
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Selective and benign alkylation of sulfido-oxo stannate clusters with propyl, pentyl, or hexyl substituents†
Gina
Stuhrmann
 a,
Jannik
Schneider
b,
Kilian
Schmidt
b and
Stefanie
Dehnen
*a
a,
Jannik
Schneider
b,
Kilian
Schmidt
b and
Stefanie
Dehnen
*a
aInstitute of Nanotechnology, Karlsruhe Institute of Technology, P.O. Box 3640, 76021, Karlsruhe, Germany. E-mail: stefanie.dehnen@kit.edu
bFachbereich Chemie, Philipps-Universität Marburg, Hans-Meerwein-Str. 4, 35043, Marburg, Germany
First published on 10th October 2023
Abstract
Chalcogenido metalate compounds that are based on tetrahedral clusters have been extensively studied in recent years due to their rich structural chemistry and uncommon chemical and physical properties. Recently it was shown that partial butylation of the inorganic cluster core by ionothermal reactions allowed access to tetrahedral sulfido-oxo stannate clusters with reasonable solubility in conventional solvents at the retainment of their opto-electronic features. We have expanded this mild alkylation approach, and herein report success in receiving the first sulfido-oxo stannate clusters that are selectively propylated, pentylated, and hexylated. This was achieved in a unique way by preparing symmetrically 1,3-substituted imidazolium bromides in preparative scale and using them as both the reaction medium and alkylatoin reagent. We discuss the effect of the organic groups attached to the cluster and present in the counterions of the products on the compounds’ structural and opto-electronic properties.
Sulfido stannate compounds, in general, exhibit a huge structural variety1,2 as a consequence of the variable oxidation states (II, III, IV) and coordination modes (tetrahedral, trigonal bipyramidal, octahedral) of the tin atoms.3 In addition, compounds comprising discrete sulfido stannate cluster molecules have attracted particular attention owing to their tunable electronic and optical properties that can be varied with the cluster size4 and exact composition of the molecules.5–7
Most of these compounds have traditionally been accessed by molten flux8–11 approaches or high temperature syntheses, before hydrothermal12 or aminothermal13 approaches were added as synthesis methods.12 The most recent technique for their preparation, however, utilizes ionic liquids under so-called ionothermal conditions, which again expanded the chemical and structural diversity of crystalline chalcogenides in general, and sulfido stannate cluster compounds in particular. Besides these benefits regarding the product spectrum, ionic liquids are also interesting in terms of ecological and economic aspects, as their reprocessing is relatively benign.14,15 Therefore, the results that were reported during the past decade have proved that the ionothermal method is a promising strategy for the synthesis of crystalline chalcogenido metalate-based compounds.16–22
