
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAn amorphous Lewis-acidic zirconium chlorofluoride as HF shuttle: C–F bond activation and formation†
Christian
Heinekamp
 ab,
Ana Guilherme
Buzanich
a,
Mike
Ahrens
b,
Thomas
Braun
*b and
Franziska
Emmerling
*ab
ab,
Ana Guilherme
Buzanich
a,
Mike
Ahrens
b,
Thomas
Braun
*b and
Franziska
Emmerling
*ab
aDepartment Materials Chemistry, Federal Institute for Material Research and Testing, Richard-Willstätter-Straße 11, 12489 Berlin, Germany. E-mail: franziska.emmerling@bam.de
bDepartment of Chemistry, Humboldt Universität zu Berlin, Brook-Taylor-Straße 2, 12489 Berlin, Germany. E-mail: thomas.braun@cms.hu-berlin.de
First published on 22nd August 2023
Abstract
An exceptional HF transfer reaction by C–F bond activation of fluoropentane and a subsequent hydrofluorination of alkynes at room temperature is reported. An amorphous Lewis-acidic Zr chlorofluoride serves as heterogeneous catalyst, which is characterised by an eightfold coordination environment at Zr including chlorine atoms. The studies are seminal in establishing sustainable fluorine chemistry.
In the past two decades, significant progress has been made in the development of heterogeneously catalysed reactions using amorphous aluminium fluorides as catalysts.1–6 The latter include a unique aluminium chlorofluoride (ACF, AlClxF3−x, with x = 0.3–0.05),7–11 which is a nanoscopic Lewis superacid. Various C–F bond activation reactions resulting either in Friedel–Crafts alkylation, dehydrofluorination or hydrodefluorination were developed. On the other hand, a hydrofluorination of alkynes using a HF-loaded ACF derivative was observed.12–14 However, only a few examples of catalytic C–F bond cleavage reactions coupled with a transfer of fluorine atoms to another organic fragment were reported, although examples for catalytic reactions, which involve the transfer of groups containing other halogen atoms are known.15–20 However, in 2010 the enantioselective addition of HF to an epoxide was reported on using a cobalt salen catalyst, in which HF was generated from benzoyl fluoride and an alcohol.21 A very recent report by Crimmin et al. imparts HF formation from perfluoroarenes and alcohols or amines followed by a catalytic hydrofluorination of alkynes using Au(I) complexes at 120 °C in toluene.22 Furthermore they described the use of BF3·OEt at 80 °C for the dehydrofluorination of fluorohexane, which was coupled with a difluorination of an alkyne.23 Note that in general, the transfer of fluorine atoms by cleavage and formation of C–F bonds will contribute to sustainability in fluorine chemistry, which is an inevitable implication due to the growing shortage of fluorspar CaF2 that has been placed among the 30 critical raw materials in the European Union in 2020.24 Herein we report on unique HF transfer reactions that involve a dehydrofluorination of fluoroalkanes coupled with a hydrofluorination of alkynes to yield olefins and difluorinated alkanes (Scheme 1). The conversions proceed at room temperature and are heterogeneously catalysed by an amorphous zirconium chlorofluoride (ZCF). The latter was characterized by a variety of methods.
 | ||
| Scheme 1 Catalytic HF transfer from fluoropentane to alkynes (R: alkyl, aryl, R′: H, alkyl); for further details see Table 1 and ESI.† | ||
ZCF was synthesized by fluorination of ZrCl4 with CFCl3.7,25 Note that ZCF was patented in 199225 and was claimed to be an Lewis superacid analogue to ACF. However, this was never confirmed by any reactivity studies and ZCF was so far not structurally characterized either.9 Our XRD data reveal that it is an X-ray amorphous material (see ESI†) with a surface area of 95 m2 g−1, as determined by N2 adsorption experiments and BET analysis. This value is low when compared to data for ACF which can exhibit a surface area of up to 334 m2 g−1.26 A type H4 hysteresis for ZCF indicates slit-like pores at the surface.27 BJH analysis shows that ZCF is mesoporous and has pores of 35 Å diameter, which is different for the microporous ACF (<15 Å).26,28 The pore volume of 0.15 cm3 g−1 exceeds the reported pore volume of ACF (0.11 cm3 g−1).26 TEM and STEM measurements confirmed the amorphous nature of the material and indicate particle sizes up to 1 μm. The homogenous distribution of chlorine atoms was confirmed by STEM/EDX experiments, which suggest a sum formula of ZrCl0.2F3.8 for ZCF (Fig. 1). The major difference in surface area (see above) between ZCF and ACF might be due to the presence of larger particles.
