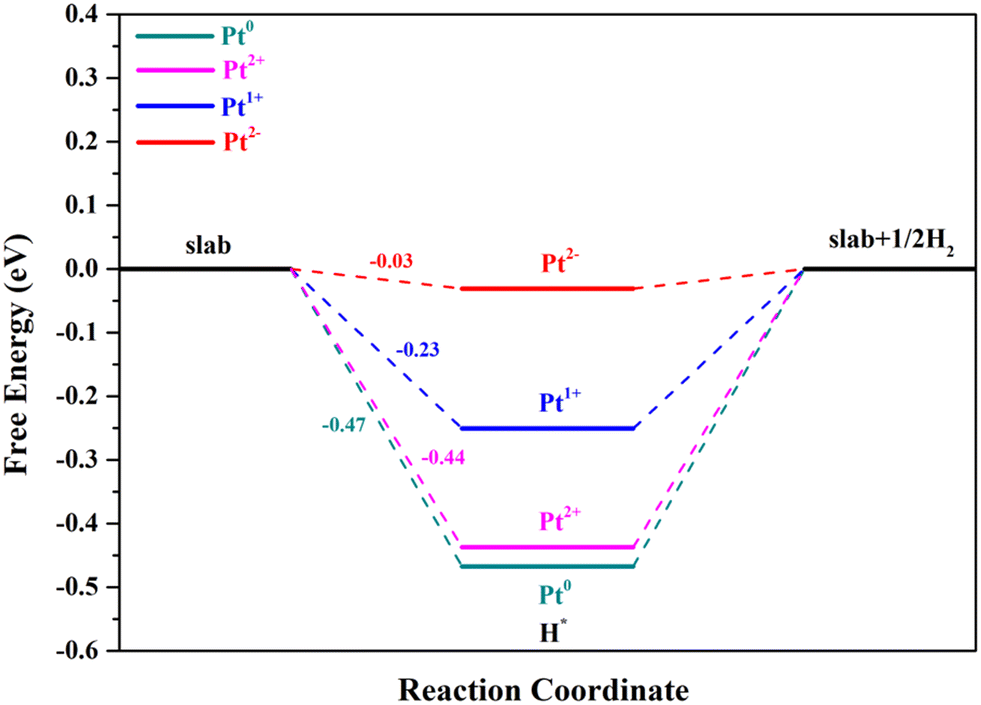 | ||
| Fig. 6 The calculated reaction pathways with the Volmer–Heyrovsky reaction mechanism of Pt2+/1+/0/2−. | ||
As for the Tafel reaction on Pt single-atoms, two H* species would combine to form H2, with the formation of two-hydrogen complexes (H–Pt–H).61 The optimized structures of Pt2+/1+/0/2−/CeO2 with two-H* adsorption are displayed in Fig. 7. The two-H* adsorption on Pt2+/0/2− will not cause the electron transfer between Pt2+/0/2− and CeO2, with the formation of (H–Pt–H)2+/0/2− states. Significantly, the Ce3+ number of Pt1+/CeO2 is 2 when the process of two-H* adsorption occurs, which means that the adsorption state of Pt1+ will turn into (H–Pt–H)0. The number of Ce3+ ions with their magnetic moment are marked in Table S1.† The adsorption of the second H atom on Pt2+/1+/0/2− is an energy decreasing process, and the ΔGH* of Pt2+, Pt1+, Pt0 and Pt2− for this step is −0.55, −1.24, −0.88 and −0.29 eV, respectively (Fig. 8). The potential-determining steps of Volmer–Tafel reactions on the Pt2+/1+/0/2− sites are all the desorption of H2, and the reaction barriers of Pt2+, Pt1+, Pt0 and Pt2− for H2 desorption are 0.99, 1.47, 1.35 and 0.32 eV, respectively. The large values of raised free energy, implying high reaction barriers, will cause a low turnover frequency and reduce the hydrogen production rate.42,62 Hence, the Volmer–Tafel reaction is more likely to take place on Pt2− than on Pt2+/1+/0. It is worth noting that Pt2− has considerable stability during 10 ps AIMD simulations at 300 K and 700 K (ESI† Movies S1 and S2).
