
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Calcium-stabilised transition metal bis(formyl) complexes: structure and bonding†
Joseph M.
Parr
,
Andrew J. P.
White
and
Mark R.
Crimmin
 *
*
Molecular Sciences Research Hub, Department of Chemistry, Imperial College London, 82 Wood Lane, White City, Shepherds Bush, London, W12 0BZ, UK. E-mail: m.crimmin@imperial.ac.uk
First published on 11th July 2023
Abstract
Reaction of a molecular calcium hydride with a series of group 9 dicarbonyl complexes [M(η5-C5Me5)(CO)2] (M = Co, Rh, Ir) led to the formation of both mono(formyl) and bis(formyl) complexes. The bis(formyl) complexes are unique. They have been characterised by multinuclear NMR spectroscopy and examples have been crystallographically characterised for the first time.
Metal bis(formyl) complexes are exceedingly rare. Only a handful of examples have been documented.1 The first metal bis(formyl) complex I was reported in 1979 from reaction of [Re(η5-C5H5)NO(CO)2][BF4] with two equiv. of LiBHEt3 (Fig. 1).2–4 This species was reported to decompose over two hours at room temperature.2 In 2010, the reaction of a pendant Lewis acid supported rhenium complex [Re(Ph2P(CH2)2B(C8H14))2(CO)4] with two equiv. of NaHBEt3 was described, the rhenium bis(formyl) complex II was identified as a reaction intermediate at −40 °C (Fig. 1).5
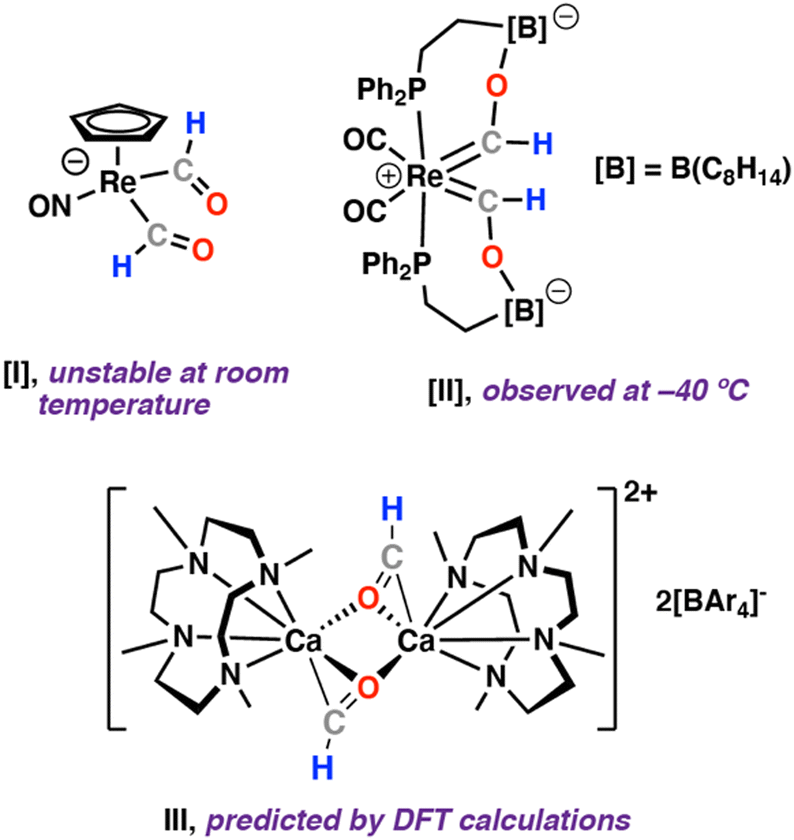 | ||
| Fig. 1 Reported metal bis(formyl) complexes and their proposed role in C–C bond formation. | ||
Interest in metal bis(formyl) complexes stems from their potential relevance in the homologation and hydrogenation of carbon monoxide (CO),6–9 key reactions underpinning the Fischer–Tropsch process.10–23 For example, II was shown to evolve into a C–C coupled product, which may derive from an intramolecular reaction of the ligands.24 Similarly, based on Density Functional Theory (DFT) calculations, III, a bimetallic calcium complex with formyl ligands bridging two calcium sites, has been proposed as an intermediate in ethene diolate formation from reaction of [Ca(μ-H)(Me4TACD)]2[BAr4]2] with CO (Me4TACD = 1,4,7,10-tetramethyl-1,4,7,10-tetraazacyclododecane; Ar = 3,5-Me2C6H3).25 We recently reported the reactions of a molecular magnesium hydride with a range of metal carbonyl complexes (Cr, Mn, Fe, Co, Rh, Ir), giving a series of mono(formyl) complexes.20