 | ||
| Fig. 1 STEM/EDX mapping of a ZCF particle. | ||
To evaluate the Lewis acidity of ZCF, it was loaded with CD3CN, which adsorbs to the Lewis-acidic sites at the surface. The adsorption resulted in a blueshift of the C≡N vibrational stretching mode to 2316 cm−1 in the DRIFTS spectrum (Fig. 2). The shift of 58 cm−1 is lower than the one observed of 68 cm−1 for the ACF·CD3CN adduct,29,30 which classifies ACF as a Lewis superacid. The DRIFTS data for ZCF are in accordance with the NH3-TPD experiments, which reveal Lewis-acidic sites of medium strength. The TPD trace (Fig. 3) shows one major NH3 desorption starting at 170 °C with a prominent desorption peak at 305 °C. There is also a smaller broad feature at 350 °C for stronger Lewis-acidic sites. TGA/DSC measurements (see ESI†) suggest the sublimation of ZrCl4 from the material at 370 °C resulting in decomposition. A comparable AlCl3 formation was reported for ACF.31
 | ||
| Fig. 2 DRIFTS spectrum of CD3CN loaded ZCF. | ||
 | ||
| Fig. 3 NH3-TPD trace of ZCF. | ||
Extended X-ray absorption fine structure spectroscopy (EXAFS) was performed at the Zr-K-edge (Fig. 4) and allowed for the investigation of the first coordination sphere at the Zr centers. It was found that the coordination environment is comparable to that in β-ZrF4.32 Taking the eightfold coordination as a basis, atomic distances between fluorine and zirconium were determined to be 2.05 Å to 2.21 Å, which is in good agreement to the bond lengths found for the β-ZrF4 model (2.04–2.19 Å). Furthermore, the EXAFS data confirmed the presence of chlorine atoms about the Zr atoms since modeling of a first coordination sphere consisting of only fluorine scattering paths led to discrepancies in the fitting results. (Fig. 4(b)). Therefore, an additional chlorine scattering path with low occupancy consistently with the STEM/EDX experiment was implemented in another fitting approach, while a fluorine scattering path was reduced in occupancy, resulting clearly in better fitting results (Fig. 4(a)). The chlorine scattering path was fitted to a distance of 2.49 Å to the absorbing Zr. Zr–Cl bond lengths are reported in the range between 2.31 Å and 2.65 Å.33 The low impact of the included chlorine scattering path on the main radial distance at approximately 2 Å (Fig. 4) implies that residual ZrCl4 is not present.
