
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA genuine germylene PGeP pincer ligand for formic acid dehydrogenation with iridium†
Marta
Fernández-Buenestado‡
,
Rosie J.
Somerville‡
,
Joaquín
López-Serrano
 * and
Jesús
Campos
*
* and
Jesús
Campos
*
Instituto de Investigaciones Químicas (IIQ), Departamento de Química Inorgánica and Centro de Innovación en Química Avanzada (ORFEO-CINQA), Consejo Superior de Investigaciones Científicas (CSIC) and Universidad de Sevilla, Avenida Américo Vespucio 49, Sevilla 41092, Spain. E-mail: joaquin.lopez@iiq.csic.es; jesus.campos@iiq.csic.es
First published on 16th June 2023
Abstract
We report an iridium system constructed around a long-tethered PGeP ligand that facilitates access to the less common germylene form, so far unreported for an ‘NHC-type’ Ge ligand. Its bonding is substantiated by computational studies and we have demonstrated its use for the catalytic dehydrogenation of formic acid, highlighting the potential of this underdeveloped type of ligand.
The coordination of heavier tetrylenes (:EX2; E = Si, Ge, Sn, Pb) to transition metals has attracted significant attention,1 in part due to their ambiphilic character and thus the possibility to develop processes based on metal/tetrylene cooperativity.2 However, in contrast to their lighter and well-known carbene congeners, heavier tetrylene complexes exhibit comparatively reduced stabilities. To circumvent this limitation, pincer-type tetrylene scaffolds have emerged and a number of catalytic applications have been developed,3 in several cases revealing direct cooperation between the transition metal and the tetrel site.4 While silylene pincer frameworks have been widely investigated,5 germylene and stannylene analogues are comparatively underdeveloped.
Within the still-limited family of pincer-type germylene ligands, PGeP compounds first reported by the group of Cabeza have shown a rich coordination chemistry.6–8 Not surprisingly, the first of the series (A in Fig. 1)9 shares its structural core with N-heterocyclic carbenes (NHCs). However, its coordination to transition metals is dominated by insertion of the germylene into M-halide bonds to yield germyl ligands, thus losing their exciting ambiphilic character.9 Although the halide substituent on A can be exchanged by other anionic groups,10 its removal to recover the germylene form has not been documented, most likely due to reduced stability. Tobita and Watanabe disclosed a nickel PGeP complex that retains the germylene ambiphilicity (B, Fig. 1), though the ligand presents a tripodal coordination due to the double bond character of the Ge![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) Ni bond.11 The use of an alternative dipyrromethane backbone by Cabeza and co-workers recently provided access to the first pincer-type PGeP complex in which the germanium center maintains its electrophilic germylene character after binding to palladium(0) (C, Fig. 1).8b This enabled cooperative reactivity between Pd and Ge, which was demonstrated for a number of substrates.
Ni bond.11 The use of an alternative dipyrromethane backbone by Cabeza and co-workers recently provided access to the first pincer-type PGeP complex in which the germanium center maintains its electrophilic germylene character after binding to palladium(0) (C, Fig. 1).8b This enabled cooperative reactivity between Pd and Ge, which was demonstrated for a number of substrates.
 | ||
| Fig. 1 Representative examples of PGeP-based transition metal complexes (IiPr = 1,3-diisopropyl-4,5-dimethylimidazol-2-ylidene). | ||
On these bases, we aimed to design a PGeP system that while sharing the classic NHC core, would still have an accessible germylene form and thus maintain the potential for cooperativity. To circumvent previous stability limitations, we hypothesized that the use of longer arms for the pincer ligand would be beneficial by providing higher coordination flexibility and larger P–M–P bond angles (i.e. reduced distortion from a square-planar geometry). Yamashita and co-workers have demonstrated the catalytic proficiency of iridium complexes constructed around long-tethered PBP12 and PAlP13 ligands. Encouraged by their work, we have now synthesized the analogous long-tethered PGeP germylene and investigated its coordination chemistry towards iridium, demonstrating that the longer arms ensure the formation of a genuine PGeP-type iridium-germylene structure, which we have fully characterized by experimental and computational methods. The catalytic usefulness of the system towards formic acid dehydrogenation is illustrated.
