
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A guide to modern methods for poly(thio)ether synthesis using Earth-abundant metals
Robert C.
Ferrier
Jr
 *a,
Gouree
Kumbhar
a,
Shaylynn
Crum-Dacon
a and
Nathaniel A.
Lynd
b
*a,
Gouree
Kumbhar
a,
Shaylynn
Crum-Dacon
a and
Nathaniel A.
Lynd
b
aMichigan State University, Department of Chemical Engineering and Materials Science, East Lansing MI, USA. E-mail: ferrier5@msu.edu
bUniversity of Texas-Austin, McKetta Department of Chemical Engineering, Austin, TX, USA
First published on 12th September 2023
Abstract
Polyethers and polythioethers have a long and storied history dating back to the start of polymer science as a distinct field. As such, these materials have been utilized in a wide range of commercial applications and fundamental studies. The breadth of their material properties and the contexts in which they are applied is ultimately owed to their diverse monomer pre-cursors, epoxides and thiiranes, respectively. The facile polymerization of these monomers, both historically and contemporaneously, across academia and industry, has occurred through the use of Earth-abundant metals as catalysts and/or initiators. Despite this, polymerization methods for these monomers are underutilized compared to other monomer classes like cyclic olefins, vinyls, and (meth)acrylates. We feel a focused review that clearly outlines the benefits and shortcomings of extant synthetic methods for poly(thio)ethers along with their proposed mechanisms and quirks will help facilitate the utilization of these methods and by extension the unique polymer materials they create. Therefore, this Feature Article briefly describes the applications of poly(thio)ethers before discussing the feature-set of each poly(thio)ether synthetic method and qualitatively scoring them on relevant metrics (e.g., ease-of-use, molecular weight control, etc.) to help would-be poly(thio)ether-makers find an appropriate synthetic approach. The article is concluded with a look ahead at the future of poly(thio)ether synthesis with Earth-abundant metals.
 Robert C. Ferrier, Jr. | Robert C. Ferrier, Jr. grew up in Norristown, PA and received a BS in Physics and MS in MSE from Drexel University in 2009 working with Chris Y. Li, and a PhD in CBE from the University of Pennsylvania in 2015 working with Russell J. Composto. Robert then moved to UT-Austin as a postdoctoral fellow in Nathaniel A. Lynd's lab before moving to Michigan State University as an Assistant Professor in the ChEMS Department in 2018. Robert's group combines concepts in polymer physics, chemistry, and nanocomposites to engineer materials that solve challenges in energy, environment, and health. |
 Gouree Kumbhar | Gouree Kumbhar received her bachelors degree in Chemical Engineering from Institute of Chemical Technology (India) in 2018. She recently received her PhD in Chemical Engineering at Michigan State University working with Dr Robert C. Ferrier Jr. At MSU, her research was focused on design and engineering of charged polyethers for separation membranes and to study polyelectrolyte self-assembly. Her expertise lies in the synthesis of organoaluminium catalysts for epoxide polymerization, novel polymer chemistry synthesis, and materials design. She currently works for the Dow Chemical Company in Houston, TX. |
 Shaylynn Crum-Dacon | Shaylynn Crum-Dacon graduated from Tuskegee University with a Bachelor of Science in Chemical Engineering with a concentration in biochemical applications. After graduating, she worked at Los Alamos National Laboratory where she participated in research that primarily focused on additive manufacturing electrochemically active materials as well as investigated alternatives to nuclear waste containment materials. In 2018, she entered the Chemical Engineering doctoral program at Michigan State University where she, currently, researches polyether-based solid electrolytes for lithium batteries. |
 Nathaniel A. Lynd | Nathaniel A. Lynd is originally from Michigan and received a BS in Chemistry from Michigan State University, where he worked with the late Prof. Gregory L. Baker. Nate completed his PhD at the University of Minnesota under Marc A. Hillmyer. In 2007, Nate pursued postdoctoral studies at UC-Santa Barabara under Glenn H. Fredrickson, Craig J. Hawker, and the late Edward J. Kramer. Eventually, Nate took a position as staff scientist at Lawrence Berkeley National Laboratory in the Materials Sciences Division before joining the faculty of UT-Austin in the McKetta Department of Chemical in late 2015. Nate is currently the Laurence E. McMakin, Jr., Centennial Fellow in Chemical Engineering. |
1. Introduction
Polyethers and polythioethers are important classes of polymers that share a significant portion of their recent synthetic history, properties, and applications. Polyethers, such as poly(ethylene oxide) (PEO), are characterized by their C–O–C backbone which imparts flexibility (i.e., low Tg), bio-compatibility, and ionic conductivity. Polythioethers, such as poly(propylene sulfide) (PPS), are characterized by their C–S–C backbone which imparts flexibility, bio-compatibility, and oxidative degradation resistance. Poly(thio)ethers are used in a wide variety of commercial and academic contexts ranging from cosmetics to drug delivery to energy storage. These polymers are most often produced from the ring opening polymerization (ROP) of multi-member oxygen or sulfur containing hetero-cycles. While 4-, 5-, and higher-member ring structures can be used as a monomer feed-stock for poly(thio)ethers, three-membered oxygen, (i.e., epoxides or oxiranes), and sulfur (i.e., episulfides or thiiranes) containing heterocycles are most often used owing to their polymerization efficiency and availability, as can be seen in Fig. 1. These monomers can be polymerized via a number of different mechanisms, which are unified by their incorporation of Earth-abundant metals such as aluminum, iron, and magnesium as a catalyst and/or initiator. As of this writing, there is no single method that can quickly and easily produce poly(thio)ethers with arbitrary composition, (micro)structure, molecular weight, and end group. As such, several different approaches are often used depending on the monomer, targeted molecular weight, and desired (micro)structure for the given application. This Feature Article reviews the long and storied history of poly(thio)ether synthesis using Earth-abundant metals, discusses mechanistic aspects as well as features and quirks of contemporary synthetic methods, highlights the authors' specific contributions to the field, and expounds on the future of poly(thio)ether synthesis. We anticipate this article will help those who may want to synthesize a poly(thio)ether but do not know where to start as well as those who want an approachable look at the entire field.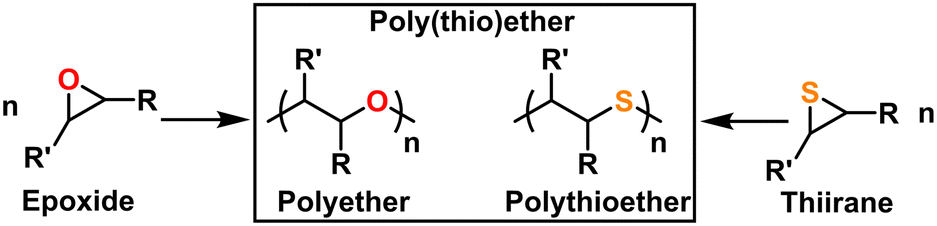 | ||
| Fig. 1 Common monomers and polymer structures for poly(thio)ethers. | ||
2. How do we define an Earth-abundant metal?
We consider a broad definition of Earth-abundant metals consisting of a subset of the alkaline Earth metals (e.g., Mg), alkali metals (e.g., K, Na), first row transition metals (e.g., Fe, Cr, Co, Zn), and post-transition metals (e.g., Al, Sn). The metals covered here take various forms and each polymerization technique uses a different subset of metals, which is summarized in Table 1. We do not intend to give an exhaustive review of all Earth-abundant metals used in a particular synthetic scheme. Instead, we will provide an overview as well as recent notable examples. To assist readers who want to go more in depth, we will point out focused reviews where appropriate.| Technique | Metal Form | Notes |
|---|---|---|
| AROP | K, Na, Cs cations | Counter-ions to oxyanions or thiolates |
| MAROP | AlR3 and [XAlR3]− [NOct4]+ | AlR3 activates monomer, X = Cl or Br |
| CROP | Cationic Al, Zn, Mg complexes | Various bulky ligands (e.g., salen, salpen, etc.) |
| Vandenberg | Bis-(μ-oxo) dialkyl aluminum (BOD) | Exact structure is unknown |
| DMC | MX2 and K3M′(CN6) | M = Zn, Fe, Co, Ni; M = Co, Fe, Cr, Ir |
| Inoue | Zn- or Al-porphyrin complex | Substitutions on the metal possible (e.g., –Cl, –OR, etc.) |
| Coates | Co- or Cr-salalen complex | Mono- or bi-metallic, substitutions on the metal possible |
| NAl adduct | R3N:AlR3 and BOD or mono-(μ-oxo)bisalkyl aluminum (MOB) | MOB acts as both a catalyst and initiator. BOD acts as initiator. |
