
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInfrared spectroscopic and theoretical investigations of novel iridium oxyfluorides†
Yan
Lu
,
Robert
Medel
,
Guohai
Deng
and
Sebastian
Riedel
 *
*
Freie Universität Berlin, Department for Chemistry and Biochemistry, Fabeckstr. 34/36, Berlin, Germany. E-mail: s.riedel@fu-berlin.de
First published on 8th June 2023
Abstract
The iridium oxyfluorides (OIrF, OIrF2 and FOIrF) were prepared for the first time by the reaction of IR-laser ablated iridium atoms and OF2, isolated in solid neon and argon matrices. The assignments of the main vibrational absorptions of these products were supported by a combined analysis of IR-matrix-isolation spectroscopy with 18OF2 substitution and quantum-chemical calculations. The OIrF molecule exhibits triple bond character. In contrast to terminal oxyl radical species OPtF2 and OAuF2, a much lower spin-density contribution at the oxygen atom was found in OIrF2.
Iridium, one of the rarest transition metal elements in the earth's crust, is the only confirmed chemical element capable of attaining the highest oxidation state +IX in the iridium tetroxide cation [IrO4]+.1 In general, apart from oxides, the family of such compounds with transition metals in their highest oxidation states includes fluoride and oxyfluoride molecules.1,2 The +I to +VI oxidation states of Ir have been experimentally observed in binary iridium fluorides, with most studies focusing on the characterization of the IrF6 molecule.3–6 However, less is known about iridium oxyfluorides from both theoretical and experimental perspectives.5–10 The IrOF5 molecule was predicted by us as an alternative target for oxidation state +VII.7 To the best of our knowledge, iridium oxyfluorides have not yet been experimentally verified, although various methods and approaches have been proposed to produce them over the years, typically based on hexafluorides as reactants.5,6,8–10
The generation of iridium oxyfluorides was first proposed by Ruff and Fischer in 1929 in the reaction of iridium hexafluoride with the alkali present in the glass.5 However, the produced iridium oxyfluoride in this reaction was later identified to the complex salts of quinquevalent iridium in 1956.6,9 In the following decades, some other methods were also tried out to prepare iridium oxyfluorides, such as fluorination of iridium dioxide,6 oxygen–fluorine exchange10 and hydrolysis reactions.8 Unfortunately, there were no iridium oxyfluorides observed, which were proposed to be extremely unstable intermediate products, while the methods have been used successfully in the preparation of other transition metal oxyfluorides (ReOF4, OsOF4, UOF4, etc.).8,10 Therefore, due to their highly reactive nature, it is still a challenge to develop a useful procedure to prepare the so far experimentally unknown class of iridium oxyfluorides.
An established quite efficient and facile route to produce oxyfluoride molecules under cryogenic conditions in rare gas matrices is the reaction of laser-ablated metal atoms with OF2.11–17 The successful preparation and identification of such compounds indicate that synthesizing iridium oxyfluorides might be possible as well. In this work, we describe for the first-time the preparation of molecular iridium oxyfluorides (OIrF, OIrF2 and FOIrF) via the reaction of laser-ablated iridium atoms with OF2 in excess neon and argon at cryogenic temperatures. The matrix-isolation infrared spectroscopic identification of these new oxyfluorides is supported by isotope labelling of 18OF2 and quantum-chemical calculations.
The infrared spectra from the reactions of laser-ablated iridium atoms with either 16OF2 or 18OF2 in solid neon (5 K) followed by annealing and irradiation are shown in Fig. 1. The spectra from analogous experiments in solid argon (12 K) are shown in Fig. S1 in the ESI.† The observed and calculated vibrational frequencies are summarized in Table 1 and Tables S1–S6 (ESI†).