Herein we describe the reactions of a molecular calcium hydride (1)26,27 with a series of Group 9 carbonyl complexes [M(η5-C5Me5)(CO)2] (M = Co, Rh, Ir).28 By altering the reaction conditions both mono(formyl) (2) and bis(formyl) (3) heterometallic complexes could be generated. These new species have been characterised by standard spectroscopic techniques and their electronic structure investigated using DFT methods. The reaction of 1 with [M(η5-C5Me5)(CO)2] (M = Co, Rh, Ir) in C6D6 solution led to the formation of two products 2-M and 3-M (Scheme 1). Taking M = Co as an example, 2-Co and 3-Co were formed in a 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 ratio after one hour at 25 °C. Repeating the reaction using 1·THF in place of 1 improved selectivity for 2-Co. Ultimately allowing its isolation in 72% from recrystallisation from n-hexane solution. Isolated samples of 2-Co are remarkably stable and C6D6 solutions can be heated to 60 °C for a week with little or no loss of the mono(formyl) complex. Nevertheless, re-exposing 2-Co to the reaction conditions does not lead to clean formation of 3-Co. The formation of mono(formyl) and bis(formyl) complexes is presumed to occur by stepwise hydrometallation of the carbonyl ligands. DFT calculations are consistent with these steps being exergonic.
:2 ratio after one hour at 25 °C. Repeating the reaction using 1·THF in place of 1 improved selectivity for 2-Co. Ultimately allowing its isolation in 72% from recrystallisation from n-hexane solution. Isolated samples of 2-Co are remarkably stable and C6D6 solutions can be heated to 60 °C for a week with little or no loss of the mono(formyl) complex. Nevertheless, re-exposing 2-Co to the reaction conditions does not lead to clean formation of 3-Co. The formation of mono(formyl) and bis(formyl) complexes is presumed to occur by stepwise hydrometallation of the carbonyl ligands. DFT calculations are consistent with these steps being exergonic.
 | ||
| Scheme 1 Synthetic procedure for the formation of 2 and 3 (Ar = 2,6-diisopropylphenyl). | ||
Similar reactions between 1·THF and [Rh(η5-C5Me5)(CO)2] or [Ir(η5-C5Me5)(CO)2] led to formation of 2-Rh-THF and 2-Ir-THF respectively. 1H NMR spectroscopic data supports the retention of a single equivalent of THF in the complex, likely bound to one of the calcium centres. Bis(formyl) complexes 3-Rh and 3-Ir were again observed as side-products in these reactions. These species are less stable than 3-Co and could not be easily separated from unreacted starting material and by-products. Nevertheless, for M = Rh and Ir, crystals of 3-Rh and 3-Ir formed on multiple occasions, allowing unambiguous confirmation of the bis(formyl) complex as the minor product (Fig. 2).