We first synthesized the targeted PGeP germylene by a similar approach to that described for its borane and alane PEP (E = B, Al) analogues. Thus, reaction of bis-amido germylene Ge[N(SiMe3)2]2 with the bis-phosphino phenylenediamine precursor 1 in toluene (55 °C, 16 h) afforded the pincer-type germylene 2 in 90% isolated yield as a waxy orange solid. At variance with its group 13 counterparts, the need for external base was avoided owing to the basicity of the silylamido ligands on the Ge(II) precursor. A single 31P{1H} NMR resonance was recorded at 26.2 ppm, only slightly shifted compared to 1 (27.2 ppm) and thus suggesting no interaction with germanium.
Next, we treated germylene 2 with one equivalent of Vaska's complex [IrCl(CO)(PPh3)2] in THF, leading to the isolation of germyl compound 3 in 55% yield via insertion of germanium across the Ir–Cl bond, with displacement of the two triphenyl phosphines. Although the two phosphine donor atoms of 3 are observed as a single broad 31P{1H} NMR resonance at 25 °C (57.7 ppm), lowering the temperature to −15 °C led to two doublets (2JPP = 233 Hz) at 62.6 and 54.2 ppm due to the asymmetry of the complex, with the two (CH2)3 arms being located at opposite sides of the Ir-square plane (Fig. 2). To access a germylene pincer complex we treated 3 with NaBArF (BArF = [B(C6H3-3,5-(CF3)2)4]−) as a chloride abstractor. As hypothesized, the formation of the germylene structure in 4, with a planar arrangement, leads to a sharp 31P{1H} resonance at 64.8 ppm.
 | ||
| Fig. 2 ORTEP diagrams for complexes 3 and 4. Hydrogen atoms and counteranion (in 4) have been excluded, and tert-butyl groups are represented in wireframe format for clarity. Thermal ellipsoids are set at 50% probability. | ||
We confirmed the abstraction of the chloride by X-ray diffraction studies after growing crystals of 4. The solid-state structures of compounds 3 and 4 are depicted in Fig. 2 and present some striking differences. The Ir–Ge bond distance decreases upon chloride removal (2.510(1), 3; 2.435(1), 4), in agreement with a stronger interaction. In turn, a slight reduction in back-bonding from iridium to the carbonyl ligand is inferred from an intense band in its IR spectrum at 1983 cm−1, shifted by almost 30 cm−1 to higher frequencies compared to germyl precursor 3 (νCO = 1955 cm−1). The planarization at germanium upon chloride abstraction becomes evident, with the sum of the three covalent bond angles around Ge in compound 4 accounting for 359.9(1)°. Furthermore, the Ge atom in 4 is almost perfectly located within the plane defined by the phenylenediamido fragment (dGe-C6N2(plane) of only 0.036 Å), while it is clearly bent out of that plane in compound 3 (dGe-C6N2(plane) = 0.420 Å) (Fig. 2 and Scheme 1).
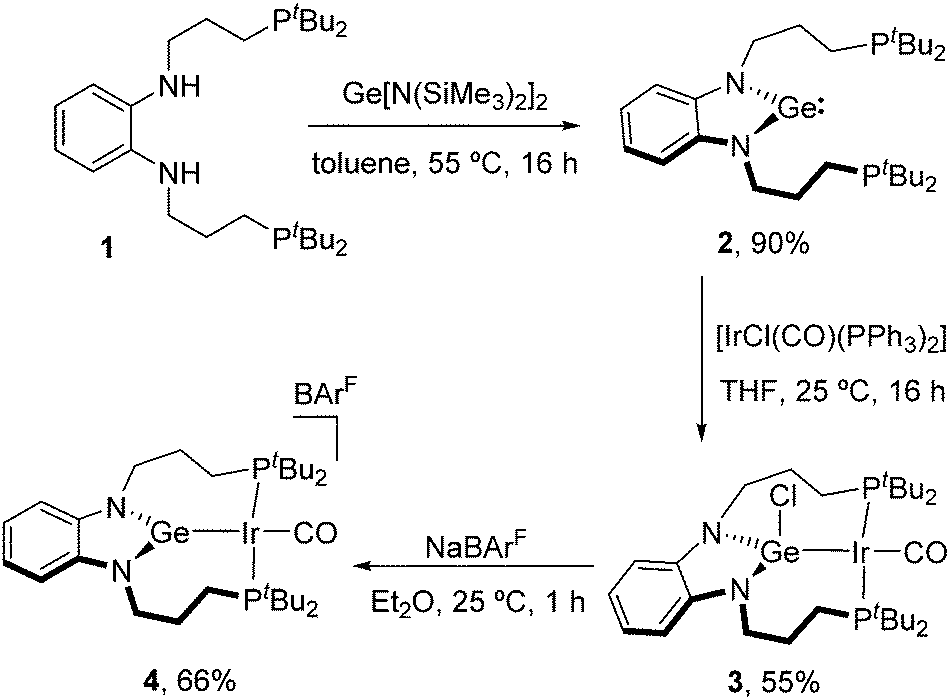 | ||
| Scheme 1 Synthesis of PGeP germylene pincer compound 2 and iridium complexes 3 and 4. | ||
We explored the bonding situation in 3 and 4 by means of DFT/NBO calculations (B3PW91-D3BJ/6-31g(d,p)+SDD//B3PW91-D3BJ/def2-TZVP). The optimized geometries of the complexes reproduce their solid-state molecular structures, specifically the angles between the N–Ge–N and iridium coordination planes, and the decrease in Ge–Ir distance on going from germyl to germylene (2.54 v. 2.44 Å, respectively). Vibrational analyses match the increase in CO stretching frequency (ca. 30 cm−1) seen by IR. Interestingly, comparing complex 3 to complex 4 the NBO charges of the Ge and Ir centers of 3 and 4 and the amount of electron density donated from the PGeP ligand to the Ir–CO fragment are quite similar. The population analysis suggests that the NHGe fragment offsets the expected changes in the donor properties of the PGeP