
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIsotopic enrichment in lanthanide coordination complexes: contribution to single-molecule magnets and spin qudit insights
Fabrice
Pointillart
 *,
Kevin
Bernot
,
Boris
Le Guennic
and
Olivier
Cador
*,
Kevin
Bernot
,
Boris
Le Guennic
and
Olivier
Cador
Univ Rennes, INSA Rennes, CNRS, ISCR (Institut des Sciences Chimiques de Rennes) – UMR6226, 35000 Rennes, France. E-mail: fabrice.pointillart@univ-rennes1.fr
First published on 12th June 2023
Abstract
Lanthanide Single-Molecule Magnets (SMMs) fascinate the scientific community due to their plethora of potential applications ranging from data storage to spintronic devices and quantum computing. This review article proposes a comprehensive description of the influence of the nuclear spin, i.e. hyperfine interaction, on the magnetic properties of lanthanide SMMs and on quantum information processing of qudit. This influence is analysed for non-Kramers and Kramers lanthanide SMMs as well as for the electronic distribution of the electron in 4f orbitals i.e. oblate and prolate ions. Then the role of magnetic interactions in isotopically enriched polynuclear Dy(III) SMMs is discussed. Finally the possible effect of superhyperfine interaction due to the nuclear spin of elements originating from the surrounding of the lanthanide centre is analyzed. The effect of nuclear spin on the dynamics of the lanthanide SMMs is demonstrated using different techniques such as magnetometry, muon spectroscopy (μ-SR), and Mössbauer and Resonance Vibrational Spectroscopies.
 Fabrice Pointillart | Fabrice Pointillart obtained his PhD in physics and chemistry of materials from Pierre et Marie Curie University in 2005, supervised by Prof. Cyrille Train and Prof. Michel Verdaguer. After postdoctoral research at the University of Florence with Prof. Roberta Sessoli he joined the University of Rennes in 2007, as a CNRS researcher. He has been awarded the CNRS Bronze Medal (2014) and Young researcher Coordination Chemistry Division (2017). His current research interests are focused on multifunctional molecular materials based on lanthanides combining magnetic and (chir)optical properties for the observation of single molecule magnet behaviour, magneto-chiral dichroism and circularly polarized luminescence. |
 Kevin Bernot | Kevin Bernot received his PhD in Inorganic Chemistry in 2007 under the supervision of Prof. O. Guillou (INSA Rennes, France) and Prof. A. Caneschi (University of Florence, Italy) working on lanthanide-radical systems. After a post-doc under the supervision of Prof R. Sessoli dedicated to single-crystal magnetometry he joined INSA Rennes in 2008. His research activities focus on lanthanides coordination chemistry for the design of molecules, material and devices with luminescent and/or magnetic properties. He has been a full professor at INSA-Rennes since 2021. |
 Boris Le Guennic | Boris Le Guennic received his PhD in Chemistry from the University of Rennes in 2002. He then successively moved to the Universities of Erlangen, Buffalo, and Bonn for postdoctoral stays in the groups of Profs. Jochen Autschbach and Markus Reiher. In 2005, he was appointed as a CNRS researcher at ENS de Lyon (France). In 2011, he moved to the Chemical Sciences Institute of Rennes (University of Rennes, France) where he is currently a Senior CNRS researcher. He has been the head of the Inorganic Theoretical Chemistry Team since 2018. His research is devoted mainly to the application of quantum chemical methodologies for the interpretation of magnetic and (chir-)optical properties in transition metal and f-element complexes, with a particular emphasis on lanthanide-based Single Molecule Magnets. |
 Olivier Cador | The research activities of Olivier Cador started in 1994 in Bordeaux (France). From its beginnings he contributed to the development of molecular magnetism under different aspects. He mainly focused on the studies of the magnetic properties of systems with various dimensionalities, from nanoparticles to isolated molecules which show quantum behaviours, and finally to magnets with atypical behaviours. Since he was recruited by the University de Rennes 1 in 2003 its research studies have oriented toward multifunctional molecular-based materials. He oriented part of its activities on Single-Molecule Magnets (SMMs); more particularly those based on lanthanides. He has been a full Professor at Université de Rennes since 2017. |
