
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRealization of nitroaromatic chromophores with intense two-photon brightness†
Bartłomiej
Sadowski
 *a,
Marzena
Kaliszewska
b,
Guillaume
Clermont
c,
Yevgen M.
Poronik
d,
Mireille
Blanchard-Desce
*c,
Piotr
Piątkowski
*b and
Daniel T.
Gryko
*d
*a,
Marzena
Kaliszewska
b,
Guillaume
Clermont
c,
Yevgen M.
Poronik
d,
Mireille
Blanchard-Desce
*c,
Piotr
Piątkowski
*b and
Daniel T.
Gryko
*d
aCentre of New Technologies, University of Warsaw, S. Banacha 2c, Warsaw 02-097, Poland. E-mail: b.sadowski@cent.uw.edu.pl
bDepartment of Chemistry, University of Warsaw, Zwirki i Wigury 101, Warsaw 02-089, Poland. E-mail: ppiatkowski@aus.edu
cUniv. Bordeaux, CNRS, Bordeaux INP, ISM, UMR 5255, Talence F-33400, France. E-mail: mireille.blanchard-desce@u-bordeaux.fr
dInstitute of Organic Chemistry, Polish Academy of Sciences, Kasprzaka 44/52, Warsaw 01-224, Poland. E-mail: dtgryko@icho.edu.pl
First published on 8th September 2023
Abstract
Strong fluorescence is a general feature of dipyrrolonaphthyridinediones bearing two nitrophenyl substituents. Methyl groups simultaneously being weakly electron-donating and inducing steric hindrance appear to be a key structural parameter that allows for significant emission enhancement, whereas Et2N groups cause fluorescence quenching. The magnitude of two-photon absorption increases if 4-nitrophenyl substituents are present while the contribution of Et2N groups is detrimental.
Recent developments in molecular photonics1 and biochemistry2 have increased the demand for novel organic materials with improved emission characteristics and enhanced resistance towards ambient conditions. Bearing in mind the stability of π-conjugated organic materials, one of the most common strategies is to decrease electron density, usually enabled by introducing an electron-deficient moiety into an aromatic backbone.3 The nitro group (−NO2) is one of the strongest electron-withdrawing moieties, and by meeting this criterion, potentially sets the stage for novel materials with intriguing optical characteristics.
Nitroaromatics have, however, long been considered as non-fluorescent, and highly emissive examples of NO2-containing chromophores have been sparsely reported in the literature (Fig. 1A).4 Yet, they are promising candidates for constructing probes5 tracking important biologically relevant species and as efficient two-photon absorbers.6
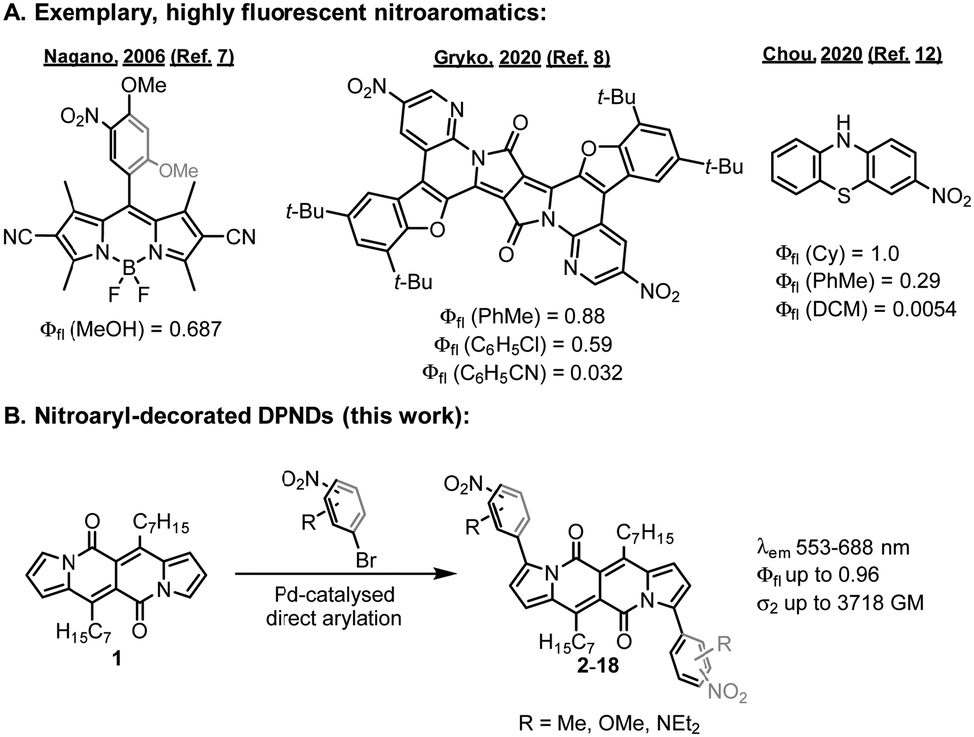 | ||
| Fig. 1 (A) Exemplary highly emissive nitroaromatics. (B) The idea of the present work. | ||
