
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCatalyst-free depolymerization of polycaprolactone to silylated monoesters and iodide derivatives using iodosilanes†
Xin
Liu
,
Marie
Kobylarski
,
Jean-Claude
Berthet
* and
Thibault
Cantat
 *
*
NIMBE, CEA Paris-Saclay, Gif-sur-Yvette Cedex 91191, France. E-mail: thibault.cantat@cea.fr; jean-claude.berthet@cea.fr
First published on 9th August 2023
Abstract
The homogeneous depolymerization of polycaprolactone (PCL) with excess iodotrimethylsilane (Me3SiI) proceeds without catalysts and selectively afforded I(CH2)5CO2SiMe3 or a mixture of I(CH2)5CO2SiMe3 and I(CH2)5CO2I depending on the solvent (CH2Cl2, MeCN). The latter mixture can undergo methanolysis or hydrolysis into the valuable ester I(CH2)5CO2Me or the acid I(CH2)5CO2H. In contrast, SiH2I2 depolymerized PCL into the fully deoxygenated species I(CH2)6I and n-hexane.
Plastics have many advantages over traditional materials (glass, wood, etc.). They are cheap, durable, easy to shape and have adjustable properties. They have strongly contributed to the post war economic rise by encouraging mass consumption with the abundance of cheap and disposable objects. The production of plastics has grown from 15 Mt in the mid-60's to ∼460 Mt in 2020 and is projected to double in the next 20 years.1,2 However, plastics also pose a serious environmental problem. About half of the plastics are used only once and then thrown away, generating 350 Mt/year of plastic waste.3,4 Most of this waste is not recycled and ends up in landfills or oceans. Out of the 9.2 billion tons of plastic produced since the beginning, less than 8% would have been recycled and around 13 Mt of waste are released into the oceans every year.5
These frightening figures underline the need to move towards a circular economy and to more sober plastic consumption. In addition to the reuse and repair of objects, the plastic industry will have to significantly reduce its negative externalities and recover the carbonaceous matter from waste. In this context, chemical recycling that is the depolymerization of plastic materials into monomers or valuable compounds useful for the chemical industry is appealing and has emerged as a long-term sustainable strategy. At present, less than 1% of plastics are chemically recycled in Europe.6 The few existing industrial processes to depolymerize plastics are based essentially on catalytic solvolysis processes (hydrolysis, aminolysis or transesterification reactions), that give back the corresponding monomers, or thermal treatments (gasification, pyrolysis) to produce syngases and hydrocarbon fractions.7 These processes are technologically advanced but their future also depends on their economic viability.
Recent fundamental advances in that field concern the reductive depolymerization of oxygenated or nitrogenated polymers using homogeneous catalysts in presence of hydride source (H2 or SiH/BH reductants).8–13 In particular, building on the previous works on the carbon–oxygen bond cleavage by hydrosilanes in homogeneous catalysis, we and others have used organosilicon hydrides (R3SiH) and various Ir(III), Mo(VI), Zr(IV) or Zn(II) or boron catalysts, to break down oxygenated plastics (polyesters, polyethers, polycarbonates) into valuable alcohols and alkanes.8,14,15 Silyl ethers and even hydrocarbons compounds derived from the true monomers were formed selectively by adjusting the reaction parameters (temperature, type and amount of the reductant, solvent). However, chemical depolymerization of oxygenated polymers with other silicon reagents, especially those with reactive Si–X bonds such as halides (X = I, Cl, Br), has not been explored to produce functionalized monomer molecules. We hypothesized that the iodosilane reagents with the weakest Si–X bond (BDE(Si–I) ≈ 70–80 kcal mol−1),16,17 could favor the formation of a high-energy Si–O bond (90–110 kcal mol−1), without the need for a catalytic activation.18
Organosilicon reagents have been studied in organic chemistry since the end of the 70's. Many studies report the capabilities of the reactive Me3SiI (Me3SiCl and Me3SiBr proved inefficient), SiH2I2 or of combined reagents (Me3SiCl–NaI, SiH2I2–I2…) to cleave carbon–oxygen bonds in ethers, esters, carbamates, ketals and alcohols, and to deoxygenate sulfoxydes (R2S![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O).19–24 Reactions of the monoesters RCO2R’ with different halosilanes led to the silylated esters and then the acyl iodide as shown in Scheme 1.
