
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Overcoming the challenges of infrared photosensitizers in photodynamic therapy: the making of redaporfin
Luis G.
Arnaut
 * and
Mariette M.
Pereira
*
* and
Mariette M.
Pereira
*
CQC-IMS, Chemistry Department, University of Coimbra, Coimbra, Portugal. E-mail: lgarnaut@ci.uc.pt
First published on 6th July 2023
Abstract
We offer a personal account of the discovery and development of a photosensitizer for photodynamic therapy (PDT) of cancer, from bench to bedside. We emphasize the more chemical aspects of drug discovery and drug development, namely the chemical landscape at the time of the discovery, the breakthrough in the field offered by stable bacteriochlorins, the challenges of synthesising a significant amount of the product with high purity for preclinical studies, the factors that relate molecular structure to pharmacology in PDT, the mechanistic interpretation of preclinical data and the management of unexpected results. Special attention is given to the implications of atropisomerism and immune responses in PDT.
1. Introduction
Photodynamic therapy (PDT, see Fig. 1) of solid tumours has a history marked by remarkable scientific and clinical successes.1–6 Although the great potential of PDT has been realized in pre-clinical and clinical studies, with the exception of topical applications, its potential is yet to be realized in the oncology market. There are at least two reasons that may explain the gap between scientific and clinical success stories and the mild market traction. The first reason is related to the nature of PDT as a combination between a drug (i.e., a photosensitizer) and a device (i.e., a light source). This places PDT between the well-established markets of pharmaceuticals and medical devices, which makes PDT resemble a niche market. Additionally, most often PDT is used only once to treat one solid tumour, as opposed to a drug that is taken regularly until reaching a cure or drug resistance, and this challenges standard pharma business models. Finally, in this first reason, it should be recognized that drug-device combinations come with two learning curves – one for learning photosensitizer pharmacology and the other for learning laser operation and dosimetry – which may be inconvenient for a given medical specialty. Nevertheless, the clinical and commercial success of PDT in the treatment of age-related macular degeneration7,8 shows that the obstacle of mastering drug-device combinations is not unsurmountable.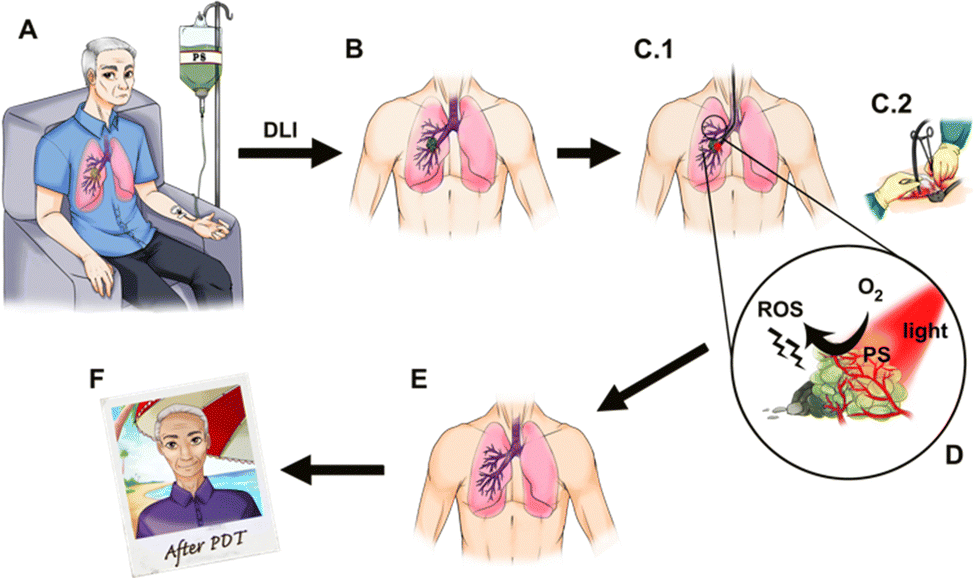 | ||
| Fig. 1 Photodynamic therapy of a patient with a lung tumour. (A) Intravenous (i.v.) administration of a photosensitizer (PS). (B) The photosensitizer accumulates in the tumour and the tumour is illuminated after a certain drug-to-light interval (DLI). (C) Light is delivered to the tumour, possibly using an optical fibre. (D) The PS molecule located in the tumour absorbs light of an appropriate wavelength and in the excited state transfers energy or an electron to nearby molecular oxygen generating reactive oxygen species (ROS) such as singlet oxygen, superoxide ion or hydroxyl radical. (E) Cell death triggered by ROS generated in the illuminated volume leads to clearance of the tumour. (F) The patient resumes normal life shortly after the treatment. Courtesy of Ana Mata. | ||
The second reason that may explain why PDT is not yet widely adopted in the treatment of solid tumours is related to the structure of photosensitizers. The first photosensitizer approved for the treatment of solid tumours – Photofrin®, which is a mixture of porphyrins of biological origin9 – has very slow clearance from the body10 and the photosensitivity of the eyes and skin of patients treated with Photofrin® requires protection from direct sunlight or bright indoor light for at least 30 days post-treatment. This limitation of Photofrin® led to the belief that patients treated with PDT have a prolonged risk of sunburn, although this limitation was overcome by more recent photosensitizers. The other major limitation of Photofrin® is its electronic absorption spectrum. Its longest wavelength absorption band has a low molar absorption coefficient (ε630 ≈ 3 × 103 M−1 cm−1) and does not occur in the spectral region where light penetrates more deeply in human tissues.11 In order to produce tissue necrosis, and destroy tumours, it is necessary to attain locally a photodynamic threshold dose within the range of 1018–1019 photons cm−3 (∼0.01 moles of photons per dm3). This dose is given by Tth = 2.3εClocLth, where Cloc is the photosensitizer local concentration and Lth is the light fluence at the maximum depth of necrosis.12,13 Photofrin® was originally identified as a promising photosensitizer because it has a natural tendency to accumulate in tumours, i.e., its Cloc is relatively high (Cloc ≈ 2 μM for a 5 mg kg−1 body weight i.v. administration)14 but higher ε and Lth are needed to treat large tumours.
The photodynamic threshold dose also teaches us that the local ROS must reach ∼10 mM within a few minutes of illumination to produce tissue necrosis. Given Cloc ≈ 2 μM, this means that each photosensitizer molecule must absorb 5000/ΦROS photons, where ΦROS is the quantum yield of ROS generation, to trigger tissue necrosis. This emphasizes the role of photosensitizers as “photocatalysts” in PDT: the photosensitizer absorbs one photon and should have a high probability of generating ROS concomitantly with its return to the ground state, where it can absorb a second photon and restart the cycle. Given that the concentration of ROS that produces necrosis is much higher than Cloc, each photosensitizer molecule must perform the cycle of light absorption and ROS generation thousands of times before it photobleaches. Hence, another condition that a good photosensitizer must obey is to have a low photodecomposition quantum yield (Φpd). A photosensitizer characterized by the data presented above and ΦROS = 0.5, should have Φpd < 10−4. This is observed in Photofrin, which has Φpd = 5 × 10−5.15
It is widely recognized that the hydrogenation of exo pyrrole double bonds of porphyrins to dihydroporphyrins (e.g., chlorins) or tetrahydroporphyrins (e.g., bacteriochlorins) preserves the same basic chromophore, characterized by the 18 π-electron delocalization.16 However, the change in beta-carbon pyrrol hybridization from sp2 to sp3 carbons introduces skeletal distortions that lift the degeneracy of frontier molecular orbitals, increases the energy of the HOMO and lowers the HOMO–LUMO gap. This enables bacteriochlorins to have absorption bands in the infrared region that are two orders of magnitude more intense than the red absorption band of porphyrins. These are the most desired spectroscopic properties for PDT photosensitizers, but they come with a challenge: that the oxidation potentials of bacteriochlorins are substantially lower compared with those of the corresponding porphyrins and consequently they are readily oxidized.17
Various researchers have investigated the use of bacteriochlorophyll derivatives and synthetic bacteriochlorins as sensitizers for PDT in animal models, but found the expected instability problems. Bacteriochlorophyll-a was rapidly degraded into bacteriopheophytin-a and other products, although some cures were obtained at short drug-to-light intervals (DLI), which suggests a predominantly vascular shutdown mechanism.18,19 Bacteriopheophorbides (see Scheme 1) were found to be inactive at the doses studied.20 More soluble bacteriochlorophyll derivatives were also rapidly metabolized and eliminated, suggesting that such photosensitizers could only be used shortly after their administration, but no treatment results were published.21 Synthetic bacteriochlorins were also investigated.22,23 By 1995–1998 the consensus in the scientific community was that known bacteriochlorins were too labile for PDT.2,3 This was also the time when we started our research program to synthesize stable bacteriochlorins for PDT at the University of Coimbra.
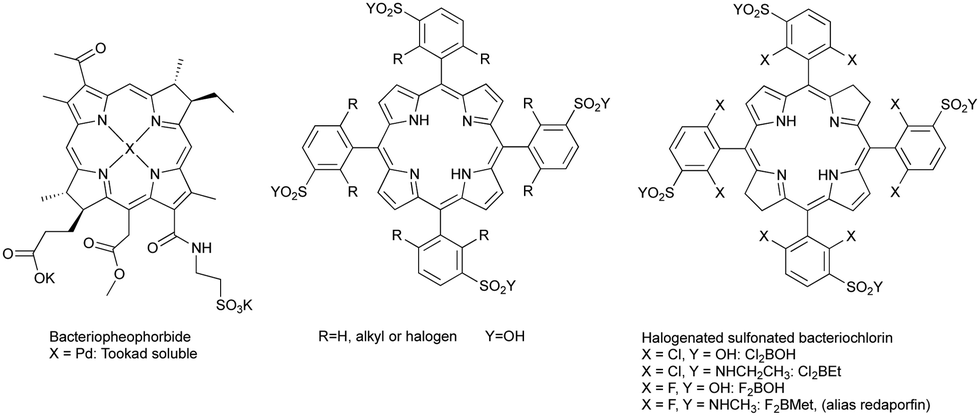 | ||
| Scheme 1 Bacteriochlorins derived from natural products, porphyrin designed in 1994 to be the precursor of synthetic bacteriochlorins and halogenated sulfonated bacteriochlorins synthesised in this research programme. | ||
The principles of PDT and the limitations of Photofrin® and of bacteriochlorins, briefly reviewed above, were determined in 1993 when Photofrin® was first approved in Canada for the treatment of bladder cancer. In 1994, impressed by Photofrin® approvals,24 but aware of its limitations, the authors of this work, under the leadership of Sebastião Formosinho, submitted to the Portuguese Science Foundation (FCT) a project (PRAXIS/QUI/2/2.1/390/94) where the abstract stated the purpose of developing “New synthesis for heterocycles, namely pyrrols, porphyrins, chlorins, and bacteriochlorins, (…) the properties of new photosensitizers will be studied in order to attempt the improvement of the efficiency of photodynamic therapy”. The target structures of the porphyrin derivatives presented in that project are presented in Scheme 1. They were designed to be the precursors of bacteriochlorins disclosed later25 and also presented in Scheme 1. The support of Sebastião Formosinho, a renowned Portuguese photochemist graduated at the University College London in 1971 with Nobel laureate George Porter, was instrumental to begin our studies on bacteriochlorins. For the records, it is also fair to say that the authors of this work were prepared to disclose redaporfin earlier in 2004, but a last-minute phone call of the then Rector of the University of Coimbra, Fernando Seabra Santos, changed their plans and prioritized the submission of a patent (PCT/EP2005/012212 priority to FR0412149, 2004).
