
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Macrocyclic peptides as inhibitors of WDR5–lncRNA interactions†
Jen-Yao
Chang
 ,
Cora
Neugebauer
,
Stefan
Schmeing
,
Gulshan
Amrahova
and
Peter
‘t Hart
*
,
Cora
Neugebauer
,
Stefan
Schmeing
,
Gulshan
Amrahova
and
Peter
‘t Hart
*
Chemical Genomics Centre of the Max Planck, Society Max Planck Institute of Molecular Physiology, Otto-Hahn-Strasse 11, 44227 Dortmund, Germany. E-mail: peter.t-hart@mpi-dortmund.mpg.de
First published on 8th August 2023
Abstract
WDR5 is an adaptor protein involved in the regulation of various epigenetic modifier complexes. Various inhibitors have been described but only as inhibitors of its protein–protein interactions. Here we describe peptidic macrocycles that act as inhibitors of the interaction between WDR5 and long non-coding RNAs. The findings provide a new strategy to modulate the biological function of WDR5 as an RNA binding epigenetic regulator.
Long non-coding RNAs (lncRNAs) are transcripts longer than 200 nucleotides but without protein coding function.1 They can have a wide variety of functions but one that has been identified for many lncRNAs is to guide proteins involved in epigenetic mechanisms to specific genomic locations.2–4 By doing so, they can promote histone modifications at these sites to either activate or repress gene transcription. Various studies have demonstrated that these lncRNA–protein interactions could be interesting targets for therapeutic intervention.5–8 However, evidence that such interactions can be targeted by chemical inhibitors is scarce as there are only very few compounds reported to do so and these only include inhibitors of EZH2–lncRNA binding.9,10 To extend the knowledge on inhibiting such interactions beyond EZH2 we focused on WDR5 as another lncRNA binding protein involved in epigenetic regulation.
As a scaffold protein WDR5 is part of the “WRAD” complex (containing WDR5, RbBP5, ASH2L, and DPY30) which can recruit the SET1/MLL histone methyltransferase to methylate histone 3 at lysine 4.11–13 The function of the MLL complex is commonly found to be exacerbated in cancer and has therefore been explored as a therapeutic target using a variety of strategies.14,15 WDR5 has a WD40 fold common for scaffolding proteins and contains two binding sites that mediate protein–protein interactions.16,17 One found on the “top” of the protein (WIN site) and one found on the “bottom” (WBM site).12 The WBM site was described to bind RbBP5, c-Myc, and KANSL but interestingly, it has also been reported to interact with long non-coding RNA (lncRNA) (Fig. 1A) that drive the activity of the MLL complex at specific genomic loci.18–21 A well-studied lncRNA that binds WDR5 via the WBM site is HOTTIP.21,22 Besides HOTTIP, which has been described to be a driver in various cancers, the WBM site binds many other lncRNAs.21–24
 | ||
| Fig. 1 (A) Left: Reported structure of WDR5 with RbBP5 peptide (PDB ID: 3P4F). Highlighted in orange (Y228, L240, and K250) and red (F266) are residues important for lncRNA binding. Right: Sequences of WBM site binding peptides of various proteins. (B) Close up of RbBP5 derived peptide bound to WDR5. Highlighted is the hydrogen bonding pattern formed by Asp376. | ||
Although a large number of WIN site inhibitors have been reported,25 the number of inhibitors for the WBM site is still limited.26–29 Furthermore, their effect on inhibition of WDR5–lncRNA interactions has not been investigated to date. Here, we report the design and optimization of a macrocyclic peptide derived from protein sequences that bind the WBM site. The binding of the peptide was studied using biophysical techniques as well as protein crystallography. Using RNA-immunoprecipitation experiments we were able to demonstrate that the cyclic peptides could indeed inhibit the interaction between WDR5 and lncRNAs confirming the role of the WBM site as an RNA-binding site.