In recent years, some strategies have been developed to improve the luminescence performance of nitroaromatics.4b From the mechanistic viewpoint, fluorescence can be successfully induced by blocking the d-PeT7 process or when the nitro group does not participate in the S0 → S1 transition.8 Applying a weaker electron-donating moiety for the construction of nitro-containing conjugates may also give rise to nitroaromatics with enhanced emission. Dyes containing nitro-functionalized thiophene,9 fluorene,10 tetrabenzofluorenes,11 or phenothiazine12 featuring enhanced emission have been described, but these approaches contain some drawbacks, such as a tendency for deexcitation via ISC (heavy atom effect of sulphur) or lack of significant spectral red-shifts of both the absorption and emission bands.
Due to its intriguing properties, the electron-deficient dipyrrolonaphthyridinedione (DPND) core13 has recently been successfully employed in the construction of dyes with red/NIR emission,14 OFETs,15 stable singlet fission (SF) materials16 and singlet oxygen (1O2) photosensitisers.17 Studying quadrupolar, centrosymmetric DPNDs possessing two nitroaryl substituents at positions 3 and 9, we discovered that creating nitroaromatics with acceptor–acceptor′–acceptor (A–A′–A) architecture constitutes a new viable strategy of preventing non-radiative deactivation.18
There is, however, still a huge gap in the knowledge of how positional isomerism of the nitro group relative to the chromophore core influences the emission characteristics of nitroaromatics.
To overcome this intrinsic limitation we now report how distinct structural modifications (different substitution patterns of nitroaryl moieties) alter the emissive properties and two-photon absorption characteristics19 of these quadrupolar, A–A′–A dyes.
DPNDs possessing A–A′–A architecture, considered in this work, were assembled by employing a Pd-catalysed direct arylation14b between the DPND 1 and the corresponding aryl bromides (Fig. 1, see ESI† for synthetic details). As a result, a series of new quadrupolar nitroaromatics 4–10 and 13–18 were synthesized and fully described (Fig. 2). The same method was used for the synthesis of the previously described dyes 2, 3, 11 and 12, which are included here for the complete assessment of substitution-dependent optical behaviour. The complete series of dyes differ in the location of the nitro groups (para or meta), the structure of the linker (phenyl or biphenyl), and the effect of further substitution with electron-donating or -withdrawing substituents.
