
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Tracking nitrite's deviation from Stokes–Einstein predictions with pulsed field gradient 15N NMR spectroscopy†
Trent R.
Graham
 *a,
Yihui
Wei
b,
Eric D.
Walter
a,
Emily T.
Nienhuis
a,
Jaehun
Chun
a,
Gregory K.
Schenter
a,
Kevin M.
Rosso
a,
Carolyn I.
Pearce
ac and
Aurora E.
Clark
b
*a,
Yihui
Wei
b,
Eric D.
Walter
a,
Emily T.
Nienhuis
a,
Jaehun
Chun
a,
Gregory K.
Schenter
a,
Kevin M.
Rosso
a,
Carolyn I.
Pearce
ac and
Aurora E.
Clark
b
aPacific Northwest National Laboratory, Richland, Washington 99354, USA. E-mail: trent.graham@pnnl.gov
bDepartment of Chemistry, University of Utah, Salt Lake City, Utah 84112, USA
cDepartment of Crop and Soil Sciences, Washington State University, Pullman, Washington 99164, USA
First published on 17th November 2023
Abstract
Predicting the behavior of oxyanions in radioactive waste stored at the Department of Energy legacy nuclear sites requires the development of novel analytical methods. This work demonstrates 15N pulsed field gradient nuclear magnetic resonance spectroscopy to quantify the diffusivity of nitrite. Experimental results, supported by molecular dynamics simulations, indicate that the diffusivity of free hydrated nitrite exceeds that of free hydrated sodium despite the greater hydrodynamic radius of nitrite. Investigations are underway to understand how the compositional and dynamical heterogeneities of the ion networks at high concentrations affect rheological and transport properties.
Radioactive wastes stored in underground tanks at the Department of Energy legacy nuclear sites, such as the Hanford Site in Washington State, are comprised in part of solutions of sodium salts of nitrite (NaNO2) and nitrate oxyanions, as well as sodium salts of hydroxide (NaOH) and aluminate.1–4 Studying the structure, stability, and dynamics of these oxyanion solutions is of benefit to ongoing activities to transport the waste out of the tanks for safe processing and long-term storage.5 Determining the intermolecular interactions of oxyanions with other constituents within concentrated alkaline electrolytes has proved challenging due to the disparate sensitivities of experimental techniques to those interactions.6 These ambiguities challenge the inclusion or omission of terms defining ion-pair formation in solubility models, such as those used to predict the stability of oxyanion-bearing electrolyte solutions.7–9 The difficulty is accentuated when attempting to predict the solubility and stability of multicomponent electrolytes approaching the complexity of radioactive waste.3
Recent research has sought to address those ambiguities through the application of hydration models to correlate the activity of water with the solubility of NaNO2 in single- and multi-component aqueous solutions.10,11 The solubility of concentrated NaNO2 in aqueous solutions of NaOH has also been measured and the solutions were characterized to assess the extent of OH− mediated ion-pair formation.12 A subsequent study correlated the vibrational properties of NO2− to the rheological and thermodynamic properties of the solution.13 However, a critical knowledge gap in understanding the properties of NO2− in aqueous solutions is information about the diffusivity of NO2−.
This work quantifies the diffusivity of dissolved, 15N-enriched NaNO2 with 15N pulsed field gradient stimulated echo nuclear magnetic resonance (PFGSTE-NMR) spectroscopy. With the addition of 23Na and 1H PFGSTE-NMR experiments, the diffusivity of NO2−, sodium ions (Na+), and water (H2O) can all be described to compare the relative diffusion coefficients of these species. The diffusion coefficient of NO2− exceeded the diffusion coefficient of Na+ across a wide range of concentrations, despite the greater hydrodynamic radius and molecular weight of NO2−. To gain insight into the experimental observation, molecular dynamics (MD) simulations of NaNO2 solutions were performed. Analysis of the trajectories yielded the pair-wise radial distribution functions (RDF), enabling estimation of the hydrodynamic radii in the absence of ion pairing, as well as mean squared displacement-derived diffusion coefficients, and PageRank-derived14 ion–ion cluster compositions. Lastly, 15N PFGSTE-NMR spectroscopy was applied to an alkaline electrolyte solution of NaOH and NaNO2 at a composition relevant to radioactive waste processing.