To identify the core binding sequence of RbBP5 a series of truncated peptides was synthesized based on the WDR5/RbBP5 cocrystal structure reported by Avdic et al. (Fig. 1A and B) and we measured their affinity for WDR5 using fluorescence polarization (FP).18 To study truncation of the C-terminus we placed the FITC fluorophore on the N-terminus (Table 1, peptides 1–4, Fig. S34, ESI†) while switching the label to the C-terminus for N-terminal truncation (Table 1, peptides 5–9, Fig. S35, ESI†). Both series demonstrated that a single amino acid truncation led to a small decrease in affinity (approx. 3-fold) but further truncation had more dramatic effects. This was confirmed by peptide 10 which was truncated by one residue on both termini and had an affinity of 10.4 μM (Table 1 and Fig. S36, ESI†). The truncation effects correlated with the binding pose observed in the crystal structure of RbBP5 bound to WDR5 where the more terminal amino acids are not visible likely due to flexibility (i.e.: Glu371 and Asp381). Residues that lead to a strong reduction in binding affinity after truncation are involved in direct interactions with WDR5. In most cases this is driven by the side chains of these residues with the exception of Asp376 and Ser379. The side chains of these residues do not directly interact with WDR5 itself but form a hydrogen bond with each other thereby stabilizing the conformation of the peptide (Fig. 1B).

|
To generate a more potent WDR5 WBM site ligand we were inspired by the intramolecular stabilization between Asp376 and Ser379 and explored the possibility of converting this inter-action into an even more stable covalent connection. To this end, we used peptide 10 as our starting point and replaced Ser379 with either L-diaminopropionic acid (Dap), L-diaminobutyric acid (Dab), or L-ornithine (Orn). These were incorporated during peptide synthesis as Alloc protected amino acids and Asp376 was incorporated with an allyl side chain protecting group (Scheme S1A, ESI†). The orthogonal protecting groups were removed, and the liberated amine and carboxylic acid connected together using PyAOP and HOAt to generate amide cyclized peptides 11–13. Evaluation of these compounds by FP demonstrated a clear effect of the macrocycle size (Table 1 and Fig. S36, ESI†). The smallest macrocycle 11 did not provide the correct conformation and had a reduced affinity in comparison to peptide 10. However, both peptide 12 and 13 with the Dab and Orn residues provided an improvement in affinity with peptide 12 being the most potent at 1.18 μM. Although the improvement in affinity was encouraging, we expected that for biochemical studies a more potent ligand would be preferred. We therefore explored the alternative peptide sequences from c-Myc (peptide 14) and KANSL (peptide 15) that were reported to bind the WBM site as well (Table 1 and Fig. S37, ESI†).19,20 When tested by FP we found that peptide 15 had a reduced affinity in comparison to peptide 1, but the c-Myc derived peptide 14 was significantly more potent. To prepare a high affinity ligand we now proceeded with this sequence and applied the two most promising cyclization strategies as identified with peptides 12 and 13. To get the highest affinity we decided to explore extending the N- as well as both the N- and C-terminus and designed peptides 16–19 (Table 1 and Fig. S38, ESI†). During synthesis of peptides 18 and 19 complications were encountered due to premature Fmoc removal after the allyl deprotection step presumably due to the free amino side chain now being able to act as the base for Fmoc deprotection. We therefore removed the Fmoc group first and replaced it with the o-nitrobenzenesulfonyl (o-Ns) group before allyl deprotection and cyclization (see Scheme S1B, ESI†). Again, the Dab containing peptide performed better in the FP assay and the full-length peptide 18 had a 3-fold improved affinity over peptide 14.
With peptide 18 being the most potent candidate we further evaluated whether we could optimize the affinity by point mutation (Table 2 and Fig. S39, ESI†). In peptide 20 we mutated Val265 to the threonine residue found in the RbBP5 peptide since its side chain hydroxyl group was involved in a hydrogen bond interaction with Lys272 from WDR5.18 Peptide 21 has a Val265 to Ile mutation in an attempt to address a small hydrophobic pocket observed in the Val binding site on WDR5 while peptide 22 contained both mutations. Unfortunately, none of the mutants led to increased affinity.