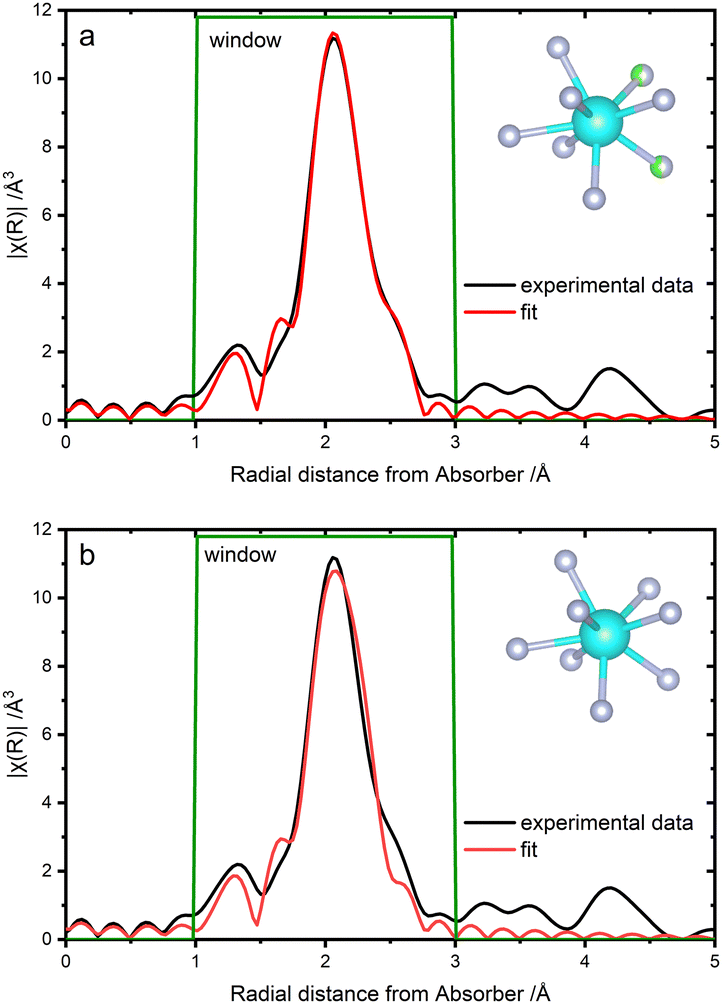 | ||
| Fig. 4 Fourier transform of the Zr-K-edge EXAFS data (black line) and fit model (red line). Magnitude of χ in real space from ZCF (a) using a fitting model including chlorine (R-factor = 0.014) and (b) using a fitting model solely based on fluorine atoms (R-factor = 0.027). | ||
Subsequently, the activity of ZCF in Friedel–Crafts and dehydrofluorination reactions of fluoropentane in C6D6 and C6D12 was studied. Treatment of ZCF with fluoropentane in C6D6 at room temperature led to the formation of Friedel–Crafts products, where the in situ formed carbeniumion species direct the obtained Friedel–Crafts product. Furthermore, only traces of the dehydrofluorination products (Scheme 2) were formed. In contrast, in C6D12 solely dehydrofluorination was observed to yield (E/Z)-pent-2-ene. Additionally, traces of the isomers 2-fluoropentane and 3-fluoropentane were formed (Scheme 2). Note that the presence of additional silanes, germanes or stannanes, which have been employed for defluorination reactions at ACF,14,34 is not necessary. Mechanistically, a carbenium ion-like species is presumably formed at the Lewis-acidic ZCF surface by abstraction of fluoride from the alkane. In a consecutive step HF is produced by deprotonation leading to the formation of the (E/Z)-pentene. Re-addition of HF to pentene at the ZCF surface can lead to 2- or 3-fluoropentane. Comparable isomerizations were described for polyfluorinated propane isomers, albeit they proceed in very low yields.35,36
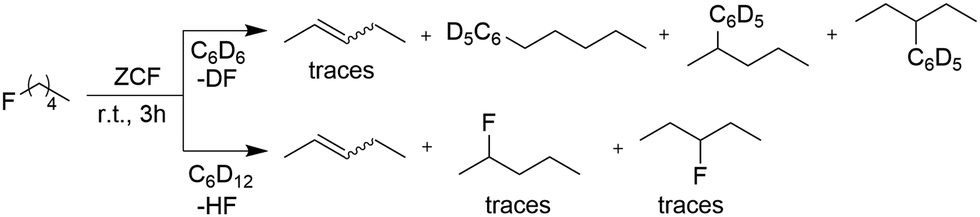 | ||
| Scheme 2 C–F bond activation of fluoropentane in C6D6 and C6D12 in the presence of ZCF. | ||
Based on this observation an HF shuttle was developed, which imparts a transfer of HF from fluoropentane to an alkyne. Thus, treatment of ZCF with hexyne and fluoropentane in C6D12 at room temperature led to the formation of the (E/Z)-pentene and the 3,3-difluorohexane as well as traces of the fluorohexene (Scheme 1). Using fluorocyclohexane gave higher conversions, possibly because of the higher stability of secondary carbenium ions (Table 1, entry 3).37 If, instead of an alkyne, styrene or trans-stilbene were used as HF acceptors, no reaction was observed.