ligand upon chloride abstraction. Focusing on 4, the NBO analysis describes the Ge–Ir bond in terms of one σ donor–acceptor interaction between the lone pair on Ge with s (s3p) character and the σ* Ir–CO bond. These results are summarized in Fig. 3. The corresponding Natural Localized Molecular Orbital (NLMO) has 71.2% Ge, 15.1% Ir, and 8.5% C character. Interestingly, while Second order perturbation theory analysis clearly accounts for Ir–CO π back-bonding by showing strong delocalization of two d lone pairs on Ir onto the two π* C–O bonds, with delocalization energies between 30 and 39 kcal mol−1, the corresponding delocalization energies onto the Ge–N linkage are below 2 kcal mol−1 (the NLMOs associated to the mentioned d orbitals tail onto the carbonyl carbon and have ca. 90% Ir, 7–8% C, but less than 1% Ge orbital contribution). This is in line with the description of the Ge–N bonds obtained from the NBO analysis as having significant π components and justifies the relative tilt of the N–Ge–N and Ir coordination planes. The single bond character of the Ge–Ir bond in 4 is further supported by its similar Wiberg index to that of the same bond in 3 (0.46 v. 0.43 respectively, compared to ca. 0.48 for the Ir–P bonds) and by the values of the electron density and related magnitudes at the bond critical points as derived from AIM analysis (ρb = 0.077 v. 0.083 a.u. for 3 and 4 respectively; see the ESI† for further details). Thus, despite retaining its germylene form upon chloride abstraction in 4, π-donation to Ge from the adjacent nitrogen atoms seem to prevent further donation from iridium.
 | ||
| Fig. 3 Relevant localized orbitals calculated for 4. Upper left figure: NLMO for the Ir–Ge σ bond; upper right: one of the NLMO accounting for Ir–CO π back donation; lower left: NBO for one of the π Ge–N bonds. The lower right figure shows the relative orientation of the N–Ge–N and iridium coordination planes, dz2 NBO on Ir and cartesian axes. | ||
We moved forward to investigate the reactivity and catalytic potential of compound 4, particularly encouraged by the accessibility of the germylene form of the PGeP ligand. In this preliminary study, we focused on the dehydrogenation of formic acid, given the interest in this transformation in the context of a possible hydrogen economy,14 the well-known proficiency of iridium complexes within the field15 and the recent report that an osmium-germylene system acts as a cooperative catalyst for this reaction.4c First, we examined the stoichiometric reactivity of compound 4 with the most relevant molecules involved in the process, namely dihydrogen, carbon dioxide and formic acid. While compound 4 did not react under CO2 atmosphere (1 bar, 25 °C, 24h), it readily evolved upon exposure to dihydrogen to yield dihydride 5 (25 °C, 1 bar; Scheme 2). The loss of symmetry is attested by two broad 31P resonances at 62.9 and 67.2 ppm that sharpen upon cooling to −40 °C and reveal coupling (2JPP = 210 Hz). Two distinctive low-frequency signals are observed in the 1H NMR spectrum at −11.50 (t, 2JHP = 14.7 Hz) and −12.54 due to the two inequivalent hydride ligands. The IR stretching frequency for the carbonyl ligand is now shifted to 2020 cm−1, as expected for reduced back-donation upon oxidation from Ir(I) to Ir(III).
 | ||
| Scheme 2 Oxidative addition of H2 over 4 towards compound 5. | ||
Attempts to isolate analytically pure samples of 5 were unsuccessful due to its instability and the reversible H2 loss upon evaporation of solvent, which partially regenerates germylene 4 alongside other unidentified species. Nonetheless, this reversibility should be beneficial in the context of catalytic hydrogen evolution from formic acid.