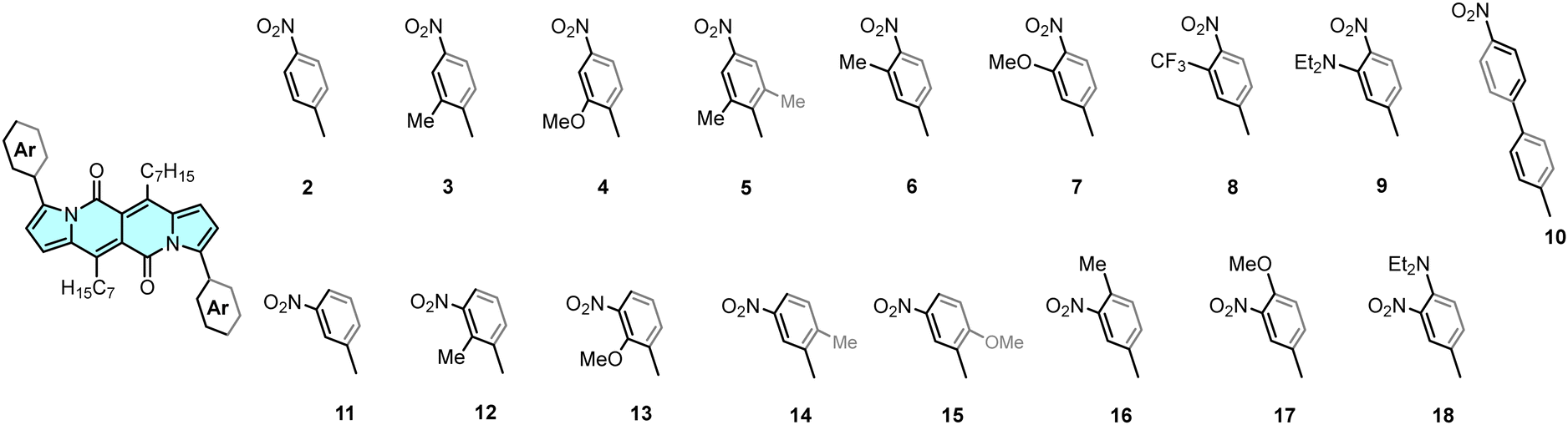 | ||
| Fig. 2 Structure of nitroaromatics studied in this work. | ||
The effect of structural variations on the linear optical properties of all DPND derivatives in solution was subsequently examined by employing 1,2-dichlorobenzene (DCB), CH2Cl2 (DCM), or MeCN (ACN) as solvents. The results are summarized in Table 1 and Fig. S2 (ESI†), and the spectral data are shown in Fig. S1–S26 (ESI†). As expected, nitroaromatics built around the DPND core display red-shifted absorption and emission bands compared with 1, resulting from π-expansion of the chromophore and the presence of groups possessing distinct electronic character.
| Cmpd | Solventa | λ abs /nm | λ fl /nm | Stokes’ shiftc/cm−1 | Φ fl | τ /ns | k r × 10−8/s−1 | k nr × 10−8/s−1 |
|---|---|---|---|---|---|---|---|---|
| a DCB = 1,2-dichlorobenzene; DCM = dichloromethane; ACN = acetonitrile. b Absorption and fluorescence maxima. c Stokes’ shifts. d Lifetimes of the emissive excited states obtained from time-correlated single photon counting (for τ ≳ 1.5 ns). e Radiative and non-radiative decay rate constants: kr = Φflτ−1 and knr = (1 − Φfl)τ−1. f The data were taken from ref. 18. | ||||||||
| 1 | DCB | 508 | 528 | 750 | 0.81 | 5.1 | 1.59 | 0.373 |
| DCM | 504 | 523 | 720 | 0.73 | 5.7 | 1.28 | 0.474 | |
| ACN | 498 | 525 | 1030 | 0.65 | 5.7 | 1.14 | 0.614 | |
| 2 | DCB | 569 | 605 | 1050 | 0.45 | 3.6 | 1.25 | 1.53 |
| DCM | 562 | 601 | 1150 | 0.41 | 3.4 | 1.21 | 1.74 | |
| ACN | 559 | 599 | 1190 | 0.07 | — | — | — | |
| 5 | DCB | 524 | 560 | 1230 | 0.96 | 4.7 | 2.04 | 0.085 |
| DCM | 521 | 560 | 1340 | 0.34 | 1.9 | 1.79 | 3.47 | |
| ACN | 518 | 564 | 1570 | 0.005 | — | — | — | |
| 9 | DCB | 555 | 602 | 1390 | 0.006 | — | — | — |
| DCM | 549 | 608 | 1760 | 0.003 | — | — | — | |
| ACN | 542 | 584 | 1330 | 0.003 | — | — | — | |
| 14 | DCB | 525 | 568 | 1440 | 0.76 | 4.9 | 1.55 | 0.490 |
| DCM | 522 | 566 | 1490 | 0.58 | 3.2 | 1.81 | 1.31 | |
| ACN | 520 | 569 | 1660 | 0.075 | — | — | — | |
| 18 | DCB | 575 | 669 | 2440 | 0.12 | 2.6 | 0.462 | 3.38 |
| DCM | 565 | 670 | 2770 | 0.02 | — | — | — | |
| ACN | 557 | 688 | 3420 | 0.04 | 2.8 | 0.143 | 3.43 | |