| Entry | R | R′ | t [h] | Conversionbc [%] |
|---|---|---|---|---|
| a 15 mg of catalyst in a JYoung NMR tube using C6D12 as solvent. b Determined from 19F NMR data based on the fluoroalkane starting compound. c Ratio of difluoroalkane: monofluoroalkene in parentheses. d ACF used as catalyst. e Performed with fluorocyclohexane. | ||||
| 1 | Et | Et | 24 | 39 (60![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) :1) |
| 2d | Et | Et | 24 | 21 (13:1) |
| 3e | Et | Et | 24 | 53 (11:1) |
| 4 | C4H9 | H | 24 | 12 (10:1) |
| 5 | 4-C6H5(CF3) | H | 168 | 67 (31:1) |
| 6 | 3,5-C6H2(CH3)3 | H | 96 | 7 (0:1) |
| 7 | 4-C6H4F | H | 168 | 8 (1:2) |
The scope of the hydrofluorination reactions using ZCF as catalyst at room temperature was then studied using dialkyl and aromatic alkynes (Scheme 1 and Table 1).
Main products were the difluoroalkanes, except for the alkynes (2,4,6-trimethylphenyl)acetylene and (p-fluorophenyl)acetylene, which yielded minor amounts of fluorinated olefin as main products. Comparison between terminal 1-hexyne and symmetrical internal 3-hexyne revealed a higher reactivity for the internal species. Regioselectivities follow the Markovnikov rule and are consistent with hydrofluorination reactions of alkynes to give difluoroalkanes at HF-loaded ACF, and with approaches that apply the HF source PVP-HF (PVP: poly[4-vinylpyridinium poly(hydrogen fluoride)]) or a combination of KHSO4-13HF and DMPU-12HF.13,38,39 At ZCF electron-rich alkynes showed a higher reactivity compared to electron-poor species, even after prolonged reaction times. Note also that, in contrast, homogeneously catalysed reactions using Au(I) and Pt(II) species as catalysts typically yield monofluoroalkenes.40–46
Mechanistically, it is feasible that the HF generated by dehydrofluorination of the fluoroalkane adds subsequently to the alkyne by hydrofluorination (Scheme 3). An alternative reaction pathway would include the direct protonation of the alkyne by the intermediate carbenium species generated by C–F bond activation, followed by a fluorination step. The observed dehydrofluorination (Scheme 2) supports the first reaction pathway, but the latter cannot be excluded. Moreover, HF was additionally immobilised on ZCF and the resulting material subsequently used for the fluorination of a phenylacetylene in a model reaction. The generation of the difluoroalkane shows that an independent alkyne fluorination at ZCF is indeed possible.13
 | ||
| Scheme 3 HF transfer by C–F bond activation at ZCF and subsequent hydrofluorination. | ||
ACF has previously been reported to form intermediate carbenium-like species for a variety of reactions.12,14 Therefore, the HF transfer between fluoropentane and 3-hexyne in C6D12 was also tested using ACF as catalyst. The conversion (21%, Table 1, entry 2) was lower when compared to the activity at ZCF. This is consistent with the fact that dehydrofluorination steps at ACF typically need to be promoted by germanes or silanes to generate intermediate germylium or silylium species.12,36,47
In conclusion a significant advancement in fluorine chemistry is reported which comprises a HF transfer process at room temperature by shuttling of HF from a fluoroalkane to alkynes by consecutive defluorination and hydrofluorination steps. A zirconium chlorofluoride (ZCF) serves as a heterogeneous catalyst, which provides Lewis-acidity for the C–F bond cleavage steps. The catalyst was characterized regarding its acidic surface properties and the coordination environment around zirconium was determined. Remarkably, the developed reaction sequence proceeds at low temperatures. It contributes to efforts for sustainability in fluorine chemistry, which is of importance due to an upcoming shortage of fluorspar as raw material. The generation of HF from a fluoroalkane and its subsequent use by coupling a dehydrofluorination step with a hydrofluorination reaction is seminal in that context.
We acknowledge financial support from the CRC 1349 “Fluorine Specific Interactions” funded by the German Research Foundation (project number 387284271). The authors thank Minh Bui for the BET and TPD experiments. The XAS measurements were done at the BAMline at BESSY II. We are thankful to Stephanos Karafiludis for helpful discussions.