PFGSTE-NMR spectroscopy implements pairs of spatially dependent magnetic field gradients to label the position of NMR-active nuclei in molecules and track their translational diffusion over millisecond timescales.15–18 While analysis of NMR-active nuclei such as 1H, 13C, and 31P (among others) is routine due to the nuclei's favorable NMR properties, many quadrupolar nuclei and spin ½ nuclei, such as 15N, are less frequently investigated due to their relatively low gyromagnetic ratio compared to 1H, poor natural abundance, or unfavorable spin–lattice and spin–spin relaxation properties.19–21 Building on others’ use of 15N PFGSTE-NMR to study isotopically enriched 15N2 diffusion in zeolites,22 this work overcomes these limitations by using aqueous solutions of isotopically enriched Na15NO2.
The 15N NMR spectrum of 98% 15N enriched Na15NO2 dissolved in H2O exhibits a single, ensemble resonance across the concentration range of 0.1 to 12 m Na15NO2. As shown in the ESI,† the 15N NMR chemical shift of NO2− increases linearly with a slope of 0.1 ppm m−1 between 0.1 and 12 m Na15NO2. As discussed previously,12 the increase in the 15N chemical shift of NO2− can be attributed to a combination of changes in the dielectric constant of the solution, along with perturbation of the local environment of the nitrogen nucleus on NO2− through changes in the hydration shell or interionic interactions underlying dynamic exchange equilibria at multiple timescales.
Fig. 1A shows the signal attenuation as a function of applied gradient strength in the 15N PFGSTE-NMR spectra of a solution of 1 m Na15NO2. For this series of spectra, the total experimental measurement time is approximately 1 day. To quantify the diffusion coefficient, the dependence of the progressive signal decoherence of the NMR resonance is related to the strength of the applied pulsed field gradients. As shown in Fig. 1B, by constructing a Stejskal–Tanner plot, the slope of the log-normal signal intensity is mathematically related via the Stejskal–Tanner equation (ESI†) to the diffusion coefficient of 15NO2.23–25 The data at all concentrations is well fit by a single linear function, facilitating the extraction of an averaged diffusion coefficient26 attributable to NO2−.
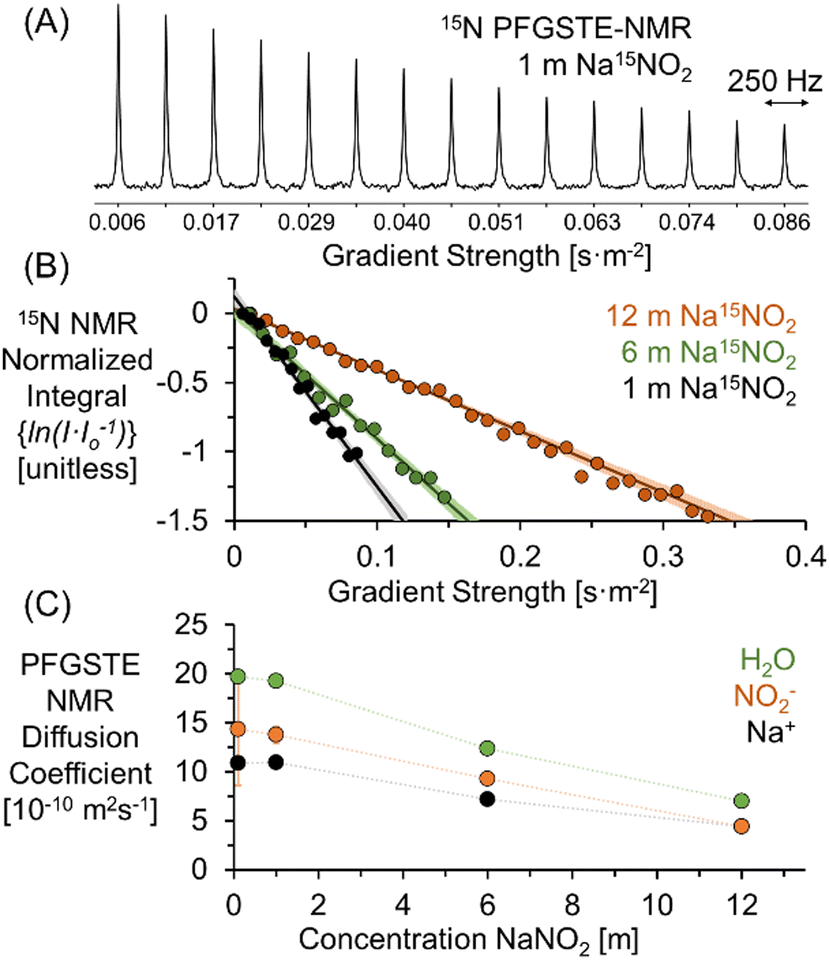 | ||