Upon inspection of Fig. 2, two series of dyes can be recognized: 2–10 bearing nitro groups at the para positions relative to the DPND core, and 11–18 where the –NO2 occupies meta positions. Regardless of the location, some trends are common to both groups. First of all, the magnitude of the response strongly depends on the presence of a substituent (Me, OMe) at the ortho position relative to the DPND core. Functionalization at this position increases the dihedral angle between the chromophore and the nitroaryl moiety, therefore decreasing conjugation, which results in a blue-shift of both the absorption and emission bands (2vs.3–4 and 11vs.12–15). Conversely, a reduced degree of rotation around the C—C single bond generally leads to an enhanced fluorescence response (Table 1 and Table S1 (ESI†), Fig. 3).
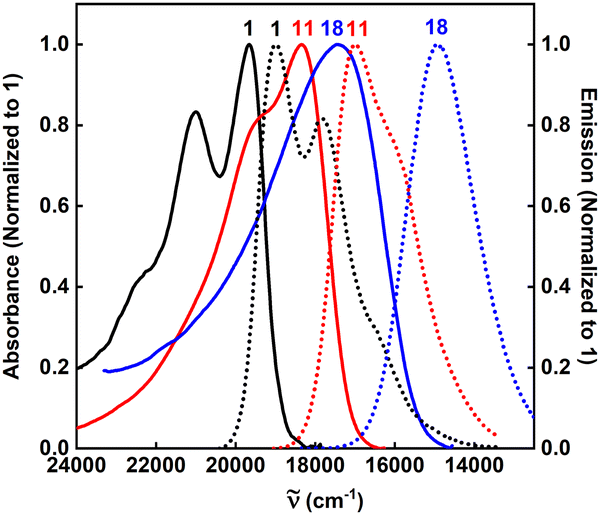 | ||
| Fig. 3 Absorption and emission spectra of 1 (black solid and dotted lines), 11 (red solid and dotted lines), and 18 (blue solid and dotted lines) dyes in DCB. The 1, 11, and 18 emission spectra were excited at 480, 515, and 535 nm, respectively. | ||
Dye 5 constitutes a special case among all the investigated nitroaromatics as it bears four ortho-located additional methyl groups that diminish the electronic coupling between substituents and the core, and effectively prevent the free rotation of aryl rings. This structural manipulation leads to a record-breaking value of φfl among all nitro-DPNDs while the maxima of both λabs and λem are further hypsochromically shifted compared to those measured for 3 and 4 and just 15–30 nm bathochromically shifted vs. parent DPND 1.
The presence of an additional weak electron-donating substituent (Me or OMe) at the ortho position relative to the NO2 group also enhances emission in all tested solvents (Table 1 and Table S1, ESI†). It marginally influences the position of both the absorption and emission bands in the case of 6 and 7 within the para series, while for the meta one, a small red-shift can be noted, especially for λem in 17. The potent effect of these structural factors, namely the presence of steric hindrance as well as an additional ortho substituent relative to the nitro group accumulates within 12, giving rise to an enormous φfl value in relatively polar CH2Cl2 (0.92).18
The incorporation of a substituent with a weak electron-accepting effect (–CF3, dye 8) or longer biaryl linkage (dye 10) does not really alter the absorption/emission characteristics as compared with 2 (Table 1 and Table S1 and Fig. S6 and S8, ESI†). At the same time, however, the presence of CF3 groups leads to a sharp decrease of φfl in polar solvents.