1. Introduction
Frederick Soddy forged the term ‘isotope’ about 110 years ago and he received the Nobel prize of chemistry a few years later (1921) for his pioneering research in this field. Historically, the concepts of isotope and radioactivity have been intimately linked because for a given chemical element a large number of nuclides (isotopes) are radioactive and disintegrate. There are thousands of nuclides, natural and artificially produced, but only about 1000 with half-lives longer than one hour and only about 250 are observationally stable. There exist plenty of applications for radioactive nuclides (dating, nuclear power plants, nuclear medicine, tracers, markers, sterilization…) and they all use disintegration processes.1–5 Stable isotopes are mostly used for identification since natural isotopic abundances are fixed for each element. The masses of the nuclei are then the key factors used to identify the different isotopes, including isotope fractionation of light elements. Indeed, for light elements, variations in the mass number even of one unit affect significantly the mass of an atom and then its inertia. For heavier elements, this fractionation becomes less obvious due to smaller variations in the mass number.6,7As far as the electronic properties are concerned, such as conductivity and/or magnetism, few efforts have involved isotopic effects.8–11 Substitution of 12C by 13C changes the ordering temperature of superconductors while replacing 16O with 18O shifts the ferromagnetic Curie temperature by 20 K, from 210 K down to 190 K, respectively.12–21 Nevertheless, all these results are related to mass effects through electron–phonon interactions. When chemists go down the periodic table this mass number effect becomes less obvious but variation of the number of neutrons changes nuclear spins which can be coupled to the electrons through hyperfine coupling. EPR spectroscopy intensively uses hyperfine (or super-hyperfine) coupling to characterize species with single electrons22,23 regardless of the metal centred or organic radicals. Historically, isotopes in molecular magnetism24–31 have never really been discussed because it was commonly considered that the contribution of nuclear spins to the global magnetism is negligible when single electrons are present.
Molecular magnetism developed principally around the concept of molecular magnets which is, strictly speaking, an electronic property that allows a chemical object to retain magnetic memory in a zero external field (magnetic remanence). The first molecular magnets were based on the concept of magnetic ordering, and therefore on interactions between electronic magnetic moments that propagate in a 3D array.32,33 In the last thirty years the so-called Single-Molecule Magnets (SMMs) have emerged.34 Their specificity is to be able to store magnetic information at the molecular level thanks to axial magnetic anisotropy that hampers microscopic magnetization to flip freely between two opposite orientations. In this prolific field of research lanthanide-based complexes have the lead because they present the largest axial magnetic anisotropy and a wide variety of coordination polyhedron geometry.35 Magnetic anisotropy is monitored by the height of the energy barrier and the magnetic moment needs to jump to flip its orientation with a barrier height of about a few thousands of wavenumbers.36–38 This barrier can also be crossed through Quantum Tunnelling of Magnetization (QTM). This of course weakens the memory since it offers a possibility for the magnetic moment to relax rapidly from one side to the other but, at the same time, it gives an opportunity to deal with quantum mechanics. There are several origins of QTM but all of them are related to the admixture of quantum states that are on both sides of the energy barrier. Hyperfine coupling is one of them39,40 and at this stage one should look at the periodic table of (stable) isotopes, their nuclear spins and how such