3. Application of poly(thio)ethers
3.1 Polyether applications
Polyethers and polythioethers share many of the same characteristics; they are low Tg polymers that are often biocompatible. Polyethers, especially PEO, have traditionally received more attention than polythioethers and so are in a slew of commercial applications. The most widespread use of polyethers is in the production of polyurethane foams through polyether polyols.1–4 Since PEO is water soluble, it is often used as an emulsifier in the cosmetic5 and food industries.6,7 Owing to their interaction with ionic species as well as gases like CO2, polyethers are employed in separations, such as polymer electrolytes for lithium ion batteries8–10 or membranes for CO2 separation.11,12 Due to their tunability and flexibility, polyethers are effective as anti-fouling coatings.13 Finally, polyethers have been transformative to the biomedical field, with PEO enabling more stable medications through bio-conjugation as well as novel methods for drug delivery.14,15 For instance, the most recent mRNA vaccines to combat the COVID-19 pandemic used lipid bound PEO to help stabilize the mRNA for delivery.16,17 Excellent reviews of polyethers by Wurm18 and Frey19 are recommended to learn more about their applications.3.2 Polythioether applications
Polythioethers have been less commercially viable partially owing to synthetic challenges.20 Traditionally, they were used in the vulcanization of rubber as well as in seals and sealants as commercial polymers like Thiokol,21,22 although these ill-defined polymers are not strictly polythioethers.23 More recently, they have found several academic applications and have been gaining traction in the biomedical space.24,25 Owing to the backbone thioether groups propensity to oxidize, polythioethers are utilized to scavenge reactive oxygen species (ROS).26 This ability has been used both in self-assembly schemes27 as well as drug delivery schemes;28 PPS in particular undergoes a hydrophobic to hydrophilic transition upon oxidation. As such, PPS is often combined with PEO to make micelles or vesicles for drug delivery as a sort of poloxamer (i.e., Pluronic™) that can be oxidatively disassembled to deliver a payload to a target.29–31 The backbone thioethers chelate a variety of metals and so can be used to incorporate metals into a polythioether matrix (i.e., for antifouling applications),32 to remove heavy metals like Hg33 or detect the presence of metals.34 Furthermore, due to the thioether backbone's interaction with Li-ions, polythioethers can be used as polymer electrolytes.35,36 Excellent reviews of sulfur containing polymers especially polythioethers were written by Tirelli24 and Plummer.204 Monomer synthesis
4.1 Epoxide synthesis
A major draw of polyether synthesis through epoxides is the diversity of their pendant groups coupled with their ease of synthesis and general availability. Epichlorohydrin (ECH), which is increasingly produced from renewable resources, is characterized by its pendant chloromethylene group which allows it to be used in a wide variety of reactions.37 In the context of epoxide synthesis, it is extremely versatile; Glycidyl ethers can easily be made by reacting an alcohol with ECH in the presence of a base (e.g., NaOH).38 Glycidyl amines can easily be synthesized from reaction with a secondary amine and then subsequent ring closing reaction with NaOH.39 Aside from ECH, epoxidation of alkenes is a common way to achieve epoxides with a variety of substituents.40,41Fig. 2 shows some general schemes involving the synthesis of epoxides.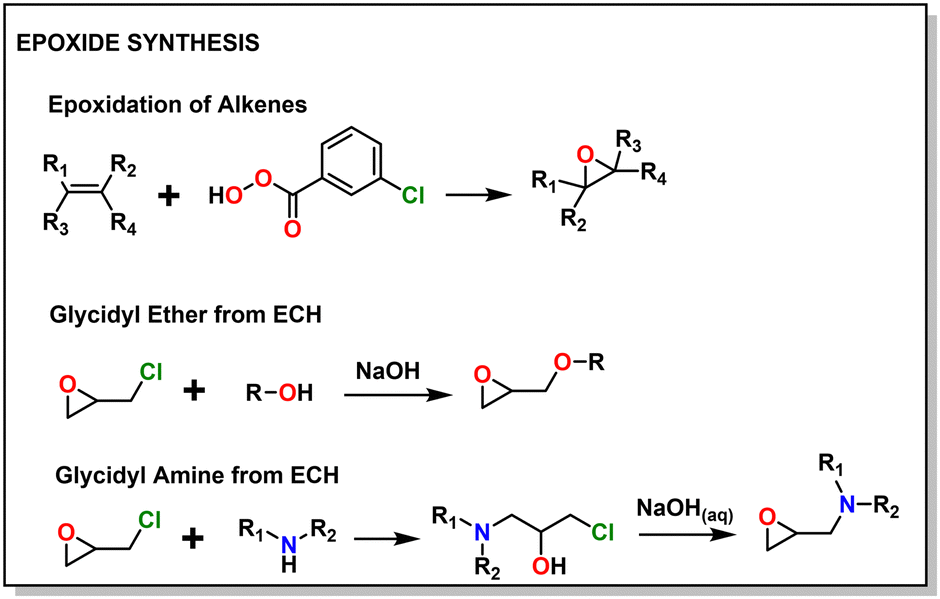 | ||
| Fig. 2 Common epoxide synthesis methods. | ||
4.2 Thiirane synthesis
Historically, thiiranes were difficult to stabilize and often resulted in the unintended formation of polymeric materials.42,43 Staudinger first synthesized stable thiiranes in 1916.44,45 Later, in 1921, Delépine provided a relatively straightforward synthetic scheme to prepare thiiranes from the reaction of chlorothioisocyanate and sodium sulfide.46,47 A more modern take utilizes an epoxide reacted with thiourea or isothiocyanate, which produces episulfide in high yield.42,43,48 Thiiranes can also be produced through thiocarbonyl ylide or aldazine N-oxide intermediates, which can preserve stereo-structure, but these are generally more difficult.49Fig. 3 shows an overview of these methods, with several other methods reported in the following ref. 45 and 48.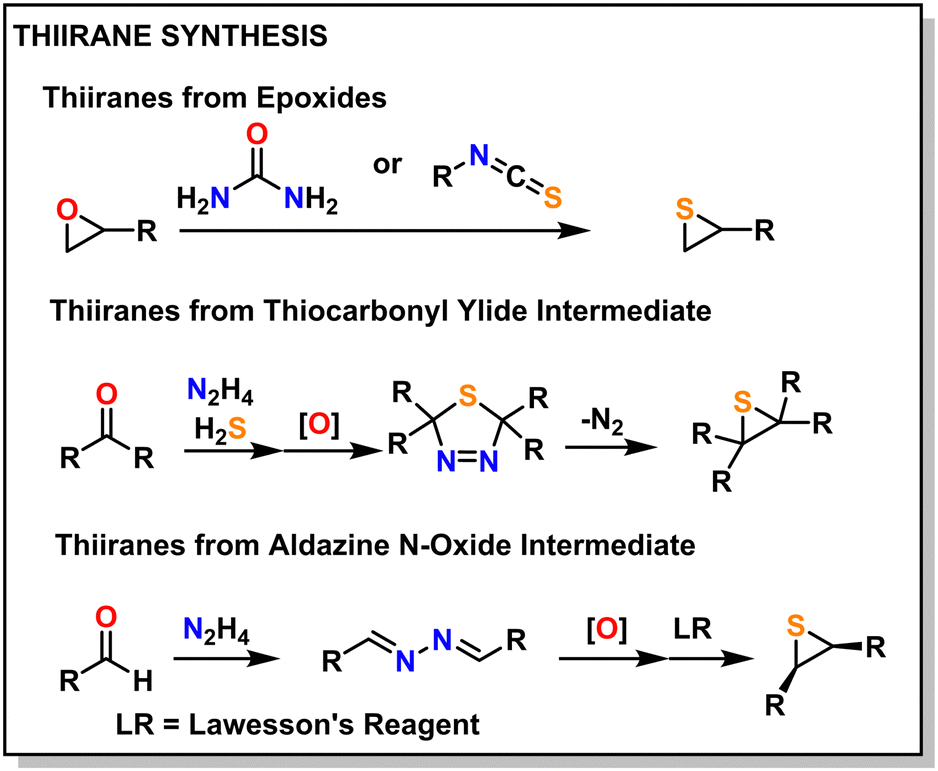 | ||
| Fig. 3 Common thiirane synthesis methods. | ||
5. Contemporary synthetic techniques for poly(thio)ethers
There are a multitude of methods to achieve poly(thio)ethers using Earth-abundant metals, each with their own pros and cons in terms of control over polymer properties. This section highlights the state-of-the-art. We opted to split this section into several sub-sections each devoted to a different class of synthetic method. To highlight the differences between methods and facilitate reader comprehension, we have provided qualitative “scores” for each of six different criteria for the polymerization method. These criteria are: molecular weight control, ease of use, monomer compatibility, (micro)structure control, kinetics, and end group control. The scores provided are based on our own assessments and are borne from each of the authors’ unique backgrounds in physics, chemistry, and chemical engineering. The scores are subjective and are meant merely to guide the reader to find a suitable synthetic method for their needs. A table summarizing the relevant techniques in terms of some of these criteria can be seen below in Table 2.| Technique | M n (kg mol−1) | Đ | Monomers | Ease of use |
|---|---|---|---|---|
| AE = alkyl epoxide, GE = glycidyl ether, AT = alkyl thiirane, GT = glycidyl thioether, O = other, R = 4 or more member ring. | ||||
| AROP | <30 | <1.2 | GE, AT, GT | Hard |
| MAROP | <200 | <1.2 | AE, GE, AT, GT, O | Easy |
| CROP | <10 | >1.5 | O, R | Medium |
| Vandenberg | 100–10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 000 |
>2.0 | AE, GE, AT, GT, O, R | Medium |
| DMC | <10 | Varies | AE, GE, O | Easy |
| Inoue | <10 | <1.2 | AE, GE, AT, GT, O, R | Medium |
| Coates | <150 | ca. 2.0 | AE, GE, O | Medium |
| NAl Adduct | <100 | <1.3 | AE, GE, AT, O | Easy |
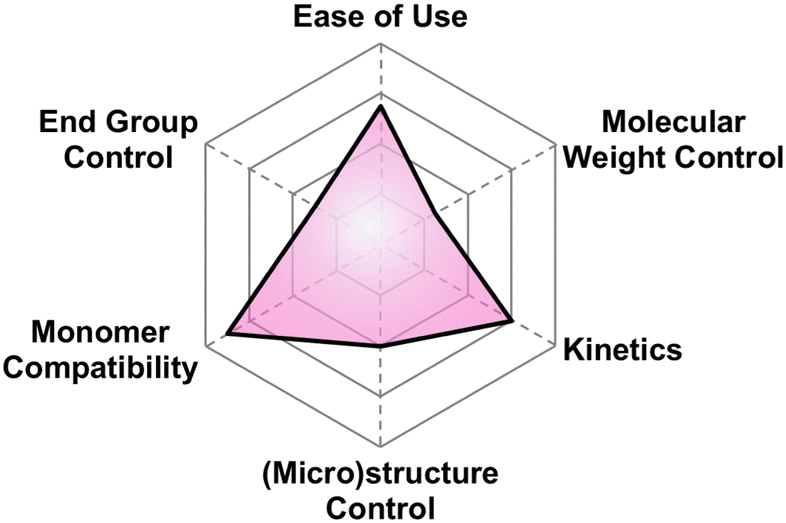
5.1 A brief explanation of evaluation categories
Here we briefly explain the categories by which we have scored each polymerization technique. These are based on our opinion and are meant to help those less versed in poly(thio)ether synthesis to find the best method for their needs. For each category we assign four different criteria each worth one point.5.2 Anionic ring opening polymerization (AROP)
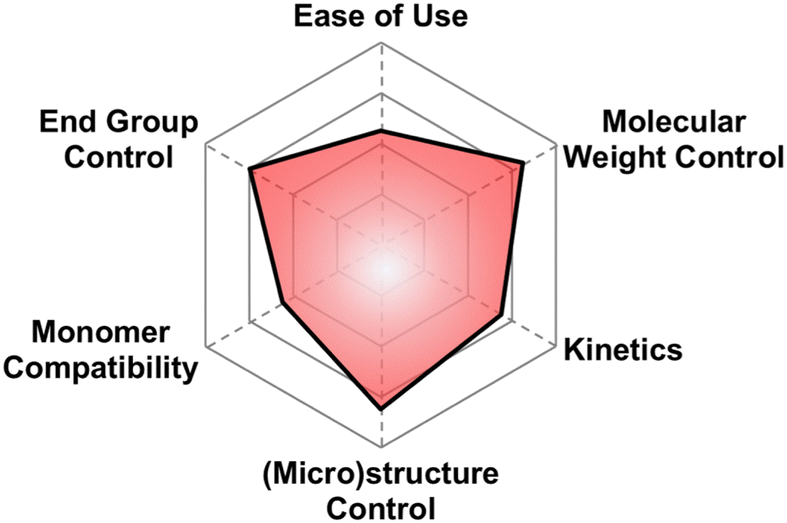 | ||
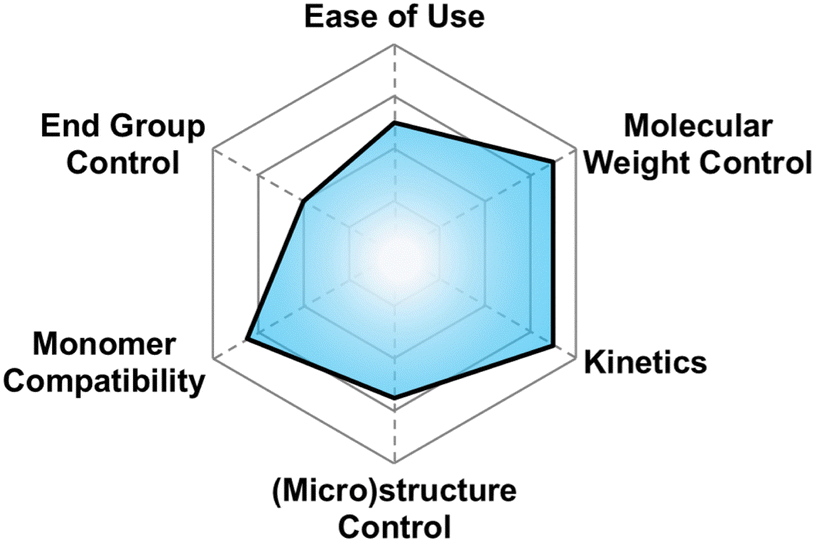
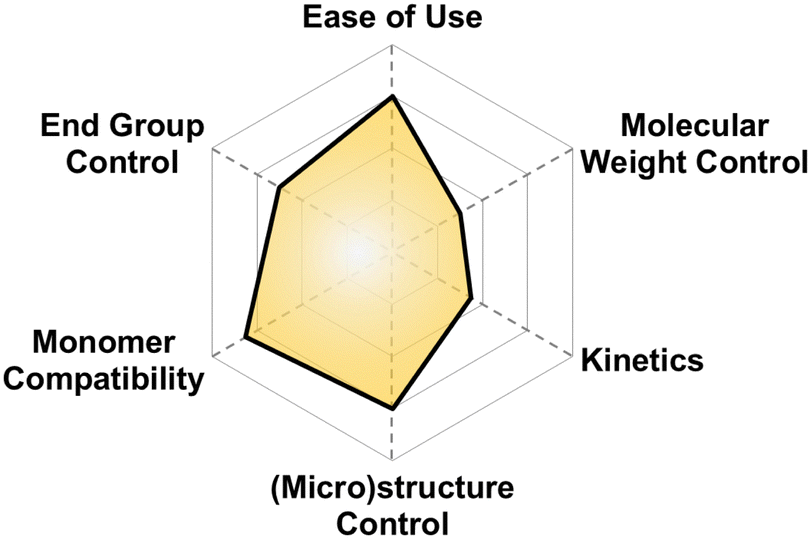
| Fig. 4 Subjective scores for AROP with Earth-abundant metals visualized via radial plot. All parameters are scored from 0–4. | ||
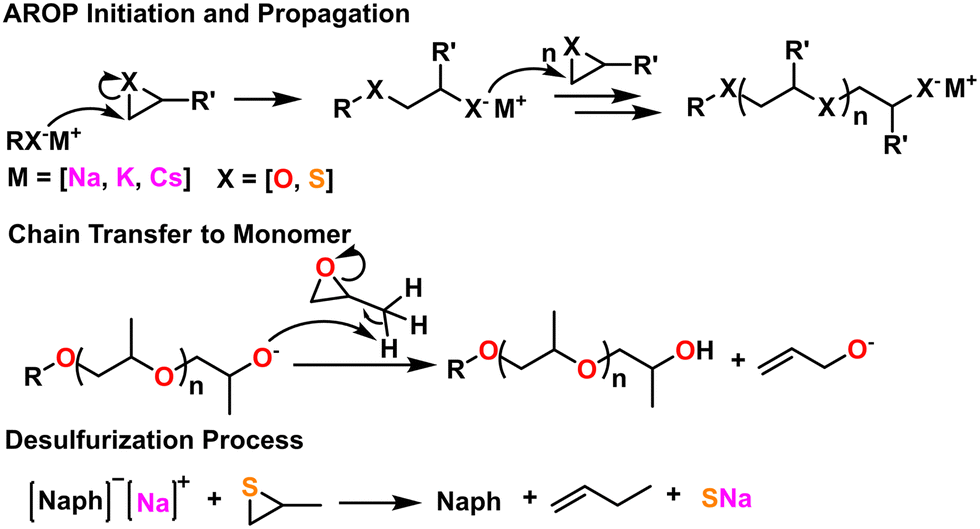 | ||
| Fig. 5 (Top) General AROP initiation and propagation mechanism. (middle) Chain transfer to monomer, a common side reaction with epoxides. (Bottom) Desulfurization process endemic to thiirane AROP. | ||
A number of side reactions can occur due to proton abstraction from substituted epoxides. Often this results in chain transfer to monomer, which limits molecular weight and bifurcates end group chemistry.63 Certain solvents, like DMSO, can also cause chain transfer.64 These side reactions can be suppressed with proper consideration of the metal complex used and identity of the counter-ion as well as the addition of compounds like crown ethers.62 This can be seen in the middle scheme in Fig. 5.
These side reactions are less problematic for substituted thiiranes as the thiolate ion is less basic than the alcoholate.24 However, the presence of disulfides, perhaps due to the oxidation of the initiating thiol, can act as transfer agents during the polymerization, lowering and broadening molecular weight.24,65 Therefore, protected thiols are sometimes used and de-protected right before the polymerization reaction. Certain Zn, Al, and Li initiators can cause “desulfurization” to occur during polymerization of substituted thiiranes, which results in the formation of disulfide bonds in the growing polymer chain and the formation of the relevant vinyl compound. This can be seen in the bottom scheme in Fig. 5.
