
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
αβ,α′β′-Diepoxyketones are mechanism-based inhibitors of nucleophilic cysteine enzymes†
Mariska
de Munnik
 a,
Jasper
Lithgow
a,
Lennart
Brewitz
a,
Kirsten E.
Christensen
b,
Robert H.
Bates
c,
Beatriz
Rodriguez-Miquel
c and
Christopher J.
Schofield
*a
a,
Jasper
Lithgow
a,
Lennart
Brewitz
a,
Kirsten E.
Christensen
b,
Robert H.
Bates
c,
Beatriz
Rodriguez-Miquel
c and
Christopher J.
Schofield
*a
aChemistry Research Laboratory, Department of Chemistry and the Ineos Oxford Institute of Antimicrobial Research, University of Oxford, 12 Mansfield Road, Oxford, OX1 3TA, UK. E-mail: christopher.schofield@chem.ox.ac.uk
bChemical Crystallography, Chemistry Research Laboratory, Department of Chemistry, University of Oxford, 12 Mansfield Road, Oxford, OX1 3TA, UK
cTres Cantos Medicines Development Campus, GlaxoSmithKline, Calle Severo Ochoa 2, Tres Cantos, Madrid, Spain
First published on 2nd October 2023
Abstract
Epoxides are an established class of electrophilic alkylating agents that react with nucleophilic protein residues. We report αβ,α′β′-diepoxyketones (DEKs) as a new type of mechanism-based inhibitors of nucleophilic cysteine enzymes. Studies with the L,D-transpeptidase LdtMt2 from Mycobacterium tuberculosis and the main protease from SARS-CoV-2 (Mpro) reveal that following epoxide ring opening by a nucleophilic cysteine, further reactions can occur, leading to irreversible alkylation.
Most covalently reacting enzyme inhibitors bear an electrophilic functional group that reacts with a nucleophile to enable covalent protein modification.1 Although many such inhibitors work by apparently simple acylation, alkylation or conjugate addition reactions, some undergo further reaction after initial covalent modification. Such mechanism-based inhibitors can be found in drugs,2–4 with one such example being inhibitors of the nucleophilic serine-β-lactamases, such as clavulanic acid.5,6
Despite the long-standing importance of covalently reacting drugs, concerns regarding potential toxicity have hindered their development. Covalently reacting drugs are, however, the subject of recent renewed interest,1,7 and are currently the basis for multiple drug development programs, including in oncology and antimicrobials.8–11 Covalent targeting of a prevalent oncogenic mutation in K-Ras (K-RasG12C) has led to development of sotorasib and adagrasib.12 Various medicinal chemistry programs targeting the main protease (Mpro) of SARS-CoV-2 have focussed on covalent reaction of the catalytic cysteine residue, with nirmatrelvir, a reversibly reacting nitrile-bearing inhibitor, being approved for COVID-19 treatment.13,14 The L,D-transpeptidase LdtMt2 of Mycobacterium tuberculosis, which is a target for TB treatment,15 is amenable to covalent inhibition via reaction with its catalytic cysteine.16–18
Epoxides are an established class of electrophilic alkylating agents, and are used to inhibit nucleophilic cysteine (and serine) proteases.1,19,20 Many epoxide inhibitors of cysteine or serine proteases contain peptide backbones, e.g. proteasome inhibitors,21–24 though the small molecule epoxide fosfomycin is a clinically important antibiotic (Fig. 1A).25,26
 | ||
| Fig. 1 αβ,α′β′-Diepoxyketones (DEKs) react with nucleophilic cysteine enzymes. (A) Examples of epoxide-bearing drugs. (B) DEK 1 was identified as a potent inhibitor of LdtMt2. (C) Symmetrical DEKs contain 3 potential sites for interactions with nucleophiles, as well as three oxygens that may react with electrophiles. Arrows in teal represent pathways consistent with mechanistic studies. (D) Synthesis of DEKs 1 and 4–11. | ||
We are interested in identifying new types of covalently reacting modulators of biological function. Recently, we reported on a high-throughput screen aiming to identify new electrophilic inhibitors of LdtMt2 and other nucleophilic enzymes.27 Here, we describe the identification of the small molecule trans,trans αβ,α′β′-diepoxyketone (DEK) 1 (Fig. 1B), and the potency and mechanism of 1 and related DEKs 4–12 for LdtMt2 and SARS-CoV-2 Mpro inhibition; the results reveal DEKs as a mechanistically interesting class of electrophile.
