
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The crystal structure and electronic properties of three novel charge transfer co-crystals TCNQFn–triphenylene (n = 0, 2, 4)†
Simon
Payne
 ab,
Iryna
Andrusenko
c,
Francesco
Papi
c,
Jason
Potticary
a,
Mauro
Gemmi
c and
Simon R.
Hall
*a
ab,
Iryna
Andrusenko
c,
Francesco
Papi
c,
Jason
Potticary
a,
Mauro
Gemmi
c and
Simon R.
Hall
*a
aComplex Functional Materials Group, School of Chemistry, University of Bristol, BS8 1TS, Bristol, UK. E-mail: simon.hall@bristol.ac.uk
bCentre for Doctoral Training in Condensed Matter Physics, H. H. Wills Physics Laboratory, University of Bristol, BS8 1TL, Bristol, UK
cElectron Crystallography, Center for Materials Interfaces, Istituto Italiano di Tecnologia, Viale Rinaldo Piaggio 34, 56025 Pontedera, Italy
First published on 4th January 2023
Abstract
The pure phase syntheses of three novel organic charge transfer complexes, obtained by combining the polyaromatic hydrocarbon triphenylene with 7,7,8,8-tetracyanoquinodimethane (TCNQ) and its fluorinated derivatives, are reported together with their crystal structures determined by 3D electron diffraction. The degree of charge transfer is estimated via examining intramolecular vibrational mode displacements between neutral TCNQFn and its complexes with triphenylene using infrared spectroscopy. The direct optical band gaps of 1.87, 1.76 and 1.70 eV for triphenylene–TCNQFn (n = 0, 2, 4) respectively, are determined via diffuse reflectance spectroscopy. The application of advanced structure determination techniques in combination with spectroscopic methods has allowed us to shed light on compelling charge transfer systems and to gain indications for the design of improved electrical organic conductors.
1 Introduction
Co-crystals are multicomponent molecular systems that consist of two or more chemical species forming a single crystal structure.1,2 In particular, organic charge transfer complexes (OCTC) are co-crystals consisting of two chemically distinct organic species, a π-electron donor and a π-electron acceptor, which interact through strong Coulomb and dipolar forces, resulting in a degree of charge transfer (CT). The versatility of organic CT complexes is apparent via the rich array of physical phenomena they exhibit, including room temperature ferroelectricity,3 semiconductivity and superconductivity.4 These donor–acceptor (DA) systems have a distinct advantage as conductive materials, over monomolecular materials, in that they offer greater charge mobility potential5 and band gap tunability.6The use of DA systems as organic field effect transistors (OFETs)7 is common and by creating molecular-level ordered hetero-junctions of segregated or mixed-stack architectures, crystals can be formed that show either n/p type or ambipolar transport respectively.8 In mixed-stack systems, the donor and acceptor molecules alternate along the stacking direction –D–A–D–A–D–, meanwhile, in segregated-stack systems, they pack into columns separately –A–A–A– and –D–D–D–.8,9 Materials of the first category are limited by simple band theory to semiconducting behavior that usually results in lower conductivity compared to segregated-stack systems.10 On the other hand, mixed-stack OCTC crystals are of interest because of the potential for a new type of mechanism for ferroelectricity, estimated to be about 20 times larger than conventional mechanisms.11 A prominent example in this regard is the tetrathiafulvalene![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :chloranil, 1:1 complex exhibiting ferroelectricity below 80 K.12 Generally, the electronic properties of OCTCs can be tuned by composition, stoichiometry, temperature or pressure.13,14 For example, by increasing acceptor electron affinity via fluorine substitution of hydrogen atoms about the aromatic core, crystal packing can be altered along with respective HOMO and LUMO energies tailoring electronic and optical properties to desired states.15 In recent years the combination of polycyclic aromatic hydrocarbons (PAHs) and halogenated acceptors have attracted considerable attention as functional materials in electronics and photonics.5,15–18 PAHs are typical examples of π-electron systems able to donate electrons,19 while 7,7,8,8-tetracyanoquinodimethane TCNQ is arguably the most investigated acceptor for the construction of CT complexes.20 It has been shown that co-crystals containing PAHs and TCNQ molecules show conducting properties with a relatively small band gap and moderate CT between the chemical components.15,19,21
:chloranil, 1:1 complex exhibiting ferroelectricity below 80 K.12 Generally, the electronic properties of OCTCs can be tuned by composition, stoichiometry, temperature or pressure.13,14 For example, by increasing acceptor electron affinity via fluorine substitution of hydrogen atoms about the aromatic core, crystal packing can be altered along with respective HOMO and LUMO energies tailoring electronic and optical properties to desired states.15 In recent years the combination of polycyclic aromatic hydrocarbons (PAHs) and halogenated acceptors have attracted considerable attention as functional materials in electronics and photonics.5,15–18 PAHs are typical examples of π-electron systems able to donate electrons,19 while 7,7,8,8-tetracyanoquinodimethane TCNQ is arguably the most investigated acceptor for the construction of CT complexes.20 It has been shown that co-crystals containing PAHs and TCNQ molecules show conducting properties with a relatively small band gap and moderate CT between the chemical components.15,19,21
In this paper we describe novel DA complexes based on the polyaromatic hydrocarbon triphenylene and TCNQFn, where n = 0, 2, 4. The effects of fluorine substitution on the TCNQ scaffold have been previously investigated in terms of molecular packing and electronic properties, in view of the possible tuning of conductivity,15 however this is the first time triphenylene as a co-former, has been investigated. Due to sub-micron crystalline domains, three-dimensional electron diffraction (3D ED), was employed to solve the crystal structures.22 Variations in the electronic and optical properties were evaluated by the degree of CT, estimated via infrared spectroscopy and the direct optical band gaps determined from diffuse reflectance spectroscopy.
2 Experimental
Materials were purchased from Fisher Scientific (triphenylene >99% purity) and Tokyo Chemical Industries (TCNQFn >98% purity) and used as received with no further purification. Co-crystals were prepared by dissolving equimolar constituents in 1 mL of toluene left at 323.15 K for 72 h before cooling to room temperature under ambient conditions. Solutions were filtered through a 0.22 μm membrane into round bottom test tubes which were cleaned with dry toluene in an attempt to reduce the number of nucleation sites, a needle hole allowed the volatile solvent to escape.Scanning electron microscopy (SEM) was performed on a JEOL SEM IT300 running an accelerating voltage of 20 kV. Electron diffraction data was recorded with a Zeiss Libra transmission electron microscope (TEM) operating at 120 kV and equipped with a LaB6 source. 3D ED was performed in STEM mode after defocusing the beam in order to have a pseudo-parallel illumination on the sample, as described in Lanza et al.23 ED patterns were collected in Kohler parallel illumination with a beam size of about 150 nm in diameter, obtained using a 5 μm C2 condenser aperture. Data were recorded by a single-electron ASI MEDIPIX detector.24 An extremely low dose illumination, corresponding to about 0.01 el Å−2 s−1, was adopted in order to avoid beam damage.