2. The synthesis of stable bacteriochlorins
The conditions for developing stable bacteriochlorins for PDT at the University of Coimbra in 1996–2000 were particularly favourable. FCT funded the project, part of the team had a strong background in the synthesis of porphyrins and the other part had the competences and techniques required to study their photochemistry. We started with halogenated porphyrins,26 followed with halogenated chlorins,27 and arrived at halogenated bacteriochlorins.28 The stability of 5,10,15,20-tetrakis(2,6-difluorophenyl)bacteriochlorin largely exceeded our best expectations: in deaerated solution it lasted for many days at elevated temperatures. Additionally, the effect of halogenation in the ortho position of the phenyl rings was shown to increase S1 → T1 intersystem crossing, with the consequent increase in triplet state quantum yields, without substantially affecting triplet lifetimes.29 Hence, fluorine atoms could both stabilize meso-tetraarylbacteriochlorins and increase their singlet oxygen quantum yield (ΦΔ).Our first approach to the synthesis of tetraphenylporphyrins was based on the nitrobenzene synthetic methodology disclosed by Pereira and Gonsalves in 1991.30 This method involves the condensation of pyrrole with mono- or di-halogenated aldehydes at the ortho positions of phenyl groups, using a mixture of acetic acid and nitrobenzene (in a 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio). In this approach, the temperature must be maintained at or above 130 °C to ensure that nitrobenzene functions simultaneously as a solvent, inducing the crystallization of halogenated arylporphyrins from the reaction mixture, and as an oxidant to convert the porphyrinogen to the corresponding porphyrins. Another relevant aspect of this methodology is that under these conditions the final halogenated porphyrins are isolated without any contamination with the corresponding chlorin, unlike previously described one-pot methods.31
:1 ratio). In this approach, the temperature must be maintained at or above 130 °C to ensure that nitrobenzene functions simultaneously as a solvent, inducing the crystallization of halogenated arylporphyrins from the reaction mixture, and as an oxidant to convert the porphyrinogen to the corresponding porphyrins. Another relevant aspect of this methodology is that under these conditions the final halogenated porphyrins are isolated without any contamination with the corresponding chlorin, unlike previously described one-pot methods.31
Using the nitrobenzene method, we obtained the following halogenated tertaphenylporphyrins (and corresponding yields): meso-tetrakis-(2-chlorophenyl)porphyrin (ToCPP, 8.5%), meso-tetrakis-(2,6-dichlorophenyl)porphyrin (TDCPP, 5%), meso-tetrakis-(2-fluorophenyl)porphyrin (ToFPP, 20%) and meso-tetrakis-(2,6-difluorophenyl)porphyrin (TDFPP, 10%) (Fig. 1).32,33 Later, improvements of this nitrobenzene synthetic methodology, using MCM 45 as a reusable Lewis acid catalyst, to activate the aldehyde, allowed us to improve the yields of mono- and di-chlorinated or fluorinated meso-aryl porphyrins.34,35 The synthesis of these porphyrins was also performed under more sustainable conditions, by using water as the solvent, and microwave irradiation (200 °C) as an alternative reaction activation technique (Scheme 2).36
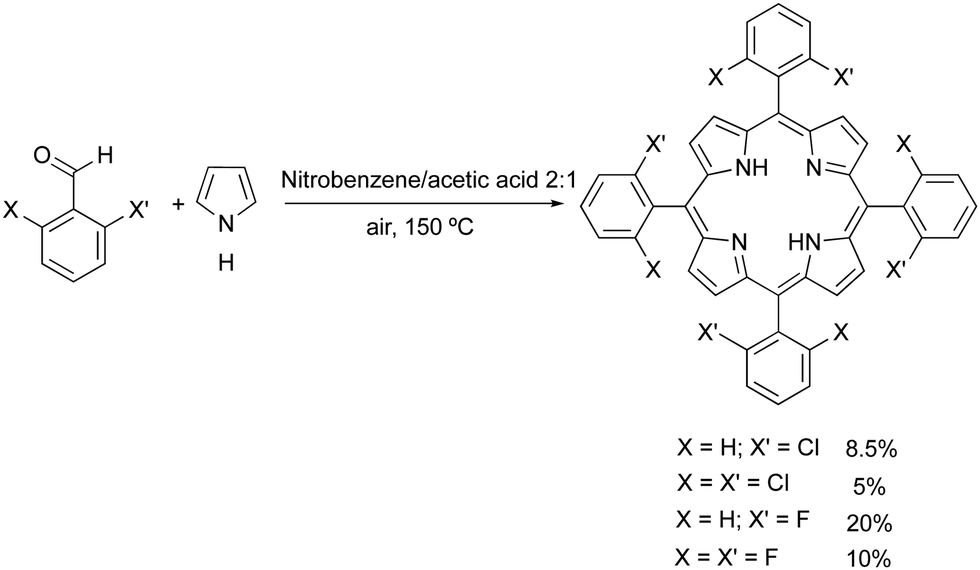 | ||
| Scheme 2 Synthesis of halogenated meso-arylporphyrins. | ||
Pursuing the original idea that fluorine or chlorine atoms in the ortho position of arylporphyrin derivatives should stabilize the structures against oxidation, we optimized the synthesis of the chlorins and bacteriochlorins through small modifications of the Withlock's method.37 This method uses p-toluenesulfonyl hydrazide, dimethylformamide (DMF) or pyridine and a base, to promote the reduction of one or two double bonds of beta pyrrolic porphyrin rings. This method always led to a complex mixture of compounds and the purification of the desired chlorin or bacteriochlorin was only possible using preparative TLC. Nevertheless, it was possible to isolate the desired chlorins27 and bacteriochorins,28 proceed with their photochemical and photophysical characterization, and demonstrate that the design principles indeed offered stable bacteriochlorins, with long-lived triplet states and high singlet oxygen quantum yields.
Although it was quite rewarding to validate the design principles, the bacteriochlorins obtained did not contain functionalities that would make them useful for PDT. In particular, they were insoluble in biocompatible solvents. The next advance had to be the introduction of polar groups in halogenated bacteriochlorins. We had abundant choices and limited resources. The choices included, for example, hydroxyl groups that proved very effective in Foscan®,38 carbonyl, ester and carboxy groups present in bacteriopheophytins, or polar amino acid residues.21 Our attention was captured by the interesting results obtained with sulfonamide porphyrins in PDT,39 and focused on the development of a method to obtain halogenated sulfonamide32 and sulfoester33 porphyrins that could be subsequently reduced to the corresponding bacteriochlorins.
Chlorosulfonation of tetraarylporphyrins proceeds in high yields and offers a variety of amphiphilic porphyrins.40 Temperature is an important variable. Chlorosulfonation of meso-tetraphenylporphyrin (TPP) could be performed at room temperature, but TDFPP and TDCPP required higher temperatures because halogens deactivate the phenyl group towards electrophilic substitution reactions. After making appropriate adjustments, we were able to prepare a diverse range of sulfonamides and sulfoesters, as shown in Fig. 2. After work-up optimization, a portfolio of amphiphilic porphyrins with a wide range of lipophilicities were isolated, as demonstrated by n-octanol/water partition coefficients with logarithmic values (logPOW) from −2.7 to higher than 4.32
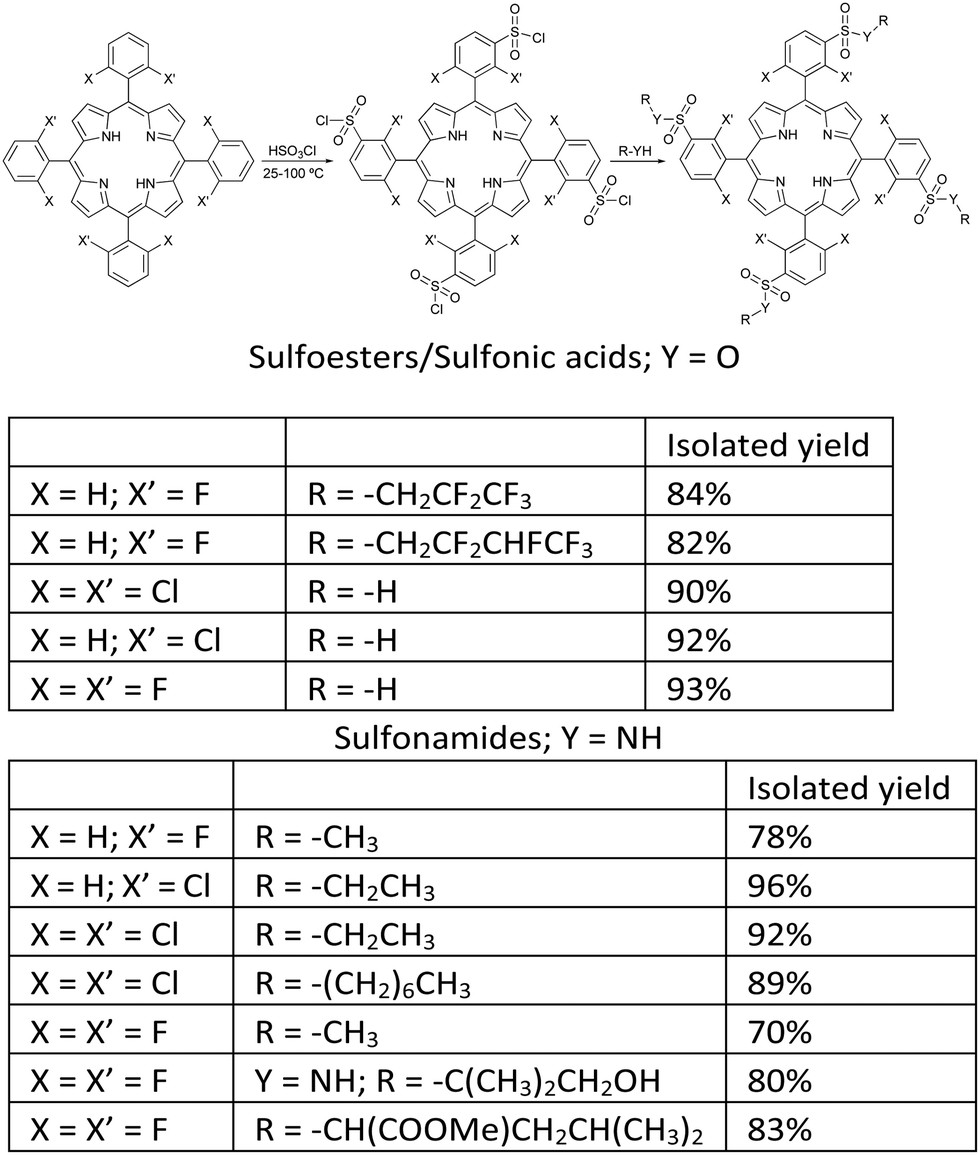 | ||
| Fig. 2 Synthesis of halogenated amphiphilic sulfonamide and sulfoester derivatives.32 | ||
The reduction of this family of porphyrins using p-toluenesulfonyl hydrazide and 1,8-diazabicyclo(5.4.0)undec-7-ene (DBU) as a hindered non-nucleophilic base, in DMF at 150 °C for approximately 24 hours, under a strictly inert atmosphere, yielded stable amphiphilic bacteriochlorin photosensitizers (Fig. 3, Path A).41,42 However, using this synthetic strategy we found that isolated yields of bacteriochlorins were difficult to reproduce and were challenged by the level of purity required for pharmaceutical drug development.