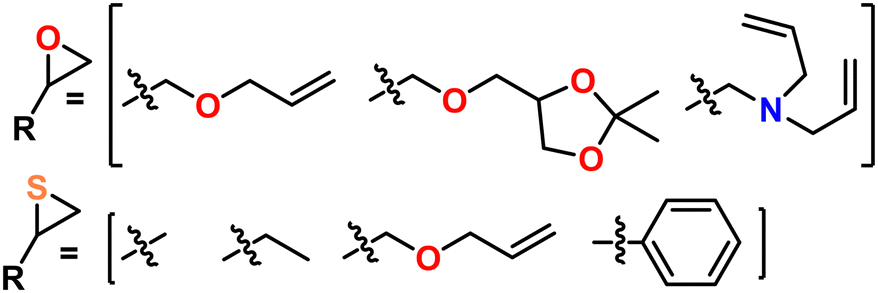 | ||
| Fig. 6 Example monomers compatible with AROP. | ||
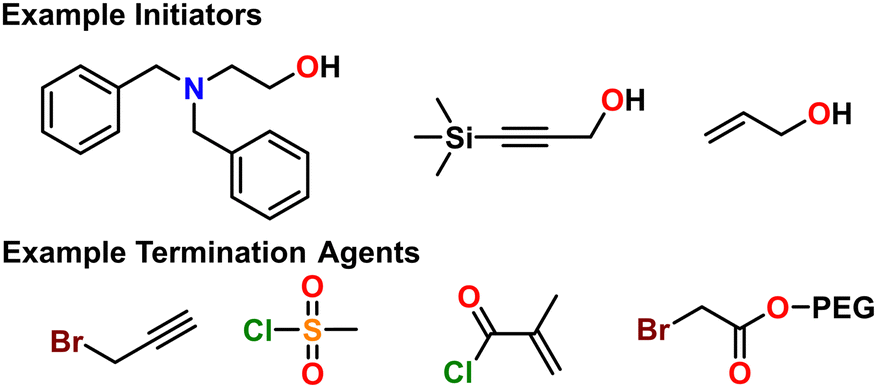 | ||
| Fig. 7 Some example initiators and termination agents for AROP, which define polymer end groups. | ||
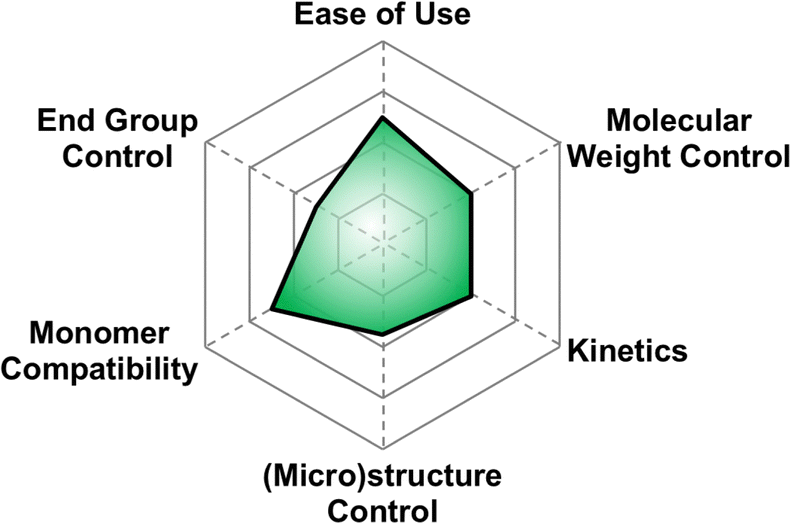
5.3 Monomer activated ring opening polymerization (MAROP)
 | ||
| Fig. 8 Subjective scores for MAROP with Earth-abundant metals visualized via radial plot. All parameters are scored from 0–4, with 4 being the best. | ||
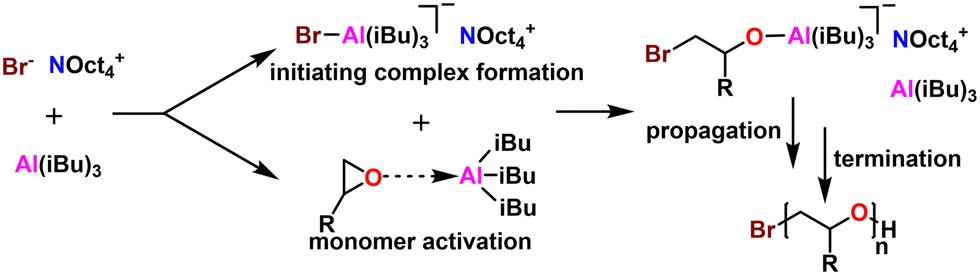 | ||
| Fig. 9 MAROP mechanistic overview. The Lewis acid serves to both form the initiator complex as well as activate the epoxide for ring opening. | ||
 | ||
| Fig. 10 Some example monomers compatible with MAROP. | ||
5.4 Cationic ring opening polymerization (CROP)
 | ||
| Fig. 11 Subjective scores for CROP with Earth-abundant metals visualized via radial plot. All parameters are scored from 0–4. | ||
An alternative route is through a Lewis acid mediated ROP using cationic metal compounds. These proceed by the activated chain end (ACE) mechanism, meaning the metal remains at the end opposite of the propagating cation. The foundational catalysts involve poorly defined Zn, Mg, and Al-based compounds and polymerized both oxygen and sulfur-based heterocycles.115,116 Defined metal complexes for CROP were first realized by Atwood and co-workers in the 1990's by using aluminum-salen complexes (cf. Fig. 12, left).117 Since then, several other Al, Mg, and Zn compounds have been applied to epoxide polymerizations. Generally, this approach produces low molecular weight polymers (Mn < 10 kg mol−1), at moderate to high Đ (Đ>1.5). Furthermore, the polymers are regio-irregular with many head to head and tail to tail defects present in the final polymer. The reader is encouraged to read the excellent review by Sarazin and Carpentier for an exhaustive list of structures related to epoxide polymerization with cationic metal compounds.118
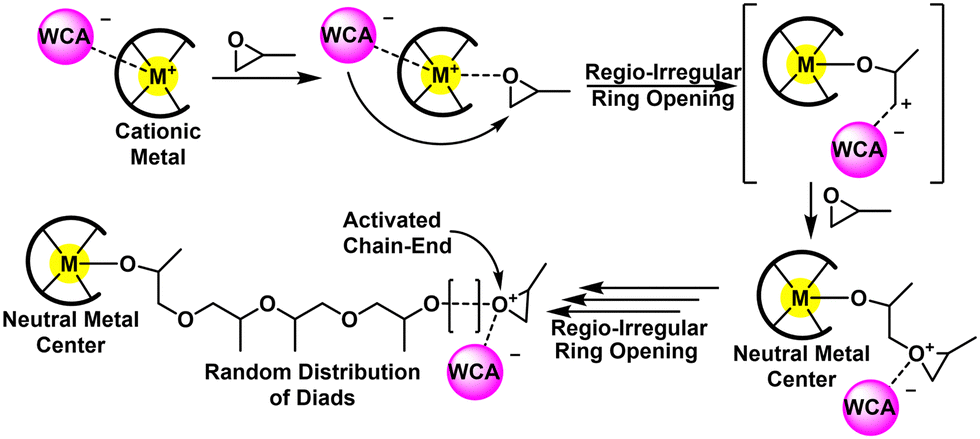 | ||
| Fig. 13 Mechanistic overview for CROP. Adapted with permission from ref. 118. Copyright 2015 American Chemical Society. | ||
5.5 Coordinative methods
There are a number of Earth-abundant metal compounds that facilitate coordinative polymerizations of epoxides and/or thiiranes. Instead of listing every single Earth-abundant metal containing compound that polymerizes epoxides/thiiranes and proceeds by a coordinative mechanism, we opted instead to break these out into several general sub-categories. Specifically, we give examples of an undefined (Vandenberg's catalyst), a weakly defined (DMC Catalyst), a well-defined (Inoue's catalyst), and a well-defined, enantioselective (Coates’ catalyst) catalyst, to give a general overview of the different types of catalysts one would encounter when synthesizing poly(thio)ethers from coordinative methods. These techniques were chosen based on their historical impact on poly(thio)ether synthesis, their use today, and their pedagogical utility for demonstrating disparate aspects of coordination polymerization. Excellent reviews by Coates on stereoselective coordination polymerization of epoxides135 and by Kuran on coordinative polymerizations of heterocycles136 can help the reader further explore this area.Why use it?. If you absolutely need to synthesize a poly(thio)ether to extremely high molecular weight and do not care too much about control, then this may be the technique for you Fig. 14.
 | ||
| Fig. 14 Subjective scores for Vandenberg's catalyst visualized via radial plot. All parameters are scored from 0–4. | ||
Overview and development. Vandenberg tells the story of the development of his eponymous catalyst in this ref. 137. We will summarize here. CC Price at the University of Pennsylvania developed a heterogeneous catalyst system, combining aluminum isopropoxide and zinc chloride enabling the polymerization of PO to high molecular weight.138 EJ Vandenberg, an industrial chemist at Hercules Inc., began to see the similarities between his catalyst for vinyl ether polymerizations and Price's catalyst and sought to see if his catalyst would polymerize epichlorohydrin (ECH). Serendipitously, the monomer he used had a small amount of water present, which resulted in a small amount of semi-crystalline PECH. The adventitious water reacted with the alkyl aluminum in his catalyst, enabling the cationic polymerization of ECH. Simplifying his catalyst system to just include the alkyl aluminum and water resulted in controlled polymerization of ECH to high molecular weight. In furtherance of the mechanistic understanding of this catalyst system, Vandenberg attempted to block a reactive site with acetyl acetonate, which he hypothesized would decrease the effectiveness of his catalyst system.139 On the contrary, he obtained high polyether quickly and in high yield. The likely active product of the reaction of two parts alkyl aluminum, one part acetylacetone, and one part water, seen in Fig. 15, is Vandenberg's catalyst, which is still used industrially today to produce a variety of commercial elastomers.
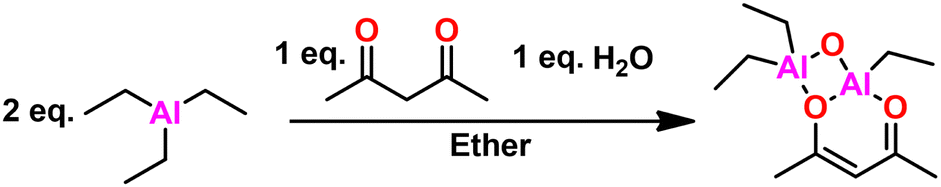 | ||
| Fig. 15 Vandenberg's catalyst synthetic scheme. | ||
Recent examples. Despite its age, Vandenberg's catalyst is still used commercially today to produce Hydrin® elastomers.137 Aside from commercial products, the catalyst is utilized in academia as well. For instance, Sanoja and Lynd employed Vandenberg's catalyst to produce crosslinked polymers and compared this with crosslinked materials made by a more controlled polymerization mechanism.140 They found significantly different mechanical degradation profiles between these materials. Lynd and Chwatko discovered the catalyst could be used to homopolymerize lactones as well as copolymerize them with epoxides.141 Statistical copolymerization between these two monomers allowed for degradable polyether materials. Finally, Beckingham, Sanoja, and Lynd used Vandenberg's catalyst to study the kinetics of epoxide copolymerizations.142
Mechanism. The reaction of water, acetyl acetonate, and trialkyl aluminum produces a whole host of side reactions, the product of any of which, or a combination, could be the active catalyst. Vandenberg and Price hypothesized a bimetallic polymerization pathway.143,144 Okuda experimentally determined this was the case for related compounds.145 We have previously investigated the mechanism for the catalyst and determined the most likely catalytic structure is a bis-(μ-oxo) di-aluminum species,146 which is consistent with the suspected structure. The epoxide is recruited through the oxygen opposite the acetyl acetonate, which causes a break in the four-membered bis-(μ-oxo) ring allowing for the epoxide to be ring opened and added. The bis-(μ-oxo) ring then recloses and a subsequent monomer can be added. We have previously mapped out the mechanism through DFT studies, the results of which are shown below in Fig. 16.
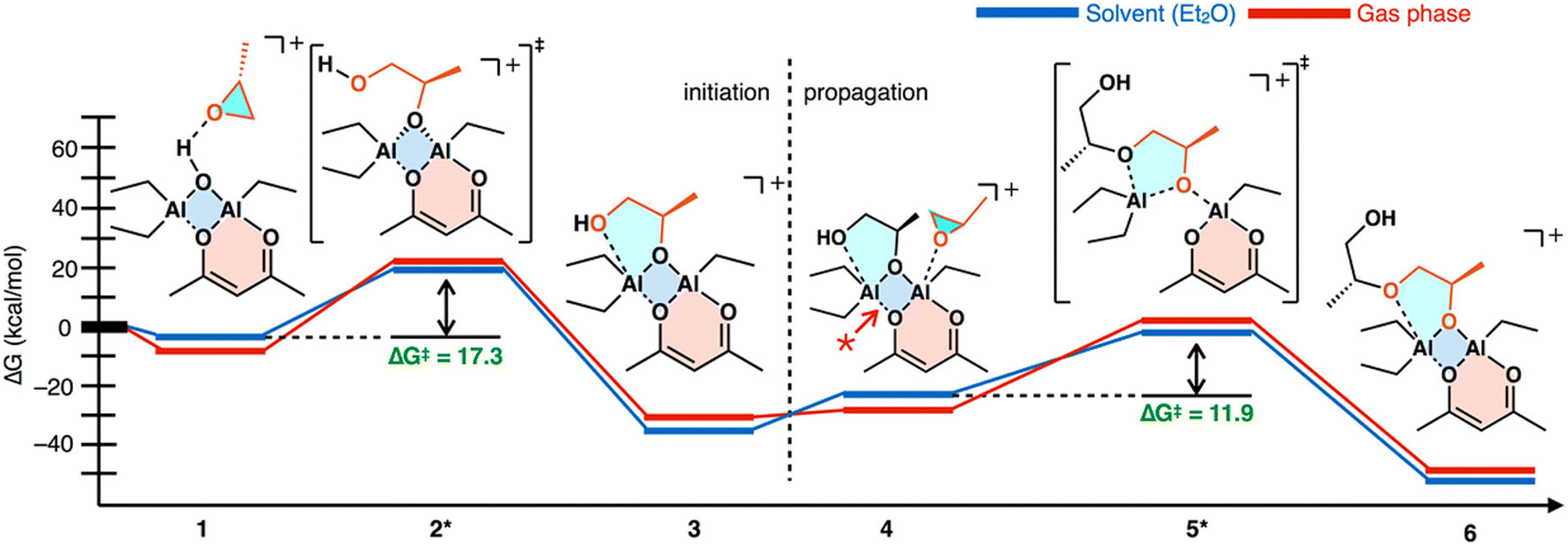 | ||
| Fig. 16 Polymerization mechanism of Vandenberg's catalyst as determined from DFT studies. Reproduced with permission from ref. 146. Copyright 2018 American Chemical Society. | ||
Kinetics. The polymerization kinetics of this catalyst are generally swift, with polymerizations often taking minutes to hours. Kinetics are first order in monomer based on our kinetic studies.142,146 Since the active catalytic species cannot be separated from the reaction, the order of reaction involving the catalyst is unknown.
Molecular weight control. Molecular weights typically range from 100 kg mol−1 to 10
000 kg mol−1.139 However, Đ are high, often greater than 2. Owing to the ill-defined nature of the catalyst, little control is possible. Further complicating control is that the activity of the catalyst varies batch to batch and so meaningful concentration/control relationships are difficult to obtain.
(Micro)structural control. Block and statistical copolymers of a variety of monomers have been demonstrated. Polymers produced with this method are isotactically enriched to about 30% and mostly regio-regular with a small amount of regio-defects.
Supported monomers. A variety of monomers are compatible with the Vandenberg catalyst. It was one of the few catalysts of its time that could polymerize ECH, a sometimes difficult to polymerize monomer, up to high molecular weight.137 Aside from ECH, a variety of glycidyl ethers,139,147 epoxides with alkyl pendants,139 epihalohydrins,148 thiiranes,115 di-substituted epoxides,149 cyclic ketals,150 oxetane,151–153 and lactones141 can be polymerized.
End group control. Owing to the ill-defined nature of the catalyst system, end group control is not possible. Polymers are very large which makes end group characterization difficult. As such, there is probably a mix of end groups.
Ease of use. The catalyst system is easy to synthesize, but does require careful monitoring and some experience with handling pyrophorics. However, polymerizations are facile and high molecular weight polymers are produced with ease at mild to moderate temperatures in bulk or in solvent. The solvents used are typically diethyl ether, heptane, benzene, or toluene. As stated above, catalyst effectiveness is batch to batch. Residual aluminum can interact with the polyether, making removal difficult.
Why use it?. If you do not want to deal with pyrophorics, but still want to polymerize a broad set of epoxide substrates to 10 kg mol−1, then this may be the technique for you Fig. 17.
 | ||
| Fig. 17 Subjective scores for DMC catalysts with Earth-abundant metals visualized via radial plot. All parameters are scored from 0–4. | ||
Overview and development. DMC catalysts are classic industrial catalyst systems that have received renewed attention in the last few decades. Originally, these catalysts were developed in the 1960's by General Tire, Inc. to produce polyether polyols for the production of polyurethane.154 The catalysts are formed from the precipitation of a metal salt of formula MX2 like ZnCl2 and a metal cyanide salt of formula K3M′(CN6) like K3FeCN6 in the presence of a complexing agent (CA), often tert-butyl alcohol, but aldehydes, ketones, ethers, amides, ureas, nitriles, and sulfides have also been identified and a co-complexing agent or initiator in the form of a multivalent alcohol.155 The metals generally consist of M = ZnII, FeII, CoII, or NiII and M′ = CoIII, FeIII, CrIII, and IrIII. An example catalyst structure is shown in Fig. 18. The resultant catalyst is often nanosized and heterogeneous in nature with its structure affected by the CA chemistry as well as the synthesis temperature.155 Epoxide polymerizations with DMC catalysts are done at around 100 °C, in an air free environment, under pressure, and in the absence of solvent. The resultant polyethers are often less than 10 kg mol−1 in size and can be narrow Đ, although this varies broadly. The DMC catalysts are also effective for the alternating polymerization of CO2 and epoxides.