In the area of chalcogenido metalate-based cluster compounds, considerable activity has taken place in the context of inorganic, anionic substructures based on large tetrahedral clusters.6,23–28 Both a smooth transfer of (super)tetrahedral clusters into functionalized nanomaterials for optoelectronic devices and also their direct application in solution are generating increasing interest in the solubility of these clusters in conventional organic solvents – without a drop of the desired optoelectronic properties, which requires a minimum of organic substituents to be present.29
Several approaches have been reported in the recent past to achieve this goal. One method that led to a notable increase in solubility was the partial replacement of sulfur with selenium atoms.30 The replacement of alkali metal counterions with organic cations also leads to a somewhat higher solubility.31 A recent approach for solubilizing large tetrahedral selenido germanate clusters is a controlled and limited aggregation of the anionic molecules into oligomers of finite size, thereby decreasing the negative charge per cluster unit, which allows for the clusters’ solubility in DMF.31 This strategy can be enhanced by forming cluster aggregates that include an alkali metal cation for additional charge compensation “from inside”: [Cs@Ge4(Ge4Se10)4]7−, which at the same time represents the largest selenido germanate cluster reported to date, shows the lowest charge per atom reported for a purely inorganic (super)tetrahedral cluster.32 There have also been recent reports about purely inorganic supertetrahedral clusters that were proven to dissolve in DMSO.30
A significant step forward regarding the solubility of chalcogenido metalate cluster compounds at the retention of optical properties, however, was the selective partial alkylation of the sulfido-oxo stannate cluster [Sn10O4S20]8−, previously known as this purely inorganic species,33 in reactive ionic liquids. This did not only turn out to be much more benign as it takes place without the use of harmful alkylation reagents (MeI or Meerwein salt), but also the only feasible way for post-synthetic alkylation of these weakly nucleophilic species. The first compound comprising an alkylated cluster obtained this way was (cat)4[Sn10O4S16(SMe)4] (cat = imidazolium-based ionic liquid; Me = methyl).3,33–36 The approach also proved suitable for tellurido mercurate clusters and a ternary Mn/Sn/Se cluster.34,37 The methyl groups reduced the negative charge of the cluster from 8− in [Sn10O4S20]8− to 4−, but the methyl groups did not increase the solubility in the desired way. As a matter of fact, the commonly used imidazolium-based ionic liquids, in which the two N atoms are functionalized by one methyl group and another alkyl chain of varying length, tend to selectively transfer the methyl group.38 Hence, in order to change the group to be transferred, it was necessary to use ionic liquids with a symmetric substitution pattern of the imidazolium ring. Upon employment of (C4C1C4Im)Br comprising a methyl group (“C1”) in 2-position, and two butyl groups (“C4”) on the N atoms in 1- and 3-position of the imidazolium ring, the cluster [Sn10O4S16(SBu)4]4− (Bu = butyl) was obtained as a salt with good solubility in conventional organic solvents like CH3CN. This was proven by means of NMR spectroscopy and mass spectrometry in solution, and also indicated that the clusters retain unchanged in solution, with their optical properties were essentially not affected by the organic ligands.39