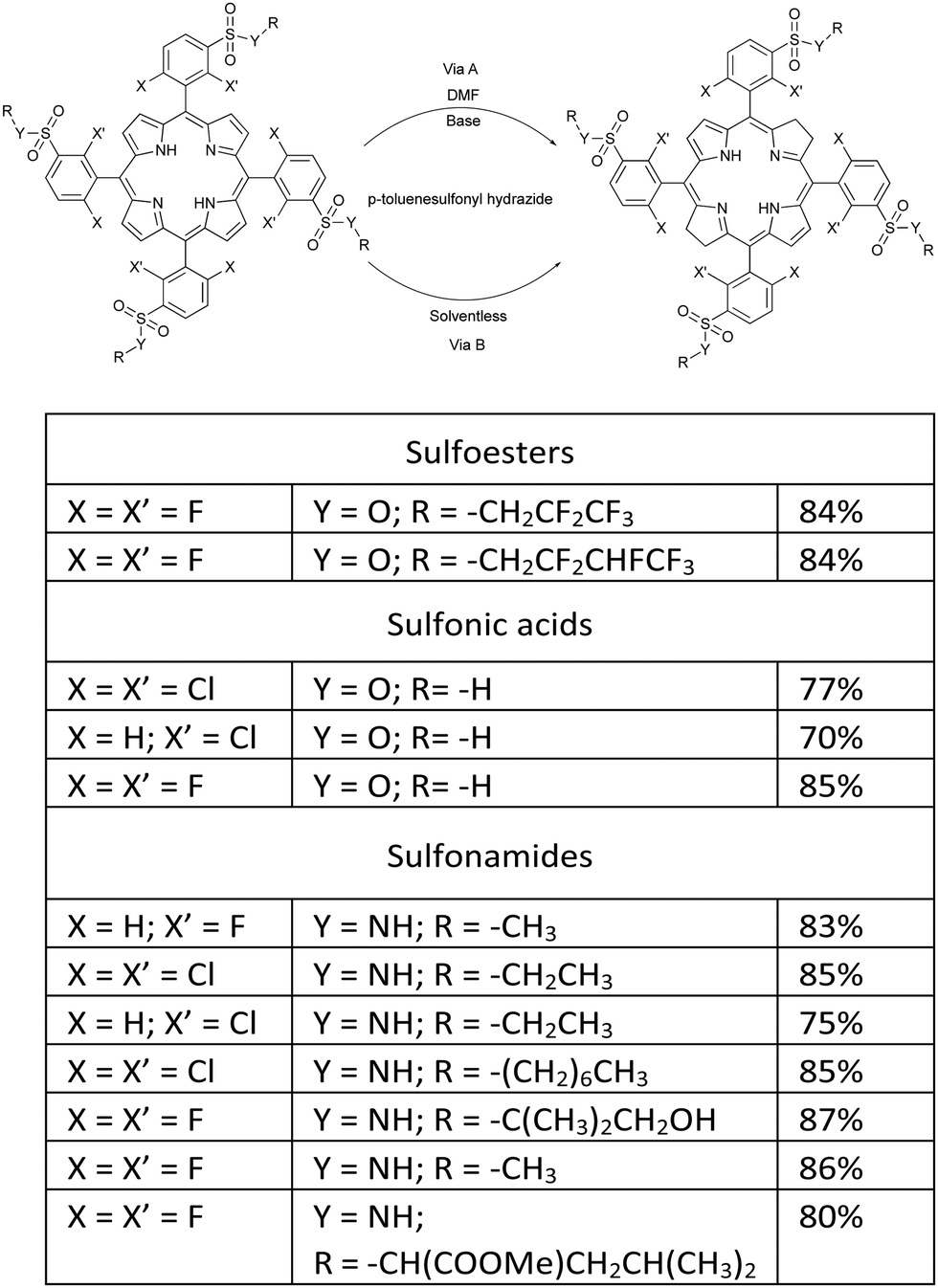 | ||
| Fig. 3 Synthetic methodologies for preparing amphiphilic bacteriochlorins through DMF/Base (Path A) and solventless (Path B) routes.41,42 | ||
Critical analysis of the complex mixture of products commonly obtained with the hydrazide/DMF/base methodology led us to hypothesize that, in 24 h in solution, isomerizations between chlorin and phlorin forms (first reduction products) and also between bacteriochlorin and isobacteriochlorin forms (second reduction products) occur in competition with the formation of bacteriochlorin, Scheme 3.
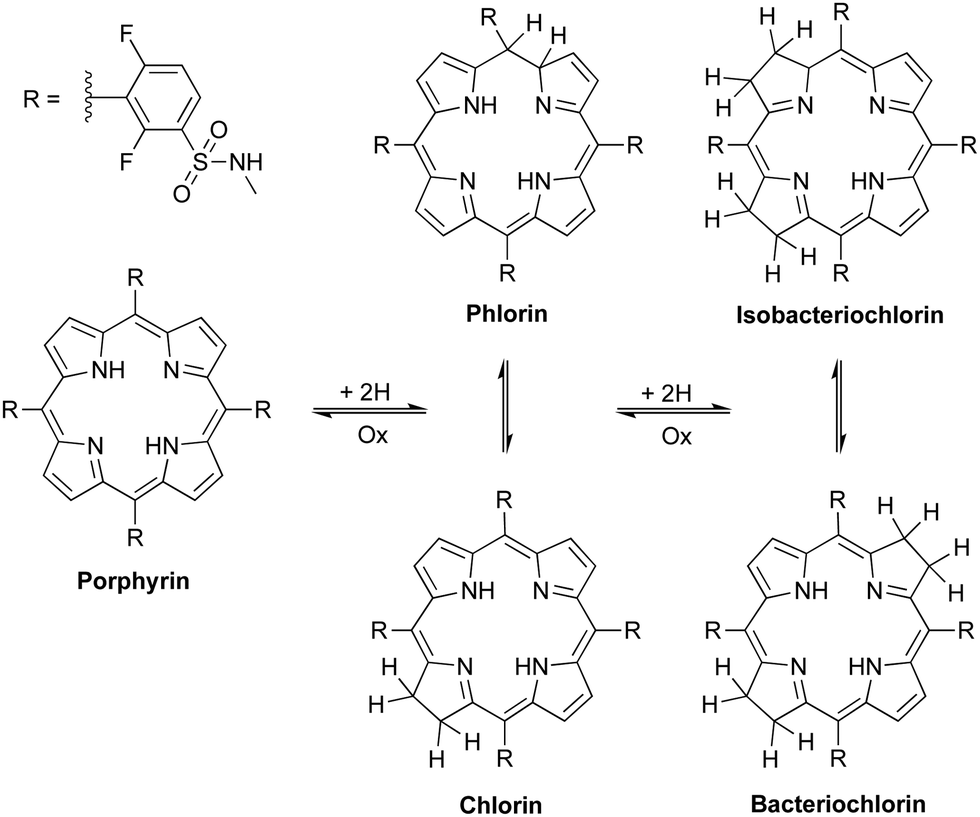 | ||
| Scheme 3 Proposed oxidation/reduction equilibria between porphyrin and its reduced forms: chlorin, phlorin, bacteriochlorin and isobacteriochlorin. | ||
In order to avoid a mixture of unreacted porphyrins, single reduction phlorins and chlorins, double reduction isobacteriochlorins and bacteriochlorins, and possibly triple reduction hexahydroporphyrins, obtained in Path A with the hydrazide/DMF/base methodology, we embarked on a systematic study to optimize the reaction conditions (time, temperature, concentration, solvent and base). In the course of these studies we had our serendipitous encounter. One day, a student instructed with testing a given experimental condition left the reduction reaction overnight, in DMF under an inert atmosphere, at 160 °C, without appropriate cooling of the condenser. DMF evaporated completely and a solid was found the next day. After recalling the safety rules of lab work to the student, rather than trashing the result and make the student run the experiment correctly, we proceeded with a detailed analysis of the product. Unexpectedly, the solid contained bacteriochlorin with an unprecedented level of purity. Porphyrin reduction was more selective in the absence of solvent, possibly because this avoided equilibria between the isomeric species depicted in Scheme 3. Paraphrasing Louis Pasteur: in the fields of observation chance favours only the prepared mind.