Conflicts of interest
There are no conflicts to declare.Notes and references
- H. Bozorgzadeh, E. Kemnitz, M. Nickkho-Amiry, T. Skapin and J. M. Winfield, J. Fluorine Chem., 2001, 107, 45–52 CrossRef CAS.
- J. L. Delattre, P. J. Chupas, C. P. Grey and A. M. Stacy, J. Am. Chem. Soc., 2001, 123, 5364–5365 CrossRef CAS PubMed.
- E. Kemnitz, U. Gross, S. Rudiger and C. S. Shekar, Angew. Chem., Int. Ed., 2003, 42, 4251–4254 CrossRef CAS PubMed.
- C. P. Marshall, T. Braun and E. Kemnitz, Catal. Sci. Technol., 2018, 8, 3151–3159 RSC.
- C. P. Marshall, G. Scholz, T. Braun and E. Kemnitz, Dalton Trans., 2019, 48, 6834–6845 RSC.
- I. K. Murwani, K. Scheurell and E. Kemnitz, Catal. Commun., 2008, 10, 227–231 CrossRef CAS.
- C. G. Krespan (DuPont), US Pat., 5,162,594, 1992 Search PubMed.
- C. G. Krespan, V. A. Petrov and B. E. Smart (DuPont), US Pat., 5,416,246A, 1995 Search PubMed.
- T. Krahl and E. Kemnitz, J. Fluorine Chem., 2006, 127, 663–678 CrossRef CAS.
- T. Krahl and E. Kemnitz, Catal. Sci. Technol., 2017, 7, 773–796 RSC.
- C. G. Krespan and V. A. Petrov, Chem. Rev., 1996, 96, 3269–3302 CrossRef CAS PubMed.
- M. Ahrens, G. Scholz, T. Braun and E. Kemnitz, Angew. Chem., Int. Ed., 2013, 52, 5328–5332 CrossRef CAS PubMed.
- M. C. Kervarec, E. Kemnitz, G. Scholz, S. Rudic, T. Braun, C. Jager, A. A. L. Michalchuk and F. Emmerling, Chem., 2020, 26, 7314–7322 CrossRef CAS PubMed.
- G. Meißner, D. Dirican, C. Jäger, T. Braun and E. Kemnitz, Catal. Sci. Technol., 2017, 7, 3348–3354 RSC.
- B. N. Bhawal and B. Morandi, ACS Catal., 2016, 6, 7528–7535 CrossRef CAS.
- B. N. Bhawal and B. Morandi, Angew. Chem., Int. Ed., 2019, 58, 10074–10103 CrossRef CAS PubMed.
- P. Yu, A. Bismuto and B. Morandi, Angew. Chem., Int. Ed., 2020, 59, 2904–2910 CrossRef CAS PubMed.
- W. Chen, J. C. L. Walker and M. Oestreich, J. Am. Chem. Soc., 2019, 141, 1135–1140 CrossRef CAS PubMed.
- D. A. Petrone, I. Franzoni, J. Ye, J. F. Rodríguez, A. I. Poblador-Bahamonde and M. Lautens, J. Am. Chem. Soc., 2017, 139, 3546–3557 CrossRef CAS PubMed.
- P. Boehm, N. Kehl and B. Morandi, Angew. Chem., Int. Ed., 2023, 62, e202214071 CrossRef CAS PubMed.
- J. A. Kalow and A. G. Doyle, J. Am. Chem. Soc., 2010, 132, 3268–3269 CrossRef CAS PubMed.
- D. Mulryan, J. Rodwell, N. A. Phillips and M. R. Crimmin, ACS Catal., 2022, 12, 3411–3419 CrossRef CAS.
- D. Mulryan, F. Rekhroukh, S. Farley and M. Crimmin, ChemRxiv, 2023, preprint DOI:10.26434/chemrxiv-2023-lxv28.
- S. Bobba, S. Carrara, J. Huisman, F. Mathieux and C. Pavel, Eur. Comm., 2020, 100 Search PubMed.
