
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUnexpected rapid P-stereomutation of phosphine oxides catalysed by chlorophosphonium salts†
Sulaiman
Al-Sulaimi
 a,
Kamalraj
Rajendran
b,
Kirill
Nikitin
*b and
Declan G.
Gilheany
*b
a,
Kamalraj
Rajendran
b,
Kirill
Nikitin
*b and
Declan G.
Gilheany
*b
aDepartment of Biological Science & Chemistry, College of Arts and Sciences, University of Nizwa, Box 33, PC 616, Nizwa, Sultanate of Oman
bSchool of Chemistry, University College Dublin, Dublin 4, Belfield, Ireland. E-mail: kirill.nikitin@ucd.ie; declan.gilheany@ucd.ie
First published on 13th September 2023
Abstract
P-Stereomutation of phosphine oxides is extremely slow. We show that it is catalysed by chlorophosphonium salts (CPS) which can directly be formed in the system in situ. The racemization of phosphine oxides at ambient conditions catalysed by 1 mol% of CPS takes 1–2 hours and can be arrested by additon of a primary alcohol. The process probably proceeds via the development of oxodiphosphonium P–O–P species.
Enantioselectivity is considered as one of the key goals in synthetic chemistry and often requires careful adjustment of conditions. Stereoselective reactions and novel approaches to chiral materials attract immense attention from the organic,1 inorganic2 and supramolecular3 communities. Rapid stereomutation of an intermediate can be critical to many successful stereoselective strategies4 but must be avoided for the product as it leads to loss of chiral information. Although understanding of stereomutation helps to maintain high enantiopurity of the product (the “know thy enemy” principle) it is fair to say that stereomutation has rarely been studied systematically.
In most cases, tri- and tetracoordinate P-stereogenic compounds such as phosphines, their oxides (PO, 1) and quaternary phosphonium salts (QPS) are configurationally very stable due to high activation barriers of typically ca. 30–40 kcal mol−1,5 making them valuable scaffolds for the design of stereoselective systems.6 Little is known however about potentially detrimental to these methodologies catalytic stereomutation of the P-center leading to racemisation. A few very recent reports indicate that such reactions, for example acid-catalysed and SET-facilitated processes, do occur under certain conditions.7 Relatively recently, we have shown that halophosphonium salts [R3P-X]+X− (XPS, X = Cl, Br) which are the key dynamic resolution intermediates in P-asymmetric Appel reaction8, undergo rapid epimerization characterized by low activation barriers ca. 10–12 kcal mol−1.9 Having proposed a possible8b mechanism for adverse parasitic effect of this epimerization in the asymmetric Appel process, its role remains to be fully understood. With that in mind, we undertook the present study and we show that dynamically racemic XPS species are very efficient catalysts of P-stereomutation of 1.
Compounds 1 are renowned for their configurational stability, and are very different from the dynamically interconverting species 2 as shown in Scheme 1. However, it was not at all obvious that, as shown here, these halogenated species e.g. mixed arylalkylchlorophosphonium salts (CPS) are not innocent bystanders and can catalyze stereomutation of the otherwise configurationally stable PO 1 under mild ambient conditions.