Following this observation, we carried out a systematic survey of porphyrin reduction with hydrazide in the total absence of oxygen, solvent and base. We found that as hydrazide melts at temperatures above 100 °C it dissolves the porphyrin, and at the same time decomposes and generates the diimide, which irreversibly reduces the porphyrin and produces the desired bacteriochlorin in high yield and purity (Path B in Fig. 3). Thermomicroscopy corroborated the mechanism of 2+2 cis-diimide reduction of the opposite porphyrin double bonds with release of N2 (Scheme 4).43
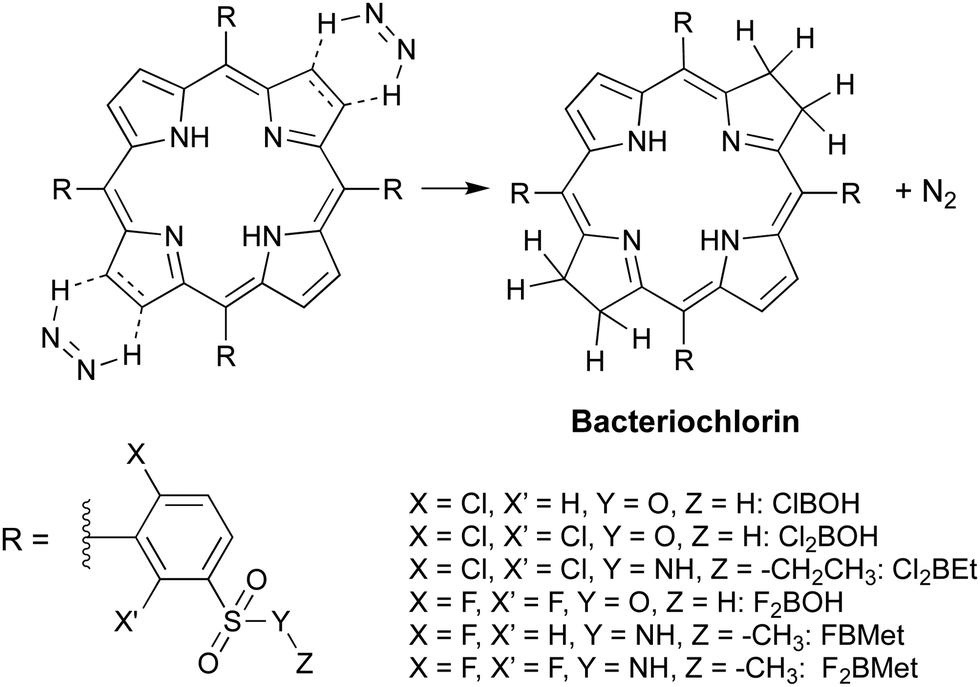 | ||
| Scheme 4 Diimide cycloaddition reduction of porphyrins to bacteriochlorins in the absence of solvent. | ||
Serendipity opened the path to a sustainable and reproductible synthetic method to synthesize a variety of stable meso-aryl bacteriochlorins with adequate purity for PDT.44,45 Meanwhile, we had access to sufficient quantities of halogenated sulfonamide bacteriochlorins to evaluate their properties and make a preliminary assessment of their efficacy.42,46 The results were very promising and in line with those obtained with the corresponding porphyrins and chlorins.47,48 At this time we were approached by a colleague of the Faculty of Pharmacy and Vice-President of a pharmaceutical company – Sérgio Simões – to start a company and contribute to studies that would identify the drug candidate and enable the approval of a clinical trial with the selected halogenated sulfonamide bacteriochlorin. The company, named Luzitin SA, started these activities in 2010 (Fig. 4). In principle, sulfonamide, sulfonic acid and sulfoester derivatives were considered, but the fast hydrolysis of the latter in biological media rapidly excluded this series of derivatives from the list.33
 | ||
| Fig. 4 Inauguration of Luzitin SA, with the Rector of the University of Coimbra speaking and Sérgio Simões sitting first on the left. Adapted with permission from the Luzitin SA photographic archive. | ||
3. Preclinical studies
The preclinical development of halogenated sulfonamide bacteriochlorins is an interesting example of a medicinal chemistry program carried out in collaboration between a start-up company and an academic laboratory. In three years, all preclinical studies were completed and the investigational medicinal product dossier was submitted by Luzitin to the regulatory agency in Portugal (INFARMED). Early in January 2014 INFARMED approved the clinical trial protocol with the drug candidate (LUZ11, alias redaporfin, or F2BMet), the first batch of the medicinal product was released for clinical trial in February 2014 and the first patient was treated in May 2014. Meanwhile, given the nature of PDT as a drug-device combination, a laser device had to be developed. The initial studies were made with a prototype, but in 2013 an agreement with Omicron-Laserage GmbH was reached to develop a laser with medical CE certification for PDT. One of the authors of this work collaborated with Omicron in that development, which was lengthier than expected, and the medical device CE mark was only obtained in February 2017.Preclinical studies aiming at regulatory approval have to follow strict guidelines, such as Administration-Distribution-Metabolism-Elimination-Toxicology (ADME-Tox) studies in GLP (Good Laboratory Practices) and synthesis in GMP (Good Manufacturing Practices). It is beyond the scope of this work to cover all of these studies. Below we discuss briefly the main tasks of drug development that require more input from medicinal chemistry.
The first task, even before the actual preclinical studies, was to assess the range of properties of the halogenated sulfonamide or sufonic acid bacteriochlorin derivatives synthesized in the programme, and to select those to be fully characterized as PDT photosensitizers. Eleven bacteriochlorins were available with high purity. Table 1 presents some of the data collected to evaluate the bacteriochlorins. Sulfonated derivatives are water soluble, which is very convenient, but they are much less photostable than the corresponding sulfonamide derivatives.
The most common strategy to select a development candidate in medicinal chemistry is to establish structure–activity relationships (SAR) using a group of compounds which have a chemical motif in common and different chemical groups bond to this core structure that impart different potencies.50 The compounds in Table 1 may be regarded as an incipient library of sulfonated bacteriochlorins that could be a building block of a SAR. However, the role of photosensitizers in PDT is to interact with light and molecular oxygen to produce ROS. This is very different from a conventional drug that interacts with a specific biological target. Considering that singlet oxygen is the most important ROS in PDT, one could naively expect that the bacteriochlorin dose required to kill 90% of the cells in vitro under a constant light dose (LD90) should be correlated with ΦΔ. Table 1 shows that this kind of correlation does not exist. In particular, F2BMet (i.e., redaporfin) has one of the lowest ΦΔ and is the most potent photosensitizer. The comparison between F2BMet and Cl2BEt is particularly striking because they have similar lipophilicities and similar Φpd but Cl2BEt has a higher ΦΔ and a lower potency than F2BMet. These intriguing relationships motivated mechanistic studies on the generation of ROS.
ROS can be generated either by energy or electron transfer from the electronically-excited photosensitizer to molecular oxygen, or by electron or hydrogen transfer to substrate molecules that subsequently react with molecular oxygen and generate ROS. A distinct feature of the “photodynamic” effect is that molecular oxygen is involved in the generation of reactive species. It became common to designate as Type II the process of energy transfer that generates singlet oxygen, and as Type I the electron transfer and hydrogen transfer processes. Interestingly, this is not the definition proposed by Christopher Foote,51 who first discovered the chemical mechanisms of photodynamic action. His recommended definition is that Type II should refer to photochemical (energy or electron transfer) reactions of the photosensitizer with molecular oxygen, whereas Type I reactions should refer to photochemical reactions of the photosensitizer with substrate molecules (excluding molecular oxygen). We use the common designation of Type I and Type II processes rather than that recommended by Foote. Hence, ΦΔ in Table 1 is associated with Type II processes.
Most photosensitizers employed in PDT make use of what we are calling the Type II (energy transfer) process. Energy transfer from the triplet state of the photosensitizer to ground-state molecular oxygen generates singlet oxygen and leaves the photosensitizer in the ground state. The introduction of chlorine atoms in the ortho positions of tetraphenylporphyrins and tetraphenylchlorins enhances their spin–orbit coupling and increases triplet quantum yields (ΦT), which in these molecules reach ΦT = 1 and enable ΦΔ ≈ 1.26,27 We were puzzled to find that the same substituents in tetraphenylbacteriochlorins did not increase ΦΔ above 0.6, although ΦT seemed to approach unity and the rate of reaction between the bacteriochlorin triplet state and molecular oxygen was faster than the analogous reaction of the porphyrin triplet state.28 The inverse correlation between the rates and efficiencies of singlet-oxygen generation is the fingerprint of charge-transfer-induced quenching.52 The observation of this inverse correlation led to the hypothesis that bacteriochlorins could lead to the full charge transfer species while interacting with molecular oxygen, which should be the superoxide ion. Using appropriate spin traps, it was possible to obtain EPR spectra that demonstrate the generation of superoxide ion in DMSO and of the hydroxyl radical in PBS.46 Using fluorescent probes, we could detect the intracellular generation of the hydroxyl radical in PDT with bacteriochlorins and show that this ROS is an important contributor to their phototoxicity.53 The “pluses” in Table 1 indicate the degree in which hydroxyl radicals were observed in PDT with the corresponding bacteriochlorins. The oxidation potentials of sulfonamide halogenated bacteriochlorins seem to be just sufficiently low to allow for electron transfer to molecular oxygen and generation of superoxide ion, and yet sufficiently high to have photostable photosensitizers.
Testing ClBOH48 and F2BOH49in vivo revealed that these water-soluble bacteriochlorins could give good results for short DLI when the photosensitizer is mostly in the vascular compartment (V-PDT), but treatment efficacies were less promising for longer DLIs. Chlorine atoms seemed to increase ΦΔ with respect to fluorine atoms, as expected from the heavy-atom effect on spin–orbit coupling and acceleration of intersystem crossing rates. However, this did not improve in vitro PDT efficacy. Cl2BEt was tested in vivo and shown to perform better than ClBOH at DLI = 24 h.54 Cl2BHep was very lipophilic and required the development of a specialized formulation, namely the encapsulation in pluronic poloxamers micelles. With this formulation, PDT with Cl2BHep was very effective at long DLI.55 F2BMet (alias LUZ11, alias redaporfin) showed good photostability, amphiphilicity and high efficacy in vitro. Preliminary in vivo studies showed great promise and this photosensitizer was selected for further formulation development56 and detailed in vivo studies.57,58 The reasons for the success of F2BMet became clear more recently. They include (i) photostability, (ii) ability to generate various types of ROS, (iii) subcellular localization in the endoplasmic reticulum, Golgi apparatus and, in part, mitochondria, and (iv) aptitude to cross cell membranes.
Having identified redaporfin as the development candidate, its large-scale synthesis became an immediate priority. A method to produce kilogram batches of redaporfin was developed and its synthesis without solvent, under GMP conditions, was outsourced. A 0.5 kg batch was prepared and released for the clinical studies (Fig. 5).
 | ||
| Fig. 5 The redaporfin batch prepared in GMP. Adapted with permission from the Luzitin SA photographic archive. | ||
A very significant part of the in vivo studies with bacteriochlorins was performed by Janusz Dabrowski as a post-doc in the authors laboratories between 2010 and 2012. His work included the development of formulations for redaporfin,56,59 and a very detailed optimization of the treatment protocol,58 which served as the basis for the clinical protocol. Redaporfin gave the most promising results in vascular-PDT (DLI = 15 min) although at DLI = 72 h it also elicited cures in BALB/c mice bearing subcutaneous CT26 tumours. The optimization of redaporfin-PDT showed that a drug dose of 0.75 mg kg−1 combined with a radiant exposure of 50 J cm−2 (130 mW cm−2 and 750 nm) at DLI = 15 min reproducibly gave ∼85% cure rates in the tumour model presented above. Based on these results, the clinical trial protocol was designed to include a dose-finding phase using a fixed light dose of 50 J cm−2, 130 mW cm−2 at 749 nm, DLI = 15 min and ascending redaporfin doses. The initial dose was a subject of much controversy with INFARMED experts, and we had to include the following dose escalations: 0.05, 0.1, 0.25, 0.5, 0.75 and 1.0 mg kg−1. The doses 0.05–0.5 mg kg−1 did not produce the desired depth of necrosis but the 0.75 mg kg−1 dose produced a necrosis depth larger than 5 mm and was considered to be the effective dose. The plasma half-live of redaporfin was determined to be 19 h.60
The perfect agreement between the effective clinical dose and the dose anticipated in preclinical studies attests for the relevance and thoroughness of the preclinical studies. However, it must be recognized that the choice of vascular-PDT facilitated this agreement. Most rewarding was the treatment in 2016 of a patient in supportive care with an extensive tumour in the mouth pavement progressing after surgery, radiotherapy and multiple lines of systemic treatment. Redaporfin-PDT in the doses described above led to the destruction of all visible tumour. Subsequently, the patient became eligible for immunotherapy and the use of an immune checkpoint blocker allowed for a sustained complete response.61 This response is still lasting at the time of this writing.