A needle of each co-crystal was gently crushed and directly loaded on a carbon-coated Cu TEM grid without any solvent or sonication. 3D ED acquisitions were performed by rotating the sample around the TEM goniometer axis in steps of 1°, with total tilt ranges up to 110°. A camera length of 180 mm was used, allowing resolution in real space up to 0.7 Å. After each tilt, a diffraction pattern was acquired and the crystal position was tracked by STEM imaging. During the experiment, the beam was precessed around the optical axis by an angle of 1°. Precession was obtained using a Nanomegas Digistar P1000 device. All data acquisitions were performed at room temperature.
3D ED data were analyzed using the software PETS2.0.25 Structure determinations were obtained by standard direct methods (SDM) for triphenylene–TCNQFn (n = 0, 2) and by simulated annealing (SA) for triphenylene–TCNQFn (n = 4), as implemented in the software SIR2014.26 Data were treated with a fully kinematical approximation, assuming that Ihkl was proportional to |Fhkl|2. Least-squares structure refinement was performed with the software SHELXL using electron atomic scattering factors.27
Geometry optimizations via the Crystal17 software package were obtained using the dispersion corrected PBE0 hybrid functional,28 Grimme's empirical energy corrections,29 bielectronic integral truncation parameters (TOLINTEG) 7 7 7 7 14, the 6-31g(d) elemental basis set and utilizing the Pack–Monkhorst method a shrinking factor 4 was used in reciprocal space and to define the sampling of k points about the “Gilat net”.30,31
The degree of CT was quantified with a powdered sample via Fourier-transformed infrared spectroscopy using a Perkin Elmer spectrum 2 FT-IR spectrometer, while direct optical band gap energies were acquired during diffuse reflectance spectroscopy using a Perkin Elmer lambda 650 series spectrophotometer and processed by applying a Kubelka–Munk transformation. Crystal structure visualisations and Hirshfeld surfaces were rendered via the CCDC Vesta (Ver. 3.3.1) and CrystalExplorer (Ver. 17.5) software packages respectively.
3 Results
All the triphenylene–TCNQFn (n = 0, 2, 4) co-crystals grown, resulted in a needle like morphology (Fig. 1). Up to 8 mm long needles of the scarlet non fluorinated complex contrast the black needles of the fluorinated systems of analogous size. SEM imaging (Fig. 2) revealed that all needle-shaped co-crystals are, in fact, polycrystalline bunches of very thin platelets (Fig. 2c insert). In the case of triphenylene–TCNQ, all 3D ED data delivered a triclinic primitive unit cell with approximate parameters a = 6.8 Å, b = 11.2 Å, c = 17.7 Å and α = 78.4°, β = 83.9°, γ = 72.9°. Such a cell would conveniently host two pairs of molecules. Examination of the main planes of 3D ED reconstructions revealed no extinction features. In all data sets, diffuse scattering was present parallel to c*. Structure solution of the triphenylene–TCNQ co-crystal was obtained ab initio in space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) (2). Ultimately, 30 out of 34 non hydrogen atoms of the structure were spotted ab initio by automatic routines (Fig. 3a and b).
(2). Ultimately, 30 out of 34 non hydrogen atoms of the structure were spotted ab initio by automatic routines (Fig. 3a and b).
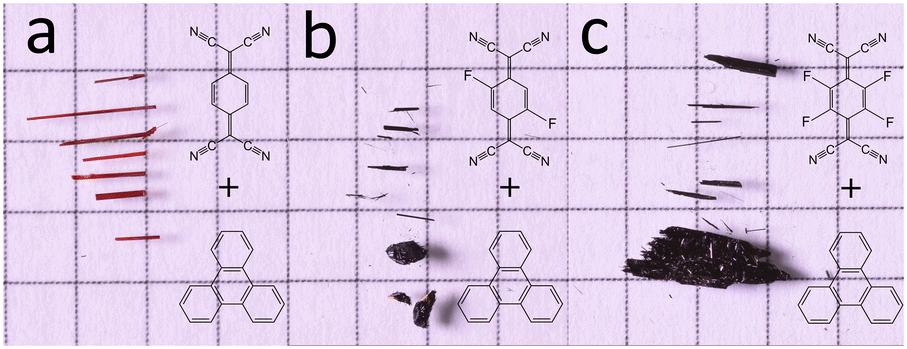 | ||
| Fig. 1 Optical images of crystalline CT complexes against 5 mm squared graph paper for scale. (a) Triphenylene–TCNQ (b) triphenylene–TCNQF2 and (c) triphenylene–TCNQF4. | ||
 | ||
| Fig. 2 SEM micrographs (a–c) and polarized light microscopy (d–f) of triphenylene–TCNQ (a and d), triphenylene–TCNQF2 (b and e) and triphenylene–TCNQF4 (c and f). | ||
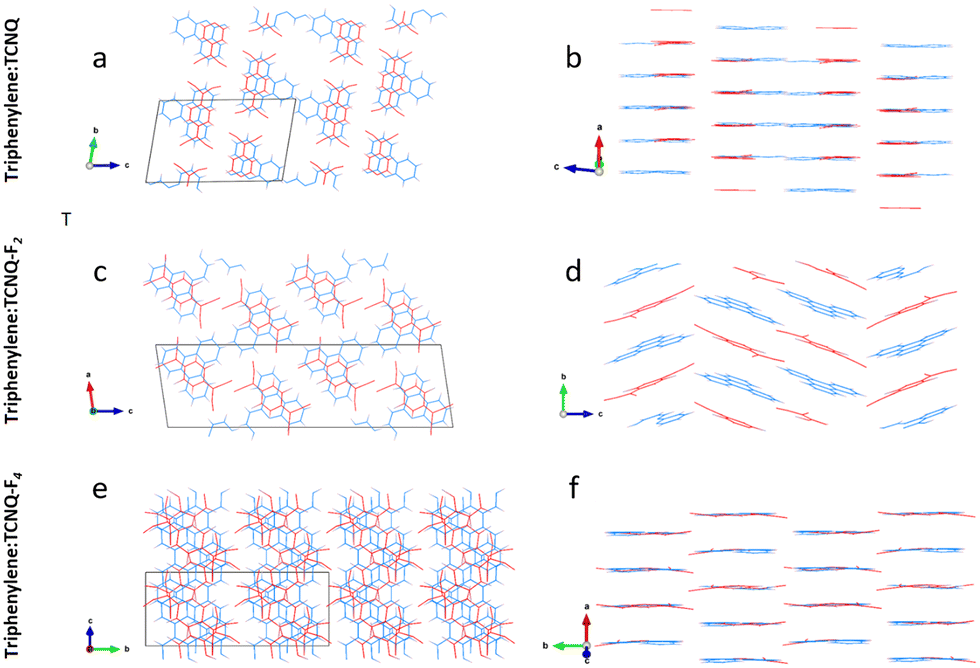 | ||
| Fig. 3 Intermolecular displacements next to an expanded unit cell view down DA stack axis: triphenylene–TCNQ (a and b), triphenylene–TCNQF2 (c and d) and triphenylene–TCNQF4 (e and f). Triphenylene donor molecules are in blue, while TCNQFn acceptor molecules are in red. | ||