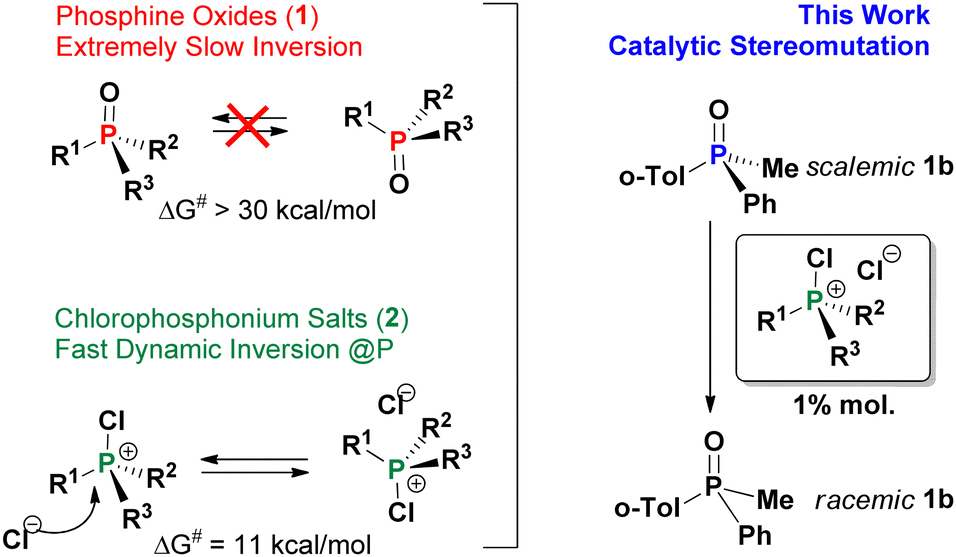 | ||
| Scheme 1 Discovery of catalytic stereomutation of configurationally stable phosphine oxides (1) in the presence of dynamically racemic chlorophosphonium salts CPS (2). | ||
In this work, we designed a set of experiments around the historically well documented structures, PAMPO (1a), PMTPO (1b) and PETPO (1c) and their respective CPS 2a–c as displayed in Scheme 2. The PO 1a–c can be prepared in high ee of either sense8b and are characterized by excellent configurational stability in the absence of catalytic agents such that no discernable drift of ee can be observed in toluene at elevated temperature.
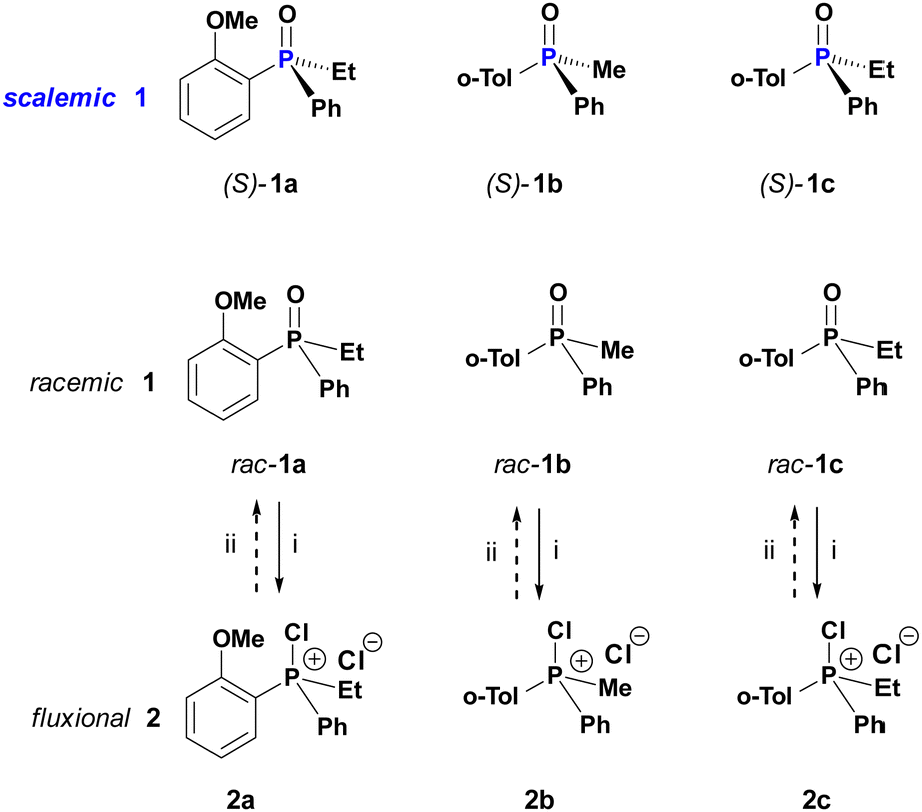 | ||
| Scheme 2 Relationships between configurationally stable phosphine oxides 1 and dynamically racemic chlorophosphonium salts CPS (2). Reagents: (i) oxalyl chloride (3); (ii) excess ethanol. | ||
In an initial landmark catalytic racemisation experiment, a sample of scalemic (S)-1b (93% ee) was treated with CPS 2a generated from 1a in DCM. Just after 1 h at room temperature, complete racemisation of the entire amount of PO 1b was observed.