| Fig. 1 (A) 15N PFGSTE-NMR spectra of 1 m Na15NO2 in H2O at 20 °C. (B) Stejskal–Tanner plots of the 15N PFGSTE-NMR spectra of 1, 6, and 12 m Na15NO2 solutions. The 95% confidence interval of the linear fit is shown in lighter hues. Note that for visual clarity, the Stejskal–Tanner analysis of the 15N PFGSTE-NMR spectra of the 0.1 m Na15NO2 solution is shown in the ESI.† (C) PFGSTE-NMR results of 1H, 23Na, and 15N diffusion coefficients quantifying the diffusion coefficient of H2O, Na+, and NO2− for Na15NO2 solutions at 20 °C. | ||
As reported in Fig. 1C, the diffusivity of H2O and Na+ can be quantitatively and specifically measured with 1H and 23Na PFGSTE-NMR spectroscopy, respectively. In combination with 15N PFGSTE-NMR spectroscopy, this allows measurement of the diffusion coefficients of the ensembles of Na+, NO2− and H2O species. These diffusivity coefficients were measured from 0.1 to 12 m Na15NO2 in H2O at 20 °C. For all concentrations, the diffusion coefficients of 23Na and 1H in solutions of Na15NO2 were compared to those measured in solutions of natural abundance NaNO2. As shown in the ESI,† across the entire measured concentration range, the 1H and 23Na diffusion coefficients were in good agreement between the enriched and unenriched samples, indicating the substitution of 15N for 14N in NO2− had little impact on the diffusivity of H2O and Na+.
As shown in Fig. 1C, a comparison of the experimentally acquired diffusion coefficients for NO2− and Na+ indicates that the diffusion coefficient of NO2− is greater than Na+ at low concentrations and that the relative diffusivities approach unity as the concentration of NaNO2 approaches saturation (∼12 m). Note that from the MD simulation partial RDF of nitrite in 1 m NaNO2, the hydration number of nitrite is about 11, which is in good agreement with the upper range found in literature (between 627 and 11.9).28 The upper bound of the range far exceeds the number of water molecules (6) solvating Na+.29
MD simulations were performed to verify that the relative diffusive coefficients for Na+ and NO2− were in agreement with simulations performed with contemporary forcefield approximations capturing the short and long-range order in NaNO2 solutions.30 The diffusion coefficients of H2O, Na+, and NO2− determined from the calculation of the mean squared displacement of these constituents in MD trajectories of solutions of NaNO2 are shown in Fig. 2. As shown in the ESI,† the mean squared displacements reached linearity with respect to simulation time across several hundred picoseconds, facilitating the extraction of the diffusion coefficient via linear regression.
