
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Transforming an esterase into an enantioselective catecholase through bioconjugation of a versatile metal-chelating inhibitor†
Laura
Fernandez-Lopez‡
 a,
Isabel
Cea-Rama‡
b,
Julia
Alvarez-Malmagro§
a,
Anna K.
Ressmann
c,
Jose L.
Gonzalez-Alfonso
a,
Cristina
Coscolín
a,
Patrick
Shahgaldian
d,
Francisco J.
Plou
a,
Jan
Modregger
c,
Marcos
Pita
a,
Julia
Sanz-Aparicio¶
b and
Manuel
Ferrer¶
*a
a,
Isabel
Cea-Rama‡
b,
Julia
Alvarez-Malmagro§
a,
Anna K.
Ressmann
c,
Jose L.
Gonzalez-Alfonso
a,
Cristina
Coscolín
a,
Patrick
Shahgaldian
d,
Francisco J.
Plou
a,
Jan
Modregger
c,
Marcos
Pita
a,
Julia
Sanz-Aparicio¶
b and
Manuel
Ferrer¶
*a
aInstituto de Catalisis y Petroleoquimica (ICP), CSIC, Madrid 28049, Spain. E-mail: mferrer@icp.csic.es
bInstituto de Quimica Fisica Rocasolano (IQFR), CSIC, Madrid 28006, Spain
cEucodis Bioscience GMBH, Wien 1030, Austria
dInstitute for Ecopreneurship, School of Life Sciences, University of Applied Sciences and Arts Northwestern Switzerland, Muttenz 4132, Switzerland
First published on 20th June 2023
Abstract
Metal complexes introduced into protein scaffolds can generate versatile biomimetic catalysts endowed with a variety of catalytic properties. Here, we synthesized and covalently bound a bipyridinyl derivative to the active centre of an esterase to generate a biomimetic catalyst that shows catecholase activity and enantioselective catalytic oxidation of (+)-catechin.
Metal complexes can be introduced, through non-covalent or covalent bioconjugation, at specific sites of protein scaffolds to generate versatile biohybrid or biomimetic catalysts. Such systems combine productively structural and functional properties of both biological macromolecules and metal-complex catalysts.1–6 Using different non-catalytic proteins as scaffolds, biomimetic catalysts supporting a broad range of catalytic activities have been designed. This includes olefin metathesis, ruthenium-catalysed deallylation, iridium-catalysed/siderophore-inspired transfer hydrogenation, dirhodium-catalysed cyclopropanation, C–H activation,1,5a,b Diels–Alder reaction and Friedel–Crafts alkylation,4a,6a,7 redox catalysis,1,4b,8 enantioselective oxidation of sulfides with H2O2,8 metal-dependent hydrolysis,8 and the hydroformylation of alkenes.1 This catalytic potential was expanded to selective olefin cyclopropanation, CO2 hydration, ester hydrolysis, nitrite reduction, enantioselective carbenoid insertion into Si–H bonds, asymmetric cyclopropanation, alkyne polymerization and metathesis,1,2b–d,3,8,9 when using nucleic acids and peptides. The capacity for C–H insertion,1,4a hydrolysis,7 oxidation of Mn2+,2a cofactor reduction,6a,b Diels–Alder reaction and Friedel–Crafts alkylation,6c,7 Hoveyda–Grubbs catalysis, Heck cross-coupling,7 selective oxidation of aromatic alkenes,10 ring-opening metathesis polymerization, dihydroxylation and epoxidation,7 and asymmetric reduction of a cyclic imine,11 was further incorporated in several enzymatic scaffolds. The environment of a catalytically competent organometallic complex or the orientation of residues into which these catalysts are introduced plays a major role in the catalytic activity of the resulting metalloenzyme.2c,d,5b,12 This is why amino acid sequences are currently being optimized with respect to the metal coordination geometry and/or the dedicated action of metal ions in protein environments.13