Analysis of this structure show that the co-former molecules are weakly coupled in dimers and form two mixed-stack columns along [100] that are reciprocally rotated by 180 and alternate along [001] (Fig. 3a and b). The molecular planes of triphenylene and TCNQ are always parallel to the (100) plane and in particular the TCNQ molecules lie along one of the three benzene fragments of the triphenylene molecule. Intra-stack dimerization of the unique unit group D–A–D is shown to have alternating face-to-face distances of 3.228 Å and 3.559 Å. In more detail, DA columns are repeating along [010] and stabilized by TCNQ interacting with triphenylene by C–H⋯N contacts along [010] (distances 2.86 Å and 3.04 Å) and along [001] (distance 3.06 Å).
Data collected on triphenylene–TCNQF2 micrometric crystals consistently indicated a primitive monoclinic cell with approximate parameters a = 9.4 Å, b = 7.18 Å, c = 32.2 Å and β = 98.4°.
Such a cell would likely contain four pairs of molecules. Upon inspection of the reciprocal space reconstruction, the extinction rules 0k0: k = 2n and h0l: l = 2n were observed, hence indicating crystallization in the space group P21/c (14). The structure of the triphenylene–TCNQF2 co-crystal was solved with the ab initio localization of 30 out of 36 non hydrogen atoms in the asymmetric unit (Fig. 3c and d).
As in the case of the non fluorinated analogue, the triphenylene–TCNQF2 structure comprises alternating DA molecules (Fig. 3c and d). Dimers are however no longer present, as D–A and A–D distances are equivalent (3.35 Å), in addition to which, TCNQF2 molecules are slightly rotated and shifted with respect to triphenylene molecules. Interestingly, DA molecules form in columns along [010]. Every second column, layers of DA molecules are kinked of about 42° along [001]. The overall structure is stabilized by lateral C–H⋯N and C–H⋯F contacts of 2.43 Å and 2.62 Å.
In the case of triphenylene–TCNQF4, all 3D ED data sets were consistent with a C-centered monoclinic cell with approximate parameters a = 17.8 Å, b = 17.4 Å, c = 9.2 Å and β = 130.3°. This cell would conveniently host four pairs of molecules. A close look on planar cuts from 3D ED reconstructions revealed that reflections h0l: l ≠ 2n were missing or relatively weak.
Then, structure solution of TCNQF4–triphenylene was performed by SA using 3D ED data in space group Cc (9). Molecular models for TCNQF4 and triphenylene were taken from literature and used as separate fragments. TCNQF4–triphenylene appears an ideal case for SA as both co-former molecules are entirely rigid. Solution revealed that the structure is actually centrosymmetric. Both TCNQF4 and triphenylene molecules fall on conceivable symmetry elements after an appropriate cell origin shift is applied, the inversion center at 4d and the two-fold axis at 4e, respectively. The subsequent refinement confirms that the correct space group is C2/c, as only half of each molecule is crystallographically independent.
The structure of triphenylene–TCNQF4 is made by mixed-stack DA dimers along [010] with intra-stack separation of 3.27 Å, separating each other by a distance of 3.40 Å (Fig. 3e and f). The TCNQF4 molecule lies along one of the three edges of the triphenylene molecule. All molecules lie in parallel planes, but alternating DA dimers are reciprocally rotated of about 60° along [010]. The structure is stabilized by lateral C–H⋯N contacts with distances 2.66–2.86 Å and by C–F⋯N distances of 3.20 Å.
A higher degree of CT is structurally dependent upon smaller interplanar distances (denser stacks) and upon the minimization of shift between in-plane molecules within each stack.19 In this regard assessing the degree of CT exhibited by these materials becomes more complex than just attributing it to fluorination of the acceptor. Any unexpected structural arrangements, noted within the Fn complexes, i.e. how the π-stack interaction and geometry of the donor triphenylene is expressed in alternating DA pairs, makes comparisons difficult.
Intermolecular bond analysis via the generation of Hirshfeld surfaces using CrystalExplorer (Ver. 17.5) for each constituent per complex shows that with the introduction of halogen bonding in the system, a stronger resultant π–π stack network is created as indicated by the increasing C⋯C interaction as quantified by the Hirshfeld surface analysis (Fig. 4).