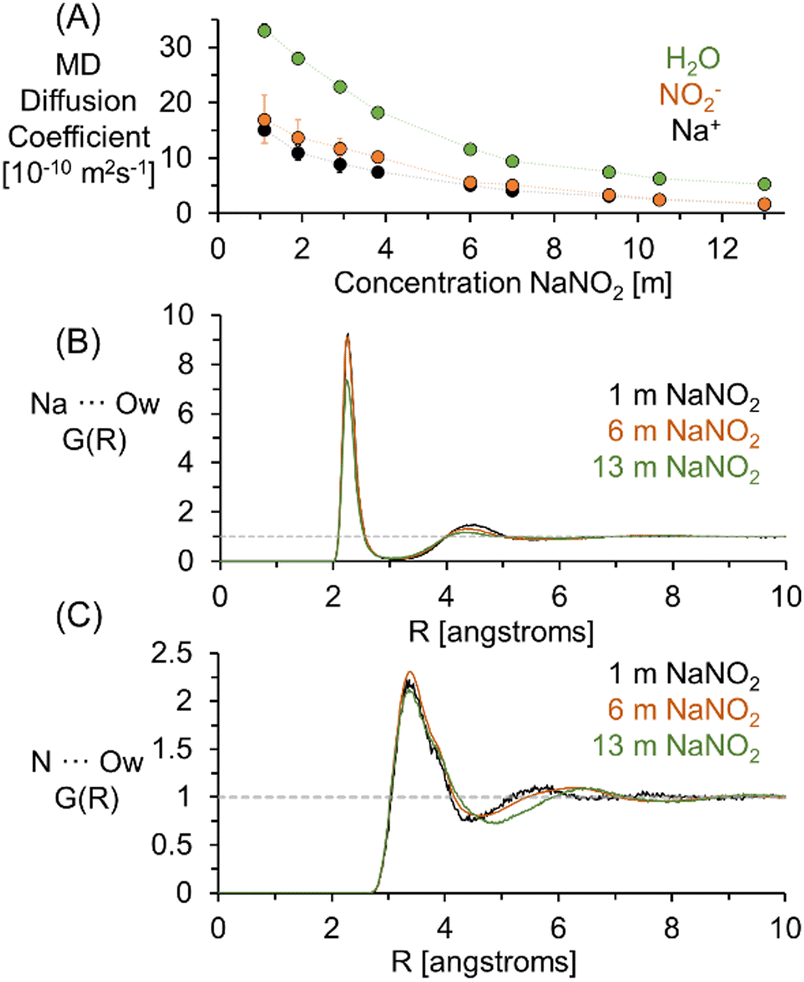 | ||
| Fig. 2 (A) Diffusion coefficients of H2O, Na+, and NO2− quantified by analysis of the MSD in MD simulations of NaNO2 solutions at a temperature of 300 K. (B) The nitrite nitrogen (N) to the oxygen of water (Ow) pairwise RDF. (C) the sodium (Na) to Ow pairwise RDF. | ||
Using both experimental measurements and MD simulations, the ratio of the Na+ to NO2− diffusion coefficients can be compared to an approximation based on the hydrodynamic radii of the free hydrated ions. The pairwise RDF between Na+ or nitrogen (N) with the solvating water oxygen is shown in Fig. 2. Based on the distance (R) to the second local minima, at 1 m NaNO2, the hydrodynamic radii are 4.1 and 2.3 angstroms for NO2− and Na+, respectively.