A possible interaction of a PO e.g. P-chiral 1 with a generic CPS 4 (Scheme 3) can result in oxodiphosphonium (POP) species 5 structurally analogous to the well-documented [Ph3P–O–PPh3]2+ dication.10 Internal nucleophilic collapse of 5 can generate the dynamically racemic species 2 in the system. If CPS 2 itself is used in place of 4 (self-racemisation), this POP-equilibrium is energy-degenerate (Keq = 1). Assuming that the POP-equilibrium is established quickly, addition of just 1 mol% of CPS 2, for example, to enantiomerically pure (S)-1 will result in 0.5 mol% of (S)-1 being converted into the fluxional 2 which, in dynamic equilibrium with PO 1, drives its complete catalytic racemisation.
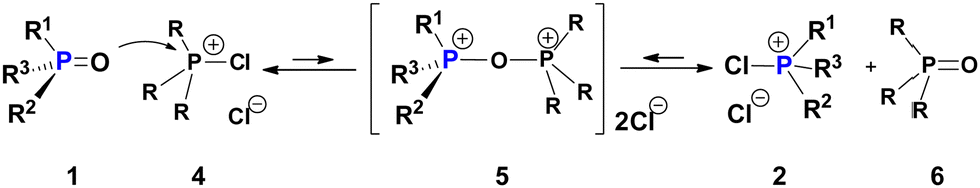 | ||
| Scheme 3 Plausible “POP-equilibrium”: chiral PO 1 (three different R-groups attached) and generic CPS 4 generate the dynamically racemic CPS 2via a minor P–O–P species 5. This explains racemisation of P-chiral PO 1. | ||
However, since our hypothesis raised a number of questions, a dedicated study of CPS-catalysed racemisation was undertaken. We focused on stereomutation of PO 1b in the presence of CPS 2b (self-racemization) or a CPS 4 derived from another structure (cross-racemization) in which case the POP-equilibrium in Scheme 3 is not anymore degenerate. The impact of the loading, temperature, solvent and CPS structure, on the racemisation of PO 1b was studied. Mechanistically, the interconversion of the dynamically racemising CPS and the corresponding phosphine oxide was established via dynamic 2D EXSY NMR experiments. The results are discussed based on the proposed mechanism involving the formation of the POP species 5.
In a first set of experiments, the amount of dynamically racemising CPS 2b was controlled by the amount of oxalyl chloride 3 added to the solution of scalemic (S)-1b (93% ee) in dry DCM under nitrogen atmosphere. The reaction mixture was stirred at ambient temperature and, at time intervals, aliquots were taken. The racemisation was arrested by adding ethanol which instantly converts catalytically active species 2b into rac-1b and analyzed by HPLC. Fig. 1(a) and (b) correspondingly show the ee values of PO 1b and the log plot for this first-order racemisation in the presence of 0.5 mol % of CPS 2b generated in situ. It can be seen that the half-life time for this pseudo-first order process is ∼23 min which corresponds to a rate constant k = 4.9 × 10−4 s−1. A two-fold increase of rate, k = 1.0 × 10−3 s−1 in the presence of double amount of 2b, 1 mol%, indicates a first-order process in the catalyst (Tables S1–S3, ESI†). The rate constant changes rapidly with the temperature, it is ca. 7 times lower at −19.3 °C (Table S6, ESI†). The rate of racemisation of PO 1b is drastically lower about 150 times in the less polar toluene compared to DCM (Table S7, ESI†). Although it is recognized that CPS structures occur in their pentacoordinate neutral form in toluene,11 the presence of excess 1b acts possibly as a highly polar additive such that dichloride 2b may exist partially in its tetrahedral ionic form, amenable to the POP equilibrium with species 5 (Scheme 3).