O).19–24 Reactions of the monoesters RCO2R’ with different halosilanes led to the silylated esters and then the acyl iodide as shown in Scheme 1.
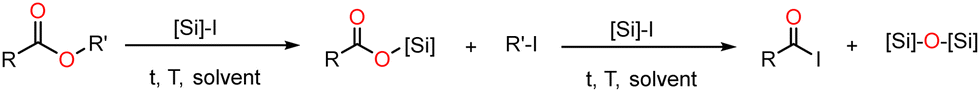 | ||
| Scheme 1 Reaction of monoesters with iodosilanes. | ||
These studies prompted us to explore the potential of two iodosilanes (Me3SiI and SiH2I2) in the depolymerization of polyesters, with the aim to produce silylated and iodo functionalized monomers of interest in organic and materials chemistry. In this communication, we focused on the depolymerization of polycaprolactone (PCL), as a model synthetic polyester. We report that under refluxing conditions and without any catalyst, Me3SiI and SiH2I2 are relevant reagents to cleanly depolymerize polycaprolactone (PCL) into value added silyl ester monomers and/or reactive iodide derivatives. Unexpectedly, we observed for the first time the ability of SiH2I2 to promote both iodide and hydride transfers to cleave C–O bonds providing diiodohexane and n-hexane.
Polycaprolactone (PCL) is a biodegradable synthetic polyester that found applications in various industrial fields such as coatings, adhesives, sealants and polyurethane elastomers.25,26 It is also broadly applied in biomedical applications, specifically in tissue engineering, sutures, drug delivery, etc. It is produced industrially mainly by ring-opening polymerization of ε-caprolactone and polycondensation of carboxylic acid methods on million tons/year with a market size valued at 415 million€ and projected to reach ca. 1 billion€ within 2030.27
Our attempts to depolymerize polyesters with Me3SiI were carried out with commercial pellets of PCL. Main results are reported in Table 1. Addition of a slight excess of Me3SiI (1.2 equiv.) to pellets of PCL in CD2Cl2 showed only 1% of conversion after 20 h at 25 °C (Table 1, entry 1). At 50 °C, the conversion reached 24% after 5 h, with clean formation of I(CH2)5CO2SiMe3 (1) (Table 1, entry 2). This yield is doubled when warming at 100 °C (46% after 5 h) (Table 1, entry 3). Increasing the temperature to 130 °C and 150 °C boosted the kinetic of depolymerization: after only 2 h, 78% and 88% of 1 were respectively obtained (Table 1, entries 4–5). The formation of 1 demonstrates, for the first time, the ability of iodosilanes to promote the depolymerization of polyesters under metal-free conditions. In presence of excess Me3SiI (2 equiv.), a CD2Cl2 solution of PCL heated at 100 °C gave 1 in a better yield (74% after 5 h) (Table 1, entry 6) than with 1.2 equiv. Me3SiI (Table 1, entry 3) and the formation of 1 is almost quantitative (>99%) after prolonged heating (40 h at 100 °C) with no side-products (Table 1, entry 6). A scale-up using this procedure (15 h at 100 °C in CH2Cl2; 2 equiv. Me3SiI) afforded 1 in 95% yield (96% purity) as a pale yellow oil after evaporation of the solvent and extraction in pentane (Table 1, entry 7). The silylated ester 1 was clearly characterized by its 1H and 13C NMR spectra and its infrared spectrum (ν(C0): 1743 cm−1) (see ESI†).