atoms have been used to produce SMMs. All the SMMs (in zero field) to date are made of 3d transition metal ions or lanthanides at the exception, to the best of our knowledge, of one Uranium-based complex.41 Most of the 3d-based zero-field SMMs are based on polynuclear complexes in which superexchange interactions take place. These interactions are orders of magnitude stronger than hyperfine coupling and therefore tend to dilute its effect. In order to avoid perturbation of the electronic system by exchange (or superexchange) interaction one should focus on SMMs based on single ions (the so-called Single-Ion Magnets even if the terminology can be a subject of debate). From a 3d ion point of view, only iron–based42,43 and cobalt-based44–46 zero-field SIM are known. Since natural cobalt is monoisotopic (100% 59Co) all the experimental data will correspond to the nuclear spin I = 7/2. This allowed the investigation of 59Co based-complexes by NMR showing the effect of magnetic nuclei on electronic effects as well as how the isotopic enrichment on the ligands affected the ligand field.47 For iron the situation is opposite since natural iron is dominated by nuclear spin free isotopes (91.7% 56Fe and 5.8% 54Fe) and only 2.1% of the nuclear spin active (I = 1/2) 57Fe. Then, in principle, one should be able to compare SMMs based on 57Fe with the ones based on 56Fe. This has been done on a polymetallic species and this pioneering work demonstrated that in the quantum regime the presence (or the absence) of nuclear spin at metal sites significantly modifies the relaxation of the magnetic moment of the molecule.48–51 Probably, the small number of mononuclear SMMs based on 3d ions limits the efforts on the effects of the isotopic enrichment on magnetic properties.42–46
The first reason why lanthanide ions are the best elements to investigate the effects of isotopic enrichment on the magnetic properties of mononuclear SMMs is the number of candidates that can be drafted. Even if we restrain ourselves to mononuclear species at oxidation number +3 that operate as a magnet in the absence of any external static field one can end with hundreds of chemical edifices in both coordination and organometallic chemistry. The second reason is that the lanthanide series offers more choice since obviously 14 ions are concerned and also because a simple look at a periodic table of stable isotopes is more coloured on the lanthanide line than on the 3d line. Table 1 lists the 14 stable lanthanide elements with the most abundant isotopes. Promethium apart, five elements are constituted of only one stable isotope (La, Pr, Tb, Ho and Tm) but all with nuclear spins different from zero. These elements can be used to probe hyperfine interaction from one single nucleus without any further isotopic separation. However, since the oxidation number +3 is by far the most stable there is no hyperfine coupling for La(III) since it is diamagnetic and, interestingly, the four others are non Kramers ions. One must consider that Lu(III) enters in the same category as La(III) with no single electrons. Eu(III) multiplet ground state (4f6, 7F0) is also diamagnetic but because orbital and spin moments compensate each other's. Ce(III) is made of two different isotopes but both are nuclear spin free with no hyperfine coupling as well. With Gd it is possible to tune the nuclear spin between 0 and 3/2 but Gd(III) is a spin pure ion with no orbital contribution and therefore with no magnetic anisotropy.
| Element | Kramers/Non-Kramersa | Z | A = mass number, in brackets the natural abundance and the nuclear spin number for Ab |
|---|---|---|---|
| a Trivalent ions. b Nuclear spin abundance rounded to the %. | |||
| La | — | 57 | A = 139 (100%, I = 7/2) |
| Ce | K | 58 | A = 140 (89%, I = 0), 142 (11%, I = 0) |
| Pr | NK | 59 | A = 141 (100%, I = 5/2) |
| Nd | K | 60 | A = 142 (27%, I = 0), A = 143 (12%, I = 7/2), A = 144 (24%, I = 0), A = 145 (8%, I = 7/2), A = 146 (17%, I = 0), A = 148 (6%, I = 0), A = 150 (6%, I = 0) |
| Sm | K | 62 | A = 144 (3%, I = 0), A = 147 (15%, I = 7/2), A = 148 (11%, I = 0), A = 149 (14%, I = 7/2), A = 150 (7%, I = 0), A = 152 (27%, I = 0), A = 154 (23%, I = 0), |