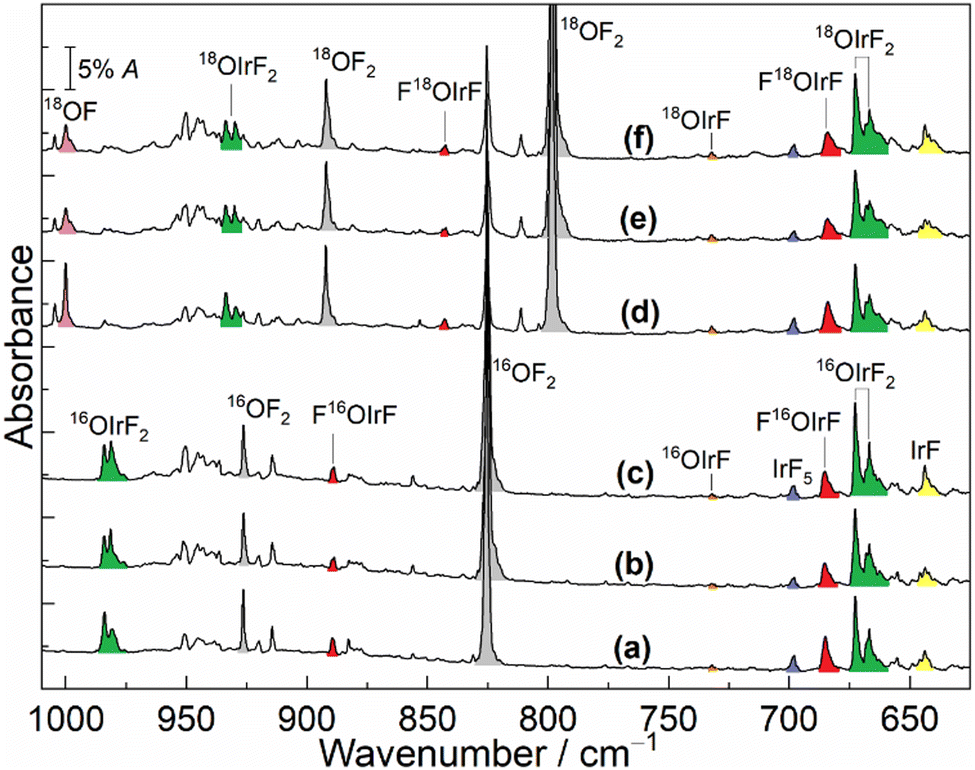 | ||
| Fig. 1 IR spectra in the neon matrix at 5 K. (a) IR spectrum of the reaction products of laser-ablated Ir atoms with 0.02% 16OF2; (b) after annealing to 9 K; (c) after full arc (>220 nm) irradiation for 15 min; (d) IR spectrum of reaction products of laser-ablated Ir atoms with 0.1% 18OF2; (e) after annealing to 9 K; (f) after full arc (>220 nm) irradiation for 15 min. | ||
| Species | Exp. | Calc. | Modes | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Ne | Ar | CCSD(T)b | ||||||||
| ν(16O) | ν(18O) | Δν | ν(16O) | ν(18O) | Δν | ν(16O) | ν(18O) | Δν | ||
| a The complete set of calculated frequencies is provided in the ESI (Tables S1–S6). For the CCSD(T) calculations no intensities are available. b Harmonic frequencies calculated at the CCSD(T)/aug-cc-pVTZ-PP level. c Bands were not observed, or too weak. | ||||||||||
| OIrF | —c | —c | —c | —c | —c | —c | 1053.4 | 998.0 | 55.4 | ν(Ir–O) |
| (1Σ+, C∞v) | 732.2 | 732.2 | 0.0 | —c | —c | —c | 741.7 | 741.4 | 0.3 | ν(Ir–F) |
| OIrF2 | 984.1/980.7 | 933.7/929.6 | 50.4/51.1 | 976.0/973.6 | 925.8/922.6 | 50.2/51.0 | 1033.1 | 978.6 | 54.5 | ν(Ir–O) |
| (2B1, C2v) | 672.6 | 672.6 | 0.0 | 663.6 | 663.6 | 0.0 | 684.1 | 684.1 | 0.0 | ν as(IrF2) |
| 666.8 | 666.8 | 0.0 | 657.5 | 657.5 | 0.0 | 682.8 | 682.8 | 0.0 | ν s(IrF2) | |
| FOIrF | 889.3 | 842.9 | 46.4 | —c | —c | —c | 938.5 | 890.6 | 47.9 | ν(Ir–O) |
| (2A′′, Cs) | 685.1 | 684.1 | 1.0 | —c | —c | —c | 698.1 | 697.9 | 0.2 | ν(Ir–F) |
| —c | —c | —c | —c | —c | —c | 464.4 | 446.1 | 18.3 | ν(O–F) | |
The diatomic OF radical is the most common species in the spectra from the reactions of OF2 and laser-ablated metal atoms and its absorption was observed at 1031.3/1028.6 cm−1 (16OF in solid Ne/Ar) and at 1000.2/997.7 cm−1 (18OF in solid Ne/Ar).12,18 The bands at 643.6 and 697.8 cm−1 were obtained in the reaction of Ir with F2 in excess neon, which have previously been assigned to IrF and IrF5, respectively.3 In addition to these known fluoride molecules, in the present work, newly formed product absorptions were observed clearly at 666.8, 672.6, 685.1, 889.3 and 984.1 cm−1 (Fig. 1(a)). The less intense absorption 685.1 cm−1 and a weak absorption at 889.3 cm−1 decreased substantially after annealing to 9 K, but both absorptions increased slightly when the sample was further exposed