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | x (equiv.) | Solvent | T (°C) | t (h) | Yield in 1 (%)b | Yield in 2 (%)b |
| a Conditions: PCL (0.28 mmol with respect to the monomer unit), solvent (0.4 mL). b Yields in 1 and 2 determined by integration of their 1H NMR signals vs. those of dodecane (standard). c Isolated yield containing 2% of 2. d Approximate ratio by 1H NMR due to H/D exchange (see ESI). e Addition of 1 equiv. I2. | ||||||
| 1 | 1.2 | CD2Cl2 | 25 | 20 | Traces | 0 |
| 2 | 1.2 | CD2Cl2 | 50 | 5 | 24 | 0 |
| 3 | 1.2 | CD2Cl2 | 100 | 5 | 46 | 0 |
| 4 | 1.2 | CD2Cl2 | 130 | 2 | 78 | 0 |
| 5 | 1.2 | CD2Cl2 | 150 | 2 | 88 | 0 |
| 6 | 2 | CD2Cl2 | 100 | 5 | 74 | 0 |
| 40 | >99 | Traces | ||||
| 7 | 2 | CH2Cl2 | 100 | 15 | 95c | — |
| 8 | 2 | CD3CN | 100 | 5 | 90 | 10 |
| 9 | 2 | CD3CN | 130 | 2 | 83 | 17 |
| 10d | 2 | CD3CN | 150 | 2 | 68 | 32 |
| 11d | 6 | CD3CN | 150 | 16 | 23 | 74 |
| 12e | 3 | CD2Cl2 | 150 | 10 | 23 | 77 |

In these reactions, the nature of the solvent plays an important role. Ethers, alcohols, amines or any compound with carbonyl functions can not be used as solvent as they can react with Me3SiI.28,29 Solvents are also not anodyne on the fate of the reaction as observed by replacing dichloromethane (bp = 40 °C, ε = 8.93) with acetonitrile which has higher boiling point and polarity (bp = 82 °C, ε = 37.5). With 2 equiv. Me3SiI at 100 °C, the depolymerization of PCL in MeCN-d3 appeared a little faster (5 h, 90% in 1) than in CD2Cl2 (5 h, 74% in 1) as evidenced in the entries 6 and 8 of Table 1. However, in these conditions, acetonitrile induced a loss of selectivity and formation of the acyl iodide I(CH2)5CO2I (2) according to the 1H and 13C NMR spectra (see ESI†). Compound 2 is obtained by the deesterification of 1 with Me3SiI and is concomitant with the release of hexamethyldisiloxane ((Me3Si)2O, observed by 1H NMR, see ESI†). Increasing the temperature to 130 °C and 150 °C for a mixture containing 2 equiv. Me3SiI sped up the formation of 2 with an increase of the 2/1 ratio from 17/83 to 32/68 (Table 1, entries 9–10).
Acyl iodides (RCO2I) are valuable compounds as they are the most reactive acyl halide species and only a few synthetic methods have been developed for their preparation.30–32 For example, Jung et al.20 briefly mentioned (but did not provide any experimental details) the thermal treatment of the mono-esters RCO2R’ with Me3SiI in chlorinated solvents (CCl4, CHCl3). However, Olah's group contradicted this claim, as the authors did not detect the formation of acyl iodide, probably due to shorter reaction times.33 In 1990, Keinan et al. demonstrated the effectiveness of Me3SiI in chloroform to convert carboxylic acids and esters RCO2R′ (R = alkyl; R′ = H, alkyl) to the corresponding silylesters (RCO2SiMe3) and that the reaction was significantly accelerated by iodine (I2), likely via the formation of the stable triiodide anion. Unfortunately, the resulting RCO2SiMe3 products were not further converted into the acyl iodide derivatives RCO2I except in a few cases, in very low amounts (<5%). Using diodosilane (SiH2I2) and I2 instead of Me3SiI/I2 enhanced the deesterification of silylesters to acyl iodides.32
To accumulate compound 2, a MeCN mixture of PCL containing an excess of Me3SiI (6 equiv.) was monitored by 1H NMR. After 16 h at 150 °C, the 1/2 ratio was equal to ca 23/74 and did not change after further heating for 17 h (Table 1, entry 11). Inspired by Keinan et al., PCL was treated with excess of a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of Me3SiI/I2 and led after 10 h at 150 °C in CD2Cl2 to a mixture of 1 and 2 in a 23/77 ratio (Table 1, entry 12). This ratio did not improve with further heating (see ESI†). In CH2Cl2, I2 effectively increased the formation of 2 but did not yield pure 2. Under these conditions, acetonitrile was not suitable, as it reacted with 2. Therefore, compound 2 could not be obtained pure by these two methods.