 | ||
| Fig. 7 The optimized structures of (a) Pt2+/CeO2, (b) Pt1+/CeO2, (c) Pt0/CeO2 and (d) Pt2−/CeO2 with two-H* adsorption. The Ce, O, Pt and H atoms are denoted by yellow, red, light blue and dark blue balls, respectively. The black color indicates the reduced Ce3+ atoms. | ||
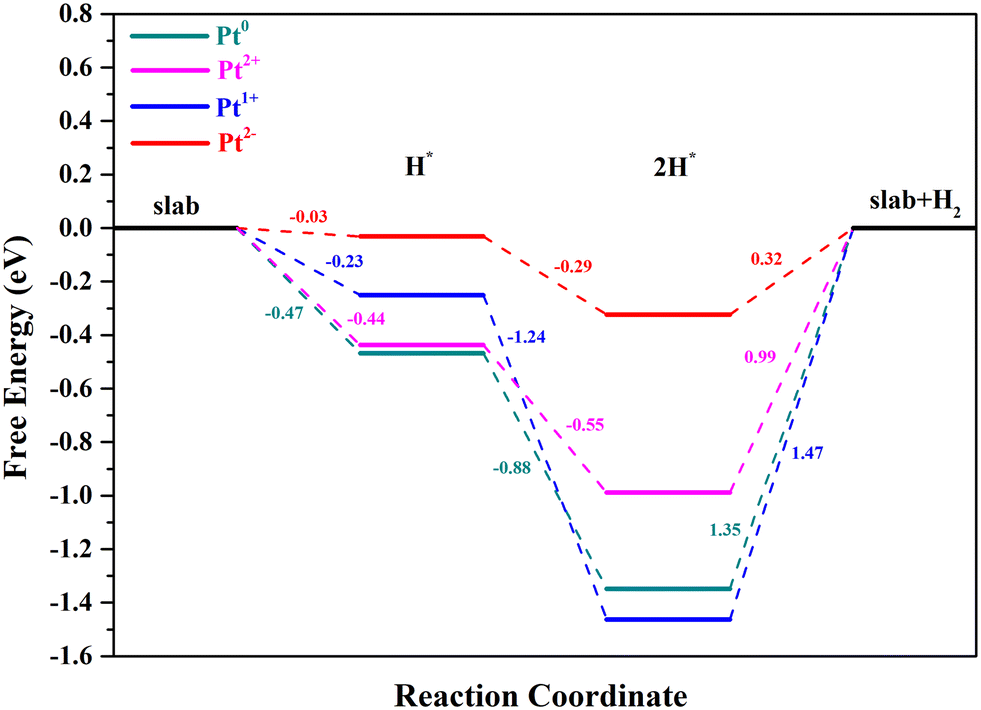 | ||
| Fig. 8 The calculated reaction pathways with the Volmer–Tafel reaction mechanism of Pt2+/1+/0/2−. | ||
4. Conclusions
We have performed systematically investigations on the structures, electronic features and HER catalysis of Pt4+/2+/1+/0/2−/CeO2 by using the density functional theory method. The Pt OSs of Pt/CeO2 are evaluated from the magnetic moment variation of the CeO2 support. Pt4+/2+ sites are constructed by Pt doping into the CeO2 lattice, and Pt0 and Pt1+/2− sites could be obtained by Pt adsorption on stoichiometric and defective CeO2 (111). As for the Volmer step of the HER, the ΔGH* values are in the following order: (Pt4+–O) > (Pt4c2+–O) > Pt0 > Pt3c2+ > Pt1+ > Pt2−, and the electron-enriched Pt2− is the most suitable for this step. Significantly, Pt2− is still the most suitable valence state for the subsequent process of H2 formation, no matter the reaction mechanism, which is the Heyrovsky or Tafel mechanism. The d-band center has been revised considering the effect of Pt OSs in this study, in which the revised d-band center could further demonstrate the importance of Pt OSs to the H* adsorption. The OSs would greatly affect the electronic structures of Pt catalysts, which will further impact their overall catalytic properties.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was funded by National Natural Science Foundation of China (No. 11874005 and No. 52202214), Sichuan Natural Science Foundation (No. 23NSFSC3565), China National Postdoctoral Program for Innovative Talents (No. BX2021053) and China Postdoctoral Science Foundation (No. 2021M700680), Youth Science Foundation of Guizhou Province Education Ministry (QJHKY[2019] 106), Science and Technology Foundation Project of Guizhou Province (QKHJC [2019] 1323) and Key field Project of Guizhou Provincial Department of Education (QKHKY[2020]048). This work was also supported by Academic Training Project of Zunyi Normal University (Grant Number: 5727-03) and Doctor Foundation of Zunyi Normal University (ZSBS[2018]10).References
- M. S. Faber and S. Jin, Energy Environ. Sci., 2014, 7, 3519–3542 RSC.