In this study, we applied the concept of covalent bioconjugation to transform an ester hydrolase enzyme into an oxidoreductase. This was achieved by exploiting the native biocatalytic activity of the enzyme to dock, within the active site of the enzyme, a metal-chelating inhibitor. We recently used the inhibitor 1 to successfully modify the active site of an esterase, which was thereby transformed into a biomimetic chemo-catalyst supporting oxidation and Friedel–Crafts alkylation reactions.14
The versatility of 1 as catalytic inhibitor is limited by the presence of two phosphoester sites that can be potentially hydrolysed (Scheme 1). The higher reactivity of the bipyridine group compared to the methyl group means that a catalytic biomimetic may not be formed in many cases. We solved this problem by synthetizing phosphodiester 2. Owing to the symmetry of the molecule, nucleophilic enzymatic hydrolysis can lead to only one hydrolytic product that retains the bipyridine moiety within the active site. To explore the capacity of 2 to transform esterases into biohybrid catalysts, we selected EH3, a highly versatile and promiscuous esterase,15,16 as model enzyme. The inhibitor 2 was produced by reacting 2,2′-bipyridine-3,3′-diol and hexyl phosphonium in N,N-dimethylformamide and N,N-diisopropylethylamine, characterized using NMR and high-resolution mass spectrometry (Fig. S1–S4, ESI†), and used to modify the esterase EH3 to yield the corresponding biomimetic (bEH3). The reaction was carried out in a 50 mM potassium phosphate buffer at pH 6.5 for 10 min and monitored by measuring the inhibition of EH3, following the hydrolysis of glyceryl tripropionate using the Phenol Red pH indicator assay.14 Incubating EH3 with 2 for 10 min resulted in complete enzyme inhibition, indicating complete bioconjugation of the EH3 active site. After the bioconjugation reaction was completed, bEH3 was dialyzed against a buffer to remove excess inhibitor. Crystals of bEH3 of sufficient quality for single-crystal X-ray crystallography were successfully obtained by co-crystallization, that is, preincubation of EH3 with 2. The crystal structure of this complex was determined (Fig. 1) using the coordinates of unliganded EH3 (PDB 6SXP).16
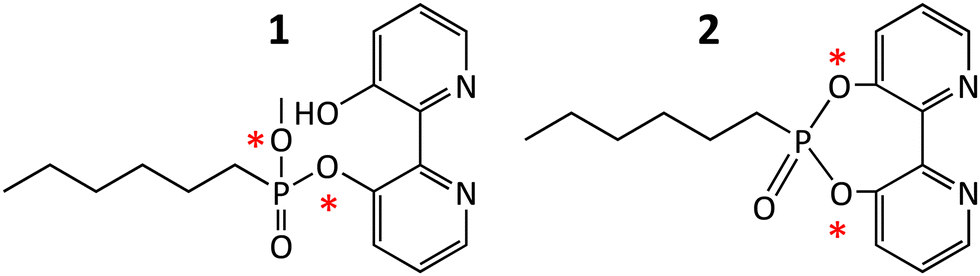 | ||
| Scheme 1 Structure of inhibitor 1 (3′-hydroxy-[2,2′-bipyridin]-3-yl methyl hexylphosphonate) and inhibitor 2 (6-hexyl-[1,3,2]dioxaphosphepino[5,4-b:6,7-b′]dipyridine 6-oxide). The Figure shows the two sites, marked with a red asterisk, that can potentially be attacked by esterases and through which the molecules that can be covalently linked to the nucleophilic serine. | ||
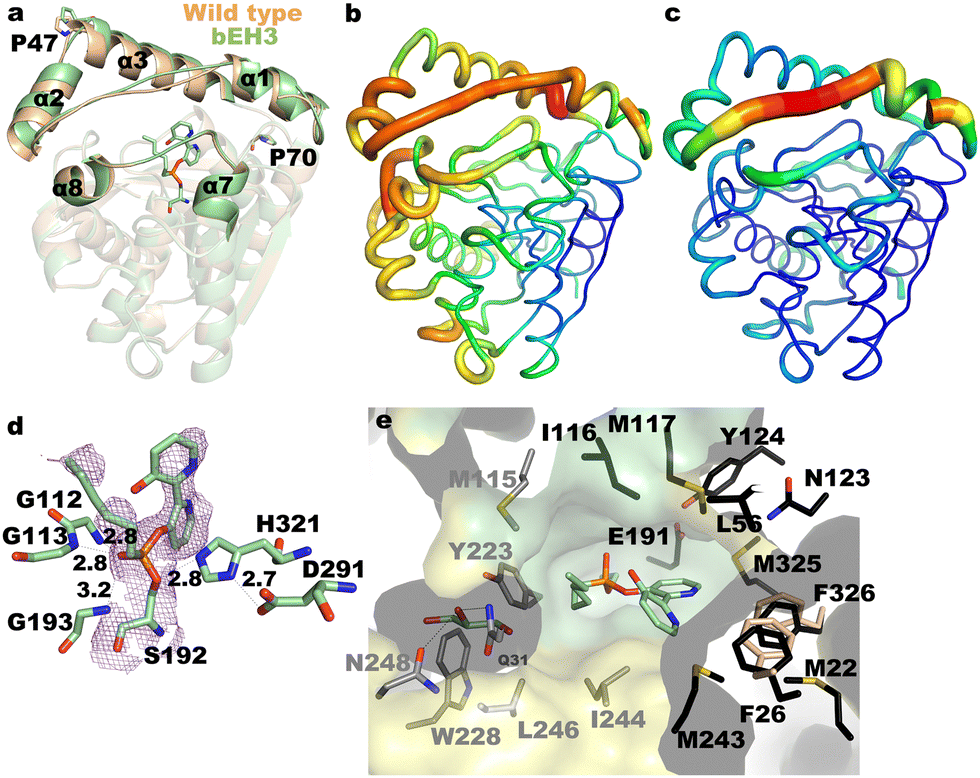 | ||