to λ > 220 nm irradiation. However, both sample annealing and irradiation resulted in an increase of the above mentioned bands at 666.8, 672.6 and 984.1 cm−1. The spectra were also recorded after sample deposition in solid argon (Fig. S1, ESI†). Apart from the known species IrF, IrF2 and IrF4,3 the newly observed bands at 657.5, 663.6 and 976.0 cm−1 in solid argon on deposition show continued growth during annealing and irradiation of the sample.
To aid in the assignment of the new bands, all the experiments in neon and argon matrices were repeated by employing the isotopically enriched reagent 18OF2 under similar conditions (Fig. 1 and Fig. S1, ESI†). According to the almost same behaviour of the absorptions during annealing and subsequent broadband irradiation, a large red-shift of 46.4 cm−1 was observed for the 889.3 cm−1 band in the neon matrix, while the more intense band 685.1 cm−1 displayed a small 16/18O-isotopic shift of 1.0 cm−1. Moreover, the band at 984.1 cm−1 showed a distinct red-shift of 50.4 cm−1, whereas no shifts were observed for the two strong bands at 666.8 and 672.6 cm−1 upon 18O substitution. In the argon matrix, there were almost no 16/18O shifts for bands at 657.5 and 663.6 cm−1. However, the higher wavenumber signal at 976.0 cm−1 displayed a noticeable isotopic shift of 50.2 cm−1.
The bands at 666.8, 672.6 and 984.1 cm−1 in the neon matrix (657.5, 663.6 and 976.0 cm−1, Ar-matrix) produced in the Ir and OF2 reactions were not observed when Ir was reacted with F2 or O2 in previous matrix-isolation investigations.3,19,20 This group of bands should be due to the different vibrational modes of iridium oxyfluoride molecules based on their identical behaviours throughout the experiments. Since the bands at 666.8 and 672.6 cm−1 in the neon matrix (657.5 and 663.6 cm−1, Ar-matrix) are located in the region of Ir–F stretching mode absorptions and show almost no 16/18O-isotopic shifts, they are assigned to the symmetric and antisymmetric F–Ir–F stretches of this new molecule, respectively. The higher wavenumber band at 984.1 cm−1 is shifted to 933.7 cm−1 with an 16O/18O frequency ratio of 1.0540, which is close to the value of diatomic IrO (1.0530),19 indicating a terminal Ir–O stretch. Hence, based on the observation of two F–Ir–F stretches and one terminal Ir–O, the new molecule is identified as OIrF2.