Symmetrical DEKs have 3 obvious positions that may react with nucleophiles and have potential to undergo further reactions (Fig. 1C). DEK 1 exhibited potent inhibition of LdtMt2, with a pIC50 of 7.2 ± 0.1, with 30 min pre-incubation (Fig. 1B). To investigate the mode of reaction of 1 with LdtMt2, we carried out protein-observed mass spectrometry employing solid-phase extraction (SPE-MS). The results reveal that 1 covalently reacts with LdtMt2, giving an initial adduct (2) with a +267 Da mass shift relative to unmodified LdtMt2 (Fig. 2B and Table S1, ESI†), corresponding to addition of one molecule of 1 to LdtMt2, which has a single cysteine (Cys354). This adduct (2) was transient, converting within 2 h into one with a mass shift of +160 Da relative to unmodified LdtMt2, provisionally assigned as 3. We proposed the reaction involves nucleophilic attack of Cys354 on the carbonyl-group adjacent carbon of one of the symmetrical epoxides, with ring opening to form 2, followed by retro-aldol fragmentation, releasing benzaldehyde (Fig. 2C). Alternatively, the reaction may proceed though reaction at the carbonyl carbon to generate a hemithioketal, after which rearrangement may occur (Fig. 2C).28
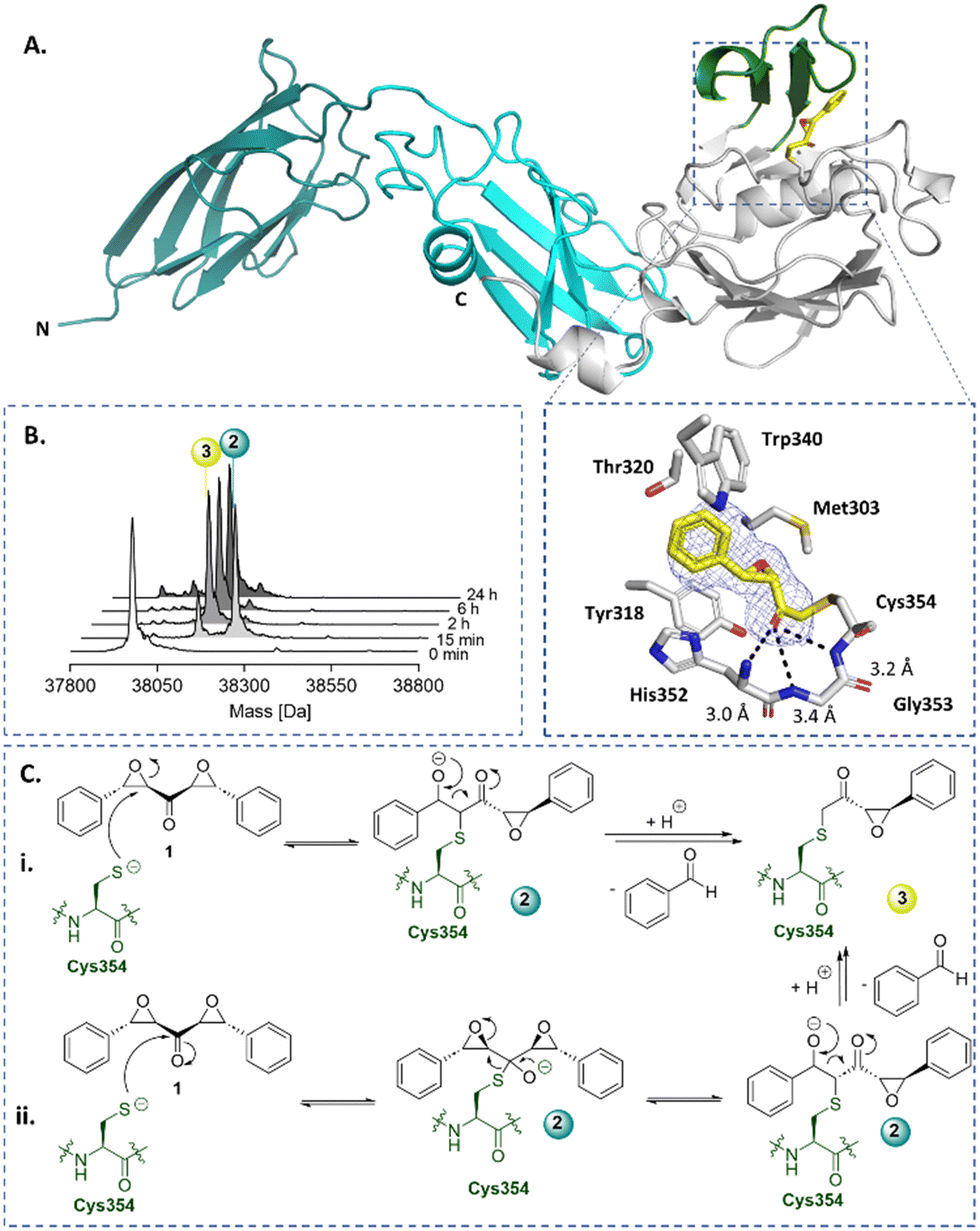 | ||
| Fig. 2 X-ray crystallography and protein-observed SPE-MS studies inform on the mechanism of DEK inhibition. (A) Views from a crystal structure derived by reaction of LdtMt2 with DEK 1 (yellow, PDB: 8BK3). The immunoglobulin-like domains are in teal and cyan. The catalytic domain is grey, with the active site lid in green. The mFo-DFc polder OMIT map is contoured at 3.0σ, carved around Cys354 bound 1 (refined as 3) and shown in blue mesh. Polar interactions are shown in black dashes. (B) Protein-observed SPE-MS experiments inform on the mechanism of reaction of 1 (20 μM) with LdtMt2 (1 μM). (C) The proposed mechanisms for reaction of Cys354 of LdtMt2 (in green) with 1via reaction with (i) the carbonyl adjacent carbon or (ii) the carbonyl carbon, followed by retro-aldol fragmentation. | ||