4. Drug development in the evolving landscape of innovative medicines
Internal and external factors, often unpredictable, add uncertainty to drug development. The most surprising internal factor that influenced the development of redaporfin was the presence of separable atropisomers. We were aware that atropisomers were likely present in redaporfin and discussed the issue with INFARMED when seeking regulatory advice. However, our perspective was that different atropisomers should have similar interactions with molecular oxygen, produce equivalent oxidative stress and have similar PDT efficacies. Nevertheless, we dedicated substantial efforts to the separation and characterization of the atropisomers present in redaporfin (Scheme 5).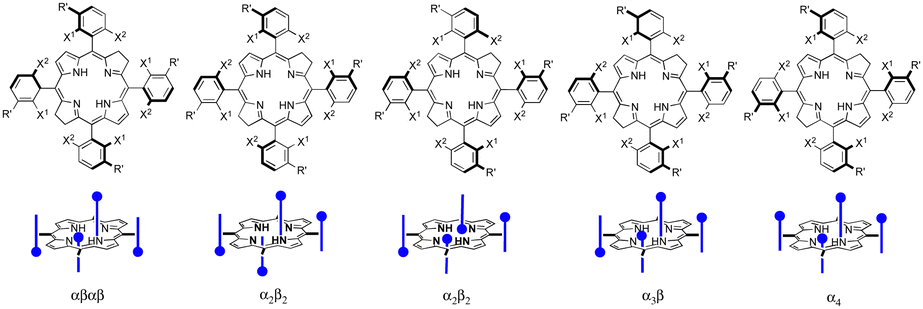 | ||
| Scheme 5 Redaporfin atropisomers. | ||
Bulky substituents in ortho positions of phenyl rings hinder the rotation of the phenyl-macrocycle single bond and allow for the separation of atropisomers at room temperature.62 In fact, this was at the origin of the separation of porphyrin atropisomers named picket-fence porphyrins, which were investigated as photosensitizers for PDT.63 Working with a mixture of atropisomers is not necessarily a major concern because the best-selling photosensitizers are mixtures: Photofrin® is a mixture of porphyrins and Visudyne® is a mixture of regioisomers of benzoporphyrin derivative monoacid ring A with similar phototoxicities,64 and each consists of a racemic mixture of two enantiomers with similar pharmacological activities.65
All redaporfin atropisomers have very similar absorption spectra and similar fluorescence, photostability and singlet oxygen quantum yields. Their logPOW are also similar. However, chromatographic separation showed an intriguing behaviour. The order of elution of the four atropisomers (αβαβ, α2β2, α3β, and α4) was the same in TLC using standard silica gel, in silica gel column chromatography, in reverse phase (C18) silica gel column chromatography and in reverse phase HPLC. Normally, a more polar atropisomer should have a higher retention in silica gel and lower retention in reverse phase silica gel. We were able to obtain the crystal structure of the atropisomer with the highest retention and identified it as atropisomer α4. Atropisomer α2β2 was identified thanks to a very well separated HPLC chromatogram exhibiting the split of the chromatographic peak in two, which were assigned to a species with two sulfonamide groups on the same side of the bacteriochlorin macrocycle separated by a methine (![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C–) group and species with such sulfonamide groups separated by a methylene (–CH2–) bridge (Scheme 5). In order to assign atropisomer α3β we synthesised the corresponding chlorin, which has only one methylene (–CH2–) bridge, and separates α3β into two peaks corresponding to a species with sulfonamide groups on both sides of the reduced pyrrole group and on the same side of the macrocycle plane, and a species with sulfonamide groups on both sides of the reduced pyrrole group but on opposite sides of the macrocycle plane. Atropisomer αβαβ had to be the remaining atropisomer.66
C–) group and species with such sulfonamide groups separated by a methylene (–CH2–) bridge (Scheme 5). In order to assign atropisomer α3β we synthesised the corresponding chlorin, which has only one methylene (–CH2–) bridge, and separates α3β into two peaks corresponding to a species with sulfonamide groups on both sides of the reduced pyrrole group and on the same side of the macrocycle plane, and a species with sulfonamide groups on both sides of the reduced pyrrole group but on opposite sides of the macrocycle plane. Atropisomer αβαβ had to be the remaining atropisomer.66
The separated and assigned atropisomers of redaporfin were then tested in vitro and in vivo. We were shocked to see that the atropisomer doses required to kill 90% of CT26 cells with a light dose of 1 J cm−2, were: αβαβ 2832 μM, α2β2 7.5 μM, α3β 2.6 μM, and α4 0.5 μM (data for αβαβ and α2β2 were extrapolated).66 The phototoxicities spanned over three orders of magnitude for photosensitizers with very similar properties. In vascular-PDT the differences were less dramatic.
The underlying mechanism that enables atropisomers to have different phototoxicities was only uncovered very recently, with an important contribution from Ligia Gomes-da-Silva working in our research unit. Sub-cellular localization studies showed that all atropisomers had similar preferences for membranous organelles (endoplasmic reticulum, Golgi, mitochondria) but the fluorescence intensities were very different. Detailed studies demonstrated that the rates of passive diffusion through the cell membrane followed the order: α4 > α3β > α2β2 > αβαβ. The difference in phototoxicity was unambiguously related to the amount of atropisomer in the cell.67 This is consistent with the in vitro phototoxicities shown in Table 1. Atropisomer α4 has all the polar groups on the same side of the macrocycle. This imparts a special amphiphilic character to this atropisomer. A bind-flip mechanism was proposed to explain its special ability to permeate cell membranes: α4 has enhanced hydrogen-bond interactions between all its sulfonamide groups and the phospholipids of the surface of the membrane that leave the apolar side of the macrocycle exposed to water, and this promotes its flip into the membrane to adopt the orientation of a surfactant (Fig. 6).
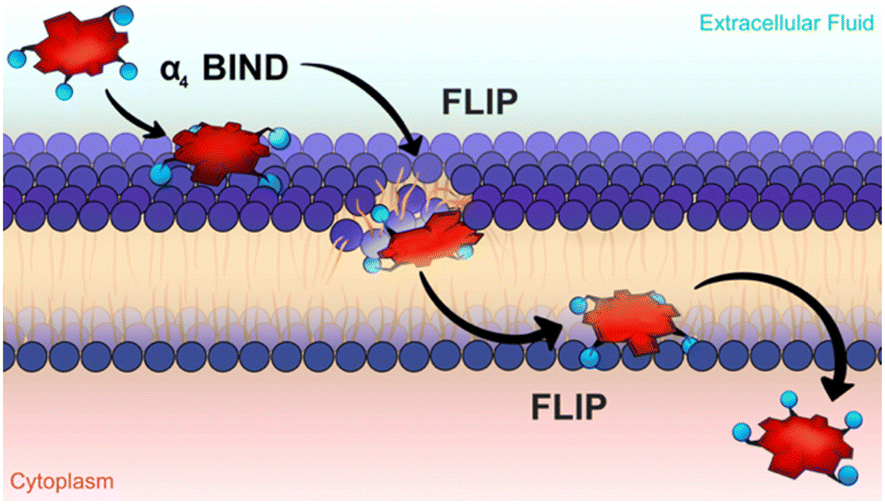 | ||
| Fig. 6 The bind-flip mechanism used by redaporfin α4 atropisomer to cross cell membranes. Courtesy of Ana Mata. | ||
Redaporfin is a macromolecule (molecular weight 1135 Da). Appropriate orientation of sulfonamide groups seems to be an interesting approach to increase passive delivery of macromolecules through cell membranes.68 This amphiphilicity can also explain the higher retention of atropisomer α4 in silica gel and in reverse phase (C18) silica gel column chromatography.
The external factor with the strongest implications in redaporfin development was the emergence of immunotherapies, in particular the clinical approval of immune-checkpoint blockade (ICB) therapies for a wide range of cancers. Negative regulators of immune activation (immune checkpoints) can be blocked by antibodies, which restore T cell proliferation and reinvigorate their antitumour functions. The most studied immunoreceptors in cancer are CTLA-4 (cytotoxic T-lymphocyte-associated protein 4) and PD-1 (programmed cell death protein 1). FDA approved the first CTLA-4 blocking antibody (ipilimumab) for the treatment of melanoma in 2011,69 and various PD-1 blocking antibodies were approved in 2014 and in subsequent years.70 In 2013, Science named immunotherapy its Breakthrough of the Year. As of that time, the development of any anticancer drug had to be positioned with respect to ICB therapies.