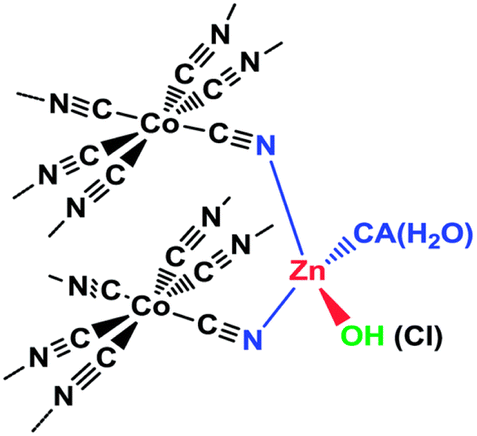 | ||
| Fig. 18 Example DMC catalyst structure. Reproduced with permission from ref. 156. Copyright 2014 Royal Society of Chemistry. | ||
Recent examples. There has been a recent resurgence of work on DMC catalysts. Lopez and co-workers157 as well as Kim and co-workers155 clarified some of the mechanistic aspects of the DMC catalysts as well as structure–activity relationships for the polymerization of epoxides. Frey and co-workers highlighted a unique aspect of the DMC catalyst, which is their proclivity to polymerize a broad set of monomer classes.158 Here, they copolymerized hexamethylcyclotrisiloxane (D3) and PO using a DMC catalyst up to 3 kg mol−1 at low Đ. The resulting copolymer exhibited a composition gradient microstructure, but incorporation of PO and D3 was apparent from the single Tg which decreased with higher incorporation of D3. This same group also prepared a statistical copolymer of PO and glycidyl methyl ether (GME) using DMC catalysts up to 4.5 kg mol−1 at low Đ. Marquez and co-workers prepared a layered DMC catalyst which crystallized.159 This catalyst showed enhanced capability over traditional catalysts to polymerize epoxyhexane. Kim and co-workers demonstrated the polymerization of glycidol with controlled branching.160
Mechanism. Owing to the heterogeneous nature of the catalyst as well as solubility issues, the polymerization mechanism is not fully known. However, DMC catalysts are thought to have a dual polymerization mechanism; an ACE-type CROP mechanism and a coordination-insertion mechanism.155 This dual mechanism is a result of the heterogeneity of the catalyst, with surface sites providing the coordination route via bound initiator and internal metal (e.g., Zn2+) sites providing the CROP due to the relative absence of bound initiator. The coordinative pathway provides regioregular polyether while the CROP pathway is regio-irregular. A scheme of both pathways can be seen in Fig. 19.
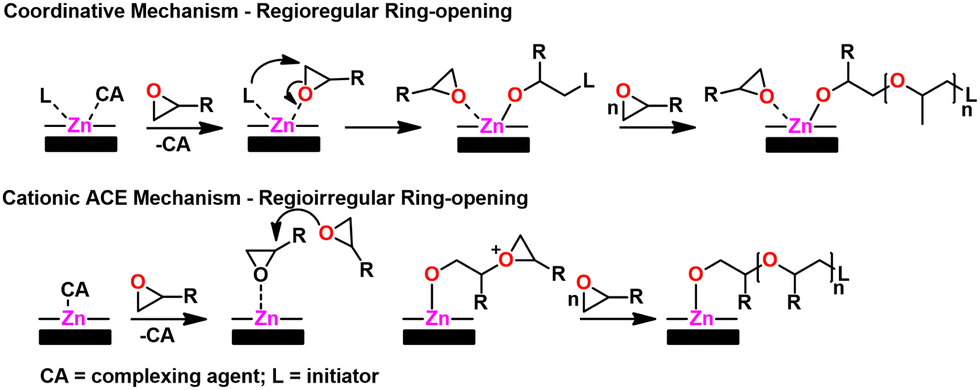 | ||
| Fig. 19 DMC catalyst mechanism. Adapted with permission from ref. 155. Copyright 2019 Elsevier. | ||
Kinetics. The kinetics of DMC catalyst mediated polymerizations of epoxides are quite fascinating.161,162 Induction times of minutes to hours are often noted; the induction time depends on the strength of the interaction between the CA and the catalyst surface with weaker interactions leading to lower induction times.155,162,163 A bell curve-like polymerization rate profile is observed; once the catalysts become active the polymerization rate increases rapidly until the acme, after which rapid catalyst deactivation occurs. The breadth, skewness, and peak height of the kinetics profile are affected by polymerization temperature, catalyst composition, and CA chemistry. To ball park the polymerization time, it takes minutes to a couple hours to produce polyethers using this method, with PO polymerizations being on the faster side and ECH on the slower side. The upside is that the system scales well and so 100s of grams of polymer can be produced in less than a couple hours.
Molecular weight control. Reported molecular weights typically cap out at around 10 kg mol−1 for all monomers studied. However, there is often residual, high molecular weight polymer owing to the suspected dual mechanism, which is mitigated through the addition of a co-CA.157,163Đ vary wildly and depend on the specific monomer, polymerization temperature, catalyst composition, CA(s), and catalyst preparation temperature. The preparation of low Đ (Đ < 1.1) PPO is well demonstrated, but other monomer substrates are less well explored and control may not be achievable.
(Micro)structural control. A wide variety of alcohols have traditionally been utilized with DMC catalysts in the polymerization of epoxides. Linear,155 star,164 and branched160 polymers have been demonstrated. These disparate architectures are achieved via the number of the alcohols of the initiator; multi-alcohol initiators generate branched structures, while one and two alcohol-containing initiators generate linear polyethers. Statistical and block copolymers have also been demonstrated. Polymers produced this way are atactic and mostly regio-regular with some regio-defects owing to the hybrid polymerization mechanism. Isotactically enriched polymer can be synthesized by using enantiopure monomer.156
Supported monomers. PO is by far the most common monomer polymerized with DMC catalysts owing to the commercial roots of this technique, however other monomers like glycidyl ethers,155,165 epoxy alkanes,155 and ECH156 have also been polymerized with this method. The catalyst composition plays a significant role in monomer compatibility and so catalysts are best tailored to the specific monomer of interest. Furthermore, this method also facilitates the copolymerization of epoxides with a variety of disparate monomers such as CO2,166,167 anhydrides,168,169 and siloxanes,158 enabling the synthesis of unique polymers.
End group control. The end group is dictated by the initiating ligand that is used.155 Several mono- and multi-functional alcohols have been demonstrated and so end group control is robust. The polymer grows from the metal center on the catalyst and once the polymerization is terminated, a terminal OH is present. This facilitates production of both heterobifunctional and telechelic polyethers. Alcohol containing CAs are initiative, which affects uniformity of end groups.
Ease of use. The preparation of the catalyst is relatively straightforward and proceeds through a mixture of the metal cyanide salt with the second metal salt in the presence of CA(s) at moderate temperatures. However catalyst structure–polymerization activity is not fully understood and so applying this method to understudied systems could be tricky.155 Pressure-resistant reactors and air free environments are required for polymerization reactions. Polymerizations proceed at mild to moderate temperatures and can be performed in bulk. The catalyst becomes inactive when exposed to air and does not remain with the polymer. Furthermore, because of the size of the inhomogeneous catalysts, separation is largely straightforward to obtain a pure polymer product.
Why use it?. If you want to polymerize basically anything to 10 kg mol−1 with good control, then this may be the technique for you Fig. 20.
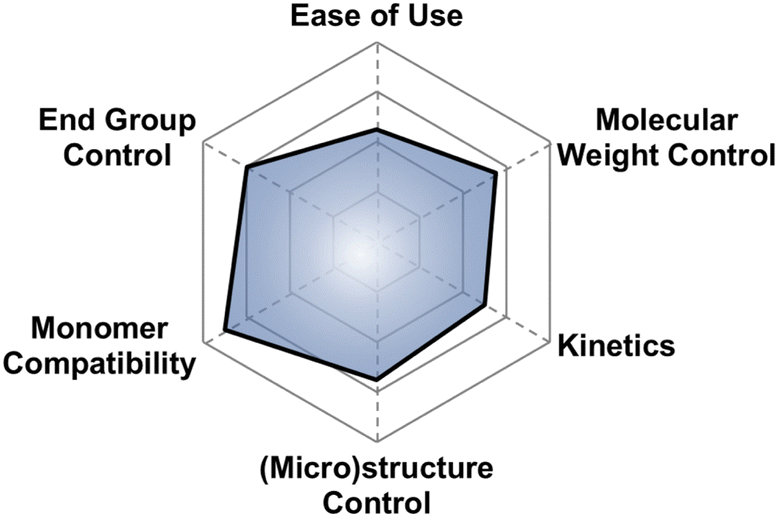 | ||
| Fig. 20 Subjective scores for Inoue's metal porphyrin catalyst visualized via radial plot. All parameters are scored from 0–4. | ||
Overview and development. The reader may find it interesting to read the story of the development of this striking class of catalysts in Inoue's own words found in this citation in English170 and this in Japanese.171 We will summarize here. In 1978, Inoue and co-workers first demonstrated a chloro-aluminum tetraphenyl porphyrin (AlCl-TPP) catalyst that allowed for the facile polymerization of epoxides172,173 to controlled molecular weight via a coordination-insertion type polymerization.174 Chemical structures of these catalysts can be seen in Fig. 21. Inoue followed this work up with several other catalysts with different substitutions in place of the Cl (e.g., –OR, –SR, etc.) as well as Zn in place of Al. The Zn based catalysts were found to be effective for polymerization of thiiranes.175
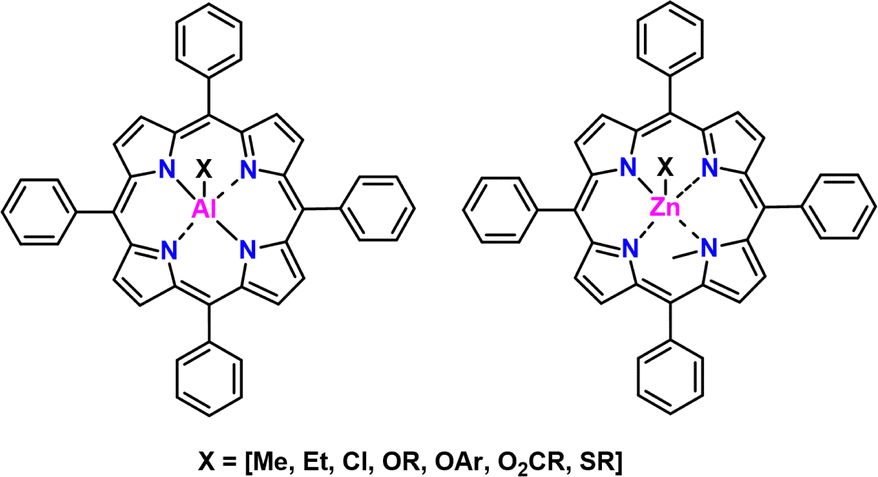 | ||
| Fig. 21 TPP catalyst structures with Al (left) and Zn (right). | ||
Inoue tested the polymerization of epoxides in the presence of several alcohols, anticipating that these would lead to a broadening of the Mn. Instead, Inoue found that these alcohols, in the presence of the TPP catalyst, underwent rapid reversible chain transfer with the growing polymer chain resulting in a narrow Đ and a single peak on the GPC. He termed these polymerizations “immortal” since they could not be killed.176 Aside from Zn and Al, other metals have recently been used in the polymerization of epoxides, like Cr, Mn, and Co.177–179
Recent examples. Due to the diversity of compatible monomer classes, this catalyst is currently heavily utilized for ring opening copolymerization (ROCOP) of epoxides with other species, rather than in pure poly(thio)ether synthesis. Honda and co-workers investigated the alternating polymerization of oxetane and CO2 with a variety of metalloporphyrins and organo-cocatalysts.180 They were able to selectively synthesize poly(trimethylene carbonate) with near 100% carbonate linkages using the AlCl-TPP catalyst along with tetra-n-butyl ammonium bromide. Wang and co-workers exploited the robustness of the metallo-porphyrin system along with clever choice of chain transfer agent for the one pot synthesis of block copolymers of poly(methyl methacrylate) (PMMA) and poly(propylene carbonate) (PPC).181 Here, the metalloporphyrin served dual roles; to initiate the alternating polymerization of propylene oxide and CO2 as well as a photoredox catalyst for photoinduced electron/energy transfer-reversible addition–fragmentation chain transfer (PET-RAFT) polymerization of vinyl monomers. The RAFT agent contained a carboxylic acid moiety that served as a chain transfer agent (CTA) for PO/CO2 polymerization, which resulted in the block copolymer. A similar approach with cyclic anhydrides as opposed to CO2 was demonstrated by the same group a few years later.182 Some excellent recent reviews have highlighted metalloporphyrins as switchable catalysts183 as well as their role in sustainable polymerization184 and we encourage the reader to check those out if interested.
Mechanism. The polymerization mechanism for the metal porphyrin catalysts is coordination-insertion, where the monomer adds at the metal center, as shown in Fig. 22a. The species bound to the metal center (e.g., Cl, –OR, –SR) initiates the polymerization. Inoue found that by adding a Lewis acidic aluminum species a rapid increase in kinetics for both epoxides185 and thiiranes170,171 was achieved. Here, the Lewis acid species activates the monomer, facilitating enchainment and ring opening while simultaneously suppressing side reactions, evoking MAROP, as seen in Fig. 22b. Adding alcohols or thiols as CTAs, results in the rapid exchange between the protic species and the propagating polymer chain, as seen in Fig. 22c. This occurs far faster than a single monomer addition, leading to immortal-type polymerizations.176 For the polymerization of thiiranes, adding thiols as a CTA resulted in this immortal character, while alcohols did not.175 Furthermore, adding chemicals that are traditionally catastrophic for epoxide polymerizations like HCl had no effect on the polymerization, further demonstrating how robust this approach is.186
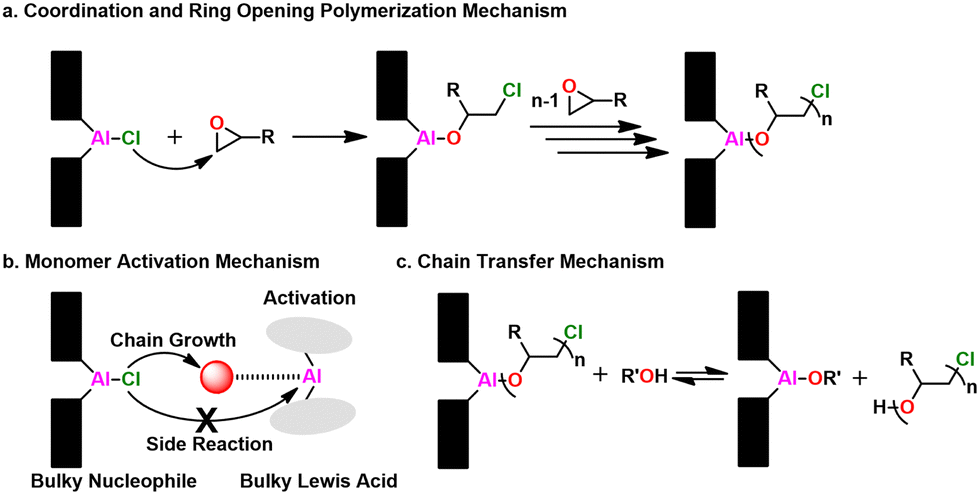 | ||
| Fig. 22 Al-porphyrin catalyst mechanism overview. (a) Coordination and ring opening, (b) Monomer activation, (c) Chain transfer. | ||
Kinetics. The polymerizations are living in nature and first order in monomer. There is a significant increase in kinetics with the addition of bulky Lewis acids that work to activate the monomer. The presence of CTA significantly slows the polymerization kinetics down. Generally, depending on monomer, CTA/TPP ratio, targeted Mn, and presence of Lewis acid, polymerizations take hours to several days.
Molecular weight control. Molecular weights are very well controlled regardless of the presence of Lewis acid and/or CTA and depends on the monomer to (initiator + CTA) ratio. For most polymerizations involving CTAs, Mn < 10 kg mol−1. However, the original report of this system demonstrated Mn > 80 kg mol−1. Đ are typically <1.2 for polymers Mn < 10 kg mol−1. Higher molecular weight may have higher Đ. Addition of CTA does not broaden the molecular weight distribution, but leads to lower overall Mn.