| Fig. 1 (a) Superimposition of unliganded EH3 (wheat, PDB Code: 6SXP) onto bEH3 (green, PDB Code: 8PC7), highlighting the structural changes at the CAP domain. Residues Pro47 and Pro70, acting as hinges, are shown as sticks. B factors for the complex (b) and the unliganded enzyme (c), with values ranging from low (blue) to high (red). (d) Close-up view of the bEH3 complex mimicking the tetrahedral intermediate, and the Polder omit map (2.5 σ cut-off) shown in violet. The polar network is shown as dashed lines, with atomic distances in Å. (e) Cross-section of the molecular surface of bEH3, with residues at the alcohol moiety coloured in black and residues at the acyl moiety coloured in grey. The cap and catalytic domains are coloured yellow and green, respectively. A glycerol molecule captured at the acyl binding site is shown. The positions of Phe26 and Phe326 in the free enzyme are shown by wheat sticks. | ||
A comparison of the structure of EH3 with that of the complexed bEH3 reveals a shift of the cap domain upon substrate entrance, particularly at helices α1, α2 and α3 (Fig. 1a). An equivalent displacement has not been observed for previously reported complexes with methyl-(R/S)-2-phenylpropanoate substrates,15 or with the EH3-butyl 4-nitrophenylhexylphosphonate complex (B-4NHP),16 which also mimics the tetrahedral intermediate of the hydrolytic reaction as 2 does. However, we previously reported that the polypeptide chain is highly flexible in the CAP domain, a peculiarity that was supported by an analysis of the B factor values and an ensemble refinement performed using the EH3 coordinates.16 This analysis revealed many potential conformations of the N-terminal segment, mostly comprising α1 and α2, with Pro47 putatively acting as a hinge. The complex reported here shows a shift of a larger CAP region that includes the long helix α3, with Pro70 being the putative hinge (Fig. 1a). Thus, the reorganization of the CAP domain observed in this complex appears to be required to accommodate the bulky bipyridyl moiety of 2 and unveils an even higher versatility of the EH3 active site cavity. In turn, this reorganization may be feasible because EH3 possesses a significantly larger catalytic cavity than its homologues, due in part to the EH3 CAP region.15,16 Nevertheless, the binding of 2 appears to destabilize EH3, as reflected by the high B factor values of 2 (Fig. 1b and c). We previously reported the catalytic residues as the nucleophile Ser192, the acidic residue Asp291 and the basic residue His321.16 The bipyridine derivative complex mimics the tetrahedral intermediate that is stabilized at the oxyanion site by Gly112, Gly113 and Gly193 (Fig. 1d). EH3 has long channels for acyl/alcohol binding that are surrounded mostly by hydrophobic residues and only a few polar residues, endowing EH3 with broad substrate specificity (Fig. 1e).15 The binding of 2 to the enzyme locates the bipyridine moiety in the alcohol-binding tunnel, resulting in appreciable reorientation of Phe326 in the catalytic domain and Phe26 at the beginning of the flexible loop α1–α2, which retracts from the catalytic site to prevent steric hindrance (Fig. 1e).