In order to selectively form further soluble clusters and probe the effect of other alkyl groups on the properties of these compounds, we aimed at an alkylation with other organic groups. For this, we prepared the symmetrically substituted ionic liquids (CxC1CxIm)Br (x = 3, 5, 6),40 and used them for the synthesis of corresponding functionalized sulfido-oxo stannate clusters [Sn10O4S16(SR)4]4− with R = propyl (Pr), pentyl (Pn), hexyl (Hex) in ionothermal reactions (Fig. S1, ESI†) that were obtained in corresponding salts (C3C1C3Im)4[Sn10O4S16(SPr)4] (1), (C5C1C5Im)4[Sn10O4S16(SPn)4] (2) and (C6C1C6Im)4[Sn10O4S16(SHex)4] (3). We present their crystal structures and the evidence of their undecomposed transfer into solution by NMR spectroscopy and mass spectrometry. In addition, we report about the opto-electronical properties.
Compounds 1 – 3 were obtained by ionothermal treatment of Na4[SnS4]·14H2O41 in (CxC1CxIm)Br (x = 3, 5, 6) at 180 °C (Scheme 1). All compounds crystallize as colorless plates (Fig. S2, ESI†). The Sn:S compositions were determined by means of μ-X-ray fluorescence spectroscopy (Fig. S3–S5; Tables S1–S3, ESI†). The structures of the single-crystalline compounds were determined by means of single-crystal X-ray diffraction (Tables S4 and S5, ESI†). Compound 1 crystallizes in the tetragonal crystal system, space group I![[4 with combining macron]](https://www.rsc.org/images/entities/char_0034_0304.gif) 2d with four formula units (Z = 4) within the unit cell (1: a, c = 19.9274(11), 26.2914(18) Å; V = 10440.3(14) Å3). Compounds 2 and 3 crystallize in the triclinic crystal system, space group P
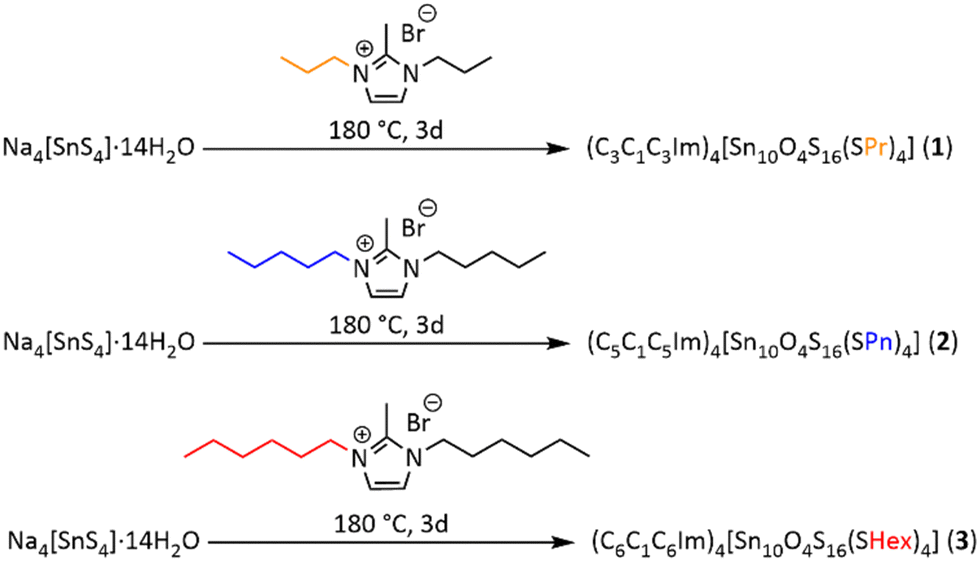
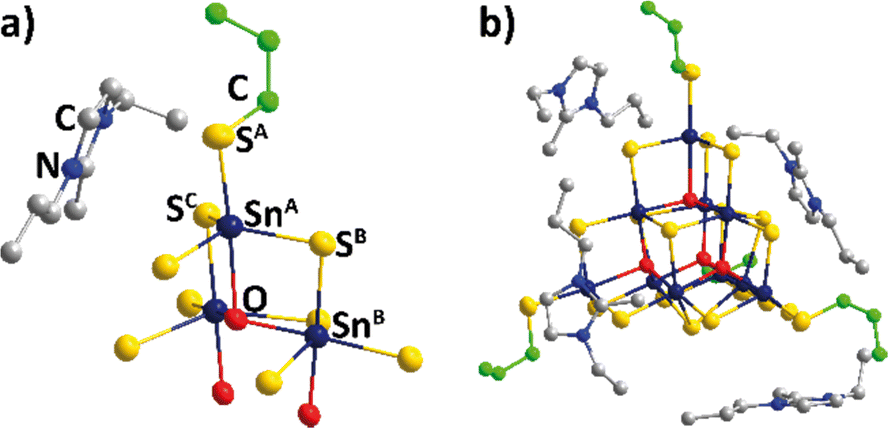
2d with four formula units (Z = 4) within the unit cell (1: a, c = 19.9274(11), 26.2914(18) Å; V = 10440.3(14) Å3). Compounds 2 and 3 crystallize in the triclinic crystal system, space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) , with two formula units (Z = 2) within the unit cell (2: a, b, c = 19.5954(4), 19.2861(5), 19.7915(5) Å; α, β, γ = 99.744(2), 112.258(2), 110.568(2) °; V = 6073.7(3) Å3; 3: a, b, c = 18.7606(17), 20.0088(19), 21.1091(19) Å; α, β, γ = 99.505(7), 115.794(7), 110.290(7) °; V = 6205.5(11) Å3). Molecular and crystal structures are depicted in Fig. 1, 2 and in Fig. S6–S9 (ESI†). Fig. 1 illustrates the molecular structure of compound 1 (for those of 2 and 3, see Fig. S7 in the ESI†).
, with two formula units (Z = 2) within the unit cell (2: a, b, c = 19.5954(4), 19.2861(5), 19.7915(5) Å; α, β, γ = 99.744(2), 112.258(2), 110.568(2) °; V = 6073.7(3) Å3; 3: a, b, c = 18.7606(17), 20.0088(19), 21.1091(19) Å; α, β, γ = 99.505(7), 115.794(7), 110.290(7) °; V = 6205.5(11) Å3). Molecular and crystal structures are depicted in Fig. 1, 2 and in Fig. S6–S9 (ESI†). Fig. 1 illustrates the molecular structure of compound 1 (for those of 2 and 3, see Fig. S7 in the ESI†).