:1 mixture of Me3SiI/I2 and led after 10 h at 150 °C in CD2Cl2 to a mixture of 1 and 2 in a 23/77 ratio (Table 1, entry 12). This ratio did not improve with further heating (see ESI†). In CH2Cl2, I2 effectively increased the formation of 2 but did not yield pure 2. Under these conditions, acetonitrile was not suitable, as it reacted with 2. Therefore, compound 2 could not be obtained pure by these two methods.
The above reactions (Table 1) revealed that the depolymerization of PCL into 1, with Me3SiI, is quite rapid (and selective in CD2Cl2) but the transformation of 1 to 2, even with the mixture Me3SiI/I2, is a more difficult step and isolation of pure 2 by this route seemed unlikely. Such a mixture of 1 and 2 can however be upgraded to the ester I(CH2)5CO2Me (3) by immersing it in dry methanol. After 3 h at 100 °C and evaporation of the alcohol, the 1H NMR spectrum showed the presence of 3 as the only species. It was isolated in a yield of 83% and the NMR data (1H and 13C, see ESI†).34 Similarly, hydrolysis of the above mixture of 1 and 2 in CH2Cl2 led after 30 min at room temperature to I(CH2)5CO2H (4) isolated after usual work-up as a white powder in an excellent yield of 97% (Scheme 2).35
 | ||
| Scheme 2 Methanolysis and hydrolysis of a mixture of 1 and 2. | ||
We also examined SiH2I2 for the transformation of PCL to 2. In presence of an excess SiH2I2 (5 equiv.) at 150 °C, 1H NMR monitoring revealed the complete denaturation of PCL into a mixture of 1′ and 2 which progressively evolved into 1,6-diodohexane (5) and n-hexane (6). The ability of SiI2H2 to engage in both iodide and hydride transfers is novel and we further confirmed this reactivity by the reduction of 2-iodobutane with SiH2I2 (see ESI†). In CD2Cl2, after 15 h at 150 °C, PCL was transformed into a mixture of 1′ and 2 without any reductive product (Table 2, entry 1). After 120 h, 1′ and 2 have been almost completely converted into 5 and 6. In MeCN, within similar conditions, an intractable brown solid deposited after 1 h and the 1H NMR spectrum was complex and not informative (Table 2, entry 2). Solvent-less reaction of PCL suspended in SiH2I2 proved interesting (Table 2, entry 3). After 4 h at 150 °C, the 1H NMR spectrum (in CD2Cl2) showed a complex mixture of 1′ (44%), 2 (40%), 5 (8%) and 6 (8%), which then evolved to only 5 (56%) and 6 (44%) after 17 h. Colorless crystals of SiI4 were obtained from this mixture,36 evidencing reduction of 5 with SiH2I2. This reaction unveiled the reductive capacity of SiH2I2 which can, without any catalyst, both cleave C–O bonds of PCL to depolymerize it into carboxylic monomers (1′ and 2) and deoxygenate the resulting acyl iodide 2 into diodohexane (5) and n-hexane (6). We also checked that in these conditions (150 °C, excess SiH2I2) both pure 1 and 2 were indeed transformed into 5 and 6 (see ESI†). This reaction is reminiscent of the reduction of aromatic acids and aromatic silylesters into benzylic trichlorosilanes (ArCH2SiCl3) by HSiCl3.37