Finally, we investigated the influence of an auxochrome with a positive mesomeric effect (–NEt2). It was found that groups featuring a strong mesomeric effect (–NO2, –NEt2) placed at the para positions relative to the DPND core mainly govern both the absorption and emission properties of the studied DPNDs. This is particularly true for the diethylamino group in 18, giving rise to a significantly red-shifted emission (ca. 670 nm) but very weak fluorescence response (Fig. 3). The emission quenching is plausibly assigned to photo-induced electron transfer (PeT) occurring in the excited state.
Inspired by the large two-photon absorption (2PA) responses of nitroaromatics bearing arylethenyl or arylethynyl π-linkers between the nitro groups and the DPND core,19 we investigated the 2PA properties of dyes 1–18 using the two-photon excited fluorescence (TPEF) method (see ESI†). The results are collected in Table 2 and Table S2, S3 (ESI†). No 2PA or very low response is observed in the spectral region corresponding to the lowest one-photon allowed excited state (i.e. above 1000 nm), due to symmetry reasons.20 In contrast, significant 2PA broad bands peaking (or shouldering) in the 800–850 nm range and even more intense 2PA bands closer to 700 nm are observed at higher energy (Fig. 4). The 2PA magnitude in the NIR1 region strongly depends on the structure of the dyes and the nature and position of the substituents (Table 2). The presence of NO2 substituents at the para position is a key structural factor: analogous dyes from the meta series show much smaller 2PA responses, as noted from the comparison of dyes 2 and 1; 3 and 12; and 4 and 13 (Fig. S27, ESI†). For the derivatives having a para NO2 group (2–10), the presence of an additional substituent generates a marked decrease of the 2PA both in the case of substituents positioned in the ortho (Fig. 4) or meta (Fig. S28, ESI†) position. For the derivatives having a meta NO2 group (11–18), the influence of additional substituents is different depending on their position (Fig. S28, ESI†). Indeed, the presence of electron-donating MeO in the para position leads to a larger 2PA response compared to derivative 11. We yet note that DPNDs 9 and 18 possessing Et2N substituents have markedly weaker two-photon absorption (Fig. 4 and Fig. S3, ESI†) most probably due to steric hindrance. Finally, adding an additional 1,4-phenylene moiety in para NO2 derivatives does not lead to an increase of the 2PA (Table 2 and Fig. S29, ESI†), indicating that the biphenyl twist may prevent extended delocalization favorable to 2PA enhancement.
| Dye | λ max12PA/nm | σ max12/GM | λ max2,max32PA/nm | σ max2,max32/GM |
|---|---|---|---|---|
| 1 | 830 | 27 | 750 | 44 |
| 2 | 820 (sh) | 707 | 755(sh)/<690 | 1204/>3718 |
| 3 | 820 | 102 | 755/690 | 176/332 |
| 4 | 840 | 270 | 755(sh)/720(sh) | 396/557 |
| 5 | 820 | 47 | 765/720 | 152/157 |