We did however obtain a small crop of crystals of 5 from a slow diffusion of pentane into a saturated dichloromethane solution. The structure of 5 (Fig. S16, ESI†) shows the rearrangement of the CO ligand to a cis-position with respect to the germylene to accommodate the two hydride ligands. Oxidation to Ir(III) results in shortening the Ir–Ge bond to 2.367(1) Å. We investigated the mechanism by computational studies, which revealed that activation at Ir takes place along the Ge–Ir–CO direction to yield the expected cis stereo isomer of 5, without direct involvement of Ge or C atom (Fig. S21, ESI†), with a barrier (ΔG‡) of 13.3 kcal mol−1 (activation along the P–Ir–P direction has a higher barrier of 22.1 kcal mol−1). We failed to locate a transition state for the activation across the Ir–Ge bond. Formation of the resulting species with one hydrogen atom on both Ge and Ir would be endergonic by 17.8 kcal mol−1 relative to 4 + H2. Furthermore, migration of the H atom on Ge to Ir to afford 5 requires an additional 8 kcal mol−1.
The stoichiometric reaction of 4 with formic acid led to dihydride 5 in around 58% spectroscopic yield. The appearance of a small 1H NMR peak at 4.55 ppm further supported the formation of H2, while the nature of other phosphorus-containing species could not be identified. However, with the formation of H2 observed by 1H NMR, we were set to investigate its production under catalytic conditions. In particular, we monitored the evolution of hydrogen after heating solutions of iridium compounds 3, 4 and [IrCl(CO)(PPh3)2] (0.02 mol%) in neat formic acid at 90 °C (Table S1, see ESI† for details). Germylene catalyst 4 exhibited superior reactivity, reaching a maximum TON of 225 (entry 3), whereas iridium germyl 3 and precursor [IrCl(CO)(PPh3)2] reached lower TONs of 98 and 50, respectively (entries 1 and 2; Table 1). The use of external bases is common in formic acid dehydrogenation catalysis.16 In this case, the addition of NEt3 increased the TON with Vaska's complex to 504, but did not substantially affect the activities of the Ge-containing catalysts (Fig. S19, ESI†). Nonetheless, we observed a dramatic increase in catalytic activity by using HCOONa as base in the presence of catalyst 3 (Fig. 4). In that case, a maximum TON of 4875 (TOF at 30 min of 9750 h−1) was recorded, two orders of magnitude superior than in the absence of additive, and also considerably higher than for catalysts 4 and [IrCl(CO)(PPh3)2], which exhibited TONs of 390 and 708 respectively under identical conditions. Although the initial TOF (30 min) values for the three complexes were comparable, the maximum activity of 3 under these conditions is clearly higher. Attempts to isolate an intermediate from stoichiometric reactions between 3 and HCOONa were unfruitful. Although the activity revealed by 3 is more than an order of magnitude below current state-of-the-art catalysts,17 it demonstrates the potential of using heavier tetrylene-based ligands. It is of note that the three catalysts exchange their superior performance depending on the conditions, with 4 being the best catalyst in the absence of external bases, Vaska's complex in the presence of NEt3 and compound 3 when using HCOONa.