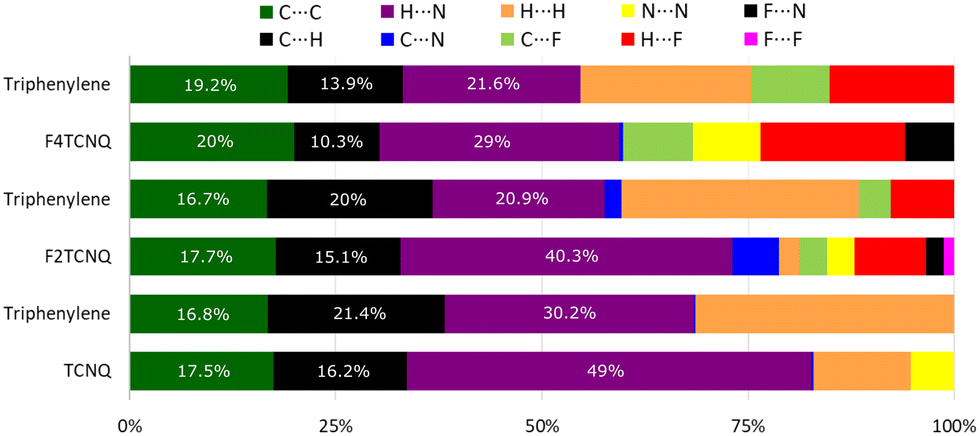 | ||
| Fig. 4 Percentage contributions to the Hirshfeld surface area for close intermolecular contacts. | ||
This coincides with a suppression of H⋯H, C⋯H and H⋯N interactions as competing C⋯F and H⋯F networks are established. Empirically, the degree of CT, ρ, for PAH–TCNQ based complexes has been estimated by extracting a ratio of selected intramolecular bond lengths via the Kistenmacher method32 such that 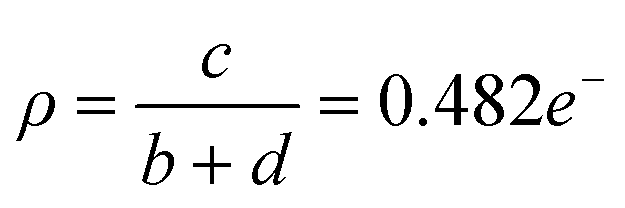 for the non fluorinated complex (Table 1).
for the non fluorinated complex (Table 1).
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C markers and mode 4 tracks C
C markers and mode 4 tracks C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N markers. Each column tracks the displacement of one peak between each TCNQFn system with a unique peak (1 cm−1) tracked for (n = 0, 2, 4). Modes for (K)–TCNQFn were taken from literature15
N markers. Each column tracks the displacement of one peak between each TCNQFn system with a unique peak (1 cm−1) tracked for (n = 0, 2, 4). Modes for (K)–TCNQFn were taken from literature15
| Mode 1 | Mode 2 | Mode 3 | Mode 4 | |
|---|---|---|---|---|
| TCNQ | 1543 | 2226 | ||
| Triphenylene–TCNQ | 1543 | 2219 | ||
| (K)–TCNQ | 1509 | |||
| TCNQF2 | 1395 | 1550 | 1575 | 2230 |
| Triphenylene–TCNQF2 | 1385 | 1547 | 1573 | 2220 |
| (K)–TCNQF2 | 1348 | 1486 | 1524 | |
| TCNQF4 | 1396 | 1550 | 1599 | 2227 |
| Triphenylene–TCNQF4 | 1389 | 1548 | 1594 | 2226 |
| (K)–TCNQF4 | 1353 | 1501 | 1540 |
To quantify the degree of CT between these three systems, one can compare the room temperature infrared absorption spectra of triphenylene, TCNQFn, triphenylene–TCNQFn and a complex in which the electron donating species has ionized. A blueshift in absorption is associated with the shortening of covalent bonds, CN and CC can be compared to a greater blueshift of these markers in ionized systems, such as potassium (K)–TCNQFn or the radical anions of TCNQFn, from which a reference can be set for a degree of CT at ρ = 1e−.
The ratio of blueshift, between triphenylene–TCNQFn and (K)–TCNQFn, can be used to estimate the degree of CT, on the assumption that bond length displacement observed via CC and CN markers, is linearly proportional to the degree of CT. Deviations in molecular configuration between triphenylene–TCNQFn cells can not be exclusively attributed to, and therefore assessed by, the degree of CT. Discrepancies are expected due to the unique set of intermolecular forces intrinsic to the structure of each complex presented. Therefore estimates from CN markers are subject to uncertainty, as the peripheral position of the CN bonds, makes them susceptible to inter-stack coupling as depicted in Fig. 5.
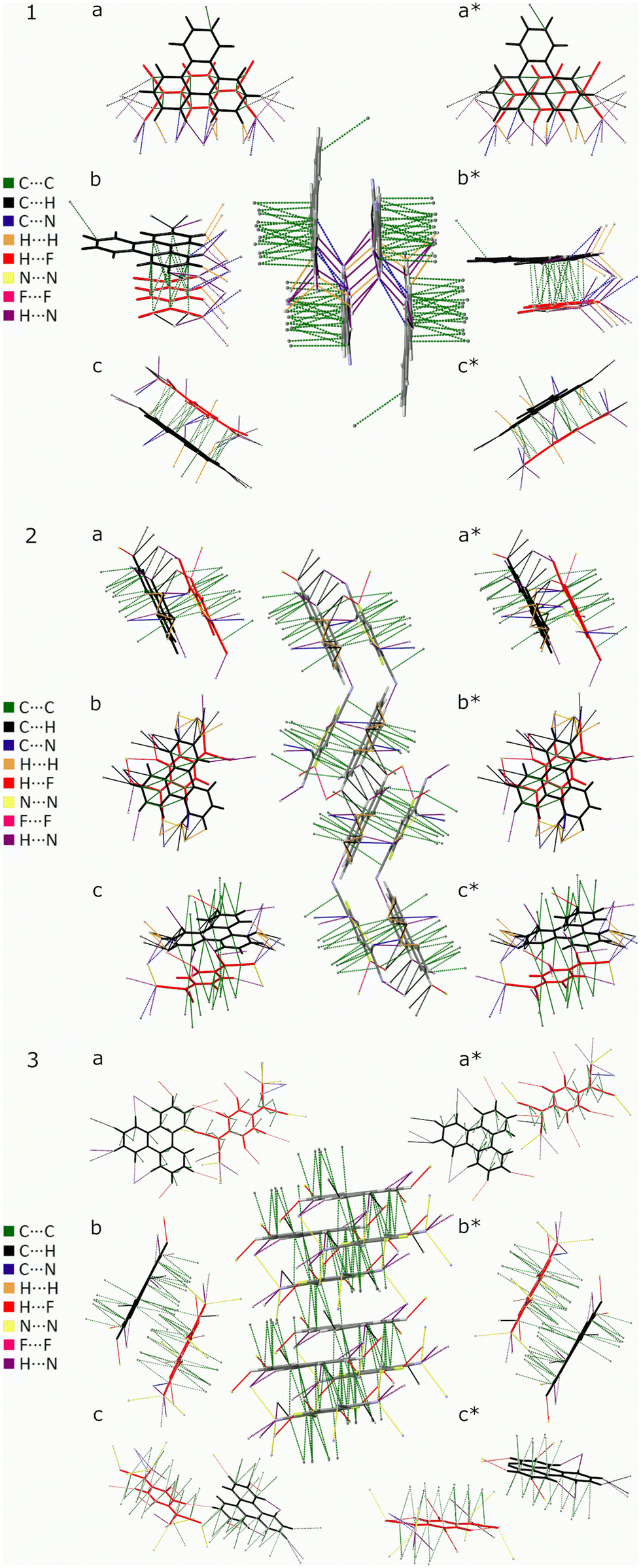 | ||
| Fig. 5 Triphenylene–TCNQFn unit cells, (1) Fn = 0, (2) Fn = 2 and (3) Fn = 4 with cell packing (centre) surrounded by asymmetric units of TCNQFn (red) and triphenylene (black) viewed down the a, b, c, a*, b* and c* cell axis. | ||
As can be seen in Fig. 6, there is only negligible shift of the charge sensitive mode at 1543 cm−1 in TCNQ; which is displaced to 1509 cm−1 in (K)–TCNQ corresponding to ρ = 1e−1. The CN mode displacement from 2226 cm−1 in TCNQ to 2219 cm−1 in triphenylene–TCNQ gives ρ = 0.38e−1 given the corresponding vibrational mode the TCNQ radical holds at 2208 cm−1. However values of ρ determined via markers in peripheral CN modes are likely inaccurate due to the interacting stack coupling C–H⋯N contacts present in system. The mode at 1543 cm−1 shows negligible CT for the complex with both markers, standing in disagreement to the empirical method. Charge sensitive modes of TCNQF2 at 1395, 1550 and 1575 cm−1 have respective wavelength absorbance shifts compared to (K)–TCNQF2 of −44, −63 and −50 cm−1. Correspondingly triphenylene–TCNQF2 observed shifts to 1385, 1547 and 1573 cm−1 yielding ρ = 0.23, ρ = 0.05 and ρ = 0.04e− respectively (Fig. 7).