As shown in Fig. 3, incorporating the hydrodynamic radii determined from MD simulations into Stokes Einstein equation (ESI†) underpredicts the diffusivity of NO2− in solution, with both the experimental and the theoretical diffusion coefficients in good agreement at concentrations under 6 m to give a D(23Na)/D(15N) of 1.3, and converging to a ratio of 1 as the concentration approaches the solubility limit of NaNO2 in H2O. Additional NMR experiments were collected for 1 m NaNO2 between 20 and 80 °C. The 1H, 23Na, and 15N diffusion coefficients exhibited exponential temperature dependence, as shown in the ESI.†
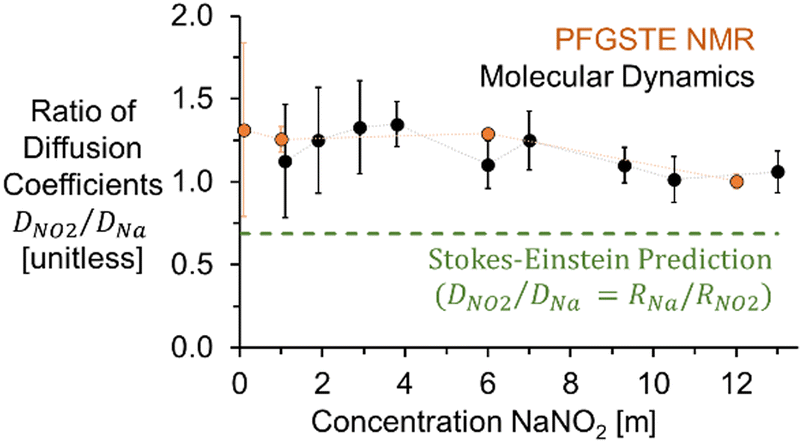 | ||
| Fig. 3 Experimental and MD simulation based ratios of diffusion coefficients (D) of NO2− and Na+ compared to Stokes–Einstein predictions using hydrodynamic radii of free ions. | ||
As shown in Fig. 4, PageRank analyses of the extents of ion–ion cluster formation of sodium nitrite from MD trajectories indicate that for 1 m NaNO2 in water, 95% of the nitrite is unassociated with sodium. The enhanced diffusion of the free nitrite relative to free sodium observed in 1 m NaNO2 is consistent with the results of others, who have suggested that greater than expected diffusion coefficients of NO2− are due to the coupling of rotational and translational diffusion modes in asymmetric polyatomic ions for both NO2− and nitrate, and there were similar observations when comparing the diffusivity of NO2− with nitrate, perchlorate, and chlorate, all of which exhibit diffusion coefficients at infinite dilution higher than predicted by Stokes–Einstein equation.31–35
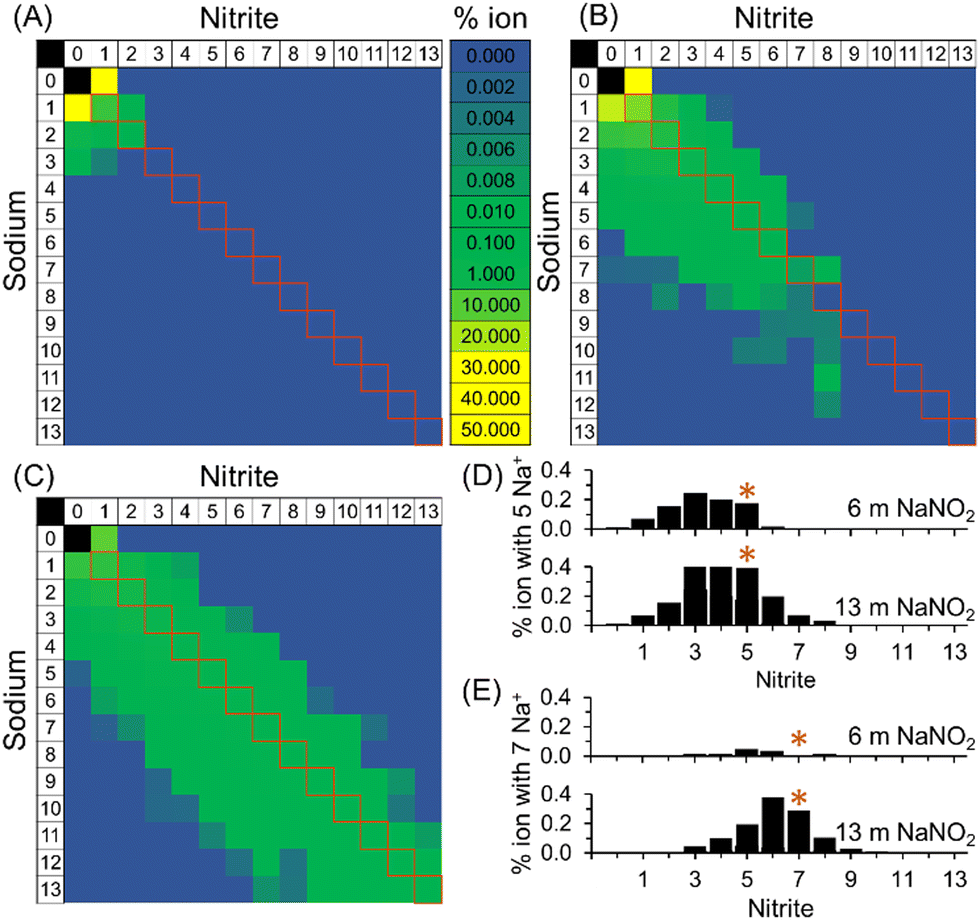 | ||
| Fig. 4 Histograms derived from PageRank analysis of MD simulations of ion–ion clusters in (A) 1, (B) 6 and (C) 13 m NaNO2. The squares highlighted in red denote the clusters where the number of anions equals the number of cations. Representative slices of the histogram are shown for (D) % ions with 5 Na+ and (E) % ions with 7 Na+. The asterisks denote where the number of anions equals cations. | ||
At high concentrations of 6 and 13 m NaNO2, PageRank analyses of the molecular dynamics trajectories indicate that the ions form ion–ion clusters. When an ion is in ion–ion clusters, its diffusion is likely restricted, with the average observed diffusion coefficient of Na+ and NO2− in these species approaching equivalence. As shown in Fig. 4, particularly at 6 and 13 m NaNO2, the relative proportions of free ions and ion–ion clusters were found to vary for cations and anions. This is highlighted in Fig. 4, where the distribution of the composition of ion–ion clusters observed with MD simulations is slightly skewed towards sodium rich ion–ion clusters. Further work is underway to develop techniques to probe the compositional and dynamical heterogeneities of these species.