|
|
|||||
|---|---|---|---|---|---|
| Entry | Catalyst | Additive | H2 (mmol)b | TON | TOF (h−1)c |
| a Conditions: neat formic acid (0.5 mL), catalyst (0.02 mol%), additive (5 mol%), 90 °C. b From monitoring gas evolution by a Man on the Moon millireactor (see ESI for details). c TOF measured after 30 min. | |||||
| 1 | Vaska | — | 0.14 | 50 | 99 |
| 2 | 3 | — | 0.25 | 98 | 195 |
| 3 | 4 | — | 2.55 | 225 | 449 |
| 4 | Vaska | NEt3 | 1.45 | 504 | 1008 |
| 5 | 3 | NEt3 | 1.25 | 452 | 904 |
| 6 | 4 | NEt3 | 0.61 | 222 | 443 |
| 7 | Vaska | HCOONa | 2.20 | 708 | 1416 |
| 8 | 3 | HCOONa | 12.35 | 4875 | 9750 |
| 9 | 4 | HCOONa | 0.96 | 390 | 779 |

 | ||
| Fig. 4 Monitoring hydrogen evolution from formic acid using catalysts [IrCl(CO)(PPh3)2], 3 and 4 without additives (dotted lines) or in the presence of HCOONa (5 mol%; solid lines). | ||
In summary, we demonstrate that the use of long-tethered arms within a PGeP framework facilitates access to its germylene form, uncommon for this type of pincer ligand and so far unreported for an ‘NHC-type’ Ge-core. This is achieved by chloride abstraction from an iridium-germyl pincer precursor, leading to a clear planarization of the GeN2C2 heterocycle. Our computational studies revealed only residual Ir → Ge back-donation due to significant π-bonding between Ge and the N atoms. The iridium complexes investigated are active precatalysts for the dehydrogenation of formic acid, with the best catalyst being highly dependent on the additive used.
This work has been supported by the European Research Council (ERC Starting Grant, CoopCat, 756575). We also thank grants PID2019-110856GA-I00 (MCIN/AEI/10.13039/501100011033) and TED2021-132225B-I00/AEI/10.13039/501100011033/by Unión Europea NextGenerationEU/PRTR, Junta de Andalucía (P18-FR-4688) and the use of CESGA computational facilities. We are extremely grateful to Prof. Javier A. Cabeza for helpful discussions.
Conflicts of interest
There are no conflicts to declare.Notes and references
- See for example: (a) M. F. Lappert and R. S. Rowe, Coord. Chem. Rev., 1990, 100, 267–292 CrossRef CAS; (b) R. Waterman, P. G. Hayes and T. D. Tilley, Acc. Chem. Res., 2007, 40(8), 712–719 CrossRef CAS PubMed; (c) L. Álvarez-Rodríguez, J. A. Cabeza, P. García-Álvarez and D. Polo, Coord. Chem. Rev., 2015, 300, 1–28 CrossRef; (d) M. Saito, Acc. Chem. Res., 2018, 51, 160–169 CrossRef CAS PubMed.
- (a) R. J. Somerville and J. Campos, Eur. J. Inorg. Chem., 2021, 3488–3498 CrossRef CAS PubMed; (b) Z. Benedek and T. Szilvási, Organometallics, 2017, 36, 1591–1600 CrossRef CAS; (c) S. Bajo, M. M. Alcaide, J. López-Serrano and J. Campos, Chem. – Eur. J., 2021, 27, 16422–16428 CrossRef CAS PubMed.
- See for example: (a) A. Brück, D. Gallego, W. Wang, E. Irran, M. Driess and J. F. Hartwig, Angew. Chem., Int. Ed., 2012, 51, 11478–11482 CrossRef PubMed; (b) Y. Dong, P. Zhang, Q. Fan, X. Du, S. Xie, H. Sun, X. Li, O. Fuhr and D. Fenske, Inorg. Chem., 2020, 59, 16489–16499 CrossRef CAS PubMed; (c) S. Bajo, C. A. Theulier and J. Campos, ChemCatChem, 2022, 14, e202200157 CAS; (d) N. Almenara, M. A. Garralda, X. Lopez, J. M. Matxain, Z. Freixa and M. A. Huertos, Angew. Chem., Int. Ed., 2022, 61, e202204558 CrossRef CAS PubMed; (e) J. A. Cabeza, J. M. Fernández-Colinas, J. García-Álvarez, P. García-Álvarez, C. J. Laglera-Gándara and M. Ramos-Martín, Chem. – Eur. J., 2022, 28, e2022008 CrossRef PubMed.
- (a) D. Gallego, A. Brück, E. Irran, F. Meier, M. Kaupp, M. Driess and J. F. Hartwig, J. Am. Chem. Soc., 2013, 135, 15617–15626 CrossRef CAS PubMed; (b) Y. Wang, A. Kostenko, S. Yao and M. Driess, J. Am. Chem. Soc., 2017, 139, 13499–13506 CrossRef CAS PubMed; (c) M. L. Buil, J. A. Cabeza, M. A. Esteruelas, S. Izquierdo, C. J. Laglera-Gámdara, A. I. Nicasio and E. Oñate, Inorg. Chem., 2021, 60, 16860–16870 CrossRef CAS PubMed.