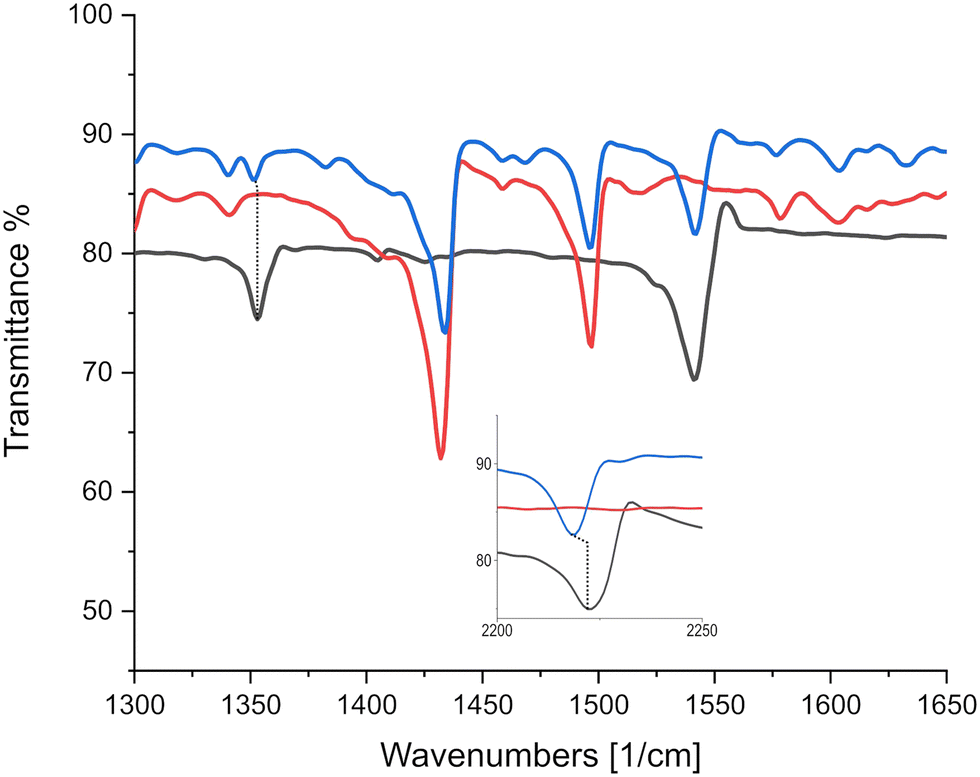 | ||
| Fig. 6 IR absorption spectra identifying negligible charge sensitive mode displacements between TCNQ and triphenylene–TCNQ. Black, red and blue lines represent TCNQ, triphenylene and the co-crystal triphenylene–TCNQ respectively. Inset displays CN markers at higher wavenumbers. | ||
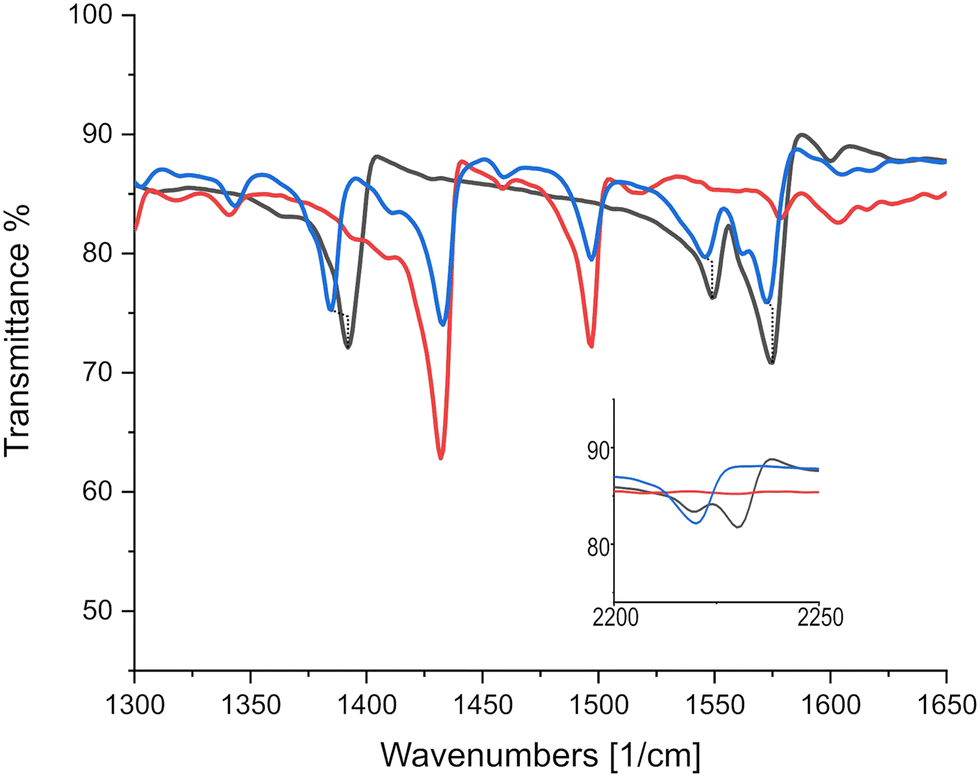 | ||
| Fig. 7 IR absorption spectra identifying significant charge sensitive mode displacements between TCNQF2 and triphenylene–TCNQF2. Black, red and blue lines represent TCNQF2, triphenylene and the triphenylene–TCNQF2 co-crystal respectively. Inset displays CN markers at higher wavenumbers. | ||
As can be seen in Fig. 8, CN markers representing ρ = 1e− correspond to a displacement from TCNQF4 at 2227 cm−1 to 2194 cm−1 in the TCNQF4 radical anion,33 with triphenylene–TCNQF4's mode displaced to 2226 cm−1 suggesting ρ = 0.03e−1. CC charge sensitive modes in TCNQF4 at 1396, 1550 and 1599 cm−1 are displaced in the complex to 1389, 1548 and 1594 cm−1, yielding ρ = 0.16, 0.04 and 0.08e−1 respectively, when compared to peak shifts in (K)–TCNQF4.