With a well-structurally-characterized conjugate in hand, catechol oxidation, an important transformation for metalloenzymes,10–14 was investigated through electrochemical characterization of EH3 and bEH3. First, an experiment was performed to compare the electrochemical response of the split components, Cu(NO3)2 and catechol (1 mM), with that of both of components on the surface of a self-assembled-monolayer (SAM)-modified electrode (Fig. 2a). As some Cu2+ was stabilized on the carboxylated SAM surface, a broad Cu2+ signal was observed, which can be attributed to the different kinetics measured for bulk Cu2+ and bound Cu2+. The redox signal of catechol is affected by the presence of Cu2+, which makes the redox reaction more irreversible. This behaviour may be attributed to the formation of ligands with the cation that stabilize catechol and shift the oxidation process to more positive values. In a second experiment, the response to immobilized EH3 and bEH3 was measured, which clearly demonstrated that 2 was located within the bEH3 structure (Fig. 2b). As this signal differed from that observed for EH3, it can be concluded that free Cu2+ was not measured. Addition of catechol produced different behaviour than that observed using the SAM-Au electrode (Fig. 2c): EH3 blocked the surface, inhibiting the catechol redox signal. However, the catechol signal could be measured using bEH3, indicative of catalytic activity for catechol oxidation/reduction.
 | ||
| Fig. 2 (a) The electrochemical response of an 3-mercaptopropionic acid (MPA)-gold electrode in a buffer with catechol (blue solid line, 1) and catechol and CuSO4 (green solid line, 2). Cyclic voltammetry (CV) curves for the control electrodes with MPA and CuSO4 (dark dashed line, 3) or only MPA (red solid line, 4) are shown. Measurements were performed at a 20 mV s−1 scan rate with a 50 mM phosphate buffer (pH = 6.5) as the electrolyte. (b) The electrochemical response of the MPA-gold electrode before (1) and after absorption of EH3 (2) and bEH3 (3). (c) The electrochemical response of the electrodes shown in (b) in the presence of 1 mM catechol. (d) The electrochemical response of the MPA-gold electrode after absorption of bEH3 (1) and bEH1AB1 (2) in the presence of 1 mM catechol. Data for bEH1AB1 are taken from Alonso et al.14 | ||
The electrochemical results provided evidence of catalytic activity of bEH3 for catechol oxidation/reduction. We recently used 1 to show that the corresponding biomimetic bEH1AB1, could also catalyse this reaction.14 The possibility of using the same bipyridine moiety to enable Cu2+ complexation in two different catalytic environments (as defined by the EH3 or EH1AB1 active sites) enables evaluation of differences in the catalytic performance. Fig. 2d shows the electrochemical response of the electrodes in the presence of bEH3 or bEH1AB1 and catechol. The starting potential is less positive and the oxidation wave is much more defined in the presence of bEH3 than in the presence of bEH1AB1. This result suggests that bEH3 catalyses the oxidation of catechol more effectively than bEH1AB1. This was confirmed by following the oxidation of 3,5-di-tert-butylcatechol to 3,5-di-tert-butyl-O-benzoquinone using high-performance liquid chromatography (Fig. S5 and S6, ESI†), which under the same assay conditions, reached 72 ± 2% (bEH1AB1) and 97 ± 1% (bEH3). The higher performance of bEH3 might reflect a favourable role played by the residues near the catalytic site, in agreement with previously reported results on the importance of the second coordination sphere provided by the biomolecular scaffold.6c This was confirmed by a structural comparison of the complexes bEH3 and bEH1AB1 (PDB 6RKY).14 As shown in Fig. 3a, the bipyridine moiety binds to the alcohol site very similarly as to the corresponding active site cavities, whereas the alkyl chain is allocated to different acyl sites. Indeed, the different topologies of the CAP domains of these two complexes result in very different clefts (Fig. 3b and c), which in the case of the bEH3 complex appears to be particularly open to the solvent, possibly facilitating access of the substrate to the metal centre and therefore enabling the oxidation/reduction reaction to occur. The close-up view of the bEH3 complex in Fig. 3d shows the residues surrounding the catalytic site that could play a role in catalysis. Remarkably, several methionines in the proximity to the bipyridine moiety could well provide a third ligand stabilizing a putative Cu2+ position. Phe26 and Phe326 could assist in substrate binding through stacking interactions. With the exception of Met117 from the catalytic domain, all these residues are located in the highly flexible CAP, which might enable the different steps of the reaction pathway. Conversely, in the bEH1AB1 complex (Fig. 3e), Phe209 is the sole residue close to the putative Cu2+ position, although two more Met residues are found within the cavity and all three residues are located in the CAP. UV/vis spectra of bEH1AB1 and bEH3 revealed subtle changes (Fig. S7, ESI†), and further mutagenic, electrochemical and/or Raman analyses, will determine the specific metal ligands in the two enzymes, whose binding environments differ (Fig. 3).