Clinical reports showing that PDT has an effect on cancer cells outside the field of illumination are very rare.71,72 However, already in 1996 it was shown that PDT with Photofrin® gave much lower sustained responses in nude mice, who have impaired T-cell functions, than in immunocompetent mice.73 We obtained similar results with redaporfin-PDT. Additionally, we observed systemic effects in a pseudo-metastatic mouse model with subcutaneous CT26 tumours and lung tumours induced by i.v. administration of CT26 cells.58 The curiosity about immune stimulation with PDT and the interest to explore virtuous combinations between PDT and ICB therapies, led one of us, together with Ligia Gomes-da-Silva, to embark on a journey that left behind the tenets of chemistry. The chances of success were heightened by a collaboration with Guido Kroemer, who had identified mechanisms of immunogenic cell death (ICD).74 The fundamental principle of ICD is that, under specific circumstances, dying cells release or expose at the surface, death-associated molecules that act as a combinatorial code to unlock distinct inflammatory and immune responses.75
A study of the mechanisms through which redaporfin-PDT kills cancer cells showed that the earliest events were oxidative damage of structures in the endoplasmic reticulum and Golgi apparatus, which were relayed to mitochondria through the intrinsic pathway of apoptosis. Redaporfin-PDT in vitro induced the typical hallmarks of ICD, namely, plasma membrane calreticulin exposure, release of adenosine triphosphate (ATP) and of high mobility group box 1 (HMGB1) protein, and the phosphorylation of eukaryotic initiation factor 2α (eIF2α).76 Conclusive evidence that redaporfin-PDT induces ICD in vitro was obtained injecting dead/dying TC1 lung cancer cells, previously treated with redaporfin-PDT, in immunocompetent mice followed by rechallenge with live/untreated TC1 cells one week later. More than half of the “vaccinated” mice rejected the tumour cells and all the remaining mice showed slower tumour growth kinetics.76 In order to connect ICD with long-term antitumour responses, we evaluated the impact of depleting neutrophils, CD4+ and CD8+ lymphocytes with appropriate monoclonal antibodies.77 We found that neutrophilia, and the underlying acute local inflammation, was important to mounting the antitumour immunity response. This is in line with previous reports linking neutrophil infiltration in the treated tumour bed with the stimulation of T-cell proliferation.78 Moreover, the depletion of CD8+, but not CD4+, lymphocytes reduced the cure rates by half. Cytotoxic CD8+ T cells (cytotoxic T lymphocytes, CTLs), of the adaptive immune system, are the most powerful effectors of anticancer immune responses.79 In fact, the role of ICBs is to inhibit suppressive immune receptors and revitalize dysfunctional CD8+ T cells. Fig. 7 schematically illustrates the various processes observed after redaporfin-PDT that relate to the stimulation of immune responses.
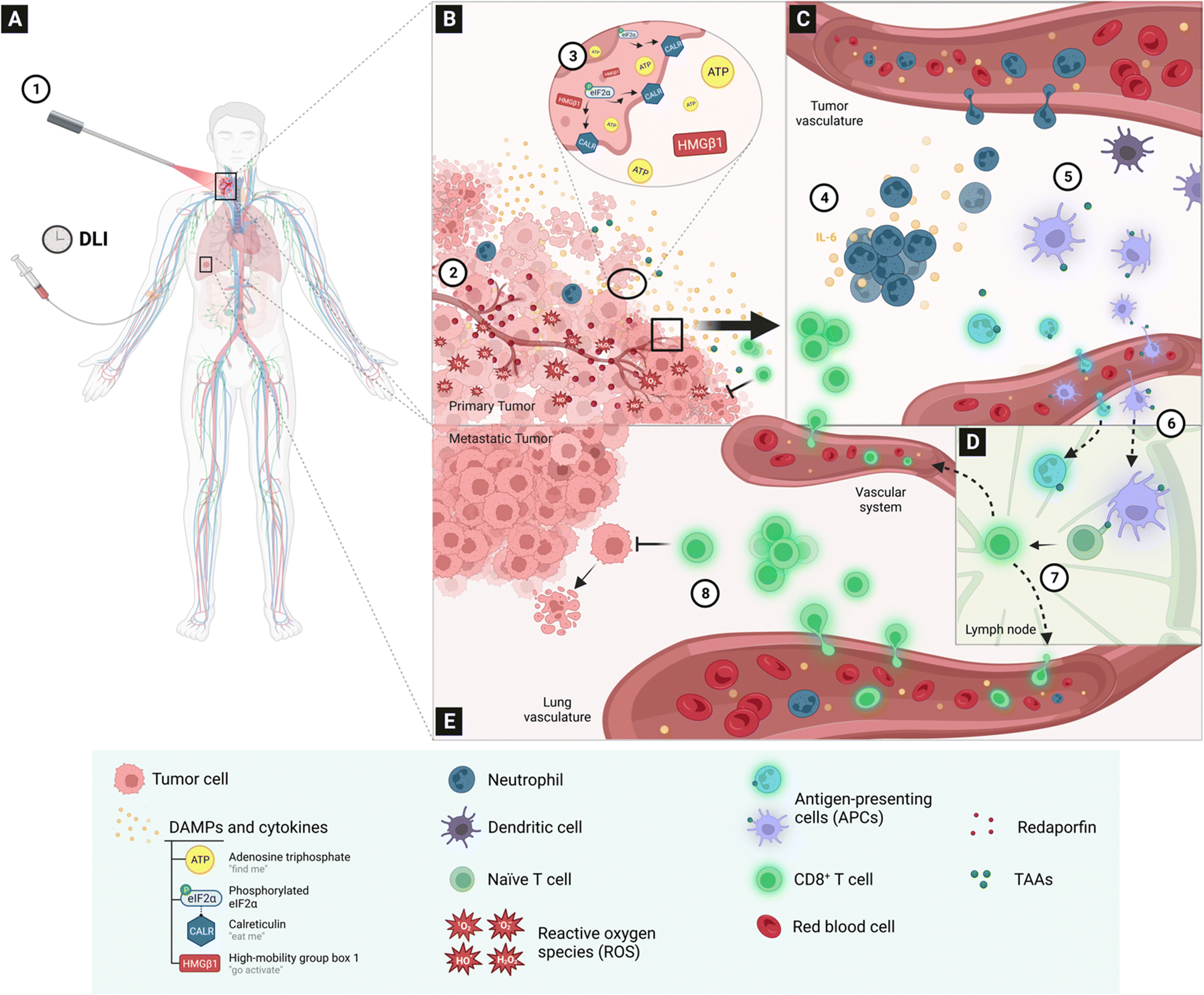 | ||
| Fig. 7 Redaporfin-PDT and the activation of the immune system, according to existing data. (A) Intravenous redaporfin administration followed, after a given drug-to-light interval (DLI), by laser (1) illumination of the primary tumour in a patient additionally having metastasis in the lungs. (B) The absorption of light by redaporfin, located in the endoplasmic reticulum, Golgi apparatus and mitochondria of tumour cells, generates reactive oxygen species (1O2, O2˙−, H2O2, OH˙) that produce oxidative stress (2); the damage caused to tumour cells triggers a spatial-temporal controlled release of specific damage-associated molecular patterns (DAMPs), namely the release of APT by the mitochondria, CALR relocation and eIF2a phosphorylation in the endoplasmic reticulum, and HMGB1 release from the nucleus (3). (C) Damage to the tumour cell and vessels in the tumour microenvironment leads to the development of a strong and acute inflammatory response within hours of the illumination, with a strong increase in the pro-inflammatory IL-6 cytokine and of neutrophils in the peripheral blood (4); inflammation attracts neutrophils and dendritic cells to the tumour microenvironment (5), where they can capture tumour-associated antigens (TAA). (D) Neutrophils and dendritic cells migrate to the lymph node (6) where they present antigens to naïve T cells, which become cytotoxic tumour-specific CD8+ T cells with co-stimulatory binding (not shown); the cytotoxic CD8+ T cells leave the lymph node (7). Cytotoxic CD8+ T cells infiltrate the tumour bed 24 h post-PDT (B) or may inhibit metastases in the lungs (E). | ||
Although the adoption of ICB therapies revolutionized oncology and the percentage of cancer patients in the United States who were eligible for treatment with ICBs approaches 50%, the percentage of patients with cancer that do not respond to ICBs is more than 87%.80 The increase in activated CD8+ cytotoxic T cells post-PDT and the revitalization of CD8+ T cells with ICB therapies provides a rational for the combination of PDT and ICB therapies. The combination between PDT and immunotherapies has been investigated by various authors and good reviews are available.81–83 Unfortunately, virtuous combinations between the two therapies are hard to achieve because various factors must be taken into consideration.
Checkpoint inhibitors, such as CTLA-4 and PD-1, are essential to maintain self-tolerance and prevent autoimmune diseases. CTLA-4 molecules are contained within intracellular vesicles in naïve T cells. Naïve T cells are activated when their T cell receptors bind to their cognate antigen presented by antigen-presenting cells (APCs) in the presence of a co-stimulatory signal. This co-stimulatory signal is the binding between CD28 expressed on the surface of the T cell with B7 molecules (B7.1, also named CD80, or B7.2, also named CD86) on APCs. APCs are immune cells that process and present antigens for recognition by T cells, and include B lymphocytes, neutrophil, dendritic cells, macrophages and other immune cells. CD28 and CTLA-4 compete for binding to B7-1 and B7-2 on APCs but CTLA-4 binds to B7-1 and B7-2 more tightly and delivers negative rather than costimulatory signals to the T cells. Hence, CTLA-4 counteracts several internal signalling nodes to impede activation and proliferation of T cells,84 and neutralizing anti-CTLA4 monoclonal antibodies enhance antitumoral immunity. Human PD-1 is expressed on T cells after T cell receptor stimulation, and binds to the B7 homologues PD-L1 and PD-L2, which are constitutively expressed on APCs and can be induced in non-hematopoietic tissues. PD-L1 (programmed cell death ligand 1, also known as B7-H1), is a transmembrane protein that down-regulates immune responses through binding to its two inhibitory receptors PD-1 and B7.1 (CD80), and is present on many cell types, including T cells, tumour cells, epithelial cells and endothelial cells, much more frequently than PD-L2 (programmed cell death ligand 2, also known as B7-DC). PD-1 restrains immune responses primarily through inhibitory signalling in CD8+ effector T cells.85 When PD-1 engages its ligands, it can induce a state of T cell dysfunction called T cell exhaustion. Tumour cells can upregulate PD-1 ligands. As the PD-1/PD-L1 pathway protects cells from T cell attack, anti-PD-1 and anti-PD-L1 antibodies can enhance the functional properties of CD8+ effector T cells at the tumour site. Activation of T cells allows T cell lymphocytes to recognize an antigen on a specific target cell. Activated CD8+ T cells gradually transform into cytotoxic T lymphocytes (CTLs), which recognize target cells and kill them.