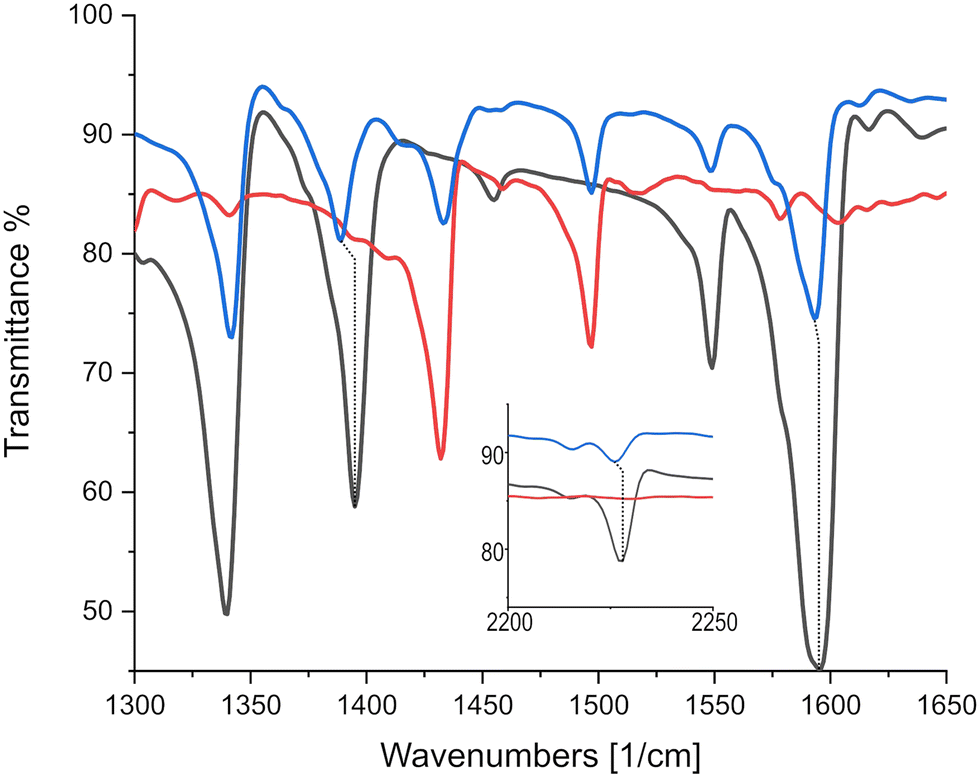 | ||
| Fig. 8 IR absorption spectra identifying significant charge sensitive mode displacements between TCNQF4 and triphneylene–TCNQF4. Black, red and blue lines represent TCNQF4, triphenylene and the triphenylene–TCNQF4 co-crystal respectively. Inset displays CN markers at higher wavenumbers. | ||
Direct optical band gaps for triphenylene–TCNQ, triphenylene–TCNQF2 and triphenylene–TCNQF4 were measured as 1.877, 1.761 and 1.704 eV indicating optoelectronic applications at respective wavelengths of 660.54 nm, 704.06 nm and 727.06 nm (Fig. 9). Whether the band gap remains the same at 1.761 eV for n = 2 in the recently reported 2,6-difluoro-7,7,8,8-tetracyanoquinodimethane molecule (where flourine is switched from a -para to an -ortho orientation)34 if co-crystallised with triphenylene, remains to be seen.