 | ||
| Scheme 1 Survey of the syntheses of compounds 1–3. | ||
 | ||
| Fig. 1 (a) Asymmetric unit of the crystal structure of compound 1, (b) anion in 1 with the four closest ionic liquid cations. Cluster atoms are represented by thermal ellipsoids (50% probability), imidazolium cations and alkyl groups are shown in balls-and-sticks mode, H atoms are omitted for clarity. | ||
 | ||
| Fig. 2 Simplified illustration of the packing of the anions in the crystal structures of 1 (a) and 2 (b) from different views. The cluster cores are shown only, with {SnS4} subunits given in polyhedral representation. Cluster atoms are represented by thermal ellipsoids (50% probability), alkyl groups and counterions are omitted for clarity. | ||
The structures are subject to some inherent crystallographic issues: The propyl, pentyl, and (especially) hexyl chains of both the anions and the cations show high conformational flexibility and disorder; thus, the alkyl groups could not be fully localized on the difference Fourier map during the refinement of the structures – despite many attempts to measure different crystals with different methods on different diffractometer types (for more details, see the ESI†). However, as outlined below, other analytical techniques and the analysis of the number of atoms that would fit into the void space served to confirm the composition. In addition, Raman spectra recorded on the single crystals (Fig. S10, ESI†) indicate the presence of S–C stretching vibrations of the S-alkyl groups at ∼735–760 cm−1, yet with decreasing relative intensity as the alkyl chains get longer. Sn–Sterminal distances, which are indicative of an alkylation of the terminal sulfur ligands, are 2.458(31) Å in 1 and range between 2.427(38) Å and 2.448(35) Å in 2 and between 2.442(31) Å and 2.448(19) Å in 3. This clearly indicates the successful alkylation, as Sn–Sterminal bonds are significantly shorter in the purely inorganic clusters (2.355–2.375 Å).33 The observed values are in good agreement with the Sn–Sterminal distances found for both the butylated (2.427(4)–2.448(4) Å)39 and the methylated (2.419(17)–2.511(11) Å) analogues.34,38
As shown in Fig. 2 and in Fig. S8 (ESI†), the alkyl chains affect the packing schemes of anions and cations in the crystals. A given cluster in 1 has four tetrahedrally arranged nearest neighbors at a distance of 11.9 Å (cluster center⋯cluster center), while the eight second-closest clusters are further apart by another ∼7 Å.
In 2, four next-neighbor clusters are found within center⋯center distances of 12.0–12.8 Å, four second-closest distances are larger by another ∼7–8 Å. The four nearest neighbors in 3 are located at center⋯center distances of 12.4–12.7 Å, while the distances to the four second-closest clusters are larger by another ∼7.5–9 Å. So, the clusters get more distant with increasing length of the alkyl chains, which correlates nicely with the optical absorption properties discussed below.
As desired, the crystals of 1, 2, and 3 are readily soluble in conventional organic solvents. Besides solubility in acetonitrile, compounds 1–3 also show solubility in DMF, DMSO, and a water:acetonitrile mixture (40![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :60). However, they dissolve most readily and most homogenously in acetonitrile, which is why all solution analytics were carried out in this solvent. This enabled us to measure 119Sn NMR, 1H, and 13C spectra from their solutions (Fig. S11–S19, ESI†). The alkylated clusters show two predominant 119Sn NMR signals at around –116 and –517 ppm (Fig. 3 and Fig. S11, S14, and S17, ESI†). Both 119Sn chemical shifts are in accordance with reported data for SnIV atoms.42 The 1H NMR spectra indicate the presence of the ionic liquid as counterions (CxC1CxIm)+ (x = 3, 5, 6) besides the dealkylated form of the ionic liquid CxCIm (x = 3, 5, 6; Fig. S12, S15, and S18, ESI†). The signals of the alkyl chains on the cluster cannot be distinguished from those attached to the counterions.39