The identity of 3 was validated by X-ray crystallography, using reported conditions,27 wherein 1 was introduced through soaking; a structure of LdtMt2 reacted with 1 was obtained (2.15 Å resolution, P1211 space group, PDB: 8BK3, Table S2, ESI†). As reported, LdtMt2 crystallised with two molecules (chains A and B) in the asymmetric unit. While this structure manifested clear additional electron density at the chain A active site, only partial density was observed at that of chain B, thus inhibitor modelling was only performed in chain A. The additional electron density in chain A supports the proposed structure of adduct 3 (Fig. 2). The carbonyl of 3 projects into the proposed oxyanion hole, formed by the backbone NH groups of His352, Gly353 and Cys354 (distances of 3.0 Å, 3.4 Å and 3.2 Å, respectively).29 Extensive hydrophobic interactions of 3 with active site residues Tyr318, His352, Trp340, Thr320, and Met303 were observed.
In aqueous solution, 1 was found to be stable for at least 12 h (Fig. S3, ESI†). Cysteine reacted with 1, apparently yielding a product analogous to adduct 3 (Fig. S4, ESI†). No evidence for reaction of 1 with serine, lysine, threonine, tyrosine, arginine, or histidine was observed by 1H NMR or LCMS under the tested conditions (Fig. S5, ESI†).
To further analyse the inhibitory potency and mechanism of the DEKs, we prepared derivatives of 1. Synthesis involved preparation of the diene ketones via solvent-free aldol condensation, mediated by lithium perchlorate and Et3N,30 followed by epoxidation using tBuOOH and KF-Al2O3,31,32 to yield stereo-isomeric mixtures of DEKs 1 and 4–11 (Fig. 1D and Table S3, ESI†).