The assignment of OIrF2 is also further supported by quantum-chemical calculations. The OIrF2 molecule is predicted to have three infrared-active absorptions above 600 cm−1 at CCSD(T) and B3LYP levels (Table 1 and Tables S1, S2, ESI†). The observed band positions at 666.8 and 672.6 cm−1 in the neon matrix (657.5 and 663.6 cm−1, Ar-matrix) are consistent with the calculated symmetric and antisymmetric F–Ir–F stretches at 682.8 and 684.1 cm−1 and no 16/18O shifts were observed at the CCSD(T) level (670.5 and 671.7 cm−1, B3LYP level). Similar to most of the other metal oxydifluorides OMF2,11,12,15–17 the predicted harmonic vibrational wavenumbers of M–O are larger than the experimental fundamentals due to neglected anharmonicity.17 However, the calculated 16/18O-isotopic shifts due to Ir–O stretching at the CCSD(T) (Δν(16/18O) = 54.5 cm−1) and B3LYP levels (Δν(16/18O) = 55.2 cm−1) are in good agreement with the observed ones of (Δν(16/18O) = 50.4 cm−1) Ne-matrix and (Δν(16/18O) = 50.2 cm−1) Ar-matrix, further supporting the assignments for OIrF2.
The set of absorptions at 685.1 and 889.3 cm−1 in the spectra belongs to another new product molecule. Both bands were detected in the neon matrix after sample deposition and decreased dramatically on sample annealing, which can be assigned to the molecule FOIrF. Surprisingly, the corresponding bands of this product were not detected in solid argon. Similar to the small 16/18O-isotope shift of 3.3 cm−1 of the Pd–F stretching in molecule FOPdF,12 the band at 685.1 cm−1 exhibits a small but significant 16/18O-isotopic shift of 1.0 cm−1 and is associated with the Ir–F stretching band of this hypofluorite. Both the band positions and 16/18O-isotopic shifts show good agreement with the predicted wavenumbers at 698.1 cm−1 and the 16/18O-isotope shift of 0.2 cm−1 at the CCSD(T) level (Table 1 and Table S4, ESI†). Moreover, while the calculated wavenumber (938.5 cm−1) for Ir–O stretching in FIrOF is higher than the experimental value (889.3 cm−1), the predicted 16/18O shift of 47.9 cm−1 is in good agreement with the experimental value (46.4 cm−1), similar to OIrF2. The 16O–F and 18O–F vibrational stretches in FIr16/18OF are computed at 464.4 and 446.1 cm−1 at the CCSD(T) level, respectively, but our search for both bands was unsuccessful in the detection range of our FTIR spectrometer (MCT-B detector, 4000–450 cm−1).
Since OMF was the major product from the reaction of hot metal atoms with OF2,11–17 the formation of the elusive OIrF in the experiments should also be considered. In contrast to the very strongly IR-active Au–F stretch and the extremely weakly IR-active Au–O stretch in OAuF,11 the B3LYP calculation predicts two absorbances for OIrF at 735.4 and 1106.0 cm−1 with an approximate intensity ratio of 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 (Table 1 and Tables S5, S6, ESI†). The Ir–F band in OIrF is tentatively assigned to a weak band at 732.2 cm−1 with almost no 16/18O-isotopic shift in solid neon, which has a large blue-shift of 88.6 cm−1 relative to the stretching vibration in IrF (643.6 cm−1, Ne-matrix).3 However, the O–Ir fundamental of this species was expected to be located at 1106.0 (Δν(16/18O) = 58.5 cm−1) or 1053.4 cm−1 (Δν(16/18O) = 55.4 cm−1) from B3LYP and CCSD(T) calculations, respectively. It is reasonable that this mode was not observed in this work because it is even weaker than the 732.2 cm−1 band. Similar to the cases of OPtF and OAuF,11,12 attempts to detect the species OIrF in argon matrices failed, which is most likely due to the stronger Ar interactions with the formed intermediate species. In argon experiments, only the OIrF2 product was produced and identified by IR spectroscopy (Fig. S1, ESI†).