- Y. P. Zhou and M. Driess, Angew. Chem., Int. Ed., 2019, 58, 3715–3728 CrossRef CAS PubMed.
- L. Álvarez-Rodríguez, J. Brugos, J. A. Cabeza, P. García-Álvarez, E. Pérez-Carreño and D. Polo, Chem. Commun., 2017, 53, 893–896 RSC.
- J. A. Cabeza, P. García-Álvarez and C. J. Laglera-Gándara, Eur. J. Inorg. Chem., 2020, 784–795 CrossRef CAS.
- (a) A. Arauzo, J. A. Cabeza, I. Fernández, P. García-Álvarez, I. García-Rubio and C. J. Laglera-Gándara, Chem. – Eur. J., 2021, 27, 4985–4992 CrossRef CAS PubMed; (b) J. A. Cabeza, P. García-Álvarez, C. J. Laglera-Gándara and E. Pérez-Carreño, Chem. Commun., 2020, 56, 14095–14097 RSC; (c) J. A. Cabeza, I. Fernández, J. M. Fernández-Colinas, P. García-Álvarez and C. J. Laglera-Gándara, Chem. – Eur. J., 2019, 25, 12423–12430 CrossRef CAS PubMed; (d) S. Bestgen, N. H. Rees and J. M. Goicoechea, Organometallics, 2018, 37, 4147–4155 CrossRef CAS.
- J. A. Cabeza and P. García-Álvarez, Chem. – Eur. J., 2023, 29, e202203096 CrossRef CAS PubMed.
- (a) L. Álvarez-Rodríguez, J. Brugos, J. A. Cabeza, P. García-Álvarez and E. Pérez-Carreño, Chem. – Eur. J., 2017, 23, 15107–15115 CrossRef PubMed; (b) J. A. Cabeza, P. García-Álvarez, C. J. Laglera-Gándara and E. Pérez-Carreño, Eur. J. Inorg. Chem., 2021, 1897–1902 CrossRef CAS.
- T. Watanabe, Y. Kasai and H. Tobita, Chem. – Eur. J., 2019, 25, 13491–13495 CrossRef CAS PubMed.
- (a) E. Huang Kwan, Y. J. Kawai, S. Kamakura and M. Yamashita, Dalton Trans., 2016, 45, 15931–15941 RSC; (b) E. H. Kwan, H. Ogawa and M. Yamashita, ChemCatChem, 2017, 9, 2457 CrossRef CAS.
- S. Morisako, S. Watanabe, S. Ikemoto, S. Muratsugu, M. Tada and M. Yamashita, Angew. Chem., Int. Ed., 2019, 58, 15031–15035 CrossRef CAS PubMed.
- (a) J. Y. Cho, H. Kim, J. E. Oh and B. Y. Park, Catalysts, 2021, 11, 1497 CrossRef CAS; (b) S. Kar, M. Rauch, G. Leitus, Y. Ben-David and D. Milstein, Nat. Catal., 2021, 4, 193–201 CrossRef CAS PubMed.
- (a) A. Matsunami, Y. Kayaki and T. Ikariya, Chem. – Eur. J., 2015, 21, 13513–13517 CrossRef CAS PubMed; (b) Y. Maenaka, T. Suenobu and S. Fukuzumi, Energy Environ. Sci., 2012, 5, 7360–7367 RSC; (c) J. J. A. Celaje, Z. Lu, E. A. Kedzie, N. J. Terrile, J. N. Lo and T. J. Williams, Nat. Commun., 2016, 7, 11308 CrossRef CAS PubMed.
- See for example: (a) M. Iglesias and L. A. Oro, Chem. Soc. Rev., 2018, 47, 2772–2808 RSC; (b) J. Guzmán, A. Urriolabeitia, V. Polo, M. Fernández-Buenestado, M. Iglesias and F. J. Fernández-Álvarez, Dalton Trans., 2022, 51, 4386–4393 RSC; (c) J. J. A. Celaje, Z. Lu, E. A. Kedzie, N. J. Terrile, J. N. Lo and T. J. Williams, Nat. Commun., 2016, 7, 11308 CrossRef CAS PubMed.
- N. Lentz and M. Albrecht, ACS Catal., 2022, 12, 12627–12631 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental synthesis and catalysis details, NMR spectra, X-ray and computational details. CCDC 2262392–2262394. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3cc02447d |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2023 |