No substantial difference in inhibition between diastereomerically pure 1 and enantiomerically pure 1 was observed. While we did not obtain the pure cis,cis diastereomer of 1, a diastereomeric mixture of 1 (∼1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 ratio of trans,trans:cis,cis stereoisomers) manifested potent, but decreased, LdtMt2 inhibition compared to diastereomerically pure 1 (pIC50 5.6 ± 0.04 compared to 6.2 ± 0.07 for diastereomerically pure 1, with 15 min preincubation, Fig. S6, ESI†). The results imply the importance of the trans,trans stereochemistry for potent LdtMt2 inhibition by the DEKs. Recrystallisation of diastereomeric mixtures from ethanol afforded the corresponding pure trans,trans diastereomers, as supported by 1H NMR analysis and small molecule X-ray diffraction (Table S4, ESI†), except for DEKs 5 and 8, which were tested as diastereomeric mixtures (trans,trans:cis,cis ratio ∼2:1 and ∼1.2:1, respectively).
:3 ratio of trans,trans:cis,cis stereoisomers) manifested potent, but decreased, LdtMt2 inhibition compared to diastereomerically pure 1 (pIC50 5.6 ± 0.04 compared to 6.2 ± 0.07 for diastereomerically pure 1, with 15 min preincubation, Fig. S6, ESI†). The results imply the importance of the trans,trans stereochemistry for potent LdtMt2 inhibition by the DEKs. Recrystallisation of diastereomeric mixtures from ethanol afforded the corresponding pure trans,trans diastereomers, as supported by 1H NMR analysis and small molecule X-ray diffraction (Table S4, ESI†), except for DEKs 5 and 8, which were tested as diastereomeric mixtures (trans,trans:cis,cis ratio ∼2:1 and ∼1.2:1, respectively).
Dose–response assays of 4–11 with LdtMt2 showed decreased potency compared to 1 (Table S3 and Fig. S1, ESI†). Determination of the second-order rate constants for covalent target inactivation (kinact/KI)33 for LdtMt2 manifested the highest rate of inhibition for 1 (kinact/KI of 484.3 ± 28.4 M−1 s−1, Table S3 and Fig. S7, ESI†). DEKs 5–7 and 9 were observed to inhibit LdtMt2, while no evidence for inhibition was observed with 4, 8 and 10. The kinetic rate constant for reactivity with GSH (kchem)27,34 was found to be below the assay limit for all DEKs (kchem of <0.08 M−1 s−1 and half-life (t1/2) >8.7 h), except 7 and 8 (kchem of 1.71 ± 0.24 and 1.11 ± 0.20 M−1 s−1, and t1/2 of 24 min and 38 min, respectively; Table S3 and Fig. S8, ESI†). DEKs therefore apparently exhibit lower intrinsic reactivity compared to the common cysteine reactive acrylate, maleimide and isothiocyanate groups (t1/2 < 1.0 min), and, with the exceptions of 7 and 8, chloroacetamide (5.8 h).35 MS studies of the reaction of GSH and 1 manifested an adduct analogous to 3 (Fig. S9, ESI†).
Protein-observed SPE-MS assays of 4–11 demonstrated covalent modification of LdtMt2 with 4–10, which manifested adducts analogous to those with 1 (Fig. S2 and Table S1, ESI†) supporting the generality of the proposed mechanism. Additional peaks of +18 Da were observed with both unfragmented and fragmented adducts of 4–10, likely due to ring opening of the second epoxide (Fig. S10, ESI†). With 1, 5, 6 and 7, over 24 h, a second fragment adduct was observed with a +56 mass shift relative to the unmodified enzyme (Fig. S10, ESI†).
DEK 1 apparently displayed a low level of β-elimination of the reacted Cys354 residue, likely to form a dehydroalanine residue (Dha, ∼5% in 24 h, as evidenced by a −34 Da mass shift relative to unmodified LdtMt2, Fig. S2 and S10, ESI†).36–38 Interestingly, the ortho-trifluoromethoxy substituents on the phenyl groups of 5 promoted Dha formation (∼30% in 24 h). Dha formation was additionally observed following reaction with 4 (∼2.5% in 24 h) and 7 (∼16% in 24 h). In the cases of 6 and 8–10, no evidence for Dha formation was observed.