| 6 | 820 | 275 | 765(sh)/720(sh) | 496/809 |
| 7 | 810 | 401 | 770/720 | 569/959 |
| 8 | 820(sh) | 288 | 755/<690 | 607/>1134 |
| 9 | 860(sh) | 0.7 | 755/720 | 4.3/5.6 |
| 10 | 840(sh) | 319 | 765(sh)/720 | 856/1384 |
| 11 | 840 | 52 | 770(sh)/730 | 115/190 |
| 12 | 820 | 47 | 760(sh)/720(sh) | 89/113 |
| 13 | 820 | 43 | 760(sh)/720 | 95/138 |
| 14 | 820 | 25 | 760(sh)/720(sh) | 47/60 |
| 15 | 880 | 31 | 820(sh)/700 | 46/162 |
| 16 | 890 | 84 | 790/730 | 272/310 |
| 17 | 890 | 89 | 770(sh)/700 | 279/513 |
| 18 | 940 | 27 | 810/755 | 86/104 |
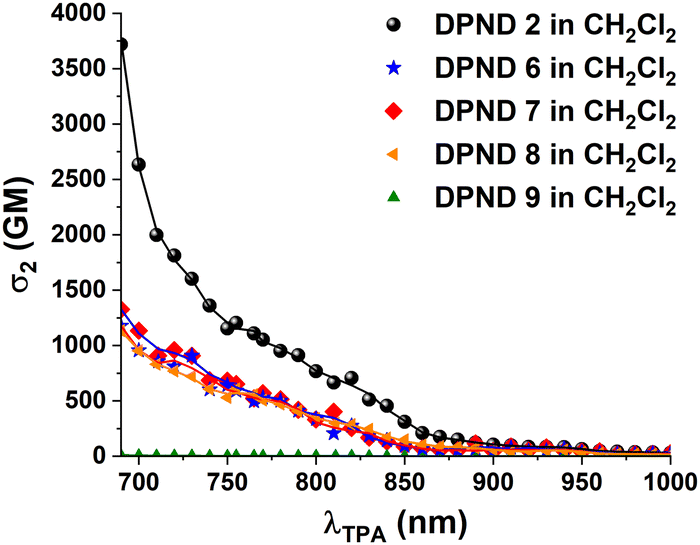 | ||
| Fig. 4 Two-photon absorption spectrum of DPND 2, 6–9 in CH2Cl2. | ||
To conclude, we found that strong fluorescence is a general feature in quadrupolar, centrosymmetric dipyrrolonaphthyridinediones possessing nitrophenyl substituents. Two-photon allowed transition to higher excited states is responsible for the main two-photon absorption band located around 700 nm. Combining the steric and electronic effect of various substituents enables the fine-tuning of both one-photon and two-photon absorption reaching emission at ca. 670 nm and a strong 2PA response (two-photon brightness >1500 GM). Careful selection of the substitution pattern allows for effective control of the optical response, which builds the foundation for designing efficient one- or two-photon absorbers based on other cross-conjugated chromophores.
Conceptualization: B. S.; investigation: B. S., G. C., M. K., P. P.; supervision: D. T. G., M. B.-D., P. P.; visualization: B. S., Y. M. P.; writing – original draft: B. S, D. T. G.; writing – review & editing: D. T. G., M. B. D, B. S.
The work was financially supported by the National Science Centre, Poland (Sonata 2021/43/D/ST4/02267 and OPUS 2020/37/B/ST4/00017) and the Foundation for Polish Science (TEAM POIR.04.04.00-00-3CF4/16-00).
Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) X. Yang, X. Lin, Y. S. Zhao and D. Yan, Chem. – Eur. J., 2018, 24, 6484–6493 CrossRef CAS PubMed; (b) A. Minotto, P. A. Haigh, G. Łukasiewicz, E. Lunedei, D. T. Gryko, I. Darwazeh and F. Cacialli, Light: Sci. Appl., 2020, 9, 70 CrossRef PubMed.
- (a) P. Kasperkiewicz, Y. Altman, M. D’Angelo, G. S. Salvesen and M. Drag, J. Am. Chem. Soc., 2017, 139, 10115–10125 CrossRef CAS PubMed; (b) W. Wang, C. P. Wildes, T. Pattarabanjird, M. I. Sanchez, G. F. Glober, G. A. Matthews, K. M. Tye and A. Y. Ting, Nat. Biotech., 2017, 35, 864–871 CrossRef CAS; (c) N. C. Payne, A. S. Kalyakina, K. Singh, M. A. Tye and R. Mazitschek, Nat. Chem. Biol., 2021, 17, 1168–1177 CrossRef CAS PubMed; (d) E. D. Cosco, B. A. Arús, A. L. Spearman, T. L. Atallah, I. Lim, O. S. Leland, J. R. Caram, T. S. Bischof, O. T. Bruns and E. M. Sletten, J. Am. Chem. Soc., 2021, 143, 6836–6846 CrossRef CAS; (e) J. B. Grimm, A. J. Sung, W. R. Legant, P. Hulamm, S. M. Matlosz, E. Betzig and L. D. Lavis, ACS Chem. Biol., 2013, 8, 1303–1310 CrossRef CAS PubMed.