(Micro)structural control. Block and statistical copolymers have been demonstrated. Alternating copolymers have been demonstrated with epoxides and CO2 or anhydrides. The functionality of the protic CTA dictates branching architectures; for instance a dihydroxyl containing CTA enables the formation of telechelic polymers or facile polymerization of triblock copolymers. Similarly, compounds with 3 or 4 protic groups result in three and four arm star polymers. Some example multi-functional groups can be seen in Fig. 23. The polymers formed with TPP catalyst are strictly regioregular and isotactically enriched, reminiscent of other coordinative systems.
 | ||
| Fig. 23 Example (multi-)functional end groups through choice of CTA. | ||
Supported monomers. Perhaps the strongest aspect of this approach is the scope of monomer substrates that can be used. A wide variety of substituted epoxides and thiiranes have been polymerized. Additionally, oxetanes, lactones, lactides, siloxanes,187 (meth)acrylates, and other monomers can be polymerized. Each class of monomers requires a slightly different catalyst.170 For instance, thiiranes polymerize from the Zn–porphyrin catalyst, while epoxides proceed from the Al-porphyrin catalsyt.
End group control. The end group is dictated by both the substitution on the metal and the identity of the CTA used. In the case of the AlCl-TPP catalyst, a completed polyether will be heterobifunctional with a chloro group on one end and a hydroxyl on the other. Al-TPP catalysts with substitutions other than Cl are a bit trickier to prepare making end group control less than facile. CTA can be added to diversify the end groups; Polymerization occurs from both the CTA as well as the aluminum porphyrin catalyst itself so a mixture of polymers with both end groups will be obtained in this way. The ratio of protic species to TPP catalyst can be increased to ensure the vast majority of polymers are capped with the targeted end group. However, this comes at the cost of Mn or kinetics. Some example functional end groups can be seen in Fig. 23.
Ease of use. Synthesis of the AlCl-TPP catalyst involves the room temperature reaction of tetraphenylporphyrin with ClEt2Al under nitrogen, while the Zn-TPP catalyst uses diethyl zinc and N-methyl tetraphenylporphyrin. The metal compounds are pyrophoric and so require caution when handling. Separation of the prepared catalyst is made by simply removing the volatile compounds under reduced pressure. Other catalysts (i.e., those with groups different than Cl) are more difficult to synthesize, and involve multi-step reactions. Polymerizations are generally performed at mild temperatures (RT - 50 °C) in bulk or solvent (often DCM). Unlike MAROP or other coordinative methods, the catalyst is generally easy to remove through extraction owing to its size.
Why use it?. If you want to make fully isotactic polyethers from a broad set of racemic epoxide substrates, then this may be the technique for you Fig. 24.
 | ||
| Fig. 24 Subjective scores for Coates' bimetallic catalyst visualized via radial plot. All parameters are scored from 0–4. | ||
Overview and development. An account in Coates’ own words of the development of this catalyst system can be found in this reference which doubles as an excellent review of stereo-selective polymerizations of epoxides.135 While exploring a cobalt catalyst for copolymerizations of PO/CO2, Coates and coworkers found one catalyst system resulted in a mixture of polyether and polycarbonate chains with the polyethers being nearly 100% isotactic in nature. This catalyst was a cobalt(III) complex oxidized by acetic acid and it polymerized PO to high Mn (> 100 kg mol−1) at moderate Đ (< 1.5) at 0 °C in bulk or select solvents with extremely high isotacticity from a racemic mixture of PO.188
The authors explored various substitutions at the metal center and periphery and saw significant effect on activity. As a result of these factors, they proposed a bimetallic polymerization mechanism and synthesized a bimetallic version of the catalyst.189,190 This bimetallic catalyst, when combined with an ionic salt, polymerizes a wide variety of mono-substituted epoxides to moderate to high Mn with moderate Đ and a high degree of isotacticity. Later, similar chromium-based catalysts were synthesized, which facilitated architectural control through chain shuttling agents (CSAs).191 In the case of the chromium catalysts with CSA, it is an immortal polymerization.192
Recent examples. Often, these catalysts are used to copolymerize epoxides with CO2 to form polycarbonates.193 In another example, Coates and co-workers explored the mechanical properties of a suite of isotactic poly(propylene oxide) (iPPO) of different structures. They found enantioenriched iPPO had similar mechanical performance to Nylon-6,6. They also observed photodegradation of the polymer under UV light and suggested this could be selectively stabilized to program in a degradation profile.194
Mechanism. The mechanism was elucidated using DFT studies and can be seen in Fig. 26.195 Briefly, they found that the stereochemistry of the binapthol linker determines the enantionmer preference during polymerization. The polymerization proceeds via coordination-insertion by a bimetallic pathway. The Cr catalyst displays an induction time which is a result of trace water.192
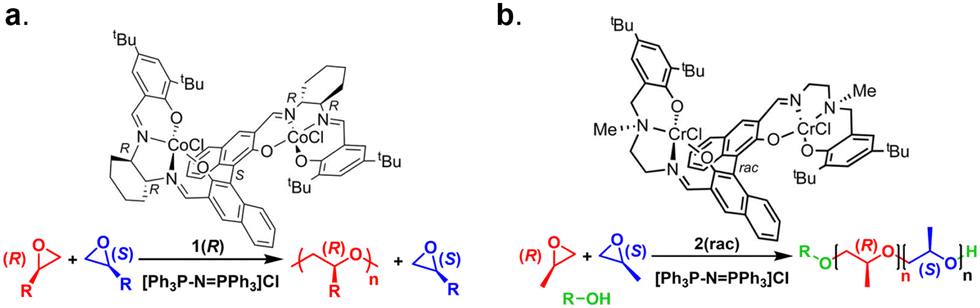 | ||
| Fig. 25 (a) Example structure of an enantioselective bimetallic cobalt catalyst and (b) racemic bimetallic chromium catalyst and their associated polymerizations. Adapted with permission from ref. 189 and 191. Copyright 2010 and 2017 American Chemical Society. | ||
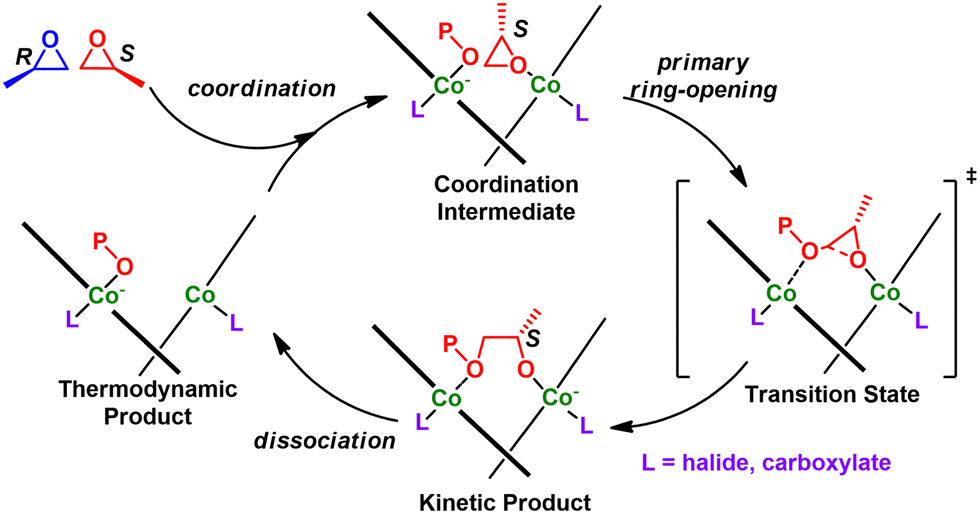 | ||
| Fig. 26 Enantioselective polymerization with double cobalt catalyst. | ||
Kinetics. The catalysts are characterized by high turn over numbers and, as such, polymerizations are swift. As an example, PO can be polymerized to 30 kg mol−1 in 15 minutes. Kinetics are affected by the specific catalyst used, the ionic salt co-catalyst, solvent, and the monomer. Most polymerizations take place in the minutes to hours range. The Cr catalyst undergoes a noticeable induction time of ca. 20% the total polymerization time depending on CSA used and substitution on the metal center.192 Polymerizations are living and, when combined with a CSA, are immortal.192
Molecular weight control. M n are controlled through monomer to (bimetallic catalyst + CSA) ratio. Moderate to high Mn (Mn up to 150 kg mol−1) can be achieved for a broad swath of epoxide substrates. There is a significant exotherm using this system so low molecular weight polymers may require additional considerations. Đ are generally moderate between 1.5 and 2 depending on the catalyst, ionic salt co-catalyst, and monomer. Lower Đ can be achieved with alkyl linked bimetallic Cr catalysts.196
(Micro)structural control. The major advantage of this method is the synthesis of polyethers with high degrees of regio- and stereo-regularity, as well as enantioselectivity of the monomer. As such, most of the polymerizations explored with this system are homopolymerizations of racemic epoxides and so limited data on statistical copolymers is extant. Stereoblock copolymers were demonstrated by polymerization of racemic PO with a racemic catalyst.194 Block coplymers were demonstrated using a telechelic polymer as a CSA. In this way, PPO-PCL-PPO terblockcopolymers were synthesized. Similarly, tri-functional CSAs resulted in 3-arm polymers.191
Supported monomers. A broad array of epoxide monomers are supported like those with alkyl, aryl, unsaturated, and fluorinated groups as well as glycidyl ethers, as can be seen in Fig. 27. Compatible epoxide substrates depend on the chemistry of the catalyst, but the catalyst shown in Fig. 25 seems to be the most general. While thiiranes have not been explored with the bimetallic version of this catalyst, a related monometallic chromium based catalyst by Nozaki has shown capability to copolymerize thiirane and CO2.197,198 As such, these catalysts may also be amenable to thiirane polymerizations.
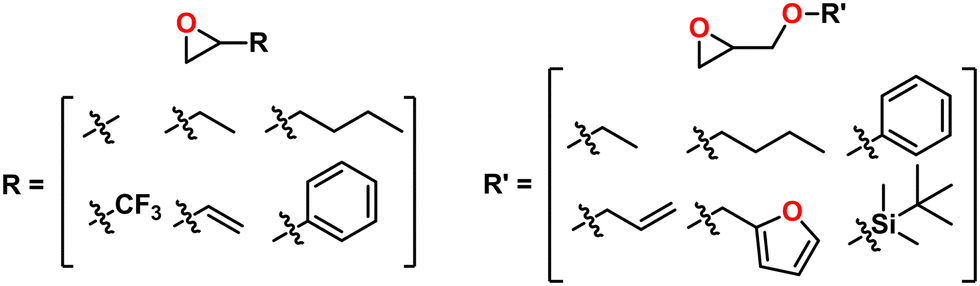 | ||
| Fig. 27 Monomers synthesized with Coates’ enantioselective cobalt catalyst. | ||
End group control. In the absence of CSA, the substitution on the metal center determines one end group, and so polymers are heterobifunctional. In the presence of CSA, one end group is determined by the CSA.191 While this provides additional end group control it comes at the cost of molecular weight and end group uniformity in a similar way to Inoue's catalyst.
Ease of use. Polymerizations are performed in air-free environments at low to moderate temperatures. Bulk polymerization is possible, but solvents (toluene or dimethoxyethane) are generally employed to mitigate the exotherm. Because the enantioselectivity is dictated by the catalyst stereostructure, only 50% of a racemic mixture of monomer can be polymerized by one catalyst. A racemic mixture of catalysts of opposite stereochemistry can mitigate this effect, but they must be synthesized separately and combined. Catalyst ligands are synthesized through a multi-step process in low-medium yields. Catalyst is often made in high yields and can be crystallized out of the reaction mixture.
5.5.5.1 Di-alkyl magnesium. Di-alkyl magnesium species were explored in the 1950s and 1960s for the polymerization of epoxides.199,200 Since then, this system has received very little attention. Recently Mejia and co-workers resurrected and modified this system and found that isotactic polymers could be synthesized from a racemic mixture of monomers, as can be seen in Fig. 28.201 They used di-n-butyl magnesium to polymerize, in the bulk, PO up to 65 kg mol−1 with Đ increasing to 2.3 with increasing Mn. Based on their kinetics studies, the polymerizations appear to be living. The end group was characterized by ESI-MS and determined to be a butyl group, suggesting two chains growing simultaneously from one Mg. Later they used this system to copolymerize CHO and CO2 which resulted in isotatically enriched polymer with a high percentage of carbonate linkages.202
 | ||
| Fig. 28 Isospecific polymerization of racemic PO with magnesium catalyst. | ||
5.5.5.2 Alkyl aluminum/phosphonic acid/Lewis base catalyst. Vandenberg's work spawned several other catalytic systems. In the 1960s and 1970s Vandenberg as well as others203 investigated a catalyst system consisting of tri-isobutyl aluminum, phosphonic acid, and trimethyl phosphine, which was found to effectively polymerize epoxides like PO, ECH, and AGE and resulted in amorphous polymers. Structurally defined catalysts based on this system were developed by Mason and co-workers in 2000.204 These catalysts consisted of methyl phosphonic acid and trialkyl aluminum and polymerized ECH to high molecular weight but with high Đ (Đ > 1.9), while PO could only be polymerized to ca. 4 kg mol−1 but at low Đ = 1.2. Later work added water or organic Lewis bases and these ill-defined catalysts achieved similar activity with high molecular weights and moderate to high Đ.205
5.5.5.3 Tin phosphate catalyst. In the 1960's and on, a few groups investigated the condensation product of organotin halides with trialkyl phosphates for the polymerization of epoxides.206,207 The product of this condensation was found to polymerize epoxides, like PO and ECH, to high molecular weight with high stereospecificity and, unlike many other examples presented in this article, they can be handled in air. Kragl and co-workers recently began to further characterize this system to better understand the factors that influence the polymerization of epoxides.208 They prepared the catalyst from butyl tin chloride and tributyl phosphate in a 1
:2 ratio. They found that they could polymerize PO up to ca. 40 kg mol−1 with 85% isotacticity. They could control the molecular weight through addition of propylene glycol. This system was further explored by Iwasa and co-workers to polymerize several other substituted epoxides up to 100 kg mol−1 but with high Đ > 2.0.209
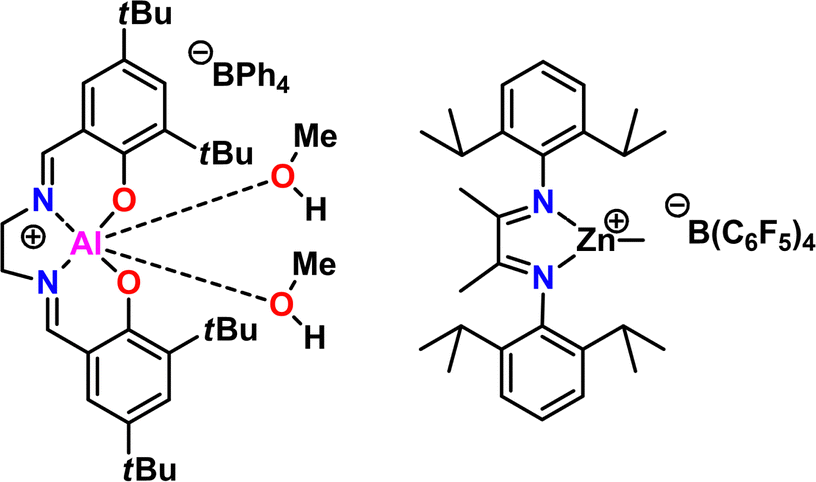
5.6 Amine-aluminum (NAl) adduct catalyst
 | ||
| Fig. 29 Subjective scores for the NAl Adduct Catalyst visualized via radial plot. All parameters are scored from 0–4. | ||
 | ||
| Fig. 30 Synthesis of the MOB catalyst/initiator for epoxide polymerizations. | ||
Kinetics were enhanced four times by replacing the triethyl aluminum species with a triisobutyl aluminum species.211 Further kinetic enhancement of nearly 100× was achieved by changing the ligand from di-benzyl amino ethanol to di-methyl amino ethanol.212 The startling increase in polymerization kinetics with the change in amine substitution suggested an important interaction between the trialkyl aluminum species and the amine. We hypothesized that a rearrangement occurred; the trialkyl aluminum species datively bound to the oxygen could associate with the amine, facilitating dimerization of two MOB species, a common occurrence for aluminum species.213 To test this, we prepared an amine aluminum (NAl) adduct from the reaction of triethyl amine and trimethyl aluminum, which we anticipated would act like a catalyst. We combined this with the BOD species that we anticipated would act like an initiator. The resulting polymerization kinetics matched the kinetics of the MOB species, lending credence to our hypothesis. We found this separate catalyst and initiator system was quite convenient; By increasing the relative concentration of the catalyst to the initiator we found a commensurate increase in kinetics. Furthermore, this allowed for the decoupling of kinetics from initiator structure, facilitating end group control. A scheme showing both approaches to epoxide polymerization can be seen in Fig. 31.