While inhibition assays with the α,β-monoepoxyketone 12 did not manifest inhibition of LdtMt2, protein-observed SPE-MS assays of LdtMt2 (1 μM) with 12 (100 μM) evidenced covalent reaction. As with DEKs 1 and 4–10, initial measurements (2 h) showed the most abundant adduct to have a mass shift of +224 Da, corresponding to the addition of a single molecule of 12. A +119 Da adduct was observed to become abundant after 6 h (Fig. S2, ESI†), indicating that the retro-aldol fragmentation is conserved between mono- and diepoxide derivatives.
While LdtMt2 contains only a single cysteine, in principle, the DEKs may alkylate other nucleophilic residues.39,40 To investigate whether the DEKs react selectively with Cys-354 of LdtMt2, we performed protein-observed SPE-MS assays with LdtMt2 that had been preincubated with ebselen, which is known to selectively and irreversibly react with Cys354.16 When 1 and 4–10 were combined with the LdtMt2-ebselen complex, no reaction was observed, evidencing that inhibition arises from at least partially, selective reaction with Cys354 (Fig. S11, ESI†).
To further investigate the reactivity of DEKs with nucleophilic cysteine enzymes, dose–response assays of 1 and 4–11 were performed with SARS-CoV-2 Mpro;41,42 note that the covalent reaction of SARS-CoV Mpro with epoxides has been reported.43 While DEKs 1 and 9 were inhibitors of Mpro (pIC50 values of 4.6 ± 0.3 and 5.9 ± 0.2, respectively), no inhibition was observed with 4–8 and 10–11 (Table S3 and Fig. S12, ESI†), providing further evidence for potential of the DEKs to react selectively.
Protein-observed SPE-MS experiments with Mpro and the DEKs 1 and 9 (Fig. S13, ESI†) manifested a +266 Da adduct (analogous to species 2, Fig. 2C), with a +160 Da adduct (analogous to species 3) becoming apparent over time. A second molecule of 1 was observed to bind to Mpro after 3 h (as evidenced by a mass shift of +266 Da relative to the +160 adduct), indicating reaction with a second residue, likely with one or more of the 12 cysteine residues of Mpro. Notably, the second adduct did not fragment by retro-aldol reaction, implying that this pathway can be promoted by the active site, likely by binding of one of the DEK-derived oxygens in the oxyanion hole of Mpro.44 Incubation of Mpro with 9 resulted in a single adduct of +186 Da, which can be assigned to a fragmented species analogous to species 3 (Fig. 2C).
As epoxide-bearing compounds may inhibit serine proteases, notably including proteasomes,45,46 we tested the ability of the DEKs to inhibit the nucleophilic serine enzyme BlaC, a class A β-lactamase of M. tuberculosis. None of compounds 1 and 4–12 exhibited inhibitory potency for BlaC (Fig. S14, ESI†).
The combined results of the reaction of DEKs with GSH, cysteine, LdtMt2 and SARS-CoV-2 Mpro, imply a conserved reaction mechanism, involving epoxide opening followed by retro-aldol reaction. Importantly, the results reveal different reactivity of the 12 Mpro cysteine residues with DEKs, indicating that selectivity for some proteins should be achievable; note that previous results showed that excess ebselen reacts covalently with all 12 cysteine residues.47
The results identify DEKs as a new class of nucleophilic cysteine reacting covalent ligands. Variations on the DEK functionality can be readily envisaged e.g., by substituting one or both epoxides for other covalently reacting electrophiles, such as aziridines or acylating agents. Notably, some natural products contain more than one epoxide, sometimes in a contiguous arrangement,48 though to our knowledge the DEK functional group has not been identified in natural products. Interestingly, DEKs have 5 hypothetical sites for reaction with nucleophiles (Fig. 1C), and they hold potential for subsequent addition of a second nucleophile. This could be useful in enabling (i) formation of cross-linked enzyme-inhibitor complexes (as can occur with other mechanism based inhibitors, e.g., certain β-lactamase inhibitors),49 (ii) labelling of an inhibited protein for analytical or diagnostic purposes, (iii) the capture of enzyme substrates, and (iv) covalent gluing of protein–protein interactions; note that epoxides are used in commonly used polyepoxide glues.50 The ability of DEKs to fragment after initial covalent reaction might be useful in releasing a functional fragment, e.g., a cytotoxic agent (the cytotoxicity of benzaldehyde in tumour cells has been reported51).