:3 (Table 1 and Tables S5, S6, ESI†). The Ir–F band in OIrF is tentatively assigned to a weak band at 732.2 cm−1 with almost no 16/18O-isotopic shift in solid neon, which has a large blue-shift of 88.6 cm−1 relative to the stretching vibration in IrF (643.6 cm−1, Ne-matrix).3 However, the O–Ir fundamental of this species was expected to be located at 1106.0 (Δν(16/18O) = 58.5 cm−1) or 1053.4 cm−1 (Δν(16/18O) = 55.4 cm−1) from B3LYP and CCSD(T) calculations, respectively. It is reasonable that this mode was not observed in this work because it is even weaker than the 732.2 cm−1 band. Similar to the cases of OPtF and OAuF,11,12 attempts to detect the species OIrF in argon matrices failed, which is most likely due to the stronger Ar interactions with the formed intermediate species. In argon experiments, only the OIrF2 product was produced and identified by IR spectroscopy (Fig. S1, ESI†).
To get further insights into the structures and electronic configurations of the new species, the iridium oxyfluorides OIrF, FOIrF and OIrF2 were calculated at the DFT and CCSD(T) levels in conjunction with scalar relativistic pseudopotentials. All possible spin states have been considered for each molecule, which further support the experimental spectral assignments for the new species. The optimized structures are presented in Fig. 2.
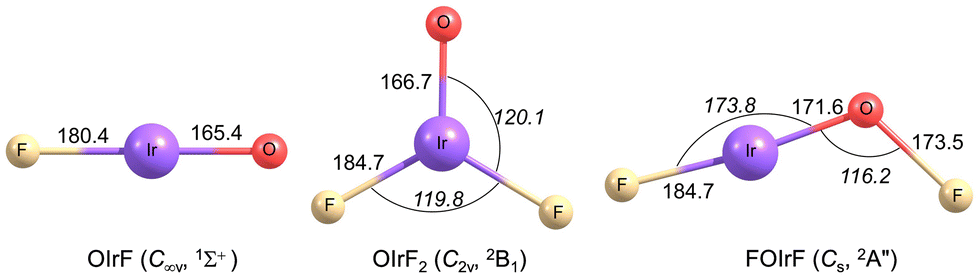 | ||
| Fig. 2 Optimized structures of OIrF, OIrF2 and FOIrF in their ground states at the CCSD(T)/aug-cc-pVTZ-PP level. Selected bond lengths in pm and angles in deg (in italics) are shown. | ||
A linear structure with a closed–shell singlet ground state for OIrF was obtained at both CCSD(T) and B3LYP levels (Table S5, ESI†). The calculated bond length of Ir–F in OIrF is 180.4 pm at the CCSD(T) level, shorter than that in free IrF (186.1 pm) and IrF2 (184.9 pm).3 Additionally, this metal–fluorine bond is much shorter than those in OPtF (189.7 pm) and OAuF (188.2 pm) (Fig. S2, ESI†).11,12 The M–F stretching frequencies in OMF follow the same trend and are confirmed by the experiments (ν(Ir–F) > ν(Au–F) > ν(Pt–F)). Moreover, the CCSD(T) M–O bond distances take large jumps from 165.4 pm for OIrF to 175.1 pm for OPtF to 181.0 pm for OAuF, and the corresponding O–M stretching wavenumbers in OMF decrease continuously from Ir (1053.4 cm−1), Pt (848.3 cm−1) to Au (767.9 cm−1).11,12 Neither of the O–M vibrational stretches in OMF (M = Ir, Pt and Au) were observed in the experiments (Table S7, ESI†).11,12 The large differences in M–O bond distances and stretching wavenumbers in OMF (M = Ir, Pt and Au) can be explained by multiple bond character for OIrF, terminal oxyl radical character for OPtF, and biradical character for OAuF.11,12 The OIrF orbital plots show (Fig. S3, ESI†) one σ bond and