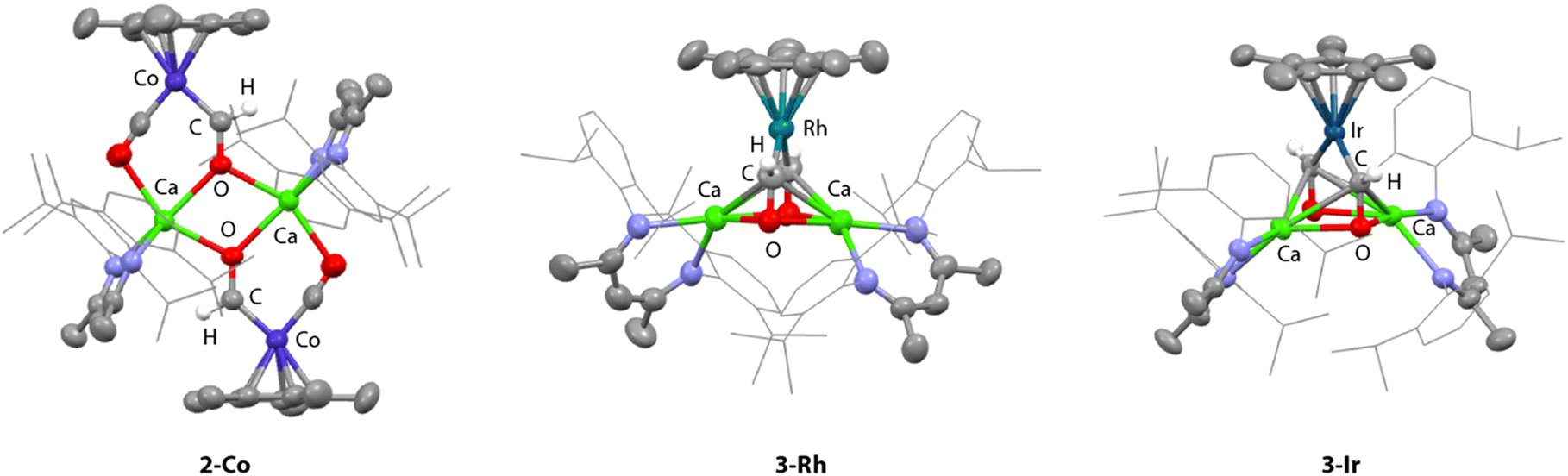 | ||
| Fig. 2 Crystal Structures of 2-Co, 3-Rh, and 3-Ir. Selected bond lengths (Å) and angles (°). 2-Co: Co–C 1.800(3), C–O 1.301(3), O–Ca 2.307(2), 2.406(2), Ca–Ca 3.766(3). 3-Rh: Rh–C 1.910(1), 19.31(8), C–O 1.290(10), 1.280(1), O–Ca 2.337(6), 2.382(6), 2.334(6), 2.349(6), Ca–C 2.680(10), 2.720(1), 2.702(9), 2.676(9, Ca–Ca 3.632(3). 3-Ir: Ir–C 1.915(7), 1.909(7), C–O 1.299(9), 1.314(9), O–Ca 2.359(5), 2.316(6), 2.358(5), 2.319(6), Ca–C 2.696(7), 2.718(9), 2.694(7), 2.724(7), Ca–Ca 3.623(2). | ||
Mono(formyl) complexes 2-Co, 2-Rh-THF and 2-Ir-THF were characterised by diagnostic resonances at δ = 13.50–13.96 ppm and δ = 247.0–298.9 ppm in the 1H and 13C NMR respectively, assigned to the formyl group. Further coupling was apparent for 2-Rh-THF, 2JRh–H = 1.7 Hz and 1JRh–C = 105.4 Hz. Infrared data reveal stretches in the region ν = 1813–1838 cm−1 for 2-Co, 2-Rh-THF and 2-Ir-THF, assigned to the isocarbonyl C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) O stretches. For 2-Ir-THF, a stretch at ν = 1658 cm−1 was observed, assigned to the formyl C
O stretches. For 2-Ir-THF, a stretch at ν = 1658 cm−1 was observed, assigned to the formyl C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretch. For 2-Co and 2-Rh-THF, the formyl CO stretch is likely obscured by the CN stretch from the β-diketiminate ligand.20 Similarly, bis (formyl) complexes 3-Co, 3-Rh and 3-Ir exhibited resonances at δ = 13.50–14.98 ppm and δ = 203.0–298.5 ppm in the 1H and 13C NMR respectively, assigned to two magnetically equivalent formyl groups.
O stretch. For 2-Co and 2-Rh-THF, the formyl CO stretch is likely obscured by the CN stretch from the β-diketiminate ligand.20 Similarly, bis (formyl) complexes 3-Co, 3-Rh and 3-Ir exhibited resonances at δ = 13.50–14.98 ppm and δ = 203.0–298.5 ppm in the 1H and 13C NMR respectively, assigned to two magnetically equivalent formyl groups.