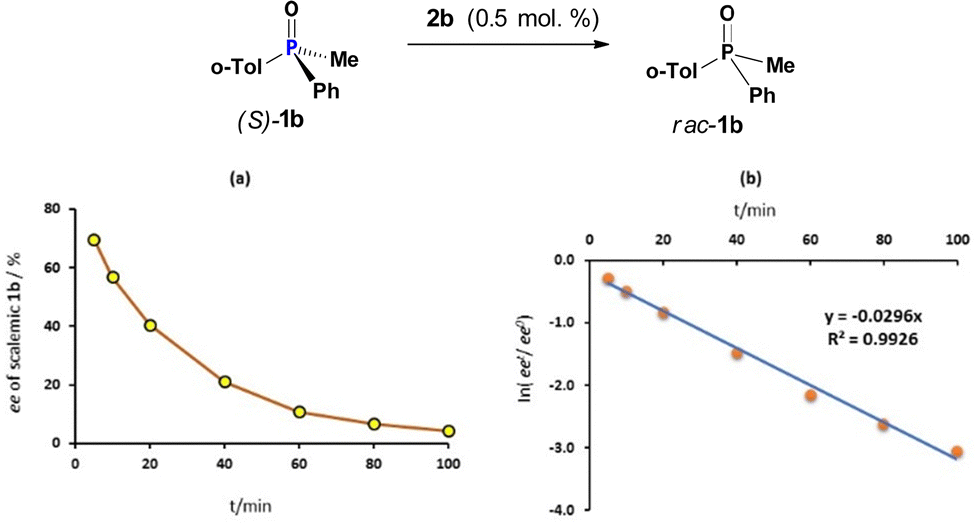 | ||
| Fig. 1 Catalytic racemisation of (S)-1b: (a) kinetic plot (0.5 mol% of 2b); (b) the corresponding log plot. | ||
Importantly, not only CPS 2 but also other related species can catalyse the racemisation of PO 1b. For example, we have earlier shown12 that P-asymmetric Appel reaction can conveniently be applied to the corresponding phosphines by using a chlorinating agent, and thus racemisation of PO 1b under similar conditions was studied. First, chlorinated 7 (section 2.6, ESI†) carrying a pentachloroacetonate enol-type anion was generated from the parent phosphine. Racemisation of PO 1b in the presence of 5 mol% of 7 (Scheme 4a) was ca. 8 times slower as compared to CPS 2b (Table S8, ESI†). The reason is that the polychlorinated counterion has a lower nucleophilicity than the chloride ion and a larger size that slows down the dynamics of POP-equilibrium.
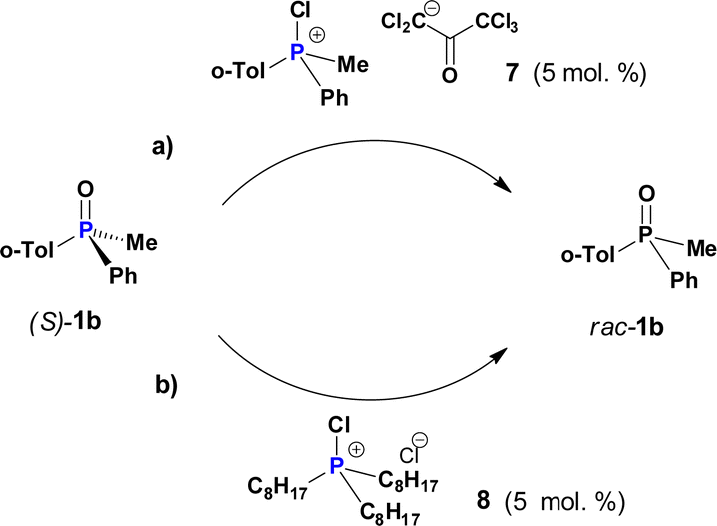 | ||
| Scheme 4 Slow racemisation of P-chiral 1b induced by: (a) mixed-group species 7; (b) tri-n-alkyl CPS 8. | ||
Having demonstrated that self-racemisation of PO 1b is efficiently catalyzed by its own CPS 2b we questioned the possibility of a cross-racemisation by using another CPS (e.g.2a, vide supra). As an example, we chose CPS 8 (Scheme 4b) comprising only n-octyl substituents. When 8 (5 mol%) was introduced, it was found that catalytic racemisation of PO 1b occurs very slowly k = 1.2 × 10−6 s−1 which is about 3 × 103 times lower than for self-racemisation using CPS 2b under equivalent conditions.