two π bonds formed by the O 2p and Ir 5d atomic orbitals, although oxygen has only two unpaired electrons. Similar to the case of the closed–shell OScF molecule,15 the terminal Ir–O bond in OIrF can be described formally as a triple bond consisting of a σ bond, a π bond and a dative bond where the oxygen 2p lone pair donates electrons into an empty Ir 5d orbital. Consistent with this character, the Ir–O bond distance (165.4 pm) in the OIrF molecule is close to the sum of proposed triple bond radii for iridium and oxygen ([IrO+], 161.5 pm).21
At the CCSD(T) level, the hypofluorite FOIrF in Cs symmetry is predicted to have a quartet ground state (4A′′), closely followed by 1.2 and 16.5 kJ mol−1 less stable 4A′/Cs and 2A′′/Cs structures, respectively. However, attempts to optimize a doublet structure for FOIrF failed at the B3LYP level, and the 2A′′/Cs state yields one imaginary frequency and the 2A′/Cs state undergoes isomerization to OIrF2 during optimization. While the energy difference favours the 4A′′/Cs structure, the vibrational wavenumbers and their 16/18O isotope shifts for the 2A′′/Cs state are in better agreement with our experimental values (Table S4, ESI†). The FIrOF molecule in the 2A′′/Cs state has a planar structure in which the FO and IrF moieties adopt a trans conformation with respect to the O–Ir bond (∠(Ir–O–F) = 116.2°; ∠(F–Ir–O) = 173.8°) at the CCSD(T) level (Fig. 2), similar to the structure of FOPdF.12
OIrF2 is optimized to have a 2B1 ground state with C2v symmetry. The 4B2/C2v state is 97.4 kJ mol−1 higher in energy than the ground state at the CCSD(T) level (Table S1, ESI†). The ∠(F–M–O) angles in OMF2 (M = Ir, Pt and Au) substantially decrease from Ir to Pt to Au at the CCSD(T) level (Fig. S4, ESI†). The M–O bond distance in OIrF2 (166.7 pm) is slightly longer than that in OIrF (165.4 pm), but is notably shorter than those in OPtF2 (172.8 pm) and OAuF2 (186.1 pm). The significant decrease in the M–O stretching wavenumber from OIrF2 and OPtF2 to OAuF2 is consistent with a significant increase in the M–O bond distance as the M–O bond transitions from a double bond for OIrF2 to a single bond with radical character on the O atom for OAuF2 (Fig. S4 and Table S8, ESI†).11,12 An analysis of the electronic structures for OIrF2 showed that the unpaired electron of the 2B1 state is found to reside in the Ir–O π*-antibonding molecular orbital (Fig. S5, ESI†). In contrast to OPtF2 and OAuF2 carrying large spin densities (≥0.96) at the oxygen atom,11,12 a much lower spin density at the oxygen atom was found in OIrF2, even slightly lower than that at the iridium center (Fig. 3). Comparing the M–F bond distances shows that there is a great increase from OIrF2 (184.6 pm) to OPtF2 (189.2 pm) and OAuF2 (190.0 pm) (Fig. S4, ESI†).