- (a) W. Zhu, Y. Wu, S. Wang, W. Li, X. Li, J. Chen, Z.-S. Wang and H. Tian, Adv. Funct. Mater., 2011, 21, 756–763 CrossRef CAS; (b) H. Qu and C. Chi, Org. Lett., 2010, 12, 3360–3363 CrossRef CAS PubMed; (c) B. Song, Q. Zhang, W.-H. Ma, X.-J. Peng, X.-M. Fu and B.-S. Wang, Dyes Pigm., 2009, 82, 396–400 CrossRef CAS.
- (a) M.-C. Chen, D.-G. Chen and P.-T. Chou, ChemPlusChem, 2021, 86, 11–27 CrossRef CAS PubMed; (b) Y. M. Poronik, B. Sadowski, K. Szychta, F. H. Quina, V. I. Vullev and D. T. Gryko, J. Mater. Chem. C, 2022, 10, 2870–2904 RSC; (c) W. Rodriguez-Cordoba, L. Gutierrez-Arzaluz, F. Cortes-Guzman and J. Peon, Chem. Commun., 2021, 57, 12218–12235 RSC.
- (a) E. Braselmann, A. J. Wierzba, J. T. Polaski, M. Chromiński, Z. E. Holmes, S.-T. Hung, D. Batan, J. R. Wheeler, R. Parker, R. Jimenez, D. Gryko, R. T. Batey and A. E. Palmer, Nat. Chem. Biol., 2018, 14, 964–971 CrossRef CAS PubMed; (b) W. Chen, H. Luo, X. Liu, J. W. Foley and X. Song, Anal. Chem., 2016, 88, 3638–3646 CrossRef CAS PubMed; (c) H. Song, Y. Zhou, H. Qu, C. Xu, X. Wang, X. Liu, Q. Zhang and X. Peng, Ind. Eng. Chem. Res., 2018, 57, 15216–15223 CrossRef CAS; (d) A. Gomes, E. Fernandes and J. L. F. C. Lima, J. Fluoresc., 2006, 16, 119–139 CrossRef CAS PubMed; (e) M. Abo, Y. Urano, K. Hanaoka, T. Terai, T. Komatsu and T. Nagano, J. Am. Chem. Soc., 2011, 133, 10629–10637 CrossRef CAS PubMed; (f) J. A. Marsden, J. J. Miller, L. D. Shirtcliff and M. M. Haley, J. Am. Chem. Soc., 2005, 127, 2464–2476 CrossRef CAS PubMed.
- (a) C.-K. Wang, P. Macak, Y. Luo and H. Ågren, J. Chem. Phys., 2001, 114, 9813–9820 CrossRef CAS; (b) W. Ma, Y. Wu, D. Gu and F. Gan, THEOCHEM, 2006, 772, 81–87 CrossRef CAS; (c) H. Myung Kim and B. Rae Cho, Chem. Commun., 2009, 153–164 RSC; (d) T. Verbiest, S. Houbrechts, M. Kauranen, K. Clays and A. Persoons, J. Mater. Chem., 1997, 7, 2175–2189 RSC.
- T. Ueno, Y. Urano, H. Kojima and T. Nagano, J. Am. Chem. Soc., 2006, 128, 10640–10641 CrossRef CAS PubMed.
- K. Skonieczny, I. Papadopoulos, D. Thiel, K. Gutkowski, P. Haines, P. M. McCosker, A. D. Laurent, P. A. Keller, T. Clark, D. Jacquemin, D. M. Guldi and D. T. Gryko, Angew. Chem., Int. Ed., 2020, 59, 16104–16113 CrossRef CAS PubMed.
- (a) A. Bolduc, Y. Dong, A. Guérin and W. G. Skene, Phys. Chem. Chem. Phys., 2012, 14, 6946–6956 RSC; (b) S. Hachiya, K. Asai and G.-I. Konishi, Tetrahedron Lett., 2013, 54, 3317–3320 CrossRef CAS; (c) J. S. Siddle, R. M. Ward, J. C. Collings, S. R. Rutter, L. Porrès, L. Applegarth, A. Beeby, A. S. Batsanov, A. L. Thompson, J. A. K. Howard, A. Boucekkine, K. Costuas, J.-F. Halet and T. B. Marder, New J. Chem., 2007, 31, 841–851 RSC.