A possible explanation of this effect is linked to the relative P–O and P–Cl bond strength in different types of phosphine oxides and CPS and the position of the POP-equilibrium. Recently, our efforts to study the relative strength of P–Cl bonds in CPS demonstrated that the strength of this bond increases by 3–4 kcal mol−1 per addition of an alkyl group attached to phosphorus atom.13 This effect, very significantly reduces the amount of mixed aryl-alkyl CPS species 2 available via the POP-equilibrium with a generic CPS 4 shown in Scheme 3. We estimate that in the case of CPS 8 used as catalyst the concentration of fluxional chiral CPS 2b present is reduced by a factor of 104 which can explain much slower cross-racemisation in this system.
Our further experiments were designed to detect the development of POP species under racemisation conditions in a system containing higher concentrations of rac-1b and the fluxional 2b (∼0.1 M each). The 31P NMR spectrum showed a signal, 0.4% intensity, at 80.7 ppm indicating possibly the new POP species 5b. This evidence was quite significant as it is consistent with the proposed catalytic cycle for rapid stereomutation of 1b in the presence of dynamically racemising CPS 2bvia POP-species 5b as depicted in Scheme 5. We postulate that either enantiomer of the dynamically racemising 2b can be attacked by (S)-1b with close to equal probability. The resulting diastereomeric POP species 5b undergo rapid ion migration followed by reversal of the nucleophilic attack. The outcome is formation of near-equal amounts of (S)-1b and (R)-1b and hence racemisation of scalemic (S)-1b.
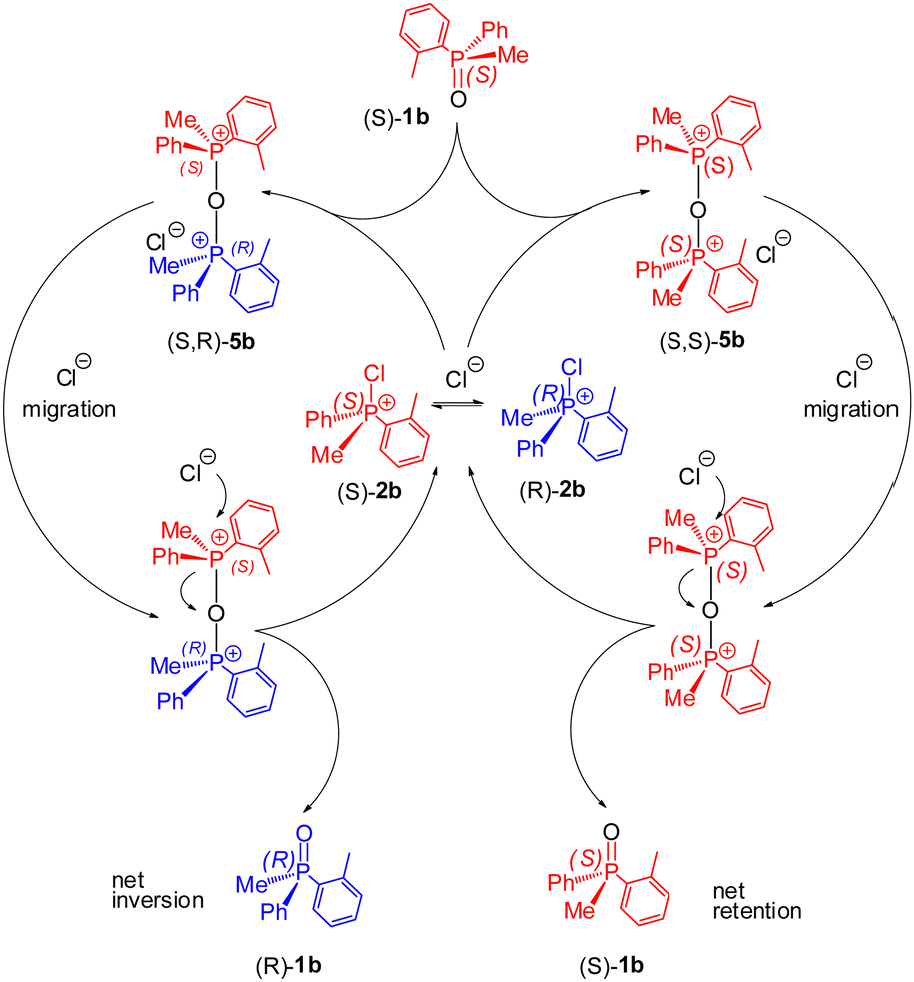 | ||
| Scheme 5 Catalytic stereomutation pathways: nucleophilic attack of P-chiral (S)-1b leads to diastereomeric POP species (S,S)-5b and (S,R)-5b which upon anion migration give both enantiomers of PO 1b. | ||
To observe the dynamics of proposed POP species 5b a 1H–1H 2D EXSY NMR experiment14 was carried out at 25 °C at 600 MHz with a mixing time of 0.100 s but no cross peak of 5b could be identified due to its relatively low concentration in the system.