| Pept. | N | K D (nM) | ΔG (kcal mol−1) | ΔH (kcal mol−1) | −TΔS (kcal mol−1) |
|---|---|---|---|---|---|
| 14 | 0.82 ± 0.01 | 805 ± 161 | −8.32 ± 0.11 | −5.67 ± 0.64 | −2.66 ± 0.77 |
| 18 | 0.95 ± 0.07 | 199 ± 64 | −9.16 ± 0.20 | −6.94 ± 0.62 | −2.23 ± 0.42 |
To explore the role of the amide cyclization linker and its effect on affinity we synthesized three variants of peptide 18 (Table 2 and Fig. S40, ESI†). In peptides 23–25 we maintained the same number of atoms in the macrocycle but shifted the amide one atom over (peptide 23), inverted its orientation (peptide 24), or both inverted and shifted it (peptide 25). These peptides highlighted the importance of the correct linker orientation as all modifications led to a complete loss of binding affinity. Since this part of the peptide does not interact with WDR5 directly this is most likely caused by the peptides adopting conformations that are not compatible with binding.
The advantage of cyclized peptides over linear peptides typically lies in a reduced flexibility that leads to a reduction in the entropic penalty paid upon binding to the protein target. We evaluated this parameter using isothermal titration calorimetry (ITC) for peptides 14 and 18. The affinities by ITC corresponded well with those determined by FP but surprisingly no clear effect of conformational stabilization was observed as the −TΔS component is not significantly different (Table 2 and Table S3, Fig. S5, S6, ESI†). Instead, a difference in enthalpy seems to be the main driver for the difference in affinity. The minimal difference in entropic penalty could be caused by the linear peptide being stabilized in its active conformation by the intramolecular hydrogen bond network formed by Asp376 (Fig. 1B) or through compensation by the displacement of bound water molecules.
To evaluate the binding mode, we cocrystallized WDR5 and an acetylated version of peptide 18 (18Ac). Peptide 18Ac was found to have an identical binding affinity as peptide 18 as measured by competitive fluorescence polarization (Fig. S41 and Table S6, ESI†). The crystal structure clearly shows that the peptide binds the WBM site (Fig. 2A–C), and that the macrocyclic component indeed mimics the bound conformation of the linear c-Myc peptide (Fig. 2B). Significant differences are observed in the acidic residues of the N-terminal part of the peptides, but the electron density in this region is weak indicating flexibility. Closer to the macrocyclic core the differences are smaller and only slight changes in side chain orientation are observed.
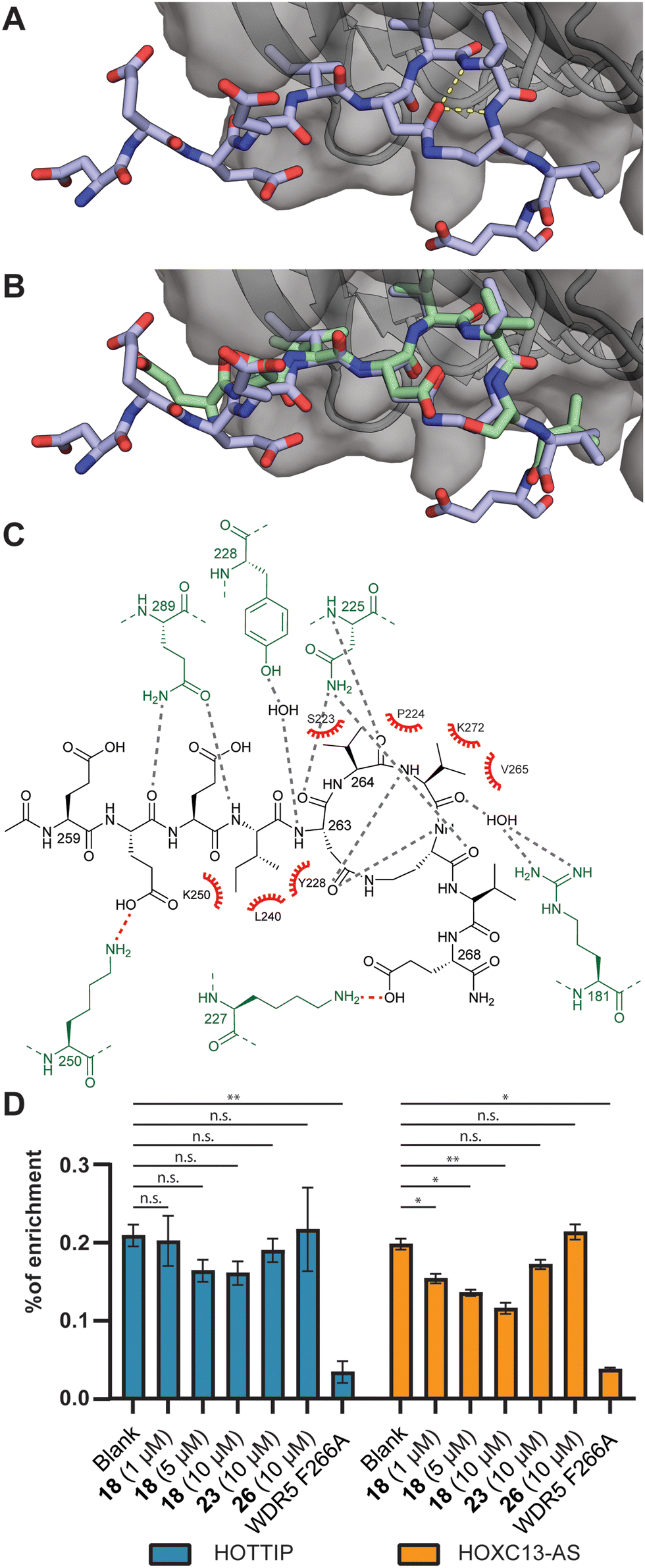 | ||