- (a) H. Kotaka, G.-i Konishi and K. Mizuno, Tetrahedron Lett., 2010, 51, 181–184 CrossRef CAS; (b) M. A. B. Larsen, A. B. Stephansen, E. Alarousu, M. Pittelkow, O. F. Mohammed and T. I. Sølling, Phys. Chem. Chem. Phys., 2018, 20, 5942–5951 RSC.
- Y. Ueda, Y. Tanigawa, C. Kitamura, H. Ikeda, Y. Yoshimoto, M. Tanaka, K. Mizuno, H. Kurata and T. Kawase, Chem. – Asian J., 2013, 8, 392–399 CrossRef CAS PubMed.
- M.-C. Chen, Y.-L. Lee, Z.-X. Huang, D.-G. Chen and P.-T. Chou, Chem. – Eur. J., 2020, 26, 7124–7130 CrossRef CAS PubMed.
- M. Grzybowski, I. Deperasińska, M. Chotkowski, M. Banasiewicz, A. Makarewicz, B. Kozankiewicz and D. T. Gryko, Chem. Commun., 2016, 52, 5108–5111 RSC.
- (a) B. Sadowski, M. Loebnitz, D. R. Dombrowski, D. H. Friese and D. T. Gryko, J. Org. Chem., 2018, 83, 11645–11653 CrossRef CAS PubMed; (b) B. Sadowski, M. F. Rode and D. T. Gryko, Chem. – Eur. J., 2018, 24, 855–864 CrossRef CAS PubMed.
- M. Grzybowski, D. T. Gryko, B. Sadowski, K. Strassel, D. Kaelblein and P. Hayoz, Polymers and compounds based on dipyrrolo[1,2-b:1′,2′-g][2,6]naphthyridine-5,11-dione, Int. Pat., WO2017/068009(A1), 2017 Search PubMed.
- (a) L. Wang, L. Lin, J. Yang, Y. Wu, H. Wang, J. Zhu, J. Yao and H. Fu, J. Am. Chem. Soc., 2020, 142, 10235–10239 CrossRef CAS PubMed; (b) L. Wang, W. Cai, J. Sun, Y. Wu, B. Zhang, X. Tian, S. Guo, W. Liang, H. Fu and J. Yao, J. Phys. Chem. Lett., 2021, 12, 12276–12282 CrossRef CAS PubMed.
- J. Morgan, Y. J. Yun and A. J.-L. Ayitou, Photochem. Photobiol., 2022, 98, 57–61 CrossRef CAS PubMed.
- B. Sadowski, M. Kaliszewska, Y. M. Poronik, M. Czichy, P. Janasik, M. Banasiewicz, D. Mierzwa, W. Gadomski, T. D. Lohrey, J. A. Clark, M. Lapkowski, B. Kozankiewicz, V. I. Vullev, A. L. Sobolewski, P. Piatkowski and D. T. Gryko, Chem. Sci., 2021, 12, 14039–14049 RSC.
- B. Sadowski, H. Kita, M. Grzybowski, K. Kamada and D. T. Gryko, J. Org. Chem., 2017, 82, 7254–7264 CrossRef CAS PubMed.
- (a) M. J. Wirth, A. Koskelo and M. J. Sanders, Appl. Spectrosc., 1981, 35, 14–21 CrossRef CAS; (b) C. W. Stark, M. Rammo, A. Trummal, M. Uudsemaa, J. Pahapill, M.-M. Sildoja, S. Tshepelevitsh, I. Leito, D. C. Young, B. Szymański, O. Vakuliuk, D. T. Gryko and A. Rebane, Angew. Chem., Int. Ed., 2022, 61, e202212581 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, 1H NMR and 13C NMR spectra, and steady-state and time-resolved spectra. See DOI: https://doi.org/10.1039/d3cc03347c |
| This journal is © The Royal Society of Chemistry 2023 |