:60). However, they dissolve most readily and most homogenously in acetonitrile, which is why all solution analytics were carried out in this solvent. This enabled us to measure 119Sn NMR, 1H, and 13C spectra from their solutions (Fig. S11–S19, ESI†). The alkylated clusters show two predominant 119Sn NMR signals at around –116 and –517 ppm (Fig. 3 and Fig. S11, S14, and S17, ESI†). Both 119Sn chemical shifts are in accordance with reported data for SnIV atoms.42 The 1H NMR spectra indicate the presence of the ionic liquid as counterions (CxC1CxIm)+ (x = 3, 5, 6) besides the dealkylated form of the ionic liquid CxCIm (x = 3, 5, 6; Fig. S12, S15, and S18, ESI†). The signals of the alkyl chains on the cluster cannot be distinguished from those attached to the counterions.39
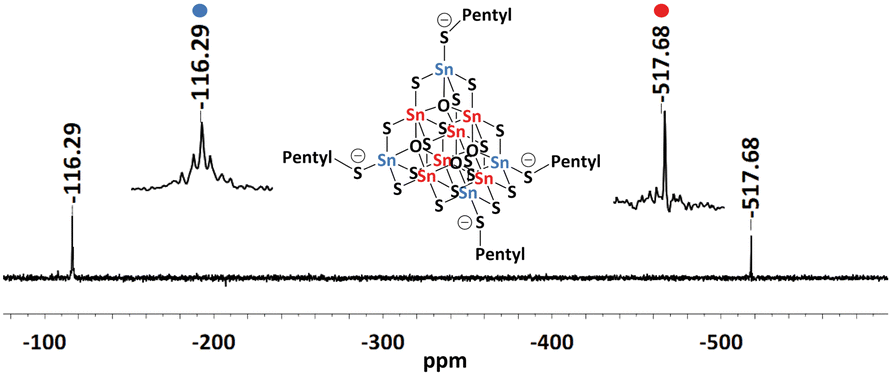 | ||
| Fig. 3 119Sn-NMR spectra of a solution of single crystals in CD3CN of compound 2 with zoom into the signals assigned to the two different Sn atomic sites. | ||
Electrospray-ionization mass spectrometry (ESI-MS; Fig. 4 and Fig. S20–S25, ESI†) in negative ion mode served to detect the alkylated anion of compound 1 as trianionic aggregate along with one (C3C1C3Im)+ cation. The anions of compounds 2 and 3 were detected as dianions along with two counterions (CxC1CxIm)+ (x = 5, 6) only, which we attribute to the size and mass of the larger alkyl chains. High-resolution spectra of the molecular peaks are shown in Fig. 4. The measured isotope patterns agree very well with the simulated ones, indicating that the clusters are transferred into the gas phase without decomposition. The overview spectra are provided in Fig. S9, S11, and S13 (ESI†). As shown in Fig. S22 and S23 (ESI†), also a monoanionic aggregate was transferred into the gas phase, though with lower relative abundance.
 | ||
| Fig. 4 High-resolution ESI(–) mass spectra of a fresh solution of single crystals of the title compounds in CH3CN. (a) 1, detected alongside one counterion as the trianionic aggregate {(C3C1C3Im)[Sn10O4S16(SPr)4]}3−, (b) 2, detected alongside two counterions as the dianionic aggregate {(C5C1C5Im)2[Sn10O4S16(SPn)4]}2−, (c) 3, detected alongside two counterions as the dianionic aggregate {(C6C1C6Im)2[Sn10O4S16(SHex)4]}2−. | ||
To investigate the influence of the different organic substituents – and the concomitant structural differences – on the opto-electronic properties, we recorded optical absorption spectra of the three solid compounds. As illustrated in Fig. 5, the measured optical bandgaps amount to 2.89 eV (1), 3.19 eV (2), and 3.27 eV (3) – close to the values observed for other related clusters.39 With larger alkyl chain length, the bandgap is slightly blue-shifted, with a maximum shift of 0.38 eV (3versus1) – in perfect agreement with the observations made for the crystal structures. The Tauc plots (Fig. S26–S28, ESI†) indicate that all transitions are of indirect, allowed nature.
 | ||
| Fig. 5 UV-visible spectra of single crystals of (a) 1, (b) 2, and (c) 3. | ||
The photoluminescence (PL) characteristics of the three compounds were investigated in CH3CN solution (Fig. S29 and S30 for the ionic liquids, ESI†). Upon excitation at λex = 249 nm, all compounds exhibit a maximum emission peak at 317 nm. The quantum yield of all three compounds is 4.3%.