| Fig. 2 (A) Crystal structure of the acetylated version of peptide 18Ac bound to WDR5 at 1.8 Å resolution (PDB ID: 8Q1N). (B) Overlay of 18Ac with the structure of the parent cMyc peptide (green, PDB ID: 4Y7R). (C) Interaction analysis of 18Ac and WDR5. WDR5 residues making polar contacts indicated in green, hydrogen bonds indicated in grey, electrostatic interactions in red and hydrophobic interactions with red spoked arcs. (D) RNA-immunoprecipitation experiments using peptide 18, 23, and a WIN-site binding peptide 26. Analysis was done by RT-qPCR. n.s.: p > 0.05, *: p ≤ 0.05, **: p ≤ 0.01. | ||
The binding of lncRNAs to WDR5 was evaluated using an RNA-immunoprecipitation assay by using FLAG-tagged WDR5 and RNA extracts isolated from U2OS cells. We investigated the interaction between WDR5 and the previously reported lncRNA binding partners HOTTIP and HOXC13-AS.21,24 The results demonstrate that increasing concentrations of peptide 18 inhibited lncRNA binding albeit more efficient for HOXC13-AS than for HOTTIP (Fig. 2D and Table S7, ESI†). The negative control peptide 23 was not able inhibit the interaction and neither was a peptide designed to bind to the WIN-site (26) rather than the lncRNA binding WBM-site.30 The F266A mutant was previously reported to have a diminished ability to bind lncRNA which was corroborated in this experiment.22,24
Therapeutic modulation of the WIN-site of WDR5 has been demonstrated to be an effective strategy for tumor therapy. However, the WBM-site remains relatively unexplored and inhibitors were only studied for their effect on protein–protein interactions.25 Here, we developed potent cyclic peptide inhibitors to evaluate the potential of blocking the WBM-site of WDR5 to inhibit lncRNA binding. After exploring various WBM site binding peptides and cyclization strategies, a cyclic peptide derived from c-Myc provided a nanomolar affinity ligand. Protein crystallography demonstrated that the macrocycle efficiently mimicked an intramolecular hydrogen bond observed in the linear bound peptide. With a potent inhibitor in hand, we set out to investigate whether the lncRNA binding capacity of WDR5 could indeed be inhibited. The RNA-immunoprecipitation assays indeed showed that dose dependent inhibition of lncRNA binding was possible for both HOTTIP and HOXC13-AS. The work described here provides insight into how targeting the WBM site of WDR5 not only affects its protein–protein interactions but also its protein–RNA interactions which will add to the therapeutic effect. As one of the first studies to investigate this property of an epigenetic regulation protein we expect that this principle can be extended to other epigenetic regulator complexes as well.