- S. Ye, F. Luo, Q. Zhang, P. Zhang, T. Xu, Q. Wang, D. He, L. Guo, Y. Zhang, C. He, X. Ouyang, M. Gu, J. Liu and X. Sun, Energy Environ. Sci., 2019, 12, 1000–1007 RSC.
- J. Zhang, E. Wang, S. Cui, S. Yang, X. Zou and Y. Gong, Nano Lett., 2022, 22, 1398–1405 CrossRef CAS PubMed.
- B. Qiao, A. Wang, X. Yang, L. F. Allard, Z. Jiang, Y. Cui, J. Liu, J. Li and T. Zhang, Nat. Chem., 2011, 3, 634–641 CrossRef CAS PubMed.
- H. Zhang, P. An, W. Zhou, Y. Guan Bu, P. Zhang, J. Dong and W. Lou Xiong, Sci. Adv., 2018, 4(1), eaao6657 CrossRef PubMed.
- J. Zhang, Y. Zhao, X. Guo, C. Chen, C.-L. Dong, R.-S. Liu, C.-P. Han, Y. Li, Y. Gogotsi and G. Wang, Nat. Catal., 2018, 1, 985–992 CrossRef CAS.
- X. Li, X. I. Pereira-Hernández, Y. Chen, J. Xu, J. Zhao, C.-W. Pao, C.-Y. Fang, J. Zeng, Y. Wang, B. C. Gates and J. Liu, Nature, 2022, 611, 284–288 CrossRef CAS PubMed.
- S. Yoon, H. Ha, J. Kim, E. Nam, M. Yoo, B. Jeong, H. Y. Kim and K. An, J. Mater. Chem. A, 2021, 9, 26381–26390 RSC.
- A. I. Boronin, E. M. Slavinskaya, A. Figueroba, A. I. Stadnichenko, T. Y. Kardash, O. A. Stonkus, E. A. Fedorova, V. V. Muravev, V. A. Svetlichnyi, A. Bruix and K. M. Neyman, Appl. Catal., A, 2021, 286, 119931 CrossRef CAS.
- K. Yuan, Y. Guo, Q.-L. Lin, L. Huang, J.-T. Ren, H.-C. Liu, C.-H. Yan and Y.-W. Zhang, J. Catal., 2021, 394, 121–130 CrossRef CAS.
- B. Song, S. Si, A. Soleymani, Y. Xin and H. E. Hagelin-Weaver, Nano Res., 2022, 15, 5922–5932 CrossRef CAS.
- Y. Gao, W. Wang, S. Chang and W. Huang, ChemCatChem, 2013, 5, 3610–3620 CrossRef CAS.
- M. Kourtelesis, T. S. Moraes, L. V. Mattos, D. K. Niakolas, F. B. Noronha and X. Verykios, Appl. Catal., A, 2021, 284, 119757 CrossRef CAS.
- H. Jeong, D. Shin, B.-S. Kim, J. Bae, S. Shin, C. Choe, J. W. Han and H. Lee, Angew. Chem., Int. Ed., 2020, 59, 20691–20696 CrossRef CAS PubMed.
- Y. Qu, B. Chen, Z. Li, X. Duan, L. Wang, Y. Lin, T. Yuan, F. Zhou, Y. Hu, Z. Yang, C. Zhao, J. Wang, C. Zhao, Y. Hu, G. Wu, Q. Zhang, Q. Xu, B. Liu, P. Gao, R. You, W. Huang, L. Zheng, L. Gu, Y. Wu and Y. Li, J. Am. Chem. Soc., 2019, 141, 4505–4509 CrossRef CAS PubMed.
- L. Liu and A. Corma, Chem. Rev., 2018, 118, 4981–5079 CrossRef CAS PubMed.
- Y. Lykhach, S. M. Kozlov, T. Skála, A. Tovt, V. Stetsovych, N. Tsud, F. Dvořák, V. Johánek, A. Neitzel, J. Mysliveček, S. Fabris, V. Matolín, K. M. Neyman and J. Libuda, Nat. Mater., 2016, 15, 284–288 CrossRef CAS PubMed.
- S. Yang, Y. J. Tak, J. Kim, A. Soon and H. Lee, ACS Catal., 2017, 7, 1301–1307 CrossRef CAS.
- F.-Y. Yu, Z.-L. Lang, L.-Y. Yin, K. Feng, Y.-J. Xia, H.-Q. Tan, H.-T. Zhu, J. Zhong, Z.-H. Kang and Y.-G. Li, Nat. Commun., 2020, 11, 490 CrossRef PubMed.