Significantly, the 1H–1H 2D EXSY experiment in the system containing equimolar amounts (50 mol% each) of rac-1b and dynamically racemic 2b clearly showed exchange cross-peaks which are an unequivocal evidence of interconversion 2b↔1b (Fig. 2). An analysis of the cross peak corresponding to the methyl group (CH3–P) gave a rate constant kex = 8.0 10−2 s−1 at 25 °C. This result is in very good agreement with the earlier observed racemisation rate constant kr = 1.0 10−3 s−1 (20.5 °C, quoted above) in the system containing only of 1 mol% of 2b. Clearly, the same set of dynamic stereomutation processes, described in Scheme 5, is responsible for slow, on the NMR-timescale, interconversion between 2b and 1b. This explains the phenomenon of racemisation of 1b in the presence of catalytic amounts of CPS in this study (2a, 2b, 7 and 8).
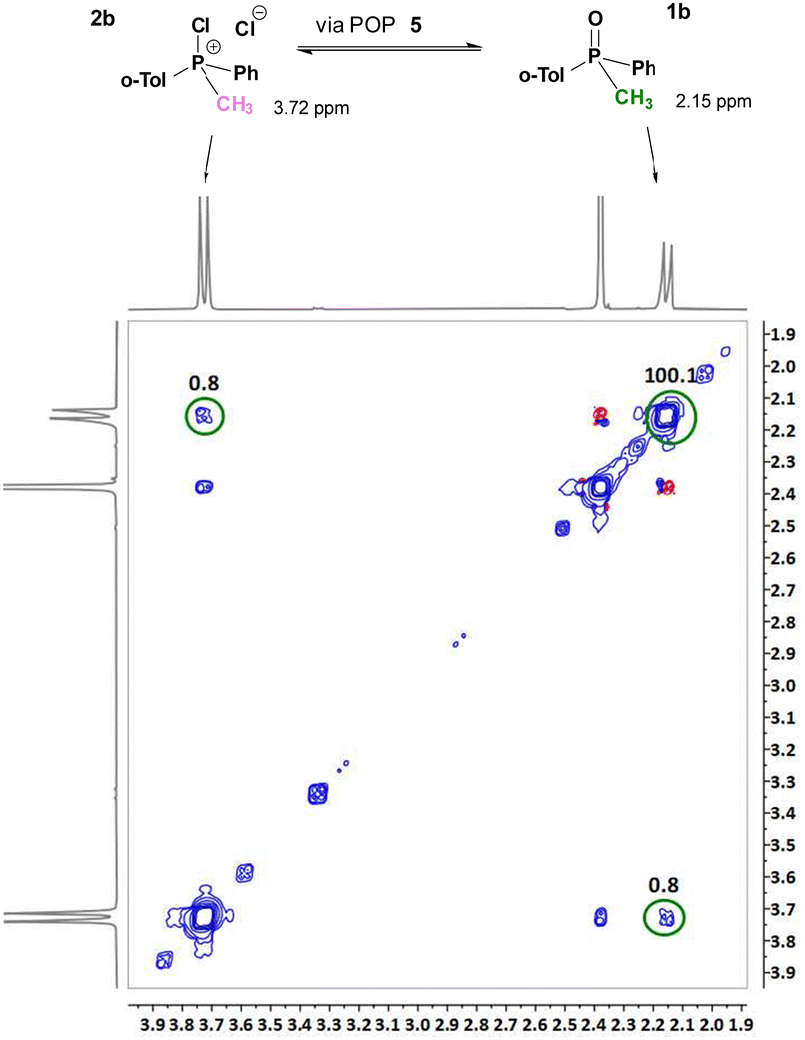 | ||
| Fig. 2 1H–1H 2D EXSY observation of dynamic interconversion between 1b and 2b: cross peak of the CH3 group is observed at δ 2.15/3.72 ppm (Tm = 0.1 s). | ||
In short, racemization of chiral PO 1 is a very slow process characterized by high activation barriers. Rapid stereomutation was directly observed for the first time due to an equilibrium involving dynamic fluxional CPS species 2. The racemisation can be conveniently stopped at any moment by adding primary alcohols. The key factors affecting racemisation rate are the structure and the loading of catalyst 2, reaction temperature, the counter ion and the solvent. It was found that achiral trialkyl-CPS can also catalyse racemisation of 1 but at a lower rate. A mechanism evoked here implies a catalytic cycle via development of the oxodiphosphonium POP species 5. This mechanistic proposal involves a Cl-anion migration followed by nucleophilic inversion of the P-centre. The interconversion of a CPS and a phosphine oxide has been directly detected by using the 2D EXSY NMR technique. These findings are consistent with the potential negative effect of CPS in our asymmetric Apple process and its involvement is critical for the asymmetric reverse Appel reaction8b too: a slow reaction of CPS with chiral auxiliary alcohol provides a window of opportunity for scalemic phosphine oxide to interact with the CPS which unavoidably leads to decreased ee of the resulting P-chiral product. From a broader perspective, this study has shown that the presence of minor by-products or intermediates can adversely affect the outcome of a stereoselective process.