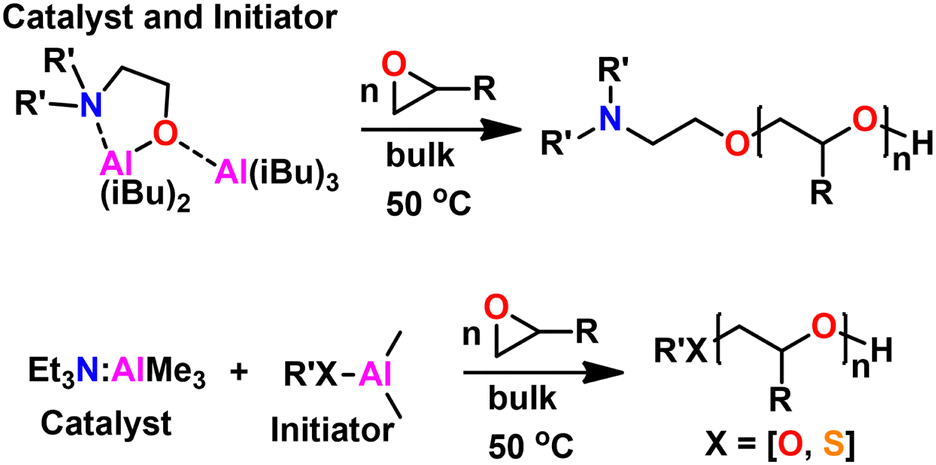 | ||
| Fig. 31 (Top) Epoxide polymerization with MOB catalyst/initiator. (Bottom) Epoxide polymerization with separate NAl adduct catalyst and initiator. | ||
Safaie, Ohno, and Ferrier later expanded this system by preparing initiators from thiol containing ligands.214 End group control was verified through ESI-MS, unequivocally demonstrating this method produces hetero-bifunctional polyethers. Furthermore, macroinitiators were prepared from polymers with thiol or alcohol end groups via reaction of trimethyl aluminum. In this way, chain extension from PEO as well as polymers prepared via RAFT like polystyrene and poly(methyl methacrylate) (PMMA) with epoxides was demonstrated.214,215 Additionally, bi-functional initiators were demonstrated through disulfide ligands, which enable the facile synthesis of telechelic polyethers and triblock copolymers. Finally, this polymerization method is compatible with episulfides as well; PS can be controllably homopolymerized up to 100 kg mol−1 and statistical and block copolymers of epoxides and PS can be prepared.215
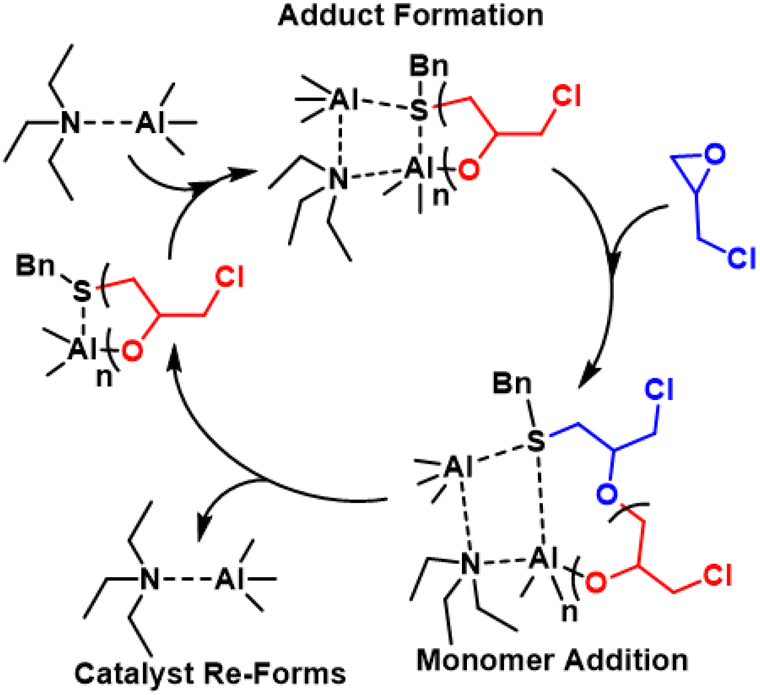 | ||
| Fig. 32 Proposed polymerization mechanism with the NAl adduct catalsyt. Reproduced with permission from ref. 219. Copyright 2023 Royal Society of Chemistry. | ||
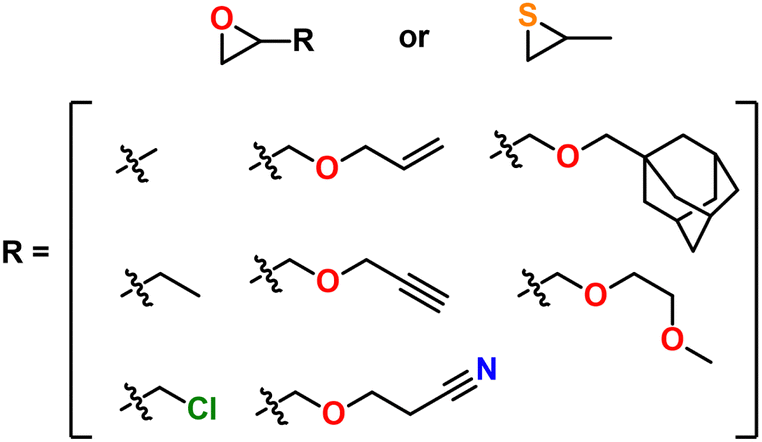 | ||
| Fig. 33 Select monomers polymerized with the NAl system so far. | ||
6 Conclusions and future directions
As the reader may have noticed, there are a multitude of polymerization techniques to synthesize poly(thio)ethers using Earth-abundant metals. All of the techniques mentioned here have seen recent developments in terms of monomer scope, (micro)structural consideration, molecular weight improvements, etc. As such, this is still a highly active field and is increasing in activity as facile techniques like MAROP and the NAl adduct catalyst have improved accessibility to these wonderful polymers. Furthermore, with the increasing importance of energy storage, CO2 sequestration, and mRNA delivery, polyethers and polythioethers have received renewed interest. The monomers remain ripe for utilization as a feedstock for a tunable polymeric materials platform. Furthermore, epoxide-based polyethers and by extension episulfide-based polythioethers have the potential to be made completely from biorenewable resources.222–224Significant challenges remain, however, in the area of accessibility. As this article demonstrated, there are several techniques to synthesize poly(thio)ethers, but none of them are as facile, broadly applicable, and accessible as techniques like RAFT polymerization for vinyl-based monomers.225 RAFT agents can be purchased from chemical storefronts and monomers require limited or no preparation to successfully perform a RAFT polymerization. As it stands, regardless of the method used to synthesize poly(thio)ethers, several chemical steps need to take place, whether it is the synthesis of the initiator, catalyst, or purification of the monomer. Simplifying synthetic processes to make them more approachable would broaden the user base for these techniques/materials. As an extension of this, efforts need to be made to make these heterocycle polymerizations more robust to air; all of the techniques described above require an air-free environment and most also require the handling of pyrophoric materials. This necessitates additional equipment that a lab that is not dedicated to polymer synthesis may not have, limiting potential utilization.
Another major concern is sustainability. As mentioned above, epoxides can be sourced from fully biorenewable resources.222–224 However, poly(thio)ether degradability is lacking; there is significant difficulty to place even some degradable units within a poly(thio)ether backbone,89 let alone fully de-polymerize it. This is a problem not only from a plastic waste point of view, but also in the biomedical applications poly(thio)ethers are generally used in.226,227 Combining epoxide/thiirane monomers with “waste” monomers like CO2 is a major area of interest and has seen rapid advancement. However, issues remain applying renewably sourced epoxides and thiiranes as well as in engineering the properties of the final materials and their degradability.228 Therefore, sustainability in the form of degradability and depolymerization should be improved.
In conclusion, there are many ways to synthesize poly(thio)ethers, which are excellent materials, applicable in several technological contexts. However, they are often overlooked for vinyl-based polymers. This is, at least partially, due to the ease and diversity of vinyl polymerization methods along with the accessibility of clear-cut tutorials that accompany them.225,229–232 With this article, we hope to have provided an informative and approachable overview of the different techniques for poly(thio)ether synthesis to garner interest from a broader scientific base. We hope this will be a positive step towards increased adoption of poly(thio)ethers, approaching that of vinyl-based polymers. We anticipate this will lead to the development of new methods and spark new ideas to help solve challenges in the field especially in the context of accessibility and sustainability.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Robert C. Ferrier Jr. acknowledges start up funding from Michigan State University. Nathaniel A. Lynd acknowledges support from the National Science Foundation, award number 2004167.Notes and references
- B. Zdovc, M. Grdadolnik, D. Pahovnik and E. Žagar, Macromolecules, 2023, 56, 3374–3382 CrossRef CAS PubMed.
- M. Pohl, E. Danieli, M. Leven, W. Leitner, B. Blümich and T. E. Müller, Macromolecules, 2016, 49, 8995–9003 CrossRef CAS.
- I. Izarra, A. Borreguero, I. Garrido, J. Rodríguez and M. Carmona, Polym. Test., 2021, 100, 107268 CrossRef CAS.
- A. Das and P. Mahanwar, Adv. Ind. Eng. Polym. Res., 2020, 3, 93–101 Search PubMed.
- C. Fruijtier-Pölloth, Toxicology, 2005, 214, 1–38 CrossRef PubMed.
- G. E. Petrowski, Adv. Food Res., 1976, 22, 309–359 CAS.
- S. I. Martínez-Monteagudo, M. Enteshari and L. Metzger, Trends Food Sci. Technol., 2019, 83, 181–191 CrossRef.
- Z. Xue, D. He and X. Xie, J. Mater. Chem. A, 2015, 3, 19218–19253 RSC.
- Y. Liu, Y. Zhu and Y. Cui, Nat. Energy, 2019, 4, 540–550 CrossRef.
- Y. Jiang, X. Yan, Z. Ma, P. Mei, W. Xiao, Q. You and Y. Zhang, Polymers, 2018, 10, 1237 CrossRef PubMed.
- Y. Han and W. W. Ho, J. Membr. Sci., 2021, 628, 119244 CrossRef CAS.
- S. L. Liu, L. Shao, M. L. Chua, C. H. Lau, H. Wang and S. Quan, Prog. Polym. Sci., 2013, 38, 1089–1120 CrossRef CAS.
- Q. Yu, Y. Zhang, H. Wang, J. Brash and H. Chen, Acta Biomater., 2011, 7, 1550–1557 CrossRef CAS PubMed.
- K. Knop, R. Hoogenboom, D. Fischer and U. Schubert, Angew. Chem., Int. Ed., 2010, 49, 6288–6308 CrossRef CAS PubMed.
- A. A. Dsouza and R. Shegokar, Expert Opin. Drug Delivery, 2016, 13, 1257–1275 CrossRef CAS PubMed.
- D. Shi, D. Beasock, A. Fessler, J. Szebeni, J. Y. Ljubimova, K. A. Afonin and M. A. Dobrovolskaia, Adv. Drug Delivery Rev., 2022, 180, 114079 CrossRef CAS PubMed.
- E. Padn-González, P. Lancaster, M. Bottini, P. Gasco, L. Tran, B. Fadeel, T. Wilkins and M. P. Monopoli, Front. Bioeng. Biotechnol., 2022, 10, 882363 CrossRef PubMed.
- R. Klein and F. R. Wurm, Macromol. Rapid Commun., 2015, 36, 1147–1165 CrossRef CAS PubMed.
- J. Herzberger, K. Niederer, H. Pohlit, J. Seiwert, M. Worm, F. R. Wurm and H. Frey, Chem. Rev., 2016, 116, 2170–2243 CrossRef CAS PubMed.
- V. B. Purohit, M. PiÄ™ta, J. Pietrasik and C. M. Plummer, Polym. Chem., 2022, 13, 4858–4878 RSC.
- R. A. Shanks and I. Kong, Advances in Elastomers I: Blends and Interpenetrating Networks, 2013, pp. 11–45 Search PubMed.
- E. M. Fettes, J. S. Jorczak and J. R. Panek, Ind. Eng. Chem., 1954, 46, 1539–1541 CrossRef CAS.
- E. M. Baker, J. Chem. Educ., 1943, 20, 427 CrossRef CAS.
- C. D. Vo, G. Kilcher and N. Tirelli, Macromol. Rapid Commun., 2009, 30, 299–315 CrossRef CAS PubMed.
- S. H. Lee, M. K. Gupta, J. B. Bang, H. Bae and H.-J. Sung, Adv. Healthcare Mater., 2013, 2, 908–915 CrossRef CAS PubMed.
- Q. Xu, C. He, C. Xiao and X. Chen, Macromol. Biosci., 2016, 16, 635–646 CrossRef CAS PubMed.
- F. Du, B. Qiao, T. D. Nguyen, M. P. Vincent, S. Bobbala, S. Yi, C. Lescott, V. P. Dravid, M. Olvera de la Cruz and E. A. Scott, Nat. Commun., 2020, 11, 4896 CrossRef CAS PubMed.
- S. Zhu, S. Li, H. Escuin-Ordinas, R. Dimatteo, W. Xi, A. Ribas and T. Segura, J. Controlled Release, 2018, 282, 156–165 CrossRef CAS PubMed.
- M. K. Gupta, T. A. Meyer, C. E. Nelson and C. L. Duvall, J. Controlled Release, 2012, 162, 591–598 CrossRef CAS PubMed.
- S. Cerritelli, D. Velluto and J. A. Hubbell, Biomacromolecules, 2007, 8, 1966–1972 CrossRef CAS PubMed.
- H. Shi, X. Zhao, J. Gao, Z. Liu, Z. Liu, K. Wang and J. Jiang, Chin. Chem. Lett., 2020, 31, 3102–3106 CrossRef CAS.
- D. Jiang, Y. Zhang, F. Zhang, Z. Liu, J. Han and X. Wu, Colloid Polym. Sci., 2016, 294, 2021–2028 CrossRef CAS.
- J. M. Desper, D. R. Powell and S. H. Gellman, J. Am. Chem. Soc., 1990, 112, 4321–4324 CrossRef CAS.
- X. Zhang, Z. Gou, Y. Zuo and W. Lin, Spectrochim. Acta, Part A, 2020, 228, 117679 CrossRef CAS PubMed.
- P. Johansson, Polymer, 2001, 42, 4367–4373 CrossRef CAS.
- J. M. Sarapas and G. N. Tew, Macromolecules, 2016, 49, 1154–1162 CrossRef CAS.
- G. Shukla and R. C. Ferrier Jr., J. Polym. Sci., 2021, 59, 2704–2718 CrossRef CAS.
- M. Gilbert, C. Henri, R. Jean-Pierre and D. Alain, Synthesis, 1983, 117–119 Search PubMed.