We are very grateful to Eidarus Salah for SARS-CoV-2 Mpro. We thank the Department of Biochemistry (Oxford) for the use of the 950 MHz spectrometer and Dr Patrick Rabe supporting NMR experiments. The project was co-funded by the Tres Cantos Open Lab Foundation (Project TC241 and project TC297). It was supported by funding from the Biotechnology and Biological Sciences Research Council (BBSRC) [BB/M011224/1] and the Wellcome Trust (106244/Z/14/Z).
Conflicts of interest
There are no conflicts to declare.Notes and references
- J. Singh, R. C. Petter, T. A. Baillie and A. Whitty, Nat. Rev. Drug. Discovery, 2011, 10, 307–317 CrossRef CAS PubMed.
- J. G. Robertson, Biochemistry, 2005, 44, 5561–5571 CrossRef CAS PubMed.
- C. T. Walsh, Annu. Rev. Biochem., 1984, 53, 493–535 CrossRef CAS PubMed.
- R. B. Silverman and M. W. Holladay, The organic chemistry of drug design and drug action, Academic press, 2014 Search PubMed.
- R. P. Brown, R. T. Aplin and C. J. Schofield, Biochemistry, 1996, 35, 12421–12432 CrossRef CAS PubMed.
- D. Sulton, D. Pagan-Rodriguez, X. Zhou, Y. Liu, A. M. Hujer, C. R. Bethel, M. S. Helfand, J. M. Thomson, V. E. Anderson, J. D. Buynak, L. M. Ng and R. A. Bonomo, J. Biol. Chem., 2005, 280, 35528–35536 CrossRef CAS PubMed.
- R. A. Bauer, Drug. Discovery Today, 2015, 20, 1061–1073 CrossRef CAS PubMed.
- J. M. Dixon, Expert Rev. Anticancer Ther., 2002, 2, 267–275 CrossRef CAS PubMed.
- D. Thomas and J. Zalcberg, Clin. Exp. Pharmacol. Physiol., 1998, 25, 887–895 CrossRef CAS PubMed.
- H. Xu, C. Faber, T. Uchiki, J. Racca and C. Dealwis, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 4028–4033 CrossRef CAS PubMed.
- D. J. Waxman and J. L. Strominger, Annu. Rev. Biochem., 1983, 52, 825–869 CrossRef CAS PubMed.
- J. Liu, R. Kang and D. Tang, Cancer Gene Ther., 2022, 29, 875–878 CrossRef CAS PubMed.
- H. Yang and J. Yang, RSC Med. Chem., 2021, 12, 1026–1036 RSC.
- D. R. Owen, C. M. N. Allerton, A. S. Anderson, L. Aschenbrenner, M. Avery, S. Berritt, B. Boras, R. D. Cardin, A. Carlo, K. J. Coffman, A. Dantonio, L. Di, H. Eng, R. Ferre, K. S. Gajiwala, S. A. Gibson, S. E. Greasley, B. L. Hurst, E. P. Kadar, A. S. Kalgutkar, J. C. Lee, J. Lee, W. Liu, S. W. Mason, S. Noell, J. J. Novak, R. S. Obach, K. Ogilvie, N. C. Patel, M. Pettersson, D. K. Rai, M. R. Reese, M. F. Sammons, J. G. Sathish, R. S. P. Singh, C. M. Steppan, A. E. Stewart, J. B. Tuttle, L. Updyke, P. R. Verhoest, L. Wei, Q. Yang and Y. Zhu, Science, 2021, 374, 1586–1593 CrossRef CAS PubMed.
- R. Gupta, M. Lavollay, J.-L. Mainardi, M. Arthur, W. R. Bishai and G. Lamichhane, Nat. Med., 2010, 16, 466–469 CrossRef CAS PubMed.