Extension of 15N PFGSTE-NMR spectroscopy to concentrated NaOH archetypical of radioactive waste solutions at the Hanford Site in Washington State requires the application of pulsed field gradients of sufficient strength to measure diffusion in solutions with viscosities much greater than that of dissolved NaNO2 in H2O.36,37 Despite this potential complication, measurement of 15N diffusion in 2 M NaOH is possible, as shown in the ESI,† resulting in a Stejskal–Tanner plot well fit by a single linear function attributable to an ensemble. For this sample emulating the concentration of NaOH found in radioactive waste at the Hanford Site, the 15N diffusion coefficient of NO2− of 10.4 ± 0.5 × 10−10 m2 s−1 was also found to be greater than the 23Na diffusion coefficient of 7.4 ± 0.3 m2 s−1, coinciding with a ratio of 1.4 ± 0.1 for DNO2/DNa The similarity between the ratio of the diffusivity coefficients in single component NaNO2 solutions and multicomponent NaOH/NaNO2 solutions may indicate that, in alkaline solutions, the composition of the ion networks is weighted towards OH− and Na+. The oxyanions predominantly exist as free ions.
In conclusion, implementing PFGSTE-NMR spectroscopy to the 15N nucleus enables direct and isotopically specific measurement of the diffusion coefficient of 15N. These measurements are possible across a wide range of concentrations, with a feasible lower concentration limit established as 0.1 m of 15N, below which the experimental time for a single measurement exceeds two days. Quantifying the diffusion coefficient of NO2− enables the determination of the relative diffusion coefficient of NO2− to Na+ in solution. In solutions of 1 M NaNO2 and 2 M NaOH in H2O of relevance to nuclear waste stored at the Hanford Site, the relative diffusion coefficient of NO2− is greater than that of Na+, in contrast to predictions based on the hydrodynamic radius of NO2− and Na+. Comparative molecular dynamics simulations of NaNO2 in H2O also led to the extraction of a greater diffusion coefficient of NO2−versus Na+. These results indicate that the coupled translational and rotational motion potentially enhance diffusion relative to Na+. Further investigations into the structure and dynamics of Na+ and NO2− are underway to understand how the compositional and dynamical heterogeneities of the ion networks affect the rheological properties across multiple length and time scales. Such studies could integrate PFGSTE-NMR spectroscopy alongside other NMR techniques, such as NMR relaxometry and observations of heteronuclear Overhauser effects between amenable nuclei.