Treatment of Na4[SnS4]·14H2O in imidazolium-based ionic liquids featuring a symmetrical substitution pattern, (CxC1CxIm)Br (with x representing the number of carbon atoms in the alkyl chains on the imidazolium N atoms), allowed for the straight-forward synthesis of salts of partially propylated (x = 3), pentylated (x = 5) or hexylated (x = 6) tetrahedral cluster anions, (CxC1CxIm)4[Sn10O4S16(SR)4]. These compounds serve to complete a series for such cluster compounds running from x = 3 to x = 6. All compounds are readily soluble in common organic solvents, which was proven by means of NMR spectroscopy and mass spectrometry. This is a consequence of lowering the total charge of the underlying inorganic cluster anion from 8− to 4− by the four alkyl groups that are attached selectively to the fourterminal sulfur atoms and the presence of organic groups. The opto-electronic properties are slightly effected by the different packing and inter-cluster distances, but the blue-shift with increasing alkyl chain length is moderate (2.89–3.27 eV). All clusters that were shown to possess an indirect allowed optical bandgap, so this approach allows to form highly soluble wide-bandgap semiconductor clusters. Further studies will be carried out to explore the potential and effect of the attachment of substituents carrying functional groups.
This work was financially supported by the German Research Foundation (Deutsche Forschungsgemeinschaft, DFG). We thank Dr Frank Tambornino for his help with the refinement of crystal structures.
Conflicts of interest
There are no conflicts to declare.Notes and references
- P. Vaqueiro, Dalton Trans., 2010, 39, 5965 RSC.
- X. Xu, W. Wang, D. Liu, D. Hu, T. Wu, X. Bu and P. Feng, J. Am. Chem. Soc., 2018, 140, 888–891 CrossRef CAS PubMed.
- A. Benkada, H. Reinsch, M. Poschmann, J. Krahmer, N. Pienack and W. Bensch, Inorg. Chem., 2019, 58, 2354–2362 CrossRef CAS PubMed.
- T. Wu, L. Wang, X. Bu, V. Chau and P. Feng, J. Am. Chem. Soc., 2010, 132, 10823–10831 CrossRef CAS PubMed.
- W.-W. Xiong, J.-R. Li, B. Hu, B. Tan, R.-F. Li and X.-Y. Huang, Chem. Sci., 2012, 3, 1200 RSC.
- Y. Wang, Z. Zhu, Z. Sun, Q. Hu, J. Li, J. Jiang and X. Huang, Chem. – Eur. J., 2020, 26, 1624–1632 CrossRef CAS PubMed.
- S. Makin and P. Vaqueiro, Molecules, 2021, 26, 5415 CrossRef CAS PubMed.
- Y. Pan, Q. Jin, J. Chen, Y. Zhang and D. Jia, Inorg. Chem., 2009, 48, 5412–5417 CrossRef CAS PubMed.
- M. J. Manos, C. D. Malliakas and M. G. Kanatzidis, Chem. – Eur. J., 2007, 13, 51–58 CrossRef CAS PubMed.
- Y. Liu, Y. Tian, F. Wei, M. S. C. Ping, C. Huang, F. Boey, C. Kloc, L. Chen, T. Wu and Q. Zhang, Inorg. Chem. Commun., 2011, 14, 884–888 CrossRef CAS.
- J. Han, Y. Liu, J. Lu, C. Tang, Y. Shen, Y. Zhang and D. Jia, J. Chem. Crystallogr., 2015, 45, 355–362 CrossRef CAS.
- Z. Wu, G. Stuhrmann and S. Dehnen, Chem. Commun., 2022, 58, 11609–11624 RSC.
- C. Donsbach and S. Dehnen, Inorg. Chem. Front., 2017, 4, 336–342 RSC.
- P. Wasserscheid and T. Welton, Ionic liquids in synthesis, Wiley-VCH, Weinheim, 2nd edn, 2008 Search PubMed.
- Z. Lei, B. Chen, Y.-M. Koo and D. R. MacFarlane, Chem. Rev., 2017, 117, 6633–6635 CrossRef PubMed.
- N.-N. Shen, B. Hu, C.-C. Cheng, G.-D. Zou, Q.-Q. Hu, C.-F. Du, J.-R. Li and X.-Y. Huang, Cryst. Growth Des., 2018, 18, 962–968 CrossRef CAS.
- I. Nußbruch and S. Dehnen, Z. Anorg. Allg. Chem., 2018, 644, 1897–1901 CrossRef.
- W.-W. Xiong, G. Zhang and Q. Zhang, Inorg. Chem. Front., 2014, 1, 292 RSC.