We recently showed that redaporfin-PDT may increase CTLA-4 and PD-L1 expressions in some cell lines.86 This adds a challenge to ICB therapy because an increase in immune checkpoint expression may exhaust the ICBs administered and dampen their effect. However, we also showed that redaporfin-PDT also increases the expression of co-stimulatory B7 (CD80) molecules. Virtuous combination between PDT and anti-CTLA-4 or anti-PD-1 monoclonal antibodies are expected for cell lines with high B7/PD-L1 or B7/CTLA-4 overexpression ratios. We found that that anti-CTLA-4 monoclonal antibodies combined favourably with redaporfin-PDT in the treatment of sub-cutaneous CT26 colon tumours and in the control of lung metastasis in orthotopic 4T1 breast tumours.86 The clinical case of advanced head and neck cancer discussed above is a combination between redaporfin-PDT and an anti-PD-1 monoclonal antibody.61
The chemistry of halogenated arylbacteriochlorins is now reasonably well understood. In science a good understanding of a system often offers firm grounds to elaborate new systems. Several examples illustrate this in the context of the development of redaporfin. We used the knowledge on halogenated arylbacteriochlorins to explore a wide diversity of fluorinated bacteriochlorins and different applications.87 Spingler and co-workers developed halogenated arylbacteriochlorins to include platinated pyridyl substituents and kill 50% of HeLa cells in vitro with 6 nM and 5 J cm−2 at 750 nm.88 The Brückner group tested related chromophores as contrast agents in photoacoustic imaging.89 Senge and co-workers synthesised strapped porphyrin to show that cis-αα atropisomers have increased cell internalizations relative to other atropisomers, confirming our findings on the dependence of cell uptake on amphiphilicity.68
5. Conclusions
There are more than ten thousand interventional clinical trials on cancer recruiting patients at this time. A survey of PDT clinical trials on cancer, covering the period between March 2013 and March 2023, identified 174 studies, excluding withdraws.90 In 2020, the estimated number of cancer deaths worldwide reached 10 million for the first time.91 The discovery and development of redaporfin is one of many comparable research programmes that contribute to better therapeutic options for patients suffering from devastating diseases. We hope that this and other research programmes will succeed. Although each drug discovery and development programme will have its own specificities, a few lessons learnt with the studies reviewed in this work may be of general interest.Base your work on the best fundamental science
Our expertise in the synthesis of tetrapyrrolic macrocycles and on photoinduced energy and electron transfer reactions served us many times to overcome problems in the preparation of photosensitizers and in the interpretation of results.Expect the unexpected
The solid-state synthesis of bacteriochlorins and the differential phototoxicities of redaporfin atropisomers were unexpected. However, they were not overlooked. Great efforts were made to understand the stories concealed in the data from the synthesis and from the atropisomers, and better synthetic methods and photosensitizers emerged.Collaborate
Two long-term collaborators were named, but the work on redaporfin involved nearly 100 co-authorships, many of them with senior researchers in other fields.Accept leaving comfort zones
Drug development is a multidisciplinary endeavour. It is impossible to become a specialist in every field but the tacit knowledge acquired in the early stages of the process is difficult to share. Drug development needs “champions” that accept leaving their comfort zones and work across the boundaries of scientific domains. We experienced detours from our comfort zones in the gram-scale manufacture of GMP batches and in the study of immune responses to PDT. Appropriately accompanied by specialists, these detours are very rewarding learning experiences.Finally, be persistent, very persistent, but also be aware of the thin line separating persistence from stubbornness.
Author contributions
LGA and MMP contributed equally to this work. MMP was responsible for the synthesis and LGA for the other components of this work.Conflicts of interest
The authors have 3 patent families licensed to Luzitin SA.Acknowledgements
We thank the Portuguese Science Foundation for continuous support (ongoing projects ROTEIRO/0152/2013, UIDB/00313/2020, PTDC/QUI-OUT/0303/2021) and the European Regional Development Fund (P2020 and CENTRO2020, reference CENTRO-01-0145-FEDER-181218).Notes and references
- I. J. MacDonald and T. J. Dougherty, J. Porphyrins phthalocyanines, 2001, 5, 105–129 CrossRef CAS
.
- E. D. Sternberg and D. Dolphin, Tetrahedron, 1998, 54, 4151–4202 CrossRef CAS
- R. Bonnett, Chem. Soc. Rev., 1995, 24, 19–33 RSC
- S. G. Bown, World J. Surg., 1983, 7, 700–709 CrossRef CAS PubMed
- D. Kessel, J. Clin. Med., 2019, 8, 1581 CrossRef CAS PubMed
- M. R. Hamblin, Photochem. Photobiol., 2020, 96, 506–516 CrossRef CAS PubMed
- U. Schmidt-Erfurth and T. Hasan, Survey Ophthalmol., 2000, 45, 195–214 CrossRef CAS PubMed
- P. Nowak-Sliwinska, A. Weiss, M. Sickenberg, A. W. Grioffioen and H. van der Bergh, J. Anal. Bioanal. Tech., 2013, S1, 007 Search PubMed
- C. J. Byrne, L. V. Marshallsay and A. D. Ward, J. Photochem. Photobiol. B: Biol., 1990, 6, 13–27 CrossRef CAS PubMed
- D. A. Bellnier, W. R. Greco, G. M. Loewen, H. Nava, A. R. Oseroff and T. J. Dougherty, Lasers Surg. Med., 2006, 38, 439–444 CrossRef PubMed
- J. M. Dabrowski and L. G. Arnaut, Photochem. Photobiol. Sci., 2015, 14, 1765–1780 CrossRef CAS PubMed
- W. R. Potter, T. S. Mang and T. J. Dougherty, Photochem. Photobiol., 1987, 46, 97–101 CrossRef CAS PubMed
- L. B. Rocha, H. T. Soares, M. I. P. Mendes, A. Cabrita, F. A. Schaberle and L. G. Arnaut, Photochem. Photobiol., 2020, 96, 692–698 CrossRef CAS PubMed
- S. L. Jacques, Surg. Clin. N. Am., 1992, 72, 531–558 CrossRef CAS PubMed
- R. Bonnett and G. Martinez, Tetrahedron, 2001, 57, 9513–9547 CrossRef CAS
-
M. Gouterman, in The Porphyrins, ed. D. Dolphin, Academic Press, New York, 1978, vol. 3, p. 1 Search PubMed
- L. G. Arnaut, Adv. Inorg. Chem., 2011, 63, 187–233 CrossRef CAS
- B. W. Henderson, W. R. Potter, A. Sumlin, B. Owczarczak, F. S. Nowakowski and T. J. Dougherty, Proc. SPIE, 1990, 1203, 211–222 CrossRef CAS
- B. W. Henderson, A. B. Sumlin, B. L. Owczarczak and T. J. Dougherty, J. Photochem. Photobiol. B: Biol., 1991, 10, 303–313 CrossRef CAS PubMed
- R. K. Pandey, F.-Y. Shiau, A. B. Sumlin, T. J. Dougherty and K. M. Smith, Bioorg. Med. Chem. Lett., 1992, 2, 491–496 CrossRef CAS
- V. Rosenbach-Belkin, L. Chen, L. Fiedor, I. Tregub, F. Pavlotsky, V. Brumfeld, Y. Salomon and A. Scherz, Photochem. Photobiol., 1996, 64, 174–181 CrossRef CAS PubMed
- R. Bonnett, R. D. White, U.-J. Winfield and M. C. Berenbaum, Biochem. J., 1989, 261, 277–280 CrossRef CAS PubMed
- K. R. Adams, M. C. Berenbaum, R. Bonnett, A. N. Nizhnik, A. Salgado and M. A. Vallés, J. Chem. Soc. Perkin Trans. 1, 1992, 1465–1470 RSC
- B. Spalding, Nat. Biotechnol., 1993, 11, 648–649 CrossRef
- L. G. Arnaut, M. M. Pereira, J. M. Dabrowski, E. F. Silva, F. A. Schaberle, A. R. Abreu, L. B. Rocha, M. M. Barsan, K. Urbanska, G. Stochel and C. M. Brett, Chem. – Eur. J., 2014, 20, 5346–5357 CrossRef CAS PubMed
- M. Pineiro, A. L. Carvalho, M. M. Pereira, A. M. R. Gonsalves, L. G. Arnaut and S. J. Formosinho, Chem.–Eur. J., 1998, 4, 2299–2307 CrossRef CAS
- M. Pineiro, M. M. Pereira, A. M. A. R. Gonsalves, L. G. Arnaut and S. J. Formosinho, J. Photochem. Photobiol. A: Chem., 2001, 138, 147–157 CrossRef CAS
- M. Pineiro, A. M. A. R. Gonsalves, M. M. Pereira, S. J. Formosinho and L. G. Arnaut, J. Phys. Chem. A, 2002, 106, 3787–3795 CrossRef CAS
- E. G. Azenha, A. C. Serra, M. Pineiro, M. M. Pereira, J. Seixas de Melo, L. G. Arnaut, S. J. Formosinho and A. M. A. R. Gonsalves, Chem. Phys., 2002, 280, 177–190 CrossRef CAS
- A. M. A. Rocha Gonsalves, J. M. T. B. Varejao and M. M. Pereira, J. Heterocycl. Chem., 1991, 28, 635–640 CrossRef
- A. D. Adler, F. R. Longo, J. D. Finarelli, J. Goldmacher, J. Assour and L. Karsakoff, J. Org. Chem., 1967, 32, 476 CrossRef CAS
- C. J. P. Monteiro, M. M. Pereira, S. M. A. Pinto, A. V. C. Simões, G. F. F. Sá, L. G. Arnaut, S. J. Formosinho, S. Simões and M. F. Wyatt, Tetrahedron, 2008, 64, 5132–5138 CrossRef CAS
- A. V. C. Simões, A. Adamowicz, J. M. Dabrowski, M. J. F. Calvete, A. A. Abreu, G. Stochel, L. G. Arnaut and M. M. Pereira, Tetrahedron, 2012, 68, 8767–8772 CrossRef
- M. Silva, A. Fernandes, S. S. Bebiano, M. J. F. Calvete, M. F. Ribeiro, H. D. Burrows and M. M. Pereira, Chem. Commun., 2014, 50, 6571–6573 RSC
- M. J. F. Calvete, L. D. Dias, C. A. Henriques, S. M. A. Pinto, R. M. B. Carrilho and M. M. Pereira, Molecules, 2017, 22, 741 CrossRef PubMed
- C. A. Henriques, S. M. A. Pinto, G. L. B. Aquino, M. Pineiro, M. J. F. Calvete and M. M. Pereira, ChemSusChem, 2014, 7, 2821–2824 CrossRef CAS PubMed
- H. W. Whitlock Jr., R. Hanauer, M. Y. Oester and B. K. Bower, J. Am. Chem. Soc., 1969, 91, 7485–7489 CrossRef
- M. C. Berenbaum, R. Bonnett, E. B. Chevretton, S. L. Akande-Adebakin and M. Ruston, Lasers Med. Sci., 1993, 8, 235–243 CrossRef
- A. J. F. N. Sobral, S. Eleouet, N. Rousset, A. M. D. Gonsalves, O. Le Meur, L. Bourre and T. Patrice, J. Porphyrins phthalocyanines, 2002, 6, 456–462 CrossRef CAS
- A. M. D. R. Gonsalves, R. A. W. Johnstone, M. M. Pereira, A. M. P. deSantAna, A. C. Serra, A. J. F. N. Sobral and P. A. Stocks, Heterocycles, 1996, 43, 829–838 CrossRef
- M. M. Pereira, L. G. Arnaut Moreira, S. J. Formosinho and C. J. P. Monteiro, PCT/EP2005/012212, 2004.