The authors sincerely thank Science Foundation Ireland (SFI) for funding this project under grant 09/IN.1/B2627. The authors are also grateful to the UCD Centre for Synthesis and Chemical Biology (CSCB) and the UCD School of Chemistry for access to their extensive analysis facilities. S. A.-S. also sincerely thanks the University of Nizwa, Oman and the Erasmus Mundus Gulf Countries Programme for scholarship funding.
Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) K. B. Sharpless, Angew. Chem., Int. Ed., 2002, 41, 2024–2032 CrossRef CAS; (b) R. Noyori, Adv. Synth. Catal., 2003, 345, 15–32 CrossRef CAS; (c) Comprehensive Chirality, ed. E. M. Carreira and H. Yamamoto, Elsevier Ltd, 2012 Search PubMed; (d) R. A. Sheldon and D. Brady, ChemSusChem, 2019, 12, 2859 CrossRef CAS PubMed; (e) G. Lonardi, R. Parolin, G. Licini and M. Orlandi, Angew. Chem., Int. Ed., 2023, 62, e202216649 CrossRef CAS PubMed.
- (a) A. Hafner, R. O. Duthaler, R. Marti, G. Rihs, P. Rothe-Streit and F. Schwarzenbach, J. Am. Chem. Soc., 1992, 114, 2321–2336 CrossRef CAS; (b) A. von Zelewsky and O. Mamula, J. Chem. Soc., Dalton Trans., 2000, 219–231 RSC; (c) W. Ma, L. Xu, A. F. de Moura, X. Wu, H. Kuang, C. Xu and N. A. Kotov, Chem. Rev., 2017, 117, 8041–8093 CrossRef CAS PubMed.
- (a) J. Crassous, Chem. Soc. Rev., 2009, 38, 830–845 RSC; (b) A. M. Castilla, W. J. Ramsay and J. R. Nitschke, Acc. Chem. Res., 2014, 47, 2063–2073 CrossRef CAS PubMed; (c) L. Lopez-Gandul, C. Naranjo, C. Sanchez, R. Rodrıguez, R. Gomez, J. Crassous and L. Sanchez, Chem. Sci., 2022, 13, 11577 RSC.
- (a) K. Dziuba, M. Lubańska and K. M. Pietrusiewicz, Synthesis, 2020, 909–916 CAS; (b) A. J. Blacker, M. J. Stirling and M. I. Page, Org. Process Res. Dev., 2007, 11, 642–648 CrossRef CAS; (c) S. J. Meek, S. J. Malcolmson, B. Li, R. R. Schrock and A. H. Hoveyda, J. Am. Chem. Soc., 2009, 131(45), 16407–16409 CrossRef CAS PubMed.