- H. Aoyama and S. Koyama (Daikin), Can. Pat., CA2070924C, 1992 Search PubMed.
- B. Calvo, C. P. Marshall, T. Krahl, J. Krohnert, A. Trunschke, G. Scholz, T. Braun and E. Kemnitz, Dalton Trans., 2018, 47, 16461–16473 RSC.
- M. Thommes, Chem. Ing. Tech., 2010, 82, 1059–1073 CrossRef CAS.
- M. Bui, K. Hoffmann, T. Braun, S. Riedel, C. Heinekamp, K. Scheurell, G. Scholz, T. Stawski and F. Emmerling, ChemCatChem, 2023, 15, e202300350 CrossRef CAS.
- L. Greb, Chem., 2018, 24, 17881–17896 CrossRef CAS PubMed.
- L. O. Müller, D. Himmel, J. Stauffer, G. Steinfeld, J. Slattery, G. Santiso-Quiñones, V. Brecht and I. Krossing, Angew. Chem., Int. Ed., 2008, 47, 7659–7663 CrossRef PubMed.
- T. Krahl, R. Stosser, E. Kemnitz, G. Scholz, M. Feist, G. Silly and J. Y. Buzare, Inorg. Chem., 2003, 42, 6474–6483 CrossRef CAS PubMed.
- S. L. Benjamin, W. Levason, D. Pugh, G. Reid and W. Zhang, Dalton Trans., 2012, 41, 12548–12557 RSC.
- B. Krebs, Z. Anorg. Allg. Chem., 1970, 378, 263–272 CrossRef CAS.
- X. Pan, M. Talavera and T. Braun, J. Fluorine Chem., 2022, 263, 110046 CrossRef CAS.
- M. C. Kervarec, C. P. Marshall, T. Braun and E. Kemnitz, J. Fluorine Chem., 2019, 221, 61–65 CrossRef CAS.
- G. Meißner, K. Kretschmar, T. Braun and E. Kemnitz, Angew. Chem., Int. Ed., 2017, 129, 16556–16559 CrossRef.
- G. A. Olah, Angew. Chem., Int. Ed., 1995, 34, 1393–1405 Search PubMed.
- G. A. Olah, J. T. Welch, Y. D. Vankar, M. Nojima, I. Kerekes and J. A. Olah, J. Org. Chem., 1979, 44, 3872–3881 CrossRef CAS.
- Z. Lu, B. S. Bajwa, S. Liu, S. Lee, G. B. Hammond and B. Xu, Green Chem., 2019, 21, 1467–1471 RSC.
- J. A. Akana, K. X. Bhattacharyya, P. Müller and J. P. Sadighi, J. Am. Chem. Soc., 2007, 129, 7736–7737 CrossRef CAS PubMed.
- R. Gauthier, M. Mamone and J.-F. Paquin, Org. Lett., 2019, 21, 9024–9027 CrossRef CAS PubMed.
- R. Gauthier, N. V. Tzouras, Z. Zhang, S. Bédard, M. Saab, L. Falivene, K. Van Hecke, L. Cavallo, S. P. Nolan and J.-F. Paquin, Chem. – Eur. J., 2022, 28, e202103886 CrossRef CAS PubMed.
- F. Nahra, S. R. Patrick, D. Bello, M. Brill, A. Obled, D. B. Cordes, A. M. Z. Slawin, D. O’Hagan and S. P. Nolan, ChemCatChem, 2015, 7, 240–244 CrossRef CAS PubMed.
- S. Sander and T. Braun, Angew. Chem., Int. Ed., 2022, 61, e202204678 CrossRef CAS PubMed.
- S. G. Rachor, R. Jaeger and T. Braun, Eur. J. Inorg. Chem., 2022, e202200158 CrossRef CAS.
- R. Gauthier and J.-F. Paquin, Chem. – Eur. J., 2023, e202301896 CrossRef PubMed.
- M. C. Kervarec, T. Braun, M. Ahrens and E. Kemnitz, Beilstein J. Org. Chem., 2020, 16, 2623–2635 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3cc03164k |
| This journal is © The Royal Society of Chemistry 2023 |