- X. Cheng, Y. Li, L. Zheng, Y. Yan, Y. Zhang, G. Chen, S. Sun and J. Zhang, Energy Environ. Sci., 2017, 10, 2450–2458 RSC.
- Y. Zhan, Y. Li, Z. Yang, X. Wu, M. Ge, X. Zhou, J. Hou, X. Zheng, Y. Lai, R. Pang, H. Duan, X. A. Chen, H. Nie and S. Huang, Adv. Sci., 2019, 6, 1801663 CrossRef PubMed.
- Z.-W. Wei, H.-J. Wang, C. Zhang, K. Xu, X.-L. Lu and T.-B. Lu, Angew. Chem., Int. Ed., 2021, 60, 16622–16627 CrossRef CAS.
- H. Tan, B. Tang, Y. Lu, Q. Ji, L. Lv, H. Duan, N. Li, Y. Wang, S. Feng, Z. Li, C. Wang, F. Hu, Z. Sun and W. Yan, Nat. Commun., 2022, 13, 2024 CrossRef CAS.
- Q. Q. Yan, D. X. Wu, S. Q. Chu, Z. Q. Chen, Y. Lin, M. X. Chen, J. Zhang, X. J. Wu and H. W. Liang, Nat. Commun., 2019, 10, 4977 CrossRef PubMed.
- Y. Shi, Z.-R. Ma, Y.-Y. Xiao, Y.-C. Yin, W.-M. Huang, Z.-C. Huang, Y.-Z. Zheng, F.-Y. Mu, R. Huang, G.-Y. Shi, Y.-Y. Sun, X.-H. Xia and W. Chen, Nat. Commun., 2021, 12, 3021 CrossRef.
- S. Fang, X. Zhu, X. Liu, J. Gu, W. Liu, D. Wang, W. Zhang, Y. Lin, J. Lu, S. Wei, Y. Li and T. Yao, Nat. Commun., 2020, 11, 1029 CrossRef CAS.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS.
- A. Bruix, J. A. Rodriguez, P. J. Ramírez, S. D. Senanayake, J. Evans, J. B. Park, D. Stacchiola, P. Liu, J. Hrbek and F. Illas, J. Am. Chem. Soc., 2012, 134, 8968–8974 CrossRef CAS PubMed.
- Y. Xuxu, W. Hengwei, L. Yue, L. Xinyu, C. Lina, G. Jian and L. Junling, Nano Res., 2019, 12, 1401–1409 CrossRef.
- F. Dvořák, M. Farnesi Camellone, A. Tovt, N.-D. Tran, F. R. Negreiros, M. Vorokhta, T. Skála, I. Matolínová, J. Mysliveček, V. Matolín and S. Fabris, Nat. Commun., 2016, 7, 10801 CrossRef.
- Y. Lu, S. Zhou, C.-T. Kuo, D. Kunwar, C. Thompson, A. S. Hoffman, A. Boubnov, S. Lin, A. K. Datye, H. Guo and A. M. Karim, ACS Catal., 2021, 11, 8701–8715 CrossRef CAS.
- D. Kunwar, S. Zhou, A. DeLaRiva, E. J. Peterson, H. Xiong, X. I. Pereira-Hernández, S. C. Purdy, R. ter Veen, H. H. Brongersma, J. T. Miller, H. Hashiguchi, L. Kovarik, S. Lin, H. Guo, Y. Wang and A. K. Datye, ACS Catal., 2019, 9, 3978–3990 CrossRef CAS.
- Y. Tang, Y.-G. Wang and J. Li, J. Phys. Chem. C, 2017, 121, 11281–11289 CrossRef CAS.
- Z. Yang, Z. Lu and G. Luo, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 76, 075421 CrossRef.
- P. Bera, K. R. Priolkar, A. Gayen, P. R. Sarode, M. S. Hegde, S. Emura, R. Kumashiro, V. Jayaram and G. N. Subbanna, Chem. Mater., 2003, 15, 2049–2060 CrossRef CAS.
- P. Bera, A. Gayen, M. S. Hegde, N. P. Lalla, L. Spadaro, F. Frusteri and F. Arena, J. Phys. Chem. B, 2003, 107, 6122–6130 CrossRef CAS.
- J. K. Nørskov, J. Rossmeisl, A. Logadottir, L. Lindqvist, J. R. Kitchin, T. Bligaard and H. Jónsson, J. Phys. Chem. B, 2004, 108, 17886–17892 CrossRef.
- A. Nilsson, L. G. M. Pettersson, B. Hammer, T. Bligaard, C. H. Christensen and J. K. Nørskov, Catal. Lett., 2005, 100, 111–114 CrossRef CAS.