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Solvent | t (h) | Yieldb (%) | |||
| 1′ | 2 | 5 | 6 | |||
| Equiv. of SiH2I2 given per monomeric fragment C6H10CO2.a Conditions: PCL (0.28 mmol with respect to the monomer unit), solvent (0.4mL).b Yields in 1′, 2, 5 and 6 determined by integration of their 1H NMR signals vs. those of the standard (dodecane or mesitylene).c NMR spectrum recorded in CD2Cl2 after evaporation of the SiH2I2. | ||||||
| 1 | CD2Cl2 | 15 | 53 | 42 | 0 | 0 |
| 120 | 1 | 2 | 35 | ∼50 | ||
| 2 | CD3CN | 1 | Degradation | |||
| 3 | Nonec | 4 | 44 | 40 | 8 | 8 |
| 17 | 0 | 0 | 56 | 44 | ||
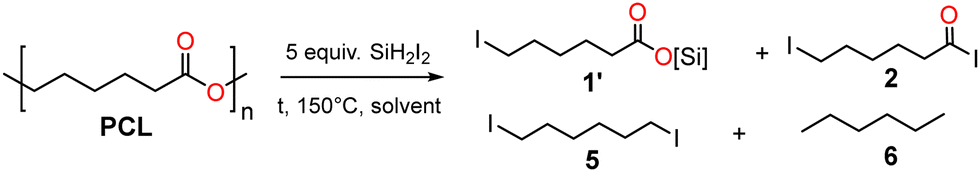
Compounds 1–6 are relevant molecules in organic chemistry and in materials science. Silyl esters are indeed convenient intermediates for the preparation of functional polymeric substrates such as easily degradable poly(silylester)s and extensively used as biodegradable surgical devices, matrices for drug delivery, etc.38–42 Acyl halide species have a wide spectrum of reactivity as electrophiles and acyl iodides are the most reactive ones.43 Compounds 1–2 and 5 are also convenient synthons toward oxygenated and nitrogenated polymers through coupling reactions with a variety of bifunctional reagents such as amino-acids, hydroxy-acids, diamines, etc.44
From the experiments and the products characterized above, a plausible mechanism to the formation of 5 from PCL can be proposed in Scheme 3. First, PCL is depolymerized by Me3SiI, Me3SiI-I2 or SiH2I2 into the silyl ester 1 or 1′ and then the acyl iodide 2. This reactivity is related to the strong oxophilic character of the [Si]–I reagents which can coordinate carbonyl groups with heterolytic cleavage of the weak Si–I bond. The silylium ion thus generated can induce further carbon–oxygen cleavages in PCL to give 1 (or 1′) and 2. The coordination of 2 to the reducing Lewis acid SiH2I2 or a hydrosilane derivative (possibly formed in these thermic conditions) strongly enhances the electrophilic character of the carbonyl moiety and favors spontaneous hydride transfer from the proximal hydrosilane. The generated alkoxysilane intermediate, that could not be detected, traps another hydrosilane to give 5 with release of the corresponding disiloxane.
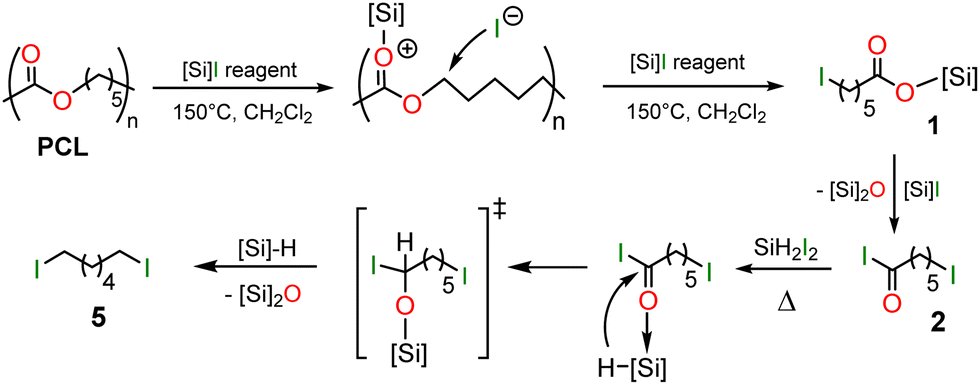 | ||
| Scheme 3 Proposed mechanism for the reductive depolymerization of PCL. | ||
Capitalizing on these results, we next targeted the depolymerization of other polyesters to show the generality of the method. First results evidenced a successful depolymerization of PET (polyethylene terephtalate) from household plastic bottles by treatment (40 h at 150 °C) with excess Me3SiI in acetonitrile (see ESI†). With this method, the depolymerization of polyesters is currently being optimized and results will be presented in a forthcoming article.