This research was supported by IDREAM (Interfacial Dynamics in Radioactive Environments and Materials), an Energy Frontier Research Center funded by the U.S. Department of Energy (DOE), Office of Science, Basic Energy Science (BES) under FWP 68932. Nuclear magnetic resonance spectroscopy was performed using facilities at the Environmental Molecular Science Laboratory (EMSL, grid.436923.9), a DOE Office of Science User Facility sponsored by the Office of Biological and Environmental Research at Pacific Northwest National Laboratory (PNNL). PNNL is a multiprogram national laboratory operated for DOE by Battelle Memorial Institute operating under Contract No. DE AC05-76RL0-1830.
Conflicts of interest
There are no conflicts to declare.Notes and references
- A. R. Felmy, J. R. Rustad, M. J. Mason and R. D. L. Bretonne, A chemical model for the major electrolyte components of the Hanford waste tanks. The binary electrolytes in the system: Na-NO3-NO2-SO4-CO3-F-PO4-OH-AI(OH)4-H2O, United States, 1994.
- R. L. Russell, H. D. Smith, D. E. Rinehart and R. A. Peterson, Development and Characterization of Gibbsite Component Simulant, United States, 2009.
- M. Dembowski, M. M. Snyder, C. H. Delegard, J. G. Reynolds, T. R. Graham, H. W. Wang, I. I. Leavy, S. R. Baum, O. Qafoku, M. S. Fountain, K. M. Rosso, S. B. Clark and C. I. Pearce, Phys. Chem. Chem. Phys., 2020, 22, 4368–4378 RSC.
- D. L. Herting, Effect of Phosphate, Fluoride, and Nitrate on Gibbsite Dissolution Rate and Solubility, United States, 2014.
- J. G. Reynolds, M. D. Britton and J. D. Belsher, J. Chem. Eng. Data, 2021, 66, 2931–2941 CrossRef CAS.
- G. Hefter, Pure Appl. Chem., 2006, 78, 1571–1586 CrossRef CAS.
- J. G. Reynolds, R. Carter and A. R. Felmy, Ind. Eng. Chem. Res., 2015, 54, 3062–3070 CrossRef CAS.
- D. A. Reynolds and D. L. Herting, Solubilities of sodium nitrate, sodium nitrite, and sodium aluminate in simulated nuclear waste, United States, 1984.
- J. G. Reynolds, ACS Omega, 2018, 3, 15149–15157 CrossRef CAS PubMed.
- J. G. Reynolds, T. R. Graham and C. I. Pearce, J. Mol. Liq., 2022, 347, 119441 CrossRef.
- J. G. Reynolds, AIChE J., 2022, 68, e17487 CrossRef CAS.
- T. R. Graham, M. Dembowski, H. W. Wang, S. T. Mergelsberg, E. T. Nienhuis, J. G. Reynolds, C. H. Delegard, Y. H. Wei, M. Snyder, I. I. Leavy, S. R. Baum, M. S. Fountain, S. B. Clark, K. M. Rosso and C. I. Pearce, Phys. Chem. Chem. Phys., 2021, 23, 112–122 RSC.
- J. G. Reynolds, T. R. Graham and C. I. Pearce, J. Mol. Liq., 2022, 360, 119441 CrossRef CAS.
- M. Hudelson, B. L. Mooney and A. E. Clark, J. Math. Chem., 2012, 50, 2342–2350 CrossRef CAS.