- M. de Munnik, C. T. Lohans, P. A. Lang, G. W. Langley, T. R. Malla, A. Tumber, C. J. Schofield and J. Brem, Chem. Commun., 2019, 55, 10214–10217 RSC.
- E. M. Steiner, G. Schneider and R. Schnell, FEBS J., 2017, 284, 725–741 CrossRef CAS PubMed.
- P. Kumar, A. Kaushik, E. P. Lloyd, S.-G. Li, R. Mattoo, N. C. Ammerman, D. T. Bell, A. L. Perryman, T. A. Zandi, S. Ekins, S. L. Ginell, C. A. Townsend, J. S. Freundlich and G. Lamichhane, Nat. Chem. Biol., 2017, 13, 54–61 CrossRef CAS PubMed.
- J. C. Powers, J. L. Asgian, Ö. D. Ekici and K. E. James, Chem. Rev., 2002, 102, 4639–4750 CrossRef CAS PubMed.
- L. Boike, N. J. Henning and D. K. Nomura, Nat. Rev. Drug. Discovery, 2022, 21, 881–898 CrossRef CAS PubMed.
- E. Weerapana, G. M. Simon and B. F. Cravatt, Nat. Chem. Biol., 2008, 4, 405–407 CrossRef CAS PubMed.
- D. Greenbaum, K. F. Medzihradszky, A. Burlingame and M. Bogyo, Chem. Biol., 2000, 7, 569–581 CrossRef CAS PubMed.
- A. Albeck and S. Kliper, Biochem. J., 2000, 346, 71–76 CrossRef CAS PubMed.
- S. Kawamura, Y. Unno, A. Asai, M. Arisawa and S. Shuto, Bioorg. Med. Chem., 2014, 22, 3091–3095 CrossRef CAS PubMed.
- A. C. Dijkmans, N. V. O. Zacarías, J. Burggraaf, J. W. Mouton, E. Wilms, C. Van Nieuwkoop, D. J. Touw, J. Stevens and I. M. C. Kamerling, Antibiotics, 2017, 6, 24 CrossRef PubMed.
- L. L. Silver, Cold Spring Harb. Perspect. Med., 2017, 7, a025262 CrossRef PubMed.
- M. de Munnik, P. A. Lang, F. De Dios Antos, M. Cacho, R. H. Bates, J. Brem, B. Rodríguez-Miquel and C. J. Schofield, Chem. Sci., 2023, 14, 7262–7278 RSC.
- A. Krantz, Methods Enzymol., 1994, 244, 656–671 CAS.
- S. B. Erdemli, R. Gupta, W. R. Bishai, G. Lamichhane, L. M. Amzel and M. A. Bianchet, Structure, 2012, 20, 2103–2115 CrossRef CAS PubMed.
- A. Arnold, M. Markert and R. Mahrwald, Synthesis, 2006, 1099–1102, DOI:10.1055/s-2006-926346,.
- W. M. Weber, L. A. Hunsaker, C. N. Roybal, E. V. Bobrovnikova-Marjon, S. F. Abcouwer, R. E. Royer, L. M. Deck and D. L. Vander Jagt, Bioorg. Med. Chem., 2006, 14, 2450–2461 CrossRef CAS PubMed.
- V. K. Yadav and K. K. Kapoor, Tetrahedron, 1996, 52, 3659–3668 CrossRef CAS.
- I. Miyahisa, T. Sameshima and M. S. Hixon, Angew. Chem., Int. Ed., 2015, 54, 14099–14102 CrossRef CAS PubMed.
- T. Sameshima, I. Miyahisa, S. Yamasaki, M. Gotou, T. Kobayashi and J. Sakamoto, Adv. Sci. Drug. Discovery, 2017, 22, 1168–1174 CrossRef CAS PubMed.
- L. Petri, P. Ábrányi-Balogh, P. R. Varga, T. Imre and G. M. Keserű, Bioorg. Med. Chem., 2020, 28, 115357 CrossRef CAS PubMed.
- T. J. Holmes and R. G. Lawton, J. Am. Chem. Soc., 1977, 99, 1984–1986 CrossRef CAS PubMed.