This work highlights the first use of a iodosilane (Me3SiI) to carry out the efficient depolymerization of a polyester, e.g. polycaprolactone, into functionalized products useful in organic chemistry and in polymer synthesis: silyl ester I(CH2)5CO2SiMe3 (1) and acyl iodide I(CH2)5CO2I (2). Using SiH2I2 also offered new possibilities in polyester deconstruction. For example, with 2, hydride transfer occurred spontaneously to deoxygenate it to diiodohexane I(CH2)6I and n-hexane. This was the first catalyst-free depolymerization of a polymer with halosilane and halo(hydro)silane. Further works are in progress to deconstruct or reductively depolymerize other pure or household oxygenated plastics.
For financial support, we acknowledge CEA, CNRS, the University Paris-Saclay, and the European Research Council (ERC Consolidator Grant Agreement no. 818260).
Conflicts of interest
There are no conflicts to declare.Notes and references
- World Economic Forum, Ellen MacArthur Foundation and McKinsey Company, The New Plastics Economy-Rethinking the future of plastics, 2016 Search PubMed.
- OECD, Global Plastics Outlook: Economic Drivers, Environmental Impacts and Policy Options, OECD Publishing, Paris, 2022 DOI:10.1787/de747aef-en.
- S. Mandard, Le Monde, 2023, 6.
- I. Tiseo, Plastic waste worldwide - statistics & facts, https://www-statista-com.translate.goog/?_x_tr_sl=en&_x_tr_tl=fr&_x_tr_hl=fr&_x_tr_pto=rq&_x_tr_hist=true#topicOverview, accessed April 11, 2023.
- H. Böll, https://eu.boell.org/sites/default/files/2021-01/Plastic%20Atlas%202019%202nd%20Edition.pdf, 2019, accessed April 11, 2023.
- The circular economy for plastics, a European overview, https://plasticseurope.org/wp-content/uploads/2022/06/PlasticsEurope-CircularityReport-<?pdb_no 2022?>2022<?pdb END?>_2804-Light.pdf, 2022, 25.
- A. R. Rahimi and J. M. García, Nat. Rev. Chem., 2017, 1, 0046 CrossRef.
- A. C. Fernandes, Green Chem., 2021, 23, 7330–7360 RSC.
- E. Feghali, L. Tauk, P. Ortiz, K. Vanbroekhoven and W. Eevers, Polym. Degrad. Stab., 2020, 179, 109241 CrossRef CAS.
- A. Ahrens, A. Bonde, H. Sun, N. K. Wittig, H. C. D. Hammershøj, G. M. F. Batista, A. Sommerfeldt, S. Frølich, H. Birkedal and T. Skrydstrup, Nature, 2023, 617, 730–737 CrossRef CAS PubMed.
- A. Kumar, N. von Wolff, M. Rauch, Y. Q. Zou, G. Shmul, Y. Ben-David, G. Leitus, L. Avram and D. Milstein, J. Am. Chem. Soc., 2020, 142, 14267–14275 CrossRef CAS PubMed.
- E. M. Krall, T. W. Klein, R. J. Andersen, A. J. Nett, R. W. Glasgow, D. S. Reader, B. C. Dauphinais, S. P. Mc Ilrath, A. A. Fischer, M. J. Carney, D. J. Hudson and N. J. Robertson, Chem. Commun., 2014, 50, 4884–4887 RSC.
- L. Wursthorn, K. Beckett, J. O. Rothbaum, R. M. Cywar, C. Lincoln, Y. Kratish and T. J. Marks, Angew. Chem., Int. Ed., 2023, 62, e202212543 CrossRef CAS PubMed.
- C. Jehanno, J. W. Alty, M. Roosen, S. De Meester, A. P. Dove, E. Y. Chen, F. A. Leibfarth and H. Sardon, Nature, 2022, 603, 803–814 CrossRef CAS PubMed.
- L. Monsigny, J.-C. Berthet and T. Cantat, ACS Sustainable Chem. Eng., 2018, 6, 10481–10488 CrossRef CAS.
- D. J. Grant and D. A. Dixon, J. Phys. Chem. A, 2009, 113, 3656–3661 CrossRef CAS PubMed.
- R. Walsh, in Bond dissociation energies in organosilicon compounds, ed. B. Arkles, G. Larson, Gelest Inc., 2008 Search PubMed.