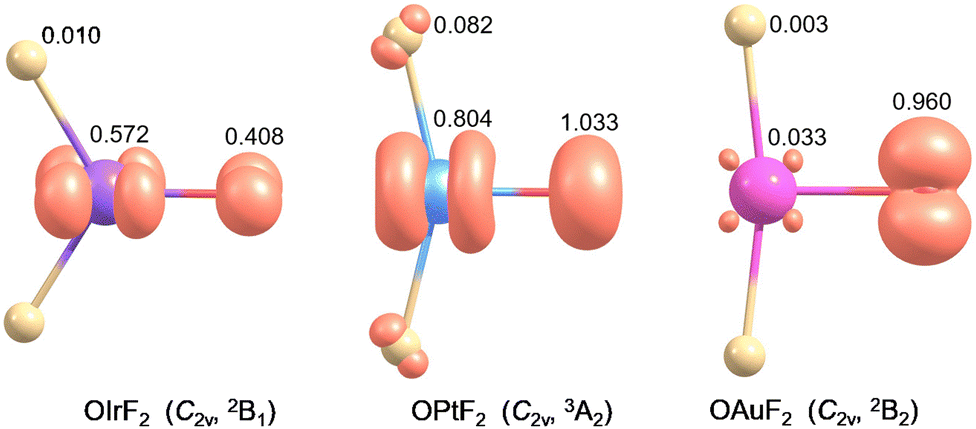 | ||
| Fig. 3 The spin density (iso-surface = 0.03 electron a.u.−3) of OIrF2, OPtF2 and OAuF2 obtained at the B3LYP/aug-cc-pVTZ-PP level. | ||
The thermochemical stability of the observed compounds was investigated (Table S9, ESI†). IR-laser ablation generates excited iridium atoms, which are expected to be inserted into the O–F bonds of OF2 to yield the hypofluorite FOIrF during deposition. The observed main product OIrF2 is likely to have formed from three channels. One is that OIrF2 is produced spontaneously from the reaction of iridium atoms with OF2, based on sample annealing, during which negligible activation energy is required and the reaction is calculated to be highly exothermic of −929.7 kJ mol−1. Another way of formation is that OIrF2 is formed by an exothermic rearrangement (FOIrF → OIrF2 ΔE = −389.0 kJ mol−1) of the initially formed FOIrF. The third way is that OIrF is initially formed from the reaction of iridium atoms and OF radicals, but it reacts rapidly further with F atoms to become OIrF2, which might explain why the OIrF bands are very weak in the experimental observations.
In conclusion, excited Ir atoms were made to react with OF2 to form the iridium oxyfluoride molecules OIrF, OIrF2 and FOIrF, which have been characterized using matrix-isolation IR spectroscopy and electronic structure calculations. The identification of these compounds was based on the metal–oxygen and metal–fluorine stretches with characteristic 16/18O isotopic shifts and support from electronic structure calculations at B3LYP and CCSD(T) levels. The B3LYP calculated molecular orbitals reveal that the linear OIrF molecule in the closed–shell singlet ground state exhibits triple Ir–O bond character. This is in accordance with the shorter bond distance and higher metal–oxygen stretching wavenumber compared with those of OPtF and OAuF. The molecule OIrF2 possesses a planar geometry with C2v symmetry in the 2B1 ground state, for which the unpaired electron is located mainly in the Ir–O π*-antibonding molecular orbital. A much lower spin-density contribution at the oxygen atom was found in OIrF2, which is in contrast to that in terminal oxyl radical species OPtF2 and OAuF2 with high spin densities at the oxygen atom, along with the sharply increasing M–O bond distance as well as the decreasing FMO bond angles from OIrF2 and OPtF2 to OAuF2.
The authors gratefully thank the Zentraleinrichtung für Datenverarbeitung (ZEDAT) of the Freie Universität Berlin for the allocation of computing resources. The authors thank for the continuous support provided by the ERC Project HighPotOx as well as the CRC 1349 (SFB 1349) Fluorine-Specific Interactions-Project-ID 387284271. Y. L. thanks the China Scholarship Council (PhD Program) for financial support.
Conflicts of interest
There are no conflicts to declare.Notes and references
- G. Wang, M. Zhou, J. T. Goettel, G. J. Schrobilgen, J. Su, J. Li, T. Schlöder and S. Riedel, Nature, 2014, 514, 475 CrossRef CAS PubMed.