- C. Bashore, P. Jaishankar, N. J. Skelton, J. Fuhrmann, B. R. Hearn, P. S. Liu, A. R. Renslo and E. C. Dueber, ACS Chem. Biol., 2020, 15, 1392–1400 CrossRef CAS PubMed.
- P. A. Lang, R. Raj, A. Tumber, C. T. Lohans, P. Rabe, C. V. Robinson, J. Brem and C. J. Schofield, Proc. Natl. Acad. Sci. U. S. A., 2022, 119, e2117310119 CrossRef CAS PubMed.
- M. Groll, K. B. Kim, N. Kairies, R. Huber and C. M. Crews, J. Am. Chem. Soc., 2000, 122, 1237–1238 CrossRef CAS.
- S. K. Grant, M. L. Moore, S. A. Fakhoury, T. A. Tomaszek Jr and T. D. Meek, Bioorg. Med. Chem. Lett., 1992, 2, 1441–1445 CrossRef CAS.
- L. Brewitz, L. Dumjahn, Y. Zhao, C. D. Owen, S. M. Laidlaw, T. R. Malla, D. Nguyen, P. Lukacik, E. Salah, A. D. Crawshaw, A. J. Warren, J. Trincao, C. Strain-Damerell, M. W. Carroll, M. A. Walsh and C. J. Schofield, J. Med. Chem., 2023, 66, 2663–2680 CrossRef CAS PubMed.
- T. R. Malla, L. Brewitz, D.-G. Muntean, H. Aslam, C. D. Owen, E. Salah, A. Tumber, P. Lukacik, C. Strain-Damerell, H. Mikolajek, M. A. Walsh and C. J. Schofield, J. Med. Chem., 2022, 65, 7682–7696 CrossRef CAS PubMed.
- T.-W. Lee, M. M. Cherney, C. Huitema, J. Liu, K. E. James, J. C. Powers, L. D. Eltis and M. N. G. James, J. Mol. Biol., 2005, 353, 1137–1151 CrossRef CAS PubMed.
- J. Lee, L. J. Worrall, M. Vuckovic, F. I. Rosell, F. Gentile, A.-T. Ton, N. A. Caveney, F. Ban, A. Cherkasov, M. Paetzel and N. C. J. Strynadka, Nat. Commun., 2020, 11, 5877 CrossRef CAS PubMed.
- L. Meng, R. Mohan, B. H. B. Kwok, M. Elofsson, N. Sin and C. M. Crews, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 10403–10408 CrossRef CAS PubMed.
- H.-J. Zhou, M. A. Aujay, M. K. Bennett, M. Dajee, S. D. Demo, Y. Fang, M. N. Ho, J. Jiang, C. J. Kirk, G. J. Laidig, E. R. Lewis, Y. Lu, T. Muchamuel, F. Parlati, E. Ring, K. D. Shenk, J. Shields, P. J. Shwonek, T. Stanton, C. M. Sun, C. Sylvain, T. M. Woo and J. Yang, J. Med. Chem., 2009, 52, 3028–3038 CrossRef CAS PubMed.
- S. T. Thun-Hohenstein, T. F. Suits, T. R. Malla, A. Tumber, L. Brewitz, H. Choudhry, E. Salah and C. J. Schofield, ChemMedChem, 2022, 17, e202100582 CrossRef CAS PubMed.
- Q. Lu, D. S. Harmalkar, Y. Choi and K. Lee, Molecules, 2019, 24, 3778 CrossRef PubMed.
- P. N. Wyrembak, K. Babaoglu, R. B. Pelto, B. K. Shoichet and R. F. Pratt, J. Am. Chem. Soc., 2007, 129, 9548–9549 CrossRef CAS PubMed.
- F.-L. Jin, X. Li and S.-J. Park, J. Ind. Eng. Chem., 2015, 29, 1–11 CrossRef CAS.
- K. Ariyoshi-Kishino, K. Hashimoto, O. Amano, J. Saitoh, M. Kochi and H. Sakagami, Anticancer Res., 2010, 30, 5069–5076 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, inhibition kinetics, mass spectrometry. CCDC 2262059. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3cc02932h |
| This journal is © The Royal Society of Chemistry 2023 |