Open Access funding provided by the Max Planck Society. We acknowledge the Ministry of Education of Taiwan for funding J.-Y. C. with a Government Scholarship for Overseas Study and the German Academic Exchange Service (DAAD) for funding G. A. with a PhD scholarship.
Conflicts of interest
There are no conflicts to declare.Notes and references
- L. Statello, C. J. Guo, L. L. Chen and M. Huarte, Nat. Rev. Mol. Cell Biol., 2021, 22, 96–118 CrossRef CAS PubMed.
- A. M. Khalil, M. Guttman, M. Huarte, M. Garber, A. Raj, D. R. Morales, K. Thomas, A. Presser, B. E. Bernstein, A. Van Oudenaarden, A. Regev, E. S. Lander and J. L. Rinn, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 11667–11672 CrossRef CAS PubMed.
- T. R. Mercer and J. S. Mattick, Nat. Struct. Mol. Biol., 2013, 20, 300–307 CrossRef CAS PubMed.
- J. T. Lee, Science, 2012, 338, 1435–1439 CrossRef CAS PubMed.
- L. Wu, P. Murat, D. Matak-Vinkovic, A. Murrell and S. Balasubramanian, Biochemistry, 2013, 52, 9519–9527 CrossRef CAS PubMed.
- A. Henrik, Cancer Lett., 2014, 419, 1–47 Search PubMed.
- G. Lavorgna, R. Vago, M. Sarmini, F. Montorsi, A. Salonia and M. Bellone, Pharmacol. Res., 2016, 110, 131–138 CrossRef CAS PubMed.
- E. J. McFadden and A. E. Hargrove, Biochemistry, 2016, 55, 1615–1630 CrossRef CAS PubMed.
- Y. Li, Y. Ren, Y. Wang, Y. Tan, Q. Wang, J. Cai, J. Zhou, C. Yang, K. Zhao, K. Yi, W. Jin, L. Wang, M. Liu, J. Yang, M. Li and C. Kang, Theranostics, 2019, 9, 4608–4623 CrossRef CAS PubMed.
- R. Pedram Fatemi, S. Salah-Uddin, F. Modarresi, N. Khoury, C. Wahlestedt and M. A. Faghihi, J. Biomol. Screening, 2015, 20, 1132–1141 CrossRef PubMed.
- P. Ernst and C. R. Vakoc, Briefings Funct. Genomics, 2012, 11, 217–226 CrossRef CAS PubMed.
- A. Guarnaccia and W. Tansey, J. Clin. Med., 2018, 7, 21 CrossRef PubMed.
- J. Wysocka, T. Swigut, T. A. Milne, Y. Dou, X. Zhang, A. L. Burlingame, R. G. Roeder, A. H. Brivanlou and C. D. Allis, Cell, 2005, 121, 859–872 CrossRef CAS PubMed.
- R. C. Rao and Y. Dou, Nat. Rev. Cancer, 2015, 15, 334–346 CrossRef CAS PubMed.
- N. L. Alicea-Velázquez, S. A. Shinsky, D. M. Loh, J. H. Lee, D. G. Skalnik and M. S. Cosgrove, J. Biol. Chem., 2016, 291, 22357–22372 CrossRef PubMed.
- F. Gori, P. Divieti and M. B. Demay, J. Biol. Chem., 2001, 276, 46515–46522 CrossRef CAS PubMed.
- X.-J. Hu, T. Li, Y. Wang, Y. Xiong, X.-H. Wu, D.-L. Zhang, Z.-Q. Ye and Y.-D. Wu, Sci. Rep., 2017, 7, 10585 CrossRef PubMed.