- (a) M. Da Silva Santos, T. Stüker, M. Flach, O. S. Ablyasova, M. Timm, B. von Issendorff, K. Hirsch, V. Zamudio-Bayer, S. Riedel and J. T. Lau, Angew. Chem., Int. Ed., 2022, 61, e202207688 CrossRef CAS PubMed; (b) S. Riedel and M. Kaupp, Coord. Chem. Rev., 2009, 253, 606 CrossRef CAS; (c) T. Vogt, A. N. Fitch and J. K. Cockcroft, Science, 1994, 263, 1265 CrossRef CAS PubMed; (d) E. G. Hope, W. Levason and J. S. Ogden, J. Chem. Soc., Dalton Trans., 1988, 61 RSC.
- Y. Lu, Y. A. Tsegaw, A. Wodyński, L. Li, H. Beckers, M. Kaupp and S. Riedel, Chem. – Eur. J., 2022, 28, e202104005 CAS.
- (a) T. Drews, J. Supel, A. Hagenbach and K. Seppelt, Inorg. Chem., 2006, 45, 3782 CrossRef CAS PubMed; (b) A. D. Richardson, K. Hedberg and G. M. Lucier, Inorg. Chem., 2000, 39, 2787 CrossRef CAS PubMed; (c) R. T. Paine and L. B. Asprey, Inorg. Chem., 1975, 14, 1111 CrossRef CAS.
- O. Ruff and J. Fischer, Z. Anorg. Allg. Chem., 1929, 179, 161 CrossRef CAS.
- P. L. Robinson and G. J. Westland, J. Chem. Soc., 1956, 4481 RSC.
- S. Riedel and M. Kaupp, Angew. Chem., Int. Ed., 2006, 45, 3708 CrossRef CAS PubMed.
- H. Selig, W. A. Sunder, F. A. Disalvo and W. E. Falconer, J. Fluorine Chem., 1978, 11, 39 CrossRef CAS.
- M. A. Hepworth and P. L. Robinson, J. Inorg. Nucl. Chem., 1957, 4, 24 CrossRef CAS.
- R. C. Burns, T. A. O’Donnell and A. B. Waugh, J. Fluorine Chem., 1978, 12, 505 CrossRef CAS.
- L. Li, T. Stüker, S. Kieninger, D. Andrae, T. Schlöder, Y. Gong, L. Andrews, H. Beckers and S. Riedel, Nat. Commun., 2018, 9, 1267 CrossRef PubMed.
- L. Li, H. Beckers, T. Stüker, T. Lindič, T. Schlöder, D. Andrae and S. Riedel, Inorg. Chem. Front., 2021, 8, 1215 RSC.
- R. Wei, Z. T. Fang, M. Vasiliu, D. A. Dixon, L. Andrews and Y. Gong, Inorg. Chem., 2019, 58, 9796 CrossRef CAS PubMed.
- L. Andrews, X. Wang, Y. Gong, T. Schlöder, S. Riedel and M. J. Franger, Angew. Chem., Int. Ed., 2012, 51, 8235 CrossRef CAS PubMed.
- Y. Gong, L. Andrews and C. W. Bauschlicher, Chem. – Eur. J., 2012, 18, 12446 CrossRef CAS PubMed.
- Y. Gong, L. Andrews and C. W. Bauschlicher, Jr., J. Phys. Chem. A, 2012, 116, 10115 CrossRef CAS PubMed.
- R. Wei, Q. N. Li, Y. Gong, L. Andrews, Z. Fang, K. S. Thanthiriwatte, M. Vasiliu and D. A. Dixon, J. Phys. Chem. A, 2017, 121, 7603 CrossRef CAS PubMed.
- A. Arkell, R. R. Reinhard and L. P. Larson, J. Am. Chem. Soc., 1965, 87, 1016 CrossRef CAS.
- Y. Gong, M. Zhou, M. Kaupp and S. Riedel, Angew. Chem., Int. Ed., 2009, 121, 8019 CrossRef.
- A. Citra and L. Andrews, J. Phys. Chem. A, 1999, 103, 4182 CrossRef CAS.
- P. Pyykkö, S. Riedel and M. Patzschke, Chem. – Eur. J., 2005, 11, 3511 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3cc02216a |
| This journal is © The Royal Society of Chemistry 2023 |