- M. M. Pereira, C. J. P. Monteiro, A. V. C. Simões, S. M. A. Pinto, L. G. Arnaut, G. F. F. Sá, E. F. F. Silva, L. B. Rocha, S. Simões and S. J. Formosinho, J. Porphyrins phthalocyanines, 2009, 13, 567–573 CrossRef CAS
- M. M. Pereira, A. A. Abreu, N. P. F. Goncalves, M. J. F. Calvete, A. V. C. Simões, C. J. P. Monteiro, L. G. Arnaut, M. E. Eusébio and J. Canotilho, Green Chem., 2012, 14, 1666–1672 RSC
- L. G. Arnaut Moreira, M. M. Pereira, S. J. Formosinho, S. Simões, G. Stochel and K. Urbanska, PCT/PT2009/000057, 2009.
- M. M. Pereira, C. J. P. Monteiro, A. V. C. Simoes, S. M. A. Pinto, A. R. Abreu, G. F. F. Sa, E. F. F. Silva, L. B. Rocha, J. M. Dabrowski, S. J. Formosinho, S. Simoes and L. G. Arnaut, Tetrahedron, 2010, 66, 9545–9551 CrossRef CAS
- E. F. F. Silva, C. Serpa, J. M. Dabrowski, C. J. P. Monteiro, L. G. Arnaut, S. J. Formosinho, G. Stochel, K. Urbanska, S. Simoes and M. M. Pereira, Chem. – Eur. J., 2010, 16, 9273–9286 CrossRef CAS PubMed
- J. M. Dabrowski, M. M. Pereira, L. G. Arnaut, C. J. P. Monteiro, A. F. Peixoto, A. Karocki, K. Urbanska and G. Stochel, Photochem. Photobiol., 2007, 83, 897–903 CrossRef CAS PubMed
- J. M. Dabrowski, M. Krzykawska, L. G. Arnaut, M. M. Pereira, C. J. P. Monteiro, S. Simoes, K. Urbanska and G. Stochel, ChemMedChem, 2011, 6, 1715–1726 CrossRef CAS PubMed
- A. F. S. Luz, B. Pucelik, M. M. Pereira, J. M. Dabrowski and L. G. Arnaut, Lasers Surg. Med., 2018, 50, 451–459 CrossRef PubMed
- J. P. Hughes, S. Rees, S. B. Kalindjan and K. L. Philpott, Br. J. Pharmacol., 2011, 162, 1239–1249 CrossRef CAS PubMed
- C. S. Foote, Photochem. Photobiol., 1991, 54, 659 CrossRef CAS PubMed
- R. Schmidt, F. Shafii, C. Schweitzer, A. Abdel-Shafi and F. Wilkinson, J. Phys. Chem. A, 2001, 105, 1811–1817 CrossRef CAS
- J. M. Dabrowski, L. G. Arnaut, M. M. Pereira, K. Urbanska, S. Simoes, G. Stochel and L. Cortes, Free Radic. Biol. Med., 2012, 52, 1188–1200 CrossRef CAS PubMed
- J. M. Dabrowski, L. G. Arnaut, M. M. Pereira, K. Urbanska and G. Stochel, Med. Chem. Commun., 2012, 3, 502–505 RSC
- B. Pucelik, L. G. Arnaut and J. M. Dabrowski, J. Clin. Med., 2020, 9, 8 CrossRef CAS PubMed
- R. Saavedra, L. B. Rocha, J. M. Dabrowski and L. G. Arnaut, ChemMedChem, 2014, 9, 390–398 CrossRef CAS PubMed
- M. Krzykawska-Serda, J. M. Dabrowski, L. G. Arnaut, M. Szczygieł, K. Urbanska, G. Stochel and M. Elas, Free Radic. Biol. Med., 2014, 73, 239–251 CrossRef CAS PubMed
- L. B. Rocha, L. C. Gomes-da-Silva, J. M. Dabrowski and L. G. Arnaut, Eur. J. Cancer, 2015, 51, 1822–1830 CrossRef CAS PubMed
- B. Pucelik, L. G. Arnaut, G. Stochel and J. M. Dabrowski, ACS Appl. Mater. Interfaces, 2016, 8, 22039–22055 CrossRef CAS PubMed
- J. Oliveira, E. Monteiro, J. Santos, J. D. Silva, L. Almeida and L. L. Santos, J. Clin. Oncol., 2017, 35, e14056 Search PubMed
- L. L. Santos, J. Oliveira, E. Monteiro, J. Santos and C. Sarmento, Case Rep. Oncol., 2018, 11, 769–776 CrossRef PubMed
- A. S. M. Ressurreição, M. Pineiro, L. G. Arnaut and A. M. A. R. Gonsalves, J. Porphyrins phthalocyanines, 2007, 11, 50–57 CrossRef
- W. J. Hagan Jr., D. C. Barber, D. G. Whitten, M. Kelly, F. Albrecht, S. L. Gibson and R. Hilf, Cancer Res., 1988, 48, 1148–1152 CAS
- A. M. Richter, A. K. Jain, A. J. Canaan, E. Waterfield, E. D. Sternberg and J. G. Levy, Biochem. Pharmacol., 1992, 43, 2349–2358 CrossRef CAS PubMed
- J.-M. Houle and A. Strong, J. Clin. Pharmacol., 2002, 42, 547–557 CrossRef CAS PubMed
-
N. P. F. Gonçalves, T. P. C. M. Santos, G. P. N. Costa, C. J. P. Monteiro, F. A. Schaberle, S. C. Alfar, A. C. R. Abreu, M. M. Pereira and L. G. Arnaut-Moreira, PCT/IB2016/051552, WO2016/151458, 2016
- C. Donohoe, F. A. Scharbele, F. M. S. Rodrigues, N. P. F. Gonçalves, C. J. Kingsbury, M. M. Pereira, M. O. Senge, L. C. Gomes-da-Silva and L. G. Arnaut, J. Am. Chem. Soc., 2022, 144, 15252–15265 CrossRef CAS PubMed
- C. Donohoe, A. Charisiadis, S. Maguire, B. Twamley, F. A. Schaberle, L. C. Gomes-da-Silva and M. O. Senge, Eur. J. Org. Chem., 2023, e202201453 CrossRef CAS
- I. Mellman, G. Coukos and G. Dranoff, Nature, 2011, 480, 480–489 CrossRef CAS PubMed
- A. Ribas and J. D. Wolchok, Science, 2018, 359, 1350–1355 CrossRef CAS PubMed
- P. S. Thong, K. W. Ong, N. S. Goh, K. W. Kho, V. Manivasager, R. Bhuvaneswari, M. Olivo and K. C. Soo, Lancer Oncol., 2007, 8, 950–952 CrossRef CAS PubMed
- E. Kabingu, A. R. Oseroff, G. E. Wilding and S. O. Gollnick, Clin. Cancer Res., 2009, 15, 4460–4466 CrossRef CAS PubMed
- M. Korbelik, G. Krosl, J. Krosl and G. J. Dougherty, Cancer Res., 1996, 56, 5647–5652 CAS
- M. Obeid, A. Tesniere, F. Ghiringhelli, G. M. Fimia, L. Apetoh, J.-L. Perfettini, M. Castedo, G. Mignot, T. Panaretakis, N. Casares, D. Métivier, N. Larochette, P. van Endert, F. Ciccosanti, M. Piacentini, L. Zitvogel and G. Kroemer, Nat. Med., 2007, 13, 54–61 CrossRef CAS PubMed
- L. Zitvogel, O. Kepp and G. Kroemer, Cell, 2010, 140, 798–804 CrossRef CAS PubMed
- L. C. Gomes-da-Silva, L. Zhao, H. Zhou, A. Sauvat, P. Liu, L. Bezu, S. Durand, M. Leduc, G. Pierron, F. Loos, B. Sveinbjornsson, O. Rekdal, G. Boncompain, F. Perez, L. G. Arnaut, O. Kepp and G. Kroemer, EMBO J., 2018, 37, e98354 CrossRef PubMed
- A. C. S. Lobo, L. C. Gomes-da-Silva, P. Rodrigues-Santos, A. Cabrita, M. Santos-Rosa and L. G. Arnaut, J. Clin. Med., 2020, 9, 104 CrossRef CAS PubMed
- P. C. Kousis, B. W. Henderson, P. G. Maier and S. O. Gollnick, Cancer Res., 2007, 67, 10501–10510 CrossRef CAS PubMed
- H. Raskov, A. Orhan, J. P. Christensen and I. Gögenur, Br. J. Cancer, 2021, 124, 359–367 CrossRef CAS PubMed
- A. Haslam and V. Prasad, JAMA Network Open, 2019, 2, e192535 CrossRef PubMed
- G. M. Cramer, E. K. Moon, K. A. Cengel and T. M. Busch, Photochem. Photobiol., 2020, 96, 954–961 CrossRef CAS PubMed
- S. Anand, T. A. Chan, T. Hasan and E. V. Maytin, Pharmaceuticals, 2021, 14, 447 CrossRef CAS PubMed
-
A. C. S. Lobo, L. C. Gomes-da-Silva and L. G. Arnaut, in Handbook of Porphyrin Science, ed. K. M. Kadish, K. M. Smith and R. Guilard, World Scientific, 2021, vol. 46, ch. 5, pp. 279–344 Search PubMed
- J. M. Fritz and M. J. Lenardo, J. Exp. Med., 2019, 216, 1244–1254 CrossRef CAS PubMed
- A. D. Waldman, J. M. Fritz and M. J. Lenardo, Nat. Rev. Immunol., 2020, 20, 651–668 CrossRef CAS PubMed
- C. S. Lobo, M. I. P. Mendes, D. A. Pereira and L. C. Gomes-da-Silva, Sci. Rep Search PubMed.
-
G. P. N. Costa, N. P. F. Gonçalves, C. J. P. Monteiro, A. C. R. Abreu, H. T. Soares, L. B. Rocha, F. A. Schaberle, M. M. Pereira and L. G. Arnaut-Moreira, PCT/IB2016/051552, WO2016/151458, 2016
- N. A. Le, V. Babu, M. Kalt, L. Schneider, F. Schumer and B. Spingler, J. Med. Chem., 2021, 64, 6792–6801 CrossRef CAS PubMed
- A. Abuteen, S. Zanganeh, J. Akhigbe, L. P. Samankumara, A. Aguirre, N. Biswal, M. Braune, A. Vollertsen, B. Röder, C. Brückner and Q. Zhu, Phys. Chem. Chem. Phys., 2013, 15, 18502–18508 RSC
- M. Penetra, L. G. Arnaut and L. C. Gomes-da-Silva, OncoImmunology, 2023, 12, 2226535 CrossRef PubMed
- H. Sung, J. Ferlay, R. L. Siegel, M. Laversanne, I. Soerjomataram, A. Jemal and F. Bray, CA Cancer J. Clin., 2021, 71, 209–249 CrossRef PubMed
| This journal is © The Royal Society of Chemistry 2023 |