- V. Avdic, P. Zhang, S. Lanouette, A. Groulx, V. Tremblay, J. Brunzelle and J. F. Couture, Structure, 2011, 19, 101–108 CrossRef CAS PubMed.
- S. Lorey, L. R. Thomas, S. W. Fesik, Z. Zhao, B. C. Grieb, W. P. Tansey, B. Cawthon, E. T. Olejniczak, Q. Wang, J. Phan, G. C. Howard, T. Clark, C. M. Eischen, S. Dey, Q. Sun, A. M. Foshage, B. Alicie and K. C. Ess, Mol. Cell, 2015, 58, 440–452 CrossRef PubMed.
- J. Dias, N. Van Nguyen, P. Georgiev, A. Gaub, J. Brettschneider, S. Cusack, J. Kadlec and A. Akhtar, Genes Dev., 2014, 28, 929–942 CrossRef CAS PubMed.
- K. C. Wang, Y. W. Yang, B. Liu, A. Sanyal, R. Corces-Zimmerman, Y. Chen, B. R. Lajoie, A. Protacio, R. A. Flynn, R. A. Gupta, J. Wysocka, M. Lei, J. Dekker, J. A. Helms and H. Y. Chang, Nature, 2011, 472, 120–126 CrossRef CAS PubMed.
- Y. W. Yang, R. A. Flynn, Y. Chen, K. Qu, B. Wan, K. C. Wang, M. Lei and H. Y. Chang, eLife, 2014, 3, e02046 CrossRef PubMed.
- S. Ghafouri-Fard, S. Dashti and M. Taheri, Biomed. Pharmacother., 2020, 127, 110158 CrossRef CAS PubMed.
- S. Subhash, K. Mishra, V. S. Akhade, M. Kanduri, T. Mondal and C. Kanduri, Nucleic Acids Res., 2018, 46, 9384–9400 CrossRef CAS PubMed.
- X. Chen, J. Xu, X. Wang, G. Long, Q. You and X. Guo, J. Med. Chem., 2021, 64, 10537–10556 CrossRef CAS PubMed.
- J. D. Macdonald, S. Chacón Simon, C. Han, F. Wang, J. G. Shaw, J. E. Howes, J. Sai, J. P. Yuh, D. Camper, B. M. Alicie, J. Alvarado, S. Nikhar, W. Payne, E. R. Aho, J. A. Bauer, B. Zhao, J. Phan, L. R. Thomas, O. W. Rossanese, W. P. Tansey, A. G. Waterson, S. R. Stauffer and S. W. Fesik, J. Med. Chem., 2019, 62, 11232–11259 CrossRef CAS PubMed.
- S. Chacon Simon, F. Wang, L. R. Thomas, J. Phan, B. Zhao, E. T. Olejniczak, J. D. Macdonald, J. G. Shaw, C. Schlund, W. G. Payne, J. Creighton, S. Stauffer, A. G. Waterson, W. P. Tansey and S. W. Fesik, J. Med. Chem., 2020, 63, 4315–4333 CrossRef CAS PubMed.
- J. Ding, G. Li, H. Liu, L. Liu, Y. Lin, J. Gao, G. Zhou, L. Shen, M. Zhao, Y. Yu, W. Guo, U. Hommel, J. Ottl, J. Blank, N. Aubin, Y. Wei, H. He, D. R. Sage, P. W. Atadja, E. Li, R. K. Jain, J. A. Tallarico, S. M. Canham, Y.-L. Chiang and H. Wang, ACS Chem. Biol., 2023, 18, 34–40 CrossRef CAS PubMed.
- Q. Han, X. Zhang, P. Ren, L. Mei, W. Lin, L. Wang, Y. Cao, K. Li and F. Bai, Acta Pharmacol. Sin., 2023, 44, 877–887 CrossRef CAS PubMed.
- H. Karatas, E. C. Townsend, D. Bernard, Y. Dou and S. Wang, J. Med. Chem., 2010, 53, 5179–5185 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3cc03221c |
| This journal is © The Royal Society of Chemistry 2023 |