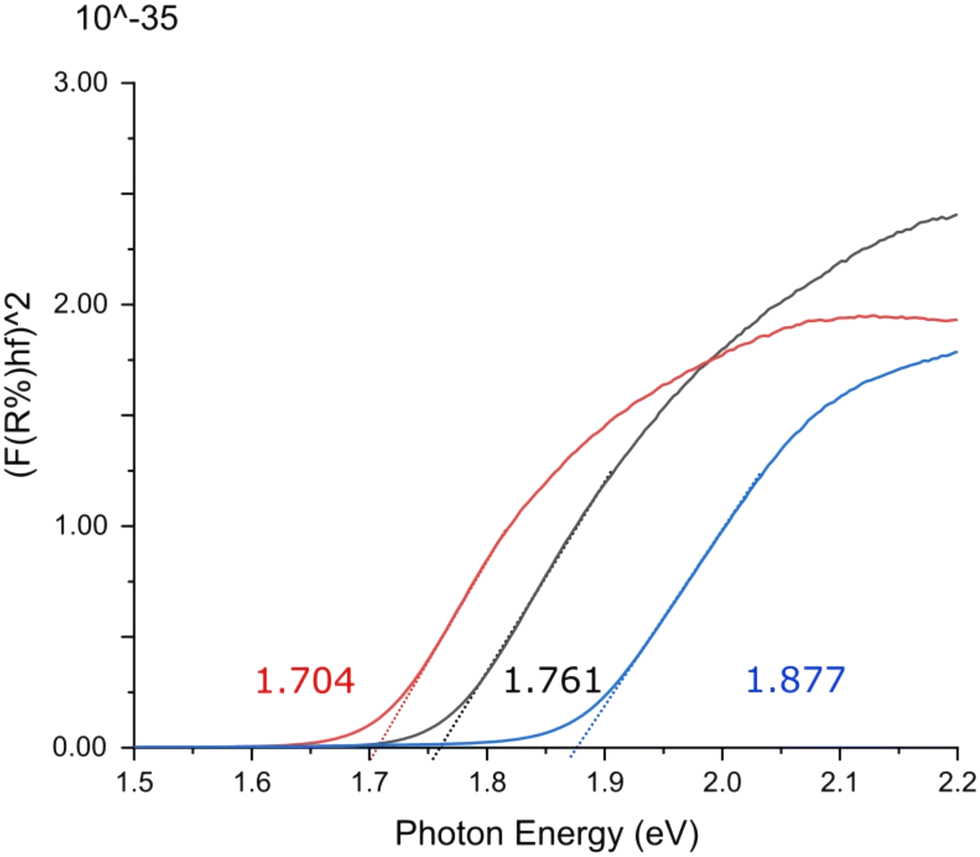 | ||
| Fig. 9 Ultraviolet-visible spectroscopic Tauc plot indicating direct optical band gaps for triphenylene–TCNQ (blue), triphenylene–TCNQF2 (black) and triphenylene–TCNQF4 (red). | ||
Comparing the apparent degree of CT, determined from vibrational mode shifts, with structural analysis of all three complexes, it can be seen that generally, a higher relative displacement reduces the degree of CT. It has been argued that an increase in the degree of CT is structurally dependent with a reduction of the face-to-face molecular displacement and a minimization of the shift between D–A molecules.35 However, in contrast to this, the triphenylene–TCNQF2 complex, has a relatively high degree of CT which coincides with the greatest average displacement. The triphenylene–TCNQF2 complex is unique in the three materials presented here in that molecules in the stack along the c-axis see a greater difference in molecular overlap on each side of a given molecule (Table 2).
| Triphenylene–TCNQ | Triphenylene–TCNQF2 | Triphenylene–TCNQF4 | |
|---|---|---|---|
| Crystallographic information | |||
| CCDC number | 2201747 | 2201748 | 2201749 |
| Formula | C30H16N4 | C30H14F2N4 | C30H12F4N4 |
| Formula weight | 432.47 | 468.46 | 504.44 |
| Crystal system | Triclinic | Primitive monoclinic | C-Centered monoclinic |
| Space group |
P |
P21c | C2c |
| a (Å) | 6.8(1) | 9.4(2) | 17.8(4) |
| b (Å) | 11.2(2) | 7.18(5) | 17.4(3) |
| c (Å) | 17.7(4) | 32.2(2) | 9.20(18) |
| α (°) | 78.4(5) | 90 | 90 |
| β (°) | 83.9(5) | 98.4(5) | 130.3(5) |
| γ (°) | 72.9(5) | 90 | 90 |
| Cell volume (Å3) | 1261(44) | 2156(50) | 2173(76) |
| Temperature (K) | 293 | 293 | 293 |
| Structure determination information | |||
| Tilt range (°) | 110 | 105 | 100 |
| Data resolution (Å) | 1.0 | 1.0 | 1.0 |
| Independent reflections (No.) | 1747 | 1641 | 1648 |
| R int | 21.2 | 23.8 | 22.1 |
| Structure refinement information | |||
| Reflections > 4σ (No.) | 712 | 955 | 477 |
| R14σ (%) | 41.04 | 30.66 | 35.33 |
| Goodness of fit | 2.925 | 1.156 | 2.461 |
| Optical information | |||
| Band gap (eV) | 1.877 | 1.761 | 1.704 |
4 Conclusions
We have reported three novel organic CT complexes based on the polyaromatic hydrocarbon triphenylene and TCNQFn (n = 0, 2, 4) with 1:1 stoichiometry. Despite crystallization on sub-micron scale, all three structures were solved using 3D electron diffraction, that already has been successfully used for the determination of nanocrystalline organic co-crystals.36,37 In particular, recently reported acetamidophenol–TCNQ co-cocrystals38,39 revealed the possibility of a new family of organic CT complexes, based on acetamidophenol molecules. Estimations of the degree of CT show that it is relatively low, but increases with the degree of fluorination of the acceptor molecule. Furthermore, the optical band gaps of these complexes show that they exhibit semiconducting behaviour. The progressive fluorination of the TCNQ scaffold prior to solution based co-crystallization therefore offers an efficient means of tuning the band gap in this co-crystal system.
Conflicts of interest
“There are no conflicts to declare”.Acknowledgements
I. A., F. P. and M. G. acknowledge the Regione Toscana for funding the purchase of the ASI TIMEPIX detector through the FELIX project (Por CREO FESR 2014–2020 action). S. R. H., J. P., and S. P. acknowledge the Engineering and Physical Sciences Research Council U.K. (Grants EP/G036780/1 and EP/L015544/1) for funding.Notes and references
- G. P. Stahly, Cryst. Growth Des., 2007, 7, 1007–1026 CrossRef CAS.
- A. D. Bond, CrystEngComm, 2007, 9, 833–834 RSC.
- A. S. Tayi, A. K. Shveyd, A. C. Sue, J. M. Szarko, B. S. Rolczynski, D. Cao, T. J. Kennedy, A. A. Sarjeant, C. L. Stern, W. F. Paxton, W. Wu, S. K. Dey, A. C. Fahrenbach, J. R. Guest, H. Mohseni, L. X. Chen, K. L. Wang, J. F. Stoddart and S. I. Stupp, Nature, 2012, 488, 485–489 CrossRef CAS PubMed.
- J. Singleton, J. Solid State Chem., 2002, 168, 675–689 CrossRef CAS.
- L. Zhu, Y. Yi, Y. Li, E. G. Kim, V. Coropceanu and J. L. Bredas, J. Am. Chem. Soc., 2012, 134, 2340–2347 CrossRef CAS.
- K. P. Goetz, D. Vermeulen, M. E. Payne, C. Kloc, L. E. McNeil and O. D. Jurchescu, J. Mater. Chem. C, 2014, 2, 3065–3076 RSC.
- J. Zhang, J. Jin, H. Xu, Q. Zhang and W. Huang, J. Mater. Chem. C, 2018, 6, 3485–3498 RSC.