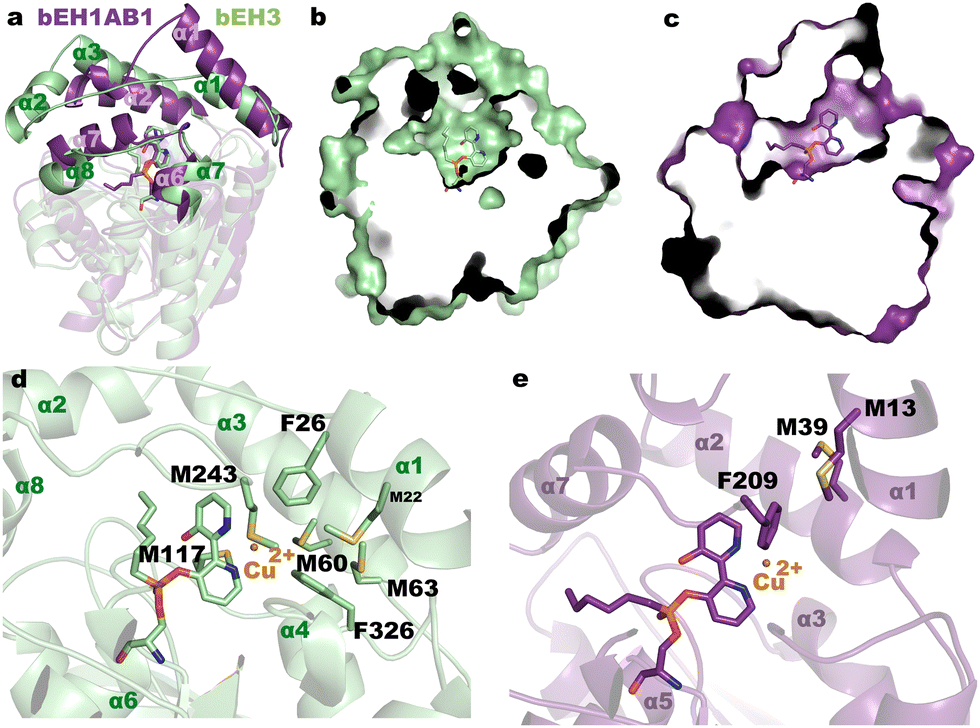 | ||
| Fig. 3 (a) Structural superimposition of the complex bEH1AB1 (violet) onto bEH3 (green). The different CAP domains shape the active site cavities shown in (b) and (c). (d) and (e) Details of the complexes showing the environment of the bipyridyl moiety and the modelled position for a putative Cu2+. The Cu2+ ion has been manually incorporated into the active sites by placing it at a suitable distance from the two nitrogen atoms found in the crystal to depict the different environment found within each enzyme. | ||
We further determine whether the differences in the topology of the active sites (Fig. 3a–c) and/or the coordination geometry of the Cu(bpy) complexes (Fig. 3d and e) play a role in the observed activity and enantioselectivity of bEH1AB1 and bEH3. On the latter point, it is well reported that the primary coordination sphere of a metal determines basic reactivity, and the secondary coordination sphere tunes the reactivity through weaker interactions. For that, we measured the conversion of (−)-catechin and (+)-catechin to the corresponding o-quinones by spectrophotometrically monitoring the product formation. Characterization of the reaction product revealed bEH3 being enantioselective for (+)-catechin; by contrast, bEH1AB1 was found to preferentially oxidize (−)-catechin (Fig. S6 and S8, ESI†). It is generally accepted that the apparent enantioselectivity (Eapp) of soluble enzymes can be calculated as the ratio between the conversion rates of the preferred chiral substrate and the non-preferred substrate obtained by testing the two enantiomers separately. Using the oxidation rates (in min−1) values for each biomimetic and enantiomer (Fig. S8, ESI†), Eapp of 91.9 ± 0.5 for bEH3 ((+), preferred) and 3.9 ± 0.7 for bEH1AB1 ((−), preferred) were obtained. It is plausible that the higher number of methionine residues in close and flexible regions of the bEH3 complex compared to that of bEH1AB1 might provide additional ligands for Cu2+, probably stabilizing the metal complex, which has higher accessibility from the solvent (Fig. 3d and e); this may promote the entrance of the substrates influencing the observed catalytic activity and enantioselectivity.