- H. Gilman, C. S. Sherman, C. C. Price, R. C. Elderfield, J. T. Maynard, R. H. Reitsema, L. Tolman, S. P. J. Massie, F. J. Marshall and L. Goldman, J. Am. Chem. Soc., 1946, 68, 1291–1293 CrossRef CAS PubMed.
- T. Katsuki and K. B. Sharpless, J. Am. Chem. Soc., 1980, 102, 5974–5976 CrossRef CAS.
- Q.-H. Xia, H.-Q. Ge, C.-P. Ye, Z.-M. Liu and K.-X. Su, Chem. Rev., 2005, 105, 1603–1662 CrossRef CAS PubMed.
- W. B. Alexander, PhD thesis, 1947 Search PubMed.
- E. E. v Tamelen, J. Am. Chem. Soc., 1951, 73, 3444–3448 CrossRef.
- H. Staudinger and F. Pfenninger, Ber. Dtsch. Chem. Ges., 1916, 49, 1941–1951 CrossRef CAS.
- W. Adam and R. M. Bargon, Chem. Rev., 2004, 104, 251–262 CrossRef CAS PubMed.
- M. Delepine, Comptes Rendus Hebdomadaires des séances de l’Académie des sciences, 1920, 171, 36 CAS.
- P. J. Marcel Delepine, Comptes rendus hebdomadaires des séances de l’Académie des sciences, 1921, 172, 158 Search PubMed.
- M. Sander, Chem. Rev., 1966, 66, 297–339 CrossRef.
- S.-m Song, J. Jin, J.-H. Choi and W.-j Chung, Nat. Commun., 2022, 13, 4818 CrossRef CAS PubMed.
- H. Staudinger and O. Schweitzer, Berichte der deutschen chemischen Gesellschaft (A and B Series), 1929, 62, 2395–2405 CrossRef.
- P. J. Flory, J. Am. Chem. Soc., 1940, 62, 1561–1565 CrossRef CAS.
- C. Marvel and E. Weil, J. Am. Chem. Soc., 1954, 76, 61–69 CrossRef CAS.
- S. Boileau, G. Champetier and P. Sigwalt, Die Makromolekulare Chemie, 1963, 69, 180–192 CrossRef CAS.
- J. Park, Y. Yu, J. W. Lee and B.-S. Kim, Macromolecules, 2022, 55, 5448–5458 CrossRef CAS.
- T. Isono, Polym. J., 2021, 53, 753–764 CrossRef CAS.
- S. Boileau and N. Illy, Prog. Polym. Sci., 2011, 36, 1132–1151 CrossRef CAS.
- A. Rehor, N. Tirelli and J. A. Hubbell, Macromolecules, 2002, 35, 8688–8693 CrossRef CAS.
- Q. J. Meisner, S. Jiang, P. Cao, T. Glossmann, A. Hintennach and Z. Zhang, J. Mater. Chem. A, 2021, 9, 25927–25933 RSC.
- J. Blankenburg, K. Maciol, C. Hahn and H. Frey, Macromolecules, 2019, 52, 1785–1793 CrossRef CAS.
- Y. Hong, J.-M. Kim, H. Jung, K. Park, J. Hong, S.-H. Choi and B.-S. Kim, Biomacromolecules, 2020, 21, 4913–4922 CrossRef CAS PubMed.
- L. Wang, G. Kilcher and N. Tirelli, Macromol. Chem. Phys., 2009, 210, 447–456 CrossRef CAS.
- A.-L. Brocas, C. Mantzaridis, D. Tunc and S. Carlotti, Prog. Polym. Sci., 2013, 38, 845–873 CrossRef CAS.
- M. Hans, H. Keul and M. Moeller, Polymer, 2009, 50, 1103–1108 CrossRef CAS.
- C. Bawn, A. Ledwith and N. McFarlane, Polymer, 1969, 10, 653–659 CrossRef CAS.
- G. Kilcher, L. Wang and N. Tirelli, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 2233–2249 CrossRef CAS.
- H. G. Buss, S. Y. Chan, N. A. Lynd and B. D. McCloskey, ACS Energy Lett., 2017, 2, 481–487 CrossRef CAS.
- C. Tonhauser, C. Schüll, C. Dingels and H. Frey, ACS Macro Lett., 2012, 1, 1094–1097 CrossRef CAS PubMed.
- C. Huin, Z. Eskandani, N. Badi, A. Farcas, V. Bennevault-Celton and P. Guégan, Carbohydr. Polym., 2013, 94, 323–331 CrossRef CAS PubMed.
- L. Wang, P. Hu and N. Tirelli, Polymer, 2009, 50, 2863–2873 CrossRef CAS.
- A. J. McGrath, W. Shi, C. G. Rodriguez, E. J. Kramer, C. J. Hawker and N. A. Lynd, Polym. Chem., 2015, 6, 1465–1473 RSC.
- B. F. Lee, M. J. Kade, J. A. Chute, N. Gupta, L. M. Campos, G. H. Fredrickson, E. J. Kramer, N. A. Lynd and C. J. Hawker, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 4498–4504 CrossRef CAS PubMed.
- V. S. Wilms and H. Frey, Polym. Int., 2013, 62, 849–859 CrossRef CAS.
- A. Lee, P. Lundberg, D. Klinger, B. F. Lee, C. J. Hawker and N. A. Lynd, Polym. Chem., 2013, 4, 5735–5742 RSC.
- P. S. Boileau and P. Sigwalt, Die Makromolekulare Chemie, 1970, 131, 7–13 CrossRef.
- A. Rakotomanga, P. Hemery, S. Boileau and B. Lotz, Eur. Polym. J., 1978, 14, 581–587 CrossRef CAS.
- I. M. Panayotov and I. V. Berlinova, Die Makromolekulare Chemie, 1972, 154, 139–149 CrossRef.
- M. Morton, R. F. Kammereck and L. J. Fetters, Br. Polym. J., 1971, 3, 120–128 CrossRef CAS.
- W. van Zoelen, H. G. Buss, N. C. Ellebracht, N. A. Lynd, D. A. Fischer, J. Finlay, S. Hill, M. E. Callow, J. A. Callow, E. J. Kramer, R. N. Zuckermann and R. A. Segalman, ACS Macro Lett., 2014, 3, 364–368 CrossRef CAS PubMed.
- G. Wang, X. Fan and J. Huang, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 5313–5321 CrossRef CAS.
- A. Gourdenne, Die Makromolekulare Chemie, 1972, 158, 261–269 CrossRef CAS.
- R. S. Nevin and E. M. Pearce, J. Polym. Sci., Part B: Polym. Lett., 1965, 3, 487–490 CrossRef CAS.
- W. Braune and J. Okuda, Angew. Chem., Int. Ed., 2003, 42, 64–68 CrossRef CAS PubMed.
- C. Billouard, S. Carlotti, P. Desbois and A. Deffieux, Macromolecules, 2004, 37, 4038–4043 CrossRef CAS.
- S. Carlotti, A. Labbé, V. Rejsek, S. Doutaz, M. Gervais and A. Deffieux, Macromolecules, 2008, 41, 7058–7062 CrossRef CAS.
- Y. Chen, J. Shen, S. Liu, J. Zhao, Y. Wang and G. Zhang, Macromolecules, 2018, 51, 8286–8297 CrossRef CAS.
- P. Walther, C. Vogler and S. Naumann, Synlett, 2020, 641–647 CAS.
- P. Walther, A. Krauß and S. Naumann, Angew. Chem., Int. Ed., 2019, 58, 10737–10741 CrossRef CAS PubMed.
- T. Gao, X. Xia, K. Tajima, T. Yamamoto, T. Isono and T. Satoh, Macromolecules, 2022, 55, 9373–9383 CrossRef CAS.
- P. Lundberg, B. F. Lee, S. A. van den Berg, E. D. Pressly, A. Lee, C. J. Hawker and N. A. Lynd, ACS Macro Lett., 2012, 1, 1240–1243 CrossRef CAS PubMed.
- P. Jung, A. D. Ziegler, J. Blankenburg and H. Frey, Angew. Chem., Int. Ed., 2019, 58, 12883–12886 CrossRef CAS PubMed.
- P. Verkoyen and H. Frey, Macromol. Rapid Commun., 2020, 41, 2000225 CrossRef CAS PubMed.
- T. Johann, H. A. Houck, T. Dinh, U. Kemmer-Jonas, F. E. Du Prez and H. Frey, Polym. Chem., 2019, 10, 4699–4708 RSC.
- M. Gervais, A.-L. Brocas, A. Deffieux, E. Ibarboure and S. Carlotti, Pure Appl. Chem., 2012, 84, 2103–2111 CrossRef CAS.
- K. Sakakibara, K. Nakano and K. Nozaki, Macromolecules, 2007, 40, 6136–6142 CrossRef CAS.
- J. Herzberger, D. Leibig, J. Langhanki, C. Moers, T. Opatz and H. Frey, Polym. Chem., 2017, 8, 1882–1887 RSC.
- T. Borke, A. Korpi, F. Pooch, H. Tenhu and S. Hietala, J. Polym. Sci., Part A: Polym. Chem., 2017, 55, 1822–1830 CrossRef CAS.
- A. Labbé, A.-L. Brocas, E. Ibarboure, T. Ishizone, A. Hirao, A. Deffieux and S. Carlotti, Macromolecules, 2011, 44, 6356–6364 CrossRef.
- K. Roos, E. Dolci, S. Carlotti and S. Caillol, Polym. Chem., 2016, 7, 1612–1622 RSC.
- H. V. Babu and K. Muralidharan, Polymer, 2014, 55, 83–94 CrossRef CAS.
- A.-L. Brocas, G. Cendejas, S. Caillol, A. Deffieux and S. Carlotti, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 2677–2684 CrossRef CAS.
- S. Carlotti, A. Labbé, V. Rejsek, S. Doutaz, M. Gervais and A. Deffieux, Macromolecules, 2008, 41, 7058–7062 CrossRef CAS.
- J. Herzberger and H. Frey, Macromolecules, 2015, 48, 8144–8153 CrossRef CAS.
- M. Gervais, A. Forens, E. Ibarboure and S. Carlotti, Polym. Chem., 2018, 9, 2660–2668 RSC.
- J. Ochs, A. Veloso, D. E. Martínez-Tong, A. Alegria and F. Barroso-Bujans, Macromolecules, 2018, 51, 2447–2455 CrossRef CAS.
- M. Gervais, A. Labbé, S. Carlotti and A. Deffieux, Macromolecules, 2009, 42, 2395–2400 CrossRef CAS.
- A.-L. Brocas, A. Deffieux, N. Le Malicot and S. Carlotti, Polym. Chem., 2012, 3, 1189–1195 RSC.
- V. Rejsek, P. Desbois, A. Deffieux and S. Carlotti, Polymer, 2010, 51, 5674–5679 CrossRef CAS.
- P. H. Plesch and P. H. Westermann, J. Polym. Sci., Part C: Polym. Symp., 1967, 16, 3837–3843 CrossRef.
- K. Matyjaszewski, S. Słomkowski and S. Penczek, J. Polym. Sci., Polym. Chem. Ed., 1979, 17, 2413–2422 CrossRef CAS.
- I. Penczek and S. Penczek, J. Polym. Sci., Part A-1: Polym. Chem., 1970, 8, 2465–2474 CrossRef CAS.
- Y. Kawakami, Y. Mizutani and Y. Yamashita, Die Makromolekulare Chemie, 1979, 180, 2279–2287 CrossRef CAS.
- P. Kubisa and S. Penczek, Prog. Polym. Sci., 1999, 24, 1409–1437 CrossRef CAS.
- S. Penczek, J. Polym. Sci., Part A: Polym. Chem., 2000, 38, 1919–1933 CrossRef CAS.
- P. Kubisa and S. Penczek, Prog. Polym. Sci., 1999, 24, 1409–1437 CrossRef CAS.
- E. J. Vandenberg, J. Polym. Sci., Part A-1: Polym. Chem., 1972, 10, 329–354 CrossRef CAS.
- S. Tsuchiya and T. Tsuruta, Die Makromolekulare Chemie, 1967, 110, 123–132 CrossRef CAS.
- D. A. Atwood, J. A. Jegier and D. Rutherford, J. Am. Chem. Soc., 1995, 117, 6779–6780 CrossRef CAS.
- Y. Sarazin and J.-F. Carpentier, Chem. Rev., 2015, 115, 3564–3614 CrossRef CAS PubMed.
- M. Bochmann, S. J. Lancaster, M. D. Hannant, A. Rodriguez, M. Schormann, D. A. Walker and T. J. Woodman, Pure Appl. Chem., 2003, 75, 1183–1195 CrossRef CAS.
- J. V. Crivello, B. Falk and M. R. Zonca Jr., J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 1630–1646 CrossRef CAS.
- J. V. Crivello, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 6435–6448 CrossRef CAS.
- J. V. Crivello, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 4331–4340 CrossRef CAS.
- J. V. Crivello, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 3036–3052 CrossRef CAS.
- Y. Miwa, H. Tsutsumi and T. Oishi, Polym. J., 2001, 33, 927–933 CrossRef CAS.
- Y. Cui, X. Liang, J. Chai, Z. Cui, Q. Wang, W. He, X. Liu, Z. Liu, G. Cui and J. Feng, Adv. Sci., 2017, 4, 1700174 CrossRef PubMed.
- K. Toyota, Polymer, 2021, 218, 123490 CrossRef CAS.
- J. R. Nair, I. Shaji, N. Ehteshami, A. Thum, D. Diddens, A. Heuer and M. Winter, Chem. Mater., 2019, 31, 3118–3133 CrossRef CAS.
- Y. Liu, H. Y. Yu and X. B. Lu, Macromol. Chem. Phys., 2019, 220, 1900377 CrossRef CAS.
- M. Haddad, M. Laghzaoui, R. Welter and S. Dagorne, Organometallics, 2009, 28, 4584–4592 CrossRef CAS.
- M. D. Hannant, M. Schormann and M. Bochmann, J. Chem. Soc., Dalton Trans., 2002, 4071–4073 RSC.
- S. Penczek, Die Makromolekulare Chemie, 1979, 3, 17–39 CrossRef CAS.
- M. P. Zussman and D. A. Tirrell, Macromolecules, 1981, 14, 1148–1153 CrossRef CAS.
- K. Aoi and M. Okada, Prog. Polym. Sci., 1996, 21, 151–208 CrossRef CAS.
- K. Weissermel, E. Fischer, K. Gutweiler, H. Hermann and H. Cherdron, Angew. Chem., Int. Ed. Engl., 1967, 6, 526–533 CrossRef CAS.
- M. I. Childers, J. M. Longo, N. J. Van Zee, A. M. LaPointe and G. W. Coates, Chem. Rev., 2014, 114, 8129–8152 CrossRef CAS PubMed.
- W. Kuran, Prog. Polym. Sci., 1998, 23, 919–992 CrossRef CAS.
- E. Vandenberg, J. Elastomers Plast., 1982, 14, 243–256 CrossRef CAS.
- M. Osgan and C. C. Price, J. Polym. Sci., 1959, 34, 153–156 CrossRef CAS.
- E. J. Vandenberg, J. Polym. Sci., 1960, 47, 486–489 CrossRef CAS.
- A. Z. Dookhith, N. A. Lynd, C. Creton and G. E. Sanoja, Macromolecules, 2022, 55, 5601–5609 CrossRef CAS.
- M. Chwatko and N. A. Lynd, Macromolecules, 2017, 50, 2714–2723 CrossRef CAS.
- B. S. Beckingham, G. E. Sanoja and N. A. Lynd, Macromolecules, 2015, 48, 6922–6930 CrossRef CAS.