- (a) R. D. Baechler and K. Mislow, J. Am. Chem. Soc., 1970, 92, 3090 CrossRef CAS; (b) R. D. Baechler and K. Mislow, J. Am. Chem. Soc., 1970, 92, 4758 CrossRef CAS; (c) R. D. Baechler and K. Mislow, J. Am. Chem. Soc., 1971, 93, 773 CrossRef; (d) K. Mislow and W. Egan, J. Am. Chem. Soc., 1971, 93, 1805 CrossRef CAS.
- (a) W. S. Knowels, Adv. Synth. Catal., 2003, 345, 3–5 CrossRef; (b) H. Ni, W.-L. Chan and Y. Lu, Chem. Rev., 2018, 118, 9344–9411 CrossRef CAS PubMed; (c) T. Werner, M. Hoffmann and S. Deshmukh, Eur. J. Org. Chem., 2014, 6630–6633 CrossRef CAS; (d) P. Rojo, A. Riera and X. Verdaguer, Coord. Chem. Rev., 2023, 215192 CrossRef CAS.
- (a) S. Humbel, C. Bertrand, C. Darcel, C. Bauduin and S. Juge, Inorg. Chem., 2003, 42, 420–427 CrossRef CAS PubMed; (b) K. D. Reichl, D. H. Ess and A. T. Radosevich, J. Am. Chem. Soc., 2013, 135, 9354–9357 CrossRef CAS PubMed; (c) J. Popp, S. Hanf and E. Hey-Hawkins, Chem. – Eur. J., 2020, 26, 5765–5769 CrossRef CAS PubMed.
- (a) K. Nikitin, D. G. Gilheany and H. Müller-Bunz, Chem. Commun., 2013, 49, 1434–1436 RSC; (b) K. Nikitin, K. Rajendran, H. Müller-Bunz and D. G. Gilheany, Angew. Chem., Int. Ed., 2014, 126, 1906–1909 CrossRef PubMed; (c) K. Rajendran, K. Nikitin and D. G. Gilheany, J. Am. Chem. Soc., 2015, 137, 9375–9381 CrossRef CAS PubMed.
- (a) E. V. Jennings, K. Nikitin, Y. Ortin and D. G. Gilheany, J. Am. Chem. Soc., 2014, 136, 16217–16226 CrossRef CAS PubMed; (b) M. W. Gillick-Healy, E. V. Jennings, H. Müller-Bunz, Y. Ortin, K. Nikitin and D. G. Gilheany, Chem. – Eur. J., 2017, 22, 2332–2339 CrossRef PubMed; (c) K. Nikitin, E. V. Jennings, S. Al Sulaimi, Y. Ortin and D. G. Gilheany, Angew. Chem., Int. Ed., 2018, 57, 1480–1484 CrossRef CAS PubMed.
- (a) J. B. Hendrickson and S. M. Schwartzman, Tetrahedron Lett., 1975, 277–280 CrossRef CAS; (b) S.-L. You, H. Razavi and J. W. Kelly, Angew. Chem., Int. Ed., 2003, 42, 83–85 CrossRef CAS PubMed.
- (a) S. M. Godfrey, C. A. Mcauli, I. Mushtaq, R. G. Pritchard and J. M. Sheffield, Dalton Trans., 1998, 3815–3818 RSC; (b) A. C. Vetter, K. Nikitin and D. G. Gilheany, Chem. Commun., 2018, 5843–5846 RSC.
- (a) E. Bergin, C. T. O’Connor, S. B. Robinson, E. M. McGarrigle, C. P. O’Mahony and D. G. Gilheany, J. Am. Chem. Soc., 2007, 129, 9566–9567 CrossRef CAS PubMed; (b) K. V. Rajendran, L. Kennedy, C. T. O'Connor, E. Bergin and D. G. Gilheany, Tetrahedron Lett., 2014, 7009–7012 Search PubMed.
- This effect has been detected first computationally and then confirmed experimentally by using various chlorinating agents.
- (a) K. Nikitin and R. O’Gara, Chem. – Eur. J., 2019, 25, 4551–4589 CrossRef CAS PubMed; (b) C. L. Perrin and T. J. Dwyer, Chem. Rev., 1990, 935–967 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3cc03719c |
| This journal is © The Royal Society of Chemistry 2023 |