- J. Zhang, W. Xu, P. Sheng, G. Zhao and D. Zhu, Acc. Chem. Res., 2017, 50, 1654–1662 CrossRef CAS PubMed.
- L. Sun, W. Zhu, F. Yang, B. Li, X. Ren, X. Zhang and W. Hu, Phys. Chem. Chem. Phys., 2018, 20, 6009–6023 RSC.
- Y. Jiang, G. Li, D. Zhu, Z. Su and M. R. Bryce, J. Mater. Chem. C, 2017, 5, 12189–12193 RSC.
- S. Horiuchi, K. Kobayashi, R. Kumai and S. Ishibashi, Chem. Lett., 2014, 43, 26–35 CrossRef CAS.
- K. Kobayashi, S. Horiuchi, R. Kumai, F. Kagawa, Y. Murakami and Y. Tokura, Phys. Rev. Lett., 2012, 108, 1–5 CrossRef PubMed.
- N. Kawai, R. Eguchi, H. Goto, K. Akaike, Y. Kaji, T. Kambe, A. Fujiwara and Y. Kubozono, J. Phys. Chem. C, 2012, 116, 7983–7988 CrossRef CAS.
- A. J. Buurma, O. D. Jurchescu, I. Shokaryev, J. Baas, A. Meetsma, G. A. De Wijs, R. A. De Groot and T. T. Palstra, J. Phys. Chem. C, 2007, 111, 3486–3489 CrossRef CAS.
- T. Salzillo, M. Masino, G. Kociok-Kohn, D. Di Nuzzo, E. Venuti, R. G. Della Valle, D. Vanossi, C. Fontanesi, A. Girlando, A. Brillante and E. Da Como, Cryst. Growth Des., 2016, 16, 3028–3036 CrossRef CAS.
- S. K. Park, S. Varghese, J. H. Kim, S. J. Yoon, O. K. Kwon, B. K. An, J. Gierschner and S. Y. Park, J. Am. Chem. Soc., 2013, 135, 4757–4764 CrossRef CAS PubMed.
- K. P. Goetz, A. Fonari, D. Vermeulen, P. Hu, H. Jiang, P. J. Diemer, J. W. Ward, M. E. Payne, C. S. Day, C. Kloc, V. Coropceanu, L. E. McNeil and O. D. Jurchescu, Nat. Commun., 2014, 5, 1–8 Search PubMed.
- N. Sikdar, K. Jayaramulu, V. Kiran, K. V. Rao, S. Sampath, S. J. George and T. K. Maji, Chem. – Eur. J., 2015, 21, 11701–11706 CrossRef CAS PubMed.
- M. A. Dobrowolski, G. Garbarino, M. Mezouar, A. Ciesielski and M. K. Cyrański, CrystEngComm, 2014, 16, 415–429 RSC.
- I. Shokaryev, A. J. Buurma, O. D. Jurchescu, M. A. Uijttewaal, G. A. De Wijs, T. T. Palstra and R. A. De Groot, J. Phys. Chem. A, 2008, 112, 2497–2502 CrossRef CAS PubMed.
- X. Chi, C. Besnard, V. K. Thorsmolle, V. Y. Butko, A. J. Taylor, T. Siegrist and A. P. Ramirez, Chem. Mater., 2004, 16, 5751–5755 CrossRef CAS.
- M. Gemmi, E. Mugnaioli, T. E. Gorelik, U. Kolb, L. Palatinus, P. Boullay, S. Hovmoller and J. P. Abrahams, ACS Cent. Sci., 2019, 5, 1315–1329 CrossRef CAS PubMed.
- A. Lanza, E. Margheritis, E. Mugnaioli, V. Cappello, G. Garau and M. Gemmi, IUCrJ, 2019, 6, 178–188 CrossRef CAS PubMed.
- J. P. Georgieva, D. Jansen, J. Sikharulidze, I. Jiang, L. Zandbergen and H. W. Abrahams, J. Instrum., 2011, 6, C01033 Search PubMed.
- M. Palatinus, L. Brazda, P. Jelinek, M. Hrda, J. Steciuk and G. Klementova, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2019, 75, 512–522 CrossRef PubMed.
- G. Burla, M. C. Caliandro, R. Carrozzini, B. Cascarano, G. L. Cuocci, C. Giacovazzo, C. Mallamo, M. Mazzone and A. Polidori, J. Appl. Crystallogr., 2015, 48, 306–309 CrossRef.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- C. Adamo and V. Barone, J. Chem. Phys., 1999, 110, 6158–6170 CrossRef CAS.
- S. Grimme, J. Comput. Chem., 2006, 27, 1787–1799 CrossRef CAS PubMed.
- G. Gilat and L. J. Raubenheimer, Phys. Rev., 1966, 144, 390–395 CrossRef CAS.
- P. M. Gill, B. G. Johnson and J. A. Pople, Chem. Phys. Lett., 1993, 209, 506–512 CrossRef CAS.
- T. J. Kistenmacher, T. J. Emge, A. N. Bloch and D. O. Cowan, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 1982, 38, 1193–1199 CrossRef.
- I. Salzmann, G. Heimel, M. Oehzelt, S. Winkler and N. Koch, Acc. Chem. Res., 2016, 49, 370–378 CrossRef CAS PubMed.
- G. Schweicher, G. Garbay, R. Jouclas, F. Vibert, F. Devaux and Y. H. Geerts, Adv. Mater., 2020, 32, 1905909 CrossRef CAS PubMed.
- M. A. Dobrowolski, G. Garbarino, M. Mezouar, A. Ciesielski and M. K. Cyrański, CrystEngComm, 2014, 16, 415–429 RSC.
- P. Brazda, L. Palatinus and M. Babor, Science, 2019, 364, 667–669 CrossRef CAS PubMed.
- I. Andrusenko, J. Potticary, S. R. Hall and M. Gemmi, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2020, 76, 1036–1044 CrossRef CAS PubMed.
- J. Hitchen, I. Andrusenko, C. L. Hall, E. Mugnaioli, J. Potticary, M. Gemmi and S. R. Hall, Cryst. Growth Des., 2022, 22, 1155–1163 CrossRef CAS.
- I. Andrusenko, J. Hitchen, E. Mugnaioli, J. Potticary, S. R. Hall and M. Gemmi, Symmetry, 2022, 14, 1–10 CrossRef.
Footnote |
| † CCDC 2201747–2201749. For crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2ce01127a |
| This journal is © The Royal Society of Chemistry 2023 |