- E. R. Cooper, C. D. Andrews, P. S. Wheatley, P. B. Webb, P. Wormald and R. E. Morris, Nature, 2004, 430, 1012–1016 CrossRef CAS PubMed.
- Y. Lin, W. Massa and S. Dehnen, J. Am. Chem. Soc., 2012, 134, 4497–4500 CrossRef CAS PubMed.
- B. Peters, C. Krampe, J. Klärner and S. Dehnen, Chem. – Eur. J., 2020, 26, 16683–16689 CrossRef CAS PubMed.
- J.-R. Li, W.-W. Xiong, Z.-L. Xie, C.-F. Du, G.-D. Zou and X.-Y. Huang, Chem. Commun., 2013, 49, 181–183 RSC.
- O. Palchik, R. G. Iyer, J. H. Liao and M. G. Kanatzidis, Inorg. Chem., 2003, 42, 5052–5054 CrossRef CAS PubMed.
- T. Wu, X. Bu, P. Liao, L. Wang, S.-T. Zheng, R. Ma and P. Feng, J. Am. Chem. Soc., 2012, 134, 3619–3622 CrossRef CAS PubMed.
- N. Zheng, X. Bu, H. Vu and P. Feng, Angew. Chem., Int. Ed., 2005, 44, 5299–5303 CrossRef CAS PubMed.
- J. Zhang, X. Bu, P. Feng and T. Wu, Acc. Chem. Res., 2020, 53, 2261–2272 CrossRef CAS PubMed.
- B. Peters, N. Lichtenberger, E. Dornsiepen and S. Dehnen, Chem. Sci., 2020, 11, 16–26 RSC.
- X. Bu, N. Zheng and P. Feng, Chem. – Eur. J., 2004, 10, 3356–3362 CrossRef CAS PubMed.
- X.-W. Hua, Y.-W. Bao and F.-G. Wu, ACS Appl. Mater. Interfaces, 2018, 10, 10664–10677 CrossRef CAS PubMed.
- M. Hao, Q. Hu, Y. Zhang, M. Luo, Y. Wang, B. Hu, J. Li and X. Huang, Inorg. Chem., 2019, 58, 5126–5133 CrossRef CAS PubMed.
- Z. Wu, I. Nußbruch, S. Nier and S. Dehnen, JACS Au, 2022, 2, 204–213 CrossRef CAS PubMed.
- Z. Wu, F. Weigend, D. Fenske, T. Naumann, J. M. Gottfried and S. Dehnen, J. Am. Chem. Soc., 2023, 145, 3802–3811 CrossRef CAS PubMed.
- W. Schiwy and B. Krebs, Angew. Chem., Int. Ed. Engl., 1975, 14, 436 CrossRef.
- B. Peters, S. Santner, C. Donsbach, P. Vöpel, B. Smarsly and S. Dehnen, Chem. Sci., 2019, 10, 5211–5217 RSC.
- J. B. Parise and Y. Ko, Chem. Mater., 1994, 6, 718–720 CrossRef CAS.
- T. Kaib, M. Kapitein and S. Dehnen, Z. Anorg. Allg. Chem., 2011, 637, 1683–1686 CrossRef CAS.
- M. Tallu, B. Peters, A. Friedrich and S. Dehnen, Inorg. Chem., 2023, 62, 13943–13952 CrossRef CAS PubMed.
- B. Peters, S. Reith and S. Dehnen, Z. Anorg. Allg. Chem., 2020, 646, 964–967 CrossRef CAS.
- B. Peters, G. Stuhrmann, F. Mack, F. Weigend and S. Dehnen, Angew. Chem., Int. Ed., 2021, 60, 17622–17628 CrossRef CAS PubMed.
- K. Boruah and R. Borah, ChemistrySelect, 2019, 4, 3479–3485 CrossRef CAS.
- W. Schiwy, S. Pohl and B. Krebs, Z. Anorg. Allg. Chem., 1973, 402, 77–86 CrossRef CAS.
- M. Gielen, A. G. Davies, K. Pannell and E. Tiekink, Tin Chemistry: Fundamentals, Frontiers, and Applications, Wiley-VCH, Weinheim, 2008 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Details in syntheses and all analyses. CCDC 2281096–2281099. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3cc03525e |
| This journal is © The Royal Society of Chemistry 2023 |