In the solid-state, 2-Co comprises a near planar tetrametallic array comprised of two Co and two Ca sites bridged by formyl and carbonyl ligands (Fig. 2). The calcium centres are five-coordinate with approximate trigonal bipyramidal geometry. The formyl ligands are located between the two calcium atoms and adopt an anti-configuration. It is likely that this parent structure is retained in solution. The observation of a single set of β-diketiminate ligand and formyl resonances for 2-Co, 2-Rh-THF and 2-Ir-THF would be consistent with retention of a time-average highly symmetric tetrametallic structure in which two halves of the molecule are related by an inversion symmetry element. An alternate plausible structure for 2-Rh-THF and 2-Ir-THF could be the monomeric THF adducts.
3-Ir crystallises in the P21/n space group. This is the first structurally characterised bis(formyl) complex reported. The formyl ligands are mutually cis- at the iridium centre. Each oxygen atom is bridged by two calcium centres, manifesting a Ca2O2 four membered ring. In contrast to 2-Co where the bridging formyl groups adopt a κ1 coordination to each metal centre, those in 3-Ir appear more side-on and approach η2 coordination to both calcium sites. The Ca–O bond lengths in 3-Ir are 2.320(5) and 2.359(5) Å compared to 2.336(2) Å in 2-Co. The Ca–C distances range 2.694–2.724(7) Å. The Ir–C and C–O bond lengths are 1.909(7)–1.915(7) and 1.299(8)–1.314(9) Å, respectively. Ir–C–O bond angles are close to 132.0(5)°, approaching the trigonal structure for a {CHO}− moiety. The C–C distance is 2.89(1) Å, well beyond the covalent radii of carbon suggesting no interaction between the carbon atoms. 3-Rh shows a very similar structure in the solid state (see ESI† for details).
The electronic structures of 3-Co, 3-Rh and 3-Ir were investigated by computational methods. Geometry optimisations were performed with DFT using the ωB97X-D29 functional and a hybrid basis set. Natural Bond Orbital (NBO),30 Extended Transition State-Natural Orbital Chemical Valence (ETS-NOCV),31 Quantum Theory of Atoms In Molecules (QTAIM)32 and Non-Covalent Interactions (NCI)33 calculations were performed.
NBO calculations revealed a consistent set of highly ionic structures, which can be best formalised as [M(η5-C5Me5)(CHO)2]2− dianions coordinated to cationic calcium fragments. Hence, across the series, NPA charges on calcium are high and positive (+1.80 to +1.81), while those on carbon are close to neutral (−0.03 to −0.07). Most of the charge resides on the oxygen atoms (−0.93 to −0.94). The Wiberg Bond Indices (WBI) for the M–C bonds increase down the group taking values of 0.95 < 0.97 < 1.08 for Co < Rh < Ir. The WBIs for the CO bonds are consistent between 1.27 and 1.31 with no discernible trends down the triad. The M–C values above 1 and CO values significantly below 2 are consistent with the formyl ligand adopting metallocarbene character. For comparison, the free dianionic complexes [M(η5-C5Me5)(CHO)2]2− (3′-M) were calculated. For 3′-M, NPA charges on carbon (0.17 to 022) and oxygen (–0.74 to –0.76) were more positive than in 3 but followed the same general trends. The close agreement of NBO data between 3 and 3′ is consistent with the proposed ionic bonding situation (Fig. 3).
 | ||
| Fig. 3 Selected NBO data comparing 3, to the parent dianions 3′, and monomeric unit of 2i.e.2′. Wiberg Bond Indices are listed in italics for each bond with descending list going down the triad Co, Rh, Ir. NPA charges are listed in bold as a range for each atomic position for the whole series of group 9 complexes. | ||
QTAIM calculations further support this bonding formulation. For 3-Ir, bond paths are identified between each calcium and oxygen atom of the bridging motif. Bond critical points show ρ(r) and ∇2ρ(r) values of 0.04 e Bohr−3 and 0.19–0.22 e Bohr−5 respectively, consistent with binding of the formyl ligand to calcium being primarily through electrostatic interactions.