In summary, we synthesized a versatile inhibitor that enables the transformation of esterases into biomimetic catecholases without manipulating the protein sequence by genetic engineering. Indeed, the introduction of Cys,6a,9 Lys,4a,6a His,4b non-natural aminoacids,6c and other mutations10 has been reported as being necessary for designing metalloenzymes. It also obviates the use of computational efforts commonly needed to find sites that can accommodate a biomolecule in protein scaffolds.9–11 We applied this molecule to an esterase and determined the structure of the biomimetics using single-crystal X-ray crystallography. The synthetic relevance and the possibility of using 2 and the resulting biomimetics for enantioselective conversions, either modulated by the orientation of the competent organometallic complex or by the nano-environment of the active site, needs to be further evaluated. Although yet to fully elucidate the nature of the copper site in the biomimetic bEH3 and how it is modulated for catecholase function, it is plausible that the catalytic cycle of the biomimetic herein designed proceeds as mononuclear copper complexes containing N4 donors,17 in contrast to natural catecholases that often have binuclear copper centres.18 In brief, the non-protonated catecholate anion first binds to the Cu2+ centre of the biomimetic. This intermediate then oxidizes the coordinated catecholate anion forming Cu+ species; these species then react with dioxygen to give a copper dioxygen complex which yields catechin quinone and H2O2, which would be decomposed to oxygen and two molecules of H2O and the catalyst is regenerated in its original active form in closing up the catalytic cycle.
We acknowledge the financial support of the European Union's Horizon 2020 (GA 101000327), and the Ministerio de Ciencia e Innovación and Agencia Estatal de Investigación (AEI) (DOI 10.13039/501100011033), and the “NextGenerationEU/PRTR” (PID2020-112758RB-I00, PDC2021-121534-I00, TED2021-130544B-I00, PID2019-105838RB-C31, PID2019-105838RB-C33). We also thank the staff of the Synchrotron Radiation Source at Alba.
Conflicts of interest
There are no conflicts to declare.Notes and references
- H. J. Davis and T. R. Ward, ACS Cent. Sci., 2019, 5, 1120–1136 CrossRef CAS PubMed.
- (a) S. K. Wilcox, C. D. Putnam, M. Sastry, J. Blankenship, W. J. Chazin, D. E. McRee and D. B. Goodin, Biochemistry, 1998, 37, 16853–16862 CrossRef CAS PubMed; (b) B. S. Der, D. R. Edwards and B. Kuhlman, Biochemistry, 2012, 51, 3933–3940 CrossRef CAS PubMed; (c) M. R. Ross, A. M. White, F. Yu, J. T. King, V. L. Pecoraro and K. J. Kubarych, J. Am. Chem. Soc., 2015, 137, 10164–10176 CrossRef CAS PubMed; (d) M. L. Zastrow and V. L. Pecoraro, J. Am. Chem. Soc., 2013, 135, 5895–5903 CrossRef CAS PubMed.
- A. Roy, D. J. Sommer, R. A. Schmitz, C. L. Brown, D. Gust, A. Astashkin and G. Ghirlanda, J. Am. Chem. Soc., 2014, 136, 17343–17349 CrossRef CAS PubMed.
- (a) Y. W. Lin, S. Nagao, M. Zhang, Y. Shomura, Y. Higuchi and S. Hirota, Angew. Chem., Int. Ed., 2015, 54, 511–515 CAS; (b) C. J. Lalaurie, V. Dufour, A. Meletiou, S. Ratcliffe, A. Harland, O. Wilson, C. Vamasiri, D. K. Shoemark, C. Williams, C. J. Arthur, R. B. Sessions, M. P. Crump, J. L. R. Anderson and P. Curnow, Sci. Rep., 2018, 8, 14564 CrossRef PubMed.
- (a) M. Jeschek, R. Reuter, T. Heinisch, C. Trindler, J. Klehr, S. Panke and T. R. Ward, Nature, 2016, 537, 661–665 CrossRef CAS PubMed; (b) A. D. Liang, J. Serrano-Plana, R. L. Peterson and T. R. Ward, Acc. Chem. Res., 2019, 52, 585–595 CrossRef CAS PubMed.