- C. C. Price and R. Spector, J. Am. Chem. Soc., 1965, 87, 2069–2070 CrossRef CAS.
- E. J. Vandenberg, J. Polym. Sci., Part A-1: Polym. Chem., 1969, 7, 525–567 CrossRef CAS.
- W. Braune and J. Okuda, Angew. Chem., Int. Ed., 2003, 42, 64–68 CrossRef CAS PubMed.
- R. C. J. Ferrier, S. Pakhira, S. E. Palmon, C. G. Rodriguez, D. J. Goldfeld, O. O. Iyiola, M. Chwatko, J. L. Mendoza-Cortes and N. A. Lynd, Macromolecules, 2018, 51, 1777–1786 CrossRef CAS.
- E. J. Vandenberg, J. Macromol. Sci., Part A: Pure Appl. Chem., 1985, 22, 619–630 CrossRef.
- J. S. Shih, J. F. Brandt, M. P. Zussman and D. A. Tirrell, J. Polym. Sci., Polym. Chem. Ed., 1982, 20, 2839–2849 CrossRef CAS.
- E. J. Vandenberg, J. Polym. Sci., 1960, 47, 489–491 CrossRef CAS.
- E. J. Vandenberg, J. Polym. Sci., Polym. Chem. Ed., 1985, 23, 951–970 CrossRef CAS.
- E. J. Vandenberg, J. C. Mullis and R. S. Juvet Jr., J. Polym. Sci., Part A: Polym. Chem., 1989, 27, 3083–3112 CrossRef CAS.
- E. J. Vandenberg, J. C. Mullis, R. S. Juvet Jr., T. Miller and R. A. Nieman, J. Polym. Sci., Part A: Polym. Chem., 1989, 27, 3113–3149 CrossRef CAS.
- E. J. Vandenberg and A. E. Robinson, Coordination Polymerization of Trimethylene Oxide, ch. 7, 1975, pp. 101–119 Search PubMed.
- J. Milgrom, Double metal cyanide complex compounds, US Patent 3,427,256, 1969 Search PubMed.
- C. H. Tran, L. T. T. Pham, Y. Lee, H. B. Jang, S. Kim and I. Kim, J. Catal., 2019, 372, 86–102 CrossRef CAS.
- R.-J. Wei, Y.-Y. Zhang, X.-H. Zhang, B.-Y. Du and Z.-Q. Fan, RSC Adv., 2014, 4, 21765–21771 RSC.
- N. Almora-Barrios, S. Pogodin, L. Bellarosa, M. García-Melchor, G. Revilla-López, M. García-Ratés, A. B. Vázquez-García, P. Hernández-Ariznavarreta and N. López, ChemCatChem, 2015, 7, 928–935 CrossRef CAS.
- R. Mohr, M. Wagner, S. Zarbakhsh and H. Frey, Macromol. Rapid Commun., 2021, 42, 2000542 CrossRef CAS PubMed.
- C. Marquez, A. Simonov, M. T. Wharmby, C. Van Goethem, I. Vankelecom, B. Bueken, A. Krajnc, G. Mali, D. De Vos and T. De Baerdemaeker, Chem. Sci., 2019, 10, 4868–4875 RSC.
- C. H. Tran, M. W. Lee, S. A. Kim, H. B. Jang and I. Kim, Macromolecules, 2020, 53, 2051–2060 CrossRef CAS.
- M. Klinger, R. Bachmann and A. Jupke, Macromol. Theory Simul., 2021, 30, 2100013 CrossRef CAS.
- S. H. Lee, I. K. Lee, J. Y. Ha, J. K. Jo, I. Park, C.-S. Ha, H. Suh and I. Kim, Ind. Eng. Chem. Res., 2010, 49, 4107–4116 CrossRef CAS.
- L.-C. Wu, A.-F. Yu, M. Zhang, B.-H. Liu and L.-B. Chen, J. Appl. Polym. Sci., 2004, 92, 1302–1309 CrossRef CAS.
- X. Zhang, J. Dong, Y. Su, E. G. Lee, Z. Duan, I. Kim and B. Liu, Polym. Chem., 2023, 14, 1263–1274 RSC.
- R. Matthes and H. Frey, Biomacromolecules, 2022, 23, 2219–2235 CrossRef CAS PubMed.
- J. Sebastian and D. Srinivas, Appl. Catal., A, 2014, 482, 300–308 CrossRef CAS.
- J. Sebastian and D. Srinivas, Appl. Catal., A, 2015, 506, 163–172 CrossRef CAS.
- J. M. Longo, M. J. Sanford and G. W. Coates, Chem. Rev., 2016, 116, 15167–15197 CrossRef CAS PubMed.
- L.-F. Hu, C.-J. Zhang, D.-J. Chen, X.-H. Cao, J.-L. Yang and X.-H. Zhang, ACS Appl. Polym. Mater., 2020, 2, 5817–5823 CrossRef CAS.
- S. Inoue, J. Polym. Sci., Part A: Polym. Chem., 2000, 38, 2861–2871 CrossRef CAS.
- S. Inoue and T. Aida, High Polymers Japan 1993, 42, 302–307.
- N. Takeda and S. Inoue, Bull. Chem. Soc. Jpn., 1978, 51, 3564–3567 CrossRef CAS.
- N. Takeda and S. Inoue, Die Makromolekulare Chemie, 1978, 179, 1377–1381 CrossRef CAS.
- T. Aida, R. Mizuta, Y. Yoshida and S. Inoue, Die Makromolekulare Chemie, 1981, 182, 1073–1079 CrossRef CAS.
- T. Aida, K. Kawaguchi and S. Inoue, Macromolecules, 1990, 23, 3887–3892 CrossRef CAS.
- S. Asano, T. Aida and S. Inoue, J. Chem. Soc., Chem. Commun., 1985, 1148–1149 RSC.
- C. Chatterjee and M. H. Chisholm, Inorg. Chem., 2012, 51, 12041–12052 CrossRef CAS PubMed.
- C. Robert, T. Ohkawara and K. Nozaki, Chem. – Eur. J., 2014, 20, 4789–4795 CrossRef CAS PubMed.
- C. Chatterjee and M. H. Chisholm, Chem. Rec., 2013, 13, 549–560 CrossRef CAS PubMed.
- M. Honda, R. Nakamura and H. Sugimoto, J. Polym. Sci., 2021, 59, 3122–3130 CrossRef CAS.
- Y. Wang, Y. Zhao, Y. Ye, H. Peng, X. Zhou, X. Xie, X. Wang and F. Wang, Angew. Chem., Int. Ed., 2018, 57, 3593–3597 CrossRef CAS PubMed.
- Y. Zhao, S. Zhu, X. Li, X. Zhao, J. Xu, B. Xiong, Y. Wang, X. Zhou and X. Xie, CCS Chem., 2022, 4, 122–131 CrossRef CAS.
- A. C. Deacy, G. L. Gregory, G. S. Sulley, T. T. D. Chen and C. K. Williams, J. Am. Chem. Soc., 2021, 143, 10021–10040 CrossRef CAS PubMed.
- M. Strianese, D. Pappalardo, M. Mazzeo, M. Lamberti and C. Pellecchia, Dalton Trans., 2021, 50, 7898–7916 RSC.
- H. Sugimoto, C. Kawamura, M. Kuroki, T. Aida and S. Inoue, Macromolecules, 1994, 27, 2013–2018 CrossRef CAS.
- T. Aida, Y. Maekawa, S. Asano and S. Inoue, Macromolecules, 1988, 21, 1195–1202 CrossRef CAS.
- K. Yoshinaga and Y. Iida, Chem. Lett., 1991, 1057–1058 CrossRef CAS.
- K. L. Peretti, H. Ajiro, C. T. Cohen, E. B. Lobkovsky and G. W. Coates, J. Am. Chem. Soc., 2005, 127, 11566–11567 CrossRef CAS PubMed.
- R. M. Thomas, P. C. B. Widger, S. M. Ahmed, R. C. Jeske, W. Hirahata, E. B. Lobkovsky and G. W. Coates, J. Am. Chem. Soc., 2010, 132, 16520–16525 CrossRef CAS PubMed.
- W. Hirahata, R. M. Thomas, E. B. Lobkovsky and G. W. Coates, J. Am. Chem. Soc., 2008, 130, 17658–17659 CrossRef CAS PubMed.
- M. I. Childers, A. K. Vitek, L. S. Morris, P. C. B. Widger, S. M. Ahmed, P. M. Zimmerman and G. W. Coates, J. Am. Chem. Soc., 2017, 139, 11048–11054 CrossRef CAS PubMed.
- B. M. Lipinski, K. L. Walker, N. E. Clayman, L. S. Morris, T. M. E. Jugovic, A. G. Roessler, Y. D. Y. L. Getzler, S. N. MacMillan, R. N. Zare, P. M. Zimmerman, R. M. Waymouth and G. W. Coates, ACS Catal., 2020, 10, 8960–8967 CrossRef CAS PubMed.
- Z. Hoštálek, R. Mundil, I. Císařová, O. Trhlíková, E. Grau, F. Peruch, H. Cramail and J. Merna, Polymer, 2015, 63, 52–61 CrossRef.
- B. M. Lipinski, L. S. Morris, M. N. Silberstein and G. W. Coates, J. Am. Chem. Soc., 2020, 142, 6800–6806 CrossRef CAS PubMed.
- S. M. Ahmed, A. Poater, M. I. Childers, P. C. B. Widger, A. M. LaPointe, E. B. Lobkovsky, G. W. Coates and L. Cavallo, J. Am. Chem. Soc., 2013, 135, 18901–18911 CrossRef CAS PubMed.
- L. S. Morris, M. I. Childers and G. W. Coates, Angew. Chem., 2018, 130, 5833–5836 CrossRef.
- K. Nakano, G. Tatsumi and K. Nozaki, J. Am. Chem. Soc., 2007, 129, 15116–15117 CrossRef CAS PubMed.
- S. Silvano, C. F. Carrozza, A. R. de Angelis, I. Tritto, L. Boggioni and S. Losio, Macromolecules, 2020, 53, 8837–8846 CrossRef CAS.
- T. Tsuruta, S. Inoue and Y. Yokota, Die Makromolekulare Chemie: Macromolecular Chemistry and Physics, 1967, 103, 164–174 CrossRef CAS.
- M. Osgan and C. C. Price, J. Polym. Sci., 1959, 34, 153–156 CrossRef CAS.
- S. Ghosh, H. Lund, H. Jiao and E. Mejía, Macromolecules, 2017, 50, 1245–1250 CrossRef CAS.
- S. Ghosh, D. Pahovnik, U. Kragl and E. Mejía, Macromolecules, 2018, 51, 846–852 CrossRef CAS.
- Y. Kida, Y. Miura, K. Shikata and K. Azuma, J. Polym. Sci., Polym. Chem. Ed., 1975, 13, 2835–2842 CrossRef CAS.
- M. R. Mason and A. M. Perkins, J. Organomet. Chem., 2000, 599, 200–207 CrossRef CAS.
- H.-Q. Xie, J.-S. Guo, G.-Q. Yu and J. Zu, J. Appl. Polym. Sci., 2001, 80, 2446–2454 CrossRef CAS.
- R. Ridenour and E. Flagg, J. Organomet. Chem., 1969, 16, 393–404 CrossRef CAS.
- J. Otera, T. Yano, E. Kunimoto and T. Nakata, Organometallics, 1984, 3, 426–431 CrossRef CAS.
- C. Schütz, T. Dwars, C. Schnorpfeil, J. Radnik, M. Menzel and U. Kragl, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 3032–3041 CrossRef.
- N. Iwasa, K. Miura, S. Kan and Y. Furukawa, Polym. Bull., 2008, 61, 207–216 CrossRef CAS.
- C. G. Rodriguez, R. C. J. Ferrier, A. Helenic and N. A. Lynd, Macromolecules, 2017, 50, 3121–3130 CrossRef CAS.
- R. C. Ferrier, J. Imbrogno, C. G. Rodriguez, M. Chwatko, P. W. Meyer and N. A. Lynd, Polym. Chem., 2017, 8, 4503–4511 RSC.
- J. Imbrogno, R. C. J. Ferrier, B. K. Wheatle, M. J. Rose and N. A. Lynd, ACS Catal., 2018, 8, 8796–8803 CrossRef CAS.
- G. Robinson, Coordination Chemistry of Aluminum, VCH, 1993 Search PubMed.
- N. Safaie, B. Rawal, K. Ohno and R. C. J. Ferrier, Macromolecules, 2020, 53, 8181–8191 CrossRef CAS.
- N. Safaie, J. Smak, D. DeJonge, S. Cheng, X. Zuo, K. Ohno and R. C. Ferrier, Polym. Chem., 2022, 13, 2803–2812 RSC.
- C. G. Rodriguez, M. Chwatko, J. Park, C. L. Bentley, B. D. Freeman and N. A. Lynd, Macromolecules, 2020, 53, 1191–1198 CrossRef CAS.
- C. Zhu, B. J. Pedretti, L. Kuehster, V. Ganesan, G. E. Sanoja and N. A. Lynd, Macromolecules, 2023, 56, 1086–1096 CrossRef CAS.
- A. A. Burkey, A. Hillsley, D. T. Harris, J. R. Baltzegar, D. Y. Zhang, W. W. Sprague, A. M. Rosales and N. A. Lynd, Biomacromolecules, 2020, 21, 3047–3055 CrossRef CAS PubMed.
- N. Safaie, A. Rodriguez, G. Jana, J. Smak, J. L. Mendoza-Cortes and R. C. Ferrier, Jr., Polym. Chem., 2023, 14, 3213–3224 RSC.
- E. L. Dias, S. T. Nguyen and R. H. Grubbs, J. Am. Chem. Soc., 1997, 119, 3887–3897 CrossRef CAS.
- N. A. Lynd, R. C. J. Ferrier and B. S. Beckingham, Macromolecules, 2019, 52, 2277–2285 CrossRef CAS.
- S. V. Mhatre, J. S. Mahajan, T. H. Epps and L. T. J. Korley, Mater. Adv., 2023, 4, 110–121 RSC.
- J. S. Mahajan, R. M. ODea, J. B. Norris, L. T. J. Korley and T. H. I. Epps, ACS Sustainable Chem. Eng., 2020, 8, 15072–15096 CrossRef CAS.
- S. J. Poland and D. J. Darensbourg, Green Chem., 2017, 19, 4990–5011 RSC.
- S. Perrier, Macromolecules, 2017, 50, 7433–7447 CrossRef CAS.
- S. Shah, T. Prematta, N. F. Adkinson and F. T. Ishmael, J. Clin. Pharmacol., 2013, 53, 352–355 CrossRef CAS PubMed.
- K. Knop, R. Hoogenboom, D. Fischer and U. Schubert, Angew. Chem., Int. Ed., 2010, 49, 6288–6308 CrossRef CAS PubMed.
- S. J. Poland and D. J. Darensbourg, Green Chem., 2017, 19, 4990–5011 RSC.
- D. Bertin, D. Gigmes, S. R. A. Marque and P. Tordo, Chem. Soc. Rev., 2011, 40, 2189–2198 RSC.
- C. J. Hawker, A. W. Bosman and E. Harth, Chem. Rev., 2001, 101, 3661–3688 CrossRef CAS PubMed.
- K. Matyjaszewski, H. Dong, W. Jakubowski, J. Pietrasik and A. Kusumo, Langmuir, 2007, 23, 4528–4531 CrossRef CAS PubMed.
- F. Lorandi, M. Fantin and K. Matyjaszewski, J. Am. Chem. Soc., 2022, 144, 15413–15430 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2023 |