ETS-NOCV calculations were performed on 3-Co, 3-Rh and 3-Ir to investigate the interaction of the formyl moiety with the transition metal centre.34 As described previously by us in a related system,20 the principal contribution (Δρ1) to the bonding is the σ-donation of the HOMO of the {CHO}− ligand to a transition-metal d-orbital (66 to 75% of ΔEORB). The secondary (Δρ2) and tertiary (Δρ3) interactions involve π back-donation from a metal-based orbital to the orthogonal π*-orbitals of the {CHO}− ligand and are largely negligible in comparison to Δρ1 (9 to 12% of ΔEORB).
Considering a simplified molecular orbital picture of selected donor–acceptor interactions, it becomes clear that both formyl ligands of 3 should be interacting with the same metal d-orbitals (Fig. 4). A consequence of this effect is apparent on comparing the electronic structures of 3 with the monomeric unit of 2, labelled 2′. The carbonyl ligand is expected to be a better π-acceptor than the formyl and compete for electron-density from the d-orbitals. As such, the back-donation component of the bonding to the mono(formyl) ligand of 2′ is expected to be weaker than that to bis(formyl) ligands of 3. The impact of the competition for electron density is apparent in the M–C WBIs to the formyl group which take values of 0.81–0.97 in 2′ but 0.94–1.08 in 3. The calculations are consistent with a degree of electronic communication between the two {CHO}− fragments when coordinated to [M(η5-C5Me5)].
 | ||
| Fig. 4 Simplified molecular orbital description of selected bonding within 3. | ||
The series of bis(formyl) complexes 3-Co, 3-Rh, and 3-Ir are potentially relevant intermediates in carbon–carbon bond formation from CO and hydride ligands. Based on DFT calculations, III has previously been proposed as a potential intermediate in ethene diolate formation through coupling of the two formyl groups bridging the calcium site.25 Monitoring solutions containing 3-Co, 3-Rh, or 3-Ir for extended periods provided no evidence for carbon–carbon bond formation. Likewise, exposing a sample of 2-Co to an atmosphere of CO resulted in decomposition products only. Given that calcium bis(formyl) complexes have previously been proposed as intermediates in ethene diolate formation, the isolation and apparent stability of 3 toward carbon–carbon bond formation is notable. We thank Imperial College London for the award of a Schrödinger's Scholarship (JMP). Pete Haycock is thanked for assistance with NMR measurements. The EPSRC is thanked for funding (EP/S036628/1).
Conflicts of interest
There are no conflicts to declare.References
- F. G. A. Stone, R. West and J. A. Gladysz, Adv. Organomet. Chem., 1982, 20, 1–35 CrossRef.
- W. Tam, W.-K. Wong and J. A. Gladysz, J. Am. Chem. Soc., 1979, 101, 1589–1591 CrossRef CAS.
- C. P. Casey, M. A. Andrews and J. E. Rinz, J. Am. Chem. Soc., 1979, 101, 741–743 CrossRef CAS.
- C. P. Casey, M. A. Andrews, D. R. Mcalister and J. E. Rinz, J. Am. Chem. Soc., 1980, 102, 1927–1933 CrossRef CAS.
- A. J. M. Miller, J. A. Labinger and J. E. Bercaw, Organometallics, 2010, 29, 4499–4516 CrossRef CAS.
- D. Unruh, K. Pabst and G. Schaub, Energy Fuels, 2010, 24, 2634–2641 CrossRef CAS.
- G. Lopez, M. Artetxe, M. Amutio, J. Alvarez, J. Bilbao and M. Olazar, Renewable Sustainable Energy Rev., 2018, 82, 576–596 CrossRef CAS.
- R. C. Baliban, J. A. Elia and C. A. Floudas, Energy Environ. Sci., 2013, 6, 267–287 RSC.
- S. Fujimori and S. Inoue, J. Am. Chem. Soc., 2022, 144, 2034–2050 CrossRef CAS PubMed.
- N. M. West, A. J. M. Miller, J. A. Labinger and J. E. Bercaw, Coord. Chem. Rev., 2011, 255, 881–898 CrossRef CAS.
- J. M. Parr and M. R. Crimmin, Angew. Chem., Int. Ed., 2023, 62, e203319203 CrossRef PubMed.