- (a) J. Bos, F. Fusetti, A. J. Driessen and G. Roelfes, Angew. Chem., Int. Ed., 2012, 51, 7472–7475 CrossRef CAS PubMed; (b) M. Basle, H. A. W. Padley, F. L. Martins, G. S. Winkler, C. M. Jäger and A. Pordea, J. Inorg. Biochem., 2021, 220, 111446 CrossRef CAS PubMed; (c) I. Drienovská, A. Rioz-Martínez, A. Draksharapu and G. Roelfes, Chem. Sci., 2015, 6, 770–776 RSC.
- W. J. Jeong, J. Yu and W. J. Song, Chem. Commun., 2020, 56, 9586–9599 RSC.
- (a) T. C. Chang, K. Vong, T. Yamamoto and K. Tanaka, Angew. Chem., Int. Ed., 2021, 60, 12446–12454 CrossRef CAS PubMed; (b) J. Tang, F. Huang, Y. Wei, H. Bian, W. Zhang and H. Liang, Dalton Trans., 2016, 45, 8061–8072 RSC.
- A. Roy, I. Sarrou, M. D. Vaughn, A. V. Astashkin and G. Ghirlanda, Biochemistry, 2013, 52, 7586–7594 CrossRef CAS PubMed.
- M. Allard, C. Dupont, V. Muñoz Robles, N. Doucet, A. Lledós, J. D. Maréchal, A. Urvoas, J. P. Mahy and R. Ricoux, ChemBioChem, 2012, 13, 240–251 CrossRef CAS PubMed.
- T. Heinisch, M. Pellizzoni, M. Dürrenberger, C. E. Tinberg, V. Köhler, J. Klehr, D. Häussinger, D. Baker and T. R. Ward, J. Am. Chem. Soc., 2015, 137, 10414–10419 CrossRef CAS PubMed.
- J. K. Kim, C. Lee, S. W. Lim, A. Adhikari, J. T. Andring, R. McKenna, C. M. Ghim and C. U. Kim, Nat. Commun., 2020, 11, 4557 CrossRef CAS PubMed.
- H. Eom and W. J. Song, J. Biol. Inorg. Chem., 2019, 24, 517–531 CrossRef CAS PubMed.
- S. Alonso, G. Santiago, I. Cea-Rama, L. Fernandez-Lopez, C. Coscolín, J. Modregger, A. K. Ressmann, M. Martínez-Martínez, H. Marrero, R. Bargiela, M. Pita, J. L. Gonzalez-Alfonso, M. L. Briand, D. Rojo, C. Barbas, F. J. Plou, P. N. Golyshin, P. Shahgaldian, J. Sanz-Aparicio, V. Guallar and M. Ferrer, Nat. Catal, 2020, 3, 319–328 CrossRef CAS.
- I. Cea-Rama, C. Coscolín, P. Katsonis, R. Bargiela, P. N. Golyshin, O. Lichtarge, M. Ferrer and J. Sanz-Aparicio, Comput. Struct. Biotechnol. J., 2021, 19, 2307–2317 CrossRef CAS PubMed.
- C. I. Giunta, I. Cea-Rama, S. Alonso, M. L. Briand, R. Bargiela, C. Coscolín, P. F. Corvini, M. Ferrer, J. Sanz-Aparicio and P. Shahgaldian, ACS Nano, 2020, 14, 17652–17664 CrossRef CAS PubMed.
- A. El-Motaleb, M. Ramadan, M. M. Ibrahim and I. M. El-Mehasseb, J. Coord. Chem., 2012, 65, 2256–2279 CrossRef.
- R. Silavi, A. Divsalar and A. A. Saboury, J. Biomol. Struct. Dyn., 2012, 30, 752–772 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Details of synthetic procedures, enzyme inhibition, metal complexation, the activity assay, oxidation reactions, NMR, HRMS, ESI-MS, and HPLC analyses can be found in the Supporting Information. See DOI: https://doi.org/10.1039/d3cc01946b |
| ‡ Equal contribution. |
| § Current address: Department of Physical Chemistry, University of Seville, 41012 Seville, Spain. |
| ¶ Equal leading contribution. |
| This journal is © The Royal Society of Chemistry 2023 |