- B. Hammer and J. K. Nørskov, in Advances in Catalysis, Academic Press, 2000, vol. 45, pp. 71–129 Search PubMed.
- S. Zhou, X. Yang, W. Pei, N. Liu and J. Zhao, Nanoscale, 2018, 10, 10876–10883 RSC.
- Y. Ouyang, C. Ling, Q. Chen, Z. Wang, L. Shi and J. Wang, Chem. Mater., 2016, 28, 4390–4396 CrossRef CAS.
- M. Qiu, Z. Fang, Y. Li, J. Zhu, X. Huang, K. Ding, W. Chen and Y. Zhang, Appl. Surf. Sci., 2015, 353, 902–912 CrossRef CAS.
- Y.-Q. Su, J.-X. Liu, I. A. W. Filot, L. Zhang and E. J. M. Hensen, ACS Catal., 2018, 8, 6552–6559 CrossRef CAS PubMed.
- A. Walsh, A. A. Sokol, J. Buckeridge, D. O. Scanlon and C. R. A. Catlow, Nat. Mater., 2018, 17, 958–964 CrossRef CAS PubMed.
- N. Daelman, M. Capdevila-Cortada and N. López, Nat. Mater., 2019, 18, 1215–1221 CrossRef CAS PubMed.
- X. Wang, J. A. van Bokhoven and D. Palagin, Phys. Chem. Chem. Phys., 2017, 19, 30513–30519 RSC.
- X.-P. Wu and X.-Q. Gong, Phys. Rev. Lett., 2016, 116, 086102 CrossRef PubMed.
- P. G. Lustemberg, Y. Pan, B. J. Shaw, D. Grinter, C. Pang, G. Thornton, R. Pérez, M. V. Ganduglia-Pirovano and N. Nilius, Phys. Rev. Lett., 2016, 116, 236101 CrossRef CAS PubMed.
- M. Nolan, S. C. Parker and G. W. Watson, Surf. Sci., 2005, 595, 223–232 CrossRef CAS.
- H.-Y. Li, H.-F. Wang, X.-Q. Gong, Y.-L. Guo, Y. Guo, G. Lu and P. Hu, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 79, 193401 CrossRef.
- W.-J. Zhu, J. Zhang, X.-Q. Gong and G. Lu, Catal. Today, 2011, 165, 19–24 CrossRef CAS.
- J. K. Nørskov, T. Bligaard, A. Logadottir, J. R. Kitchin, J. G. Chen, S. Pandelov and U. Stimming, J. Electrochem. Soc., 2005, 152, J23 CrossRef.
- J. Mahmood, F. Li, S. M. Jung, M. S. Okyay, I. Ahmad, S. J. Kim, N. Park, H. Y. Jeong and J. B. Baek, Nat. Nanotechnol., 2017, 12, 441–446 CrossRef CAS PubMed.
- C. G. Morales-Guio, L.-A. Stern and X. Hu, Chem. Soc. Rev., 2014, 43, 6555–6569 RSC.
- H. Häkkinen, M. Moseler, O. Kostko, N. Morgner, M. A. Hoffmann and B. V. Issendorff, Phys. Rev. Lett., 2004, 93, 093401 CrossRef PubMed.
- N. J. O'Connor, A. S. M. Jonayat, M. J. Janik and T. P. Senftle, Nat. Catal., 2018, 1, 531–539 CrossRef.
- P. Kuang, Y. Wang, B. Zhu, F. Xia, C.-W. Tung, J. Wu, H. M. Chen and J. Yu, Adv. Mater., 2021, 33, 2008599 CrossRef CAS PubMed.
- H. Li, L. Wang, Y. Dai, Z. Pu, Z. Lao, Y. Chen, M. Wang, X. Zheng, J. Zhu, W. Zhang, R. Si, C. Ma and J. Zeng, Nat. Nanotechnol., 2018, 13, 411–417 CrossRef CAS PubMed.
- B. Hinnemann, P. G. Moses, J. Bonde, K. P. Jørgensen, J. H. Nielsen, S. Horch, I. Chorkendorff and J. K. Nørskov, J. Am. Chem. Soc., 2005, 127, 5308–5309 CrossRef CAS PubMed.
- G. Di Liberto, L. A. Cipriano and G. Pacchioni, J. Am. Chem. Soc., 2021, 143, 20431–20441 CrossRef CAS PubMed.
- T. Wu, M. M. Melander and K. Honkala, ACS Catal., 2022, 12, 2505–2512 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ce01246d |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2023 |