- P. J. Fagan, K. G. Moloy and T. J. Marks, J. Am. Chem. Soc., 1981, 103, 6959–6962 CrossRef CAS.
- D. A. Katahlra, K. G. Moloy and T. J. Marks, Organometallics, 1982, 1, 1723–1726 CrossRef.
- J. A. Labinger, J. Organomet. Chem., 2017, 847, 4–12 CrossRef CAS.
- J. S. Mcmullen, R. Huo, P. Vasko, J. Alison and J. Hicks, Angew. Chem., Int. Ed., 2022, 62, e202215218 Search PubMed.
- R. Lalrempuia, C. E. Kefalidis, S. J. Bonyhady, B. Schwarze, L. Maron, A. Stasch and C. Jones, J. Am. Chem. Soc., 2015, 137, 8944–8947 CrossRef CAS PubMed.
- M. D. Anker, M. S. Hill, J. P. Lowe and M. F. Mahon, Angew. Chem., Int. Ed., 2015, 127, 10147–10149 Search PubMed.
- M. D. Anker, C. E. Kefalidis, Y. Yang, J. Fang, M. S. Hill, M. F. Mahon and L. Maron, J. Am. Chem. Soc., 2017, 139, 10036–10054 CrossRef CAS PubMed.
- T. J. Hadlington and T. Szilvasi, Nat. Commun., 2022, 13, 2–11 CrossRef PubMed.
- J. M. Parr, A. J. P. White and M. R. Crimmin, Chem. Sci., 2022, 13, 6592–6598 RSC.
- R. Y. Kong and M. R. Crimmin, Dalton Trans., 2020, 49, 16587–16597 RSC.
- J. M. Manriquez, D. R. McAlister, R. D. Sanner and J. E. Bercaw, J. Am. Chem. Soc., 1976, 98, 6733–6735 CrossRef CAS.
- C. P. Casey and S. M. Neumann, J. Am. Chem. Soc., 1976, 5395–5396 CrossRef CAS.
- The authors note that bis-formyl formation is reverisble and that an alternate migratory insertion mechanism via an oxymethylene intermediate is an equally likely pathway to the C–C coupled product.
- D. Schuhknecht, T. P. Spaniol, Y. Yang, L. Maron and J. Okuda, Inorg. Chem., 2020, 59, 9406–9415 CrossRef CAS PubMed.
- S. Harder and J. Brettar, Angew. Chem., Int. Ed., 2006, 45, 3474–3478 CrossRef CAS PubMed.
- A. Causero, G. Ballmann, J. Pahl, C. Färber, J. Intemann and S. Harder, Dalton Trans., 2017, 46, 1822–1831 RSC.
- S. A. Frith, J. L. Spencer, W. E. Geiger Jr. and J. Edwin, (η5-Pentamethylcyclopentadienyl)Cobalt Complexes, in Inorganic Syntheses, ed. S. Kirschner, 1985, vol. 23, pp. 15–21 Search PubMed.
- J. Da Chai and M. Head-Gordon, Phys. Chem. Chem. Phys., 2008, 10, 6615–6620 RSC.
- F. Weinhold and C. R. Landis, Chem. Educ. Res. Pract., 2001, 2, 91–104 RSC.
- M. P. Mitoraj, A. Michalak and T. Ziegler, J. Chem. Theory Comput., 2009, 5, 962–975 CrossRef CAS PubMed.
- T. A. Keith, AIMALL (Version 19.10.12), TK Gristmill Software, Overland Park KS, USA, 2019 Search PubMed.
- E. R. Johnson, S. Keinan, P. Mori-Sánchez, J. Contreras-García, A. J. Cohen and W. Yang, J. Am. Chem. Soc., 2010, 132, 6498–6506 CrossRef CAS PubMed.
- A second set of ETS-NOCV calculations split complexes 3 into anionic and cationic fragmnets by dividing at the Ca–O bond, giving consistenly lower ΔEORB values (Table S8 and Fig. S32–S34, ESI†).
Footnote |
| † Electronic supplementary information (ESI) available: Full details of experimental and computational procedures. CCDC 2265688–2265690. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3cc03009a |
| This journal is © The Royal Society of Chemistry 2023 |