- C. S. Johnson, Prog. Nucl. Magn. Reson. Spectrosc., 1999, 34, 203–256 CrossRef CAS.
- R. Evans, G. Dal Poggetto, M. Nilsson and G. A. Morris, Anal. Chem., 2018, 90, 3987–3994 CrossRef CAS PubMed.
- A. Macchioni, G. Ciancaleoni, C. Zuccaccia and D. Zuccaccia, Chem. Soc. Rev., 2008, 37, 479–489 RSC.
- R. Evans, Prog. Nucl. Magn. Reson. Spectrosc., 2020, 117, 33–69 CrossRef CAS PubMed.
- K. S. Han, J. D. Bazak, Y. Chen, T. R. Graham, N. M. Washton, J. Z. Hu, V. Murugesan and K. T. Mueller, Chem. Mater., 2021, 33, 8562–8590 CrossRef CAS.
- T. R. Graham, K. S. Han, M. Dembowski, A. J. Krzysko, X. Zhang, J. Z. Hu, S. B. Clark, A. E. Clark, G. K. Schenter, C. I. Pearce and K. M. Rosso, J. Phys. Chem. B, 2018, 122, 10907–10912 CrossRef CAS PubMed.
- T. R. Graham, J. Chun, G. K. Schenter, X. Zhang, S. B. Clark, C. I. Pearce and K. M. Rosso, Magn. Reson. Chem., 2022, 60, 226–238 CrossRef CAS PubMed.
- P. L. McDaniel, C. G. Coe, J. Karger and J. D. Moyer, J. Phys. Chem., 1996, 100, 16263–16267 CrossRef CAS.
- E. O. Stejskal and J. E. Tanner, J. Chem. Phys., 1965, 42, 288–292 CrossRef CAS.
- K. I. Momot and P. W. Kuchel, Concepts Magn. Reson., Part A, 2003, 19a, 51–64 CrossRef CAS.
- D. Sinnaeve, Concepts Magn. Reson., Part A, 2012, 40, 39–65 CrossRef.
- P. S. Pregosin, Magn. Reson. Chem., 2017, 55, 405–413 CrossRef CAS PubMed.
- S. Vchirawongkwin, C. Kritayakornupong, A. Tongraar and V. Vchirawongkwin, Dalton Trans., 2014, 43, 12164–12174 RSC.
- J. W. Smith, R. K. Lam, O. Shih, A. M. Rizzuto, D. Prendergast and R. J. Saykally, J. Chem. Phys., 2015, 143, 084503 CrossRef PubMed.
- M. Carrillo-Tripp, H. Saint-Martin and I. Ortega-Blake, J. Chem. Phys., 2003, 118, 7062–7073 CrossRef CAS.
- Y. Wei, E. T. Nienhuis, S. T. Mergelsberg, T. R. Graham, Q. Guo, G. K. Schenter, C. I. Pearce and A. E. Clark, Chem. Commun., 2023, 59, 10400–10403 RSC.
- P. Banerjee, S. Yashonath and B. Bagchi, J. Chem. Phys., 2017, 146, 164502 CrossRef PubMed.
- P. Banerjee, S. Yashonath and B. Bagchi, J. Chem. Phys., 2016, 145, 234502 CrossRef PubMed.
- P. Banerjee and B. Bagchi, J. Chem. Phys., 2018, 148, 224504 CrossRef PubMed.
- P. Banerjee and B. Bagchi, J. Chem. Phys., 2019, 150, 190901 CrossRef PubMed.
- P. Banerjee and B. Bagchi, J. Chem. Phys., 2017, 147, 164502 CrossRef PubMed.
- P. M. Sipos, G. Hefter and P. M. May, J. Chem. Eng. Data, 2000, 45, 613–617 CrossRef CAS.
- P. Sipos, A. Stanley, S. Bevis, G. Hefter and P. M. May, J. Chem. Eng. Data, 2001, 46, 657–661 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3cc04168a |
| This journal is © The Royal Society of Chemistry 2023 |