- G. A. Olah, S. C. Narang, B. G. B. Gupta and R. Malhotra, J. Org. Chem., 1979, 44, 1247–1251 CrossRef CAS.
- M. E. Jung and M. A. Lyster, J. Org. Chem., 1977, 42, 3761–3764 CrossRef CAS.
- M. E. Jung and M. A. Lyster, J. Am. Chem. Soc., 1977, 99, 968–969 CrossRef CAS.
- M. E. Jung and P. L. Ornstein, Tetrahedron Lett., 1977, 2659–2662 CrossRef CAS.
- H. R. Kricheldorf, Angew. Chem., Int. Ed. Engl., 1979, 18, 689–690 CrossRef.
- M. E. Jung and M. A. Lyster, J. Chem. Soc., Chem. Commun., 1978, 315–316 RSC.
- G. M. Blackburn and D. Ingleson, J. Chem. Soc., Chem. Commun., 1978, 870–871 RSC.
- M. A. Woodruff and D. W. Hutmacher, Prog. Polym. Sci., 2010, 35, 1217–1256 CrossRef CAS.
- M. Labet and W. Thielemans, Chem. Soc. Rev., 2009, 38, 3484–3504 RSC.
- https://www.alliedmarketresearch.com/polycaprolactone-market-A06096, accessed april 11, 2023.
- N. Ajvazi and S. Stavber, Tetrahedron Lett., 2016, 57, 2430–2433 CrossRef CAS.
- F. Wang, M. Qu, X. Lu, F. Chen, F. Chen and M. Shi, Chem. Commun., 2012, 48, 6259–6261 RSC.
- H. M. R. Hoffmann and K. Haase, Synthesis, 1981, 715–719 CrossRef CAS.
- M. F. Anssel, Preparation of acyl halides, ed. S. Patai, PATAI'S Chemistry of Functional Groups, 1972 Search PubMed.
- E. Keinan and M. Sahai, J. Org. Chem., 1990, 55, 3922–3926 CrossRef CAS.
- T. L. Ho and G. A. Olah, Proc. Natl. Acad. Sci. U. S. A., 1978, 75, 4–6 CrossRef CAS PubMed.
- S. El Fangour, A. Guy, V. Despres, J. P. Vidal, J. C. Rossi and T. Durand, J. Org. Chem., 2004, 69, 2498–2503 CrossRef CAS PubMed.
- The ester I(CH2)5CO2Me (3) and the acid I(CH2)5CO2H (4) are sold by Merck at the respective prices of 310€/g and 964€/g (06-07-2023), Iodotrimethylsilane Me3SiI is at the price 36€ for 5 g (Merck).
- E. Biehl and U. Schubert, Monatsh. Chem., 2000, 131, 813–818 CrossRef CAS.
- R. A. Benkeser, E. C. Mozdzen and C. L. Muth, J. Org. Chem., 1979, 44, 2185–2188 CrossRef CAS.
- D. K. Gilding and A. M. Reed, Polymer, 1979, 20, 1459–1464 CrossRef CAS.
- R. A. Gross and B. Kalra, Science, 2002, 297, 803–807 CrossRef CAS PubMed.
- M. Wang, D. Gan and K. L. Wooley, Macromolecules, 2001, 34, 3215–3223 CrossRef CAS.
- M. Cazacu, G. Munteanu, C. Racles, A. Vlad and M. Marcu, J. Organomet. Chem., 2006, 691, 3700–3707 CrossRef CAS.
- J. M. Weinberg, S. P. Gitto and K. L. Wooley, Macromolecules, 1998, 31, 15–21 CrossRef CAS.
- M. G. Voronkov, N. N. Vlasova, O. Y. Grigor’eva, L. I. Belousova and A. V. Vlasov, Russian J. Org. Chem., 2009, 45, 486–490 CrossRef CAS.
- F.-R. Zeng, J. Xu, L.-H. Sun, J. Ma, H. Jiang and Z.-L. Li, Polym. Chem., 2020, 11, 1211–1219 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3cc03029f |
| This journal is © The Royal Society of Chemistry 2023 |