| Eu | NK | 63 | A = 151 (48%, I = 5/2), 153 (52%, I = 5/2), |
| Gd | K | 64 | A = 154 (2%, I = 0), A = 155 (15%, I = 3/2), A = 156 (21%, I = 0), A = 157 (16%, I = 3/2), A = 158 (25%, I = 0), A = 160 (22%, I = 0), |
| Tb | NK | 65 | A = 159 (100%, I = 3/2) |
| Dy | K | 66 | A = 160 (2%, I = 0), A = 161 (19%, I = 5/2), A = 162 (25%, I = 0), A = 163 (25%, I = 5/2), A = 164 (28%, I = 0), |
| Ho | NK | 67 | A = 165 (100%, I = 7/2) |
| Er | K | 68 | A = 164 (2%, I = 0), A = 166 (33%, I = 0), A = 167 (23%, I = 7/2), A = 168 (27%, I = 0), A = 170 (15%, I = 0), |
| Tm | NK | 69 | A = 169 (100%, I = 1/2) |
| Yb | K | 70 | A = 170 (3%, I = 0), A = 171 (14%, I = 1/2), A = 172 (22%, I = 0), A = 173 (16%, I = 5/2), A = 174 (32%, I = 0), A = 176 (13%, I = 0), |
| Lu | — | 71 | A = 175 (97%, I = 7/2), A = 176 (3%, I = 7) |
This element is therefore out of the launch window of zero-field mononuclear SMMs. In the end, five Ln(III) elements possess an electronic magnetic moment with substantial magnetic anisotropy and stable isotopes with various nuclear spins: Nd, Sm, Dy, Er and Yb. These five elements are Kramers ions as far as the oxidation number +3 is retained. In the next sections we will survey the recent works dealing with isotopic features of SMMs without being exhaustive but focusing on key examples. We will target Kramers and non-Kramers ions, ligand's nuclei isotopic substitution and finally address the perspective of isotopologues for qudits applications.
2. Slow relaxation of magnetic moment as a signature of single-molecule magnet
By definition a magnet operates in the absence of an external stimulus and so, in a zero external static field. The opening of the magnetic hysteresis loop at zero field is the ultimate signature of the magnetic memory with the magnetic moment that can be oriented in two opposite directions. However, even if the terminology “permanent magnet” is widely employed one should keep in mind that the magnetic memory depends on time scale and temperature scale. This memory effect disappears at very long time scales and so cannot be viewed as a static property but more in a dynamic perspective. In this dynamic framework, the time it takes for the magnetic moment to align parallel to the external field corresponds to the relaxation time. Magnetometry in an alternating magnetic field provides an experimental tool that gives access to this kind of relaxation time in a relatively large time window: in the absence of relaxation the magnetization is in-phase and follows instantaneously the oscillation of the external field, while in the presence of relaxation processes the magnetization is delayed and an out-of-phase component appears. There are basically four different processes that govern the relaxation time τ of the magnetic moments in matter that depend on temperature (T) and external magnetic field (H).53
In the previous equation the first three terms involve coupling between spin and phonons and are temperature dependent while the last term is temperature independent and refers to the tunnelling of the magnetization between two opposite directions of the magnetic moment. The direct process implies the application of an external static field and does not operate in zero field. The thermal variations of the relaxation time in a zero external dc field is represented on Scheme 1b with the separated contribution of Raman, Orbach and QTM terms. The fastest process wins and so, QTM takes the lead at the lowest temperature and Orbach at the highest, and eventually Raman in an intermediate regime. Since hyperfine coupling between nuclear spins and electronic spins contributes to QTM we differentiated on Scheme 1b contributions from a hypothetic nuclear spin free system with I = 0 (slow) or I ≠ 0 (fast). At this stage it is important to mention that the coupling between spin and nuclei can occur through hyperfine coupling but also through superhyperfine coupling with the nuclei of ligands (Scheme 1a) for which selection of isotopes is also possible. The application of an external static field suppresses the possibility for the magnetic moment to tunnel through the barrier so the relaxation becomes much slower in this field. This creates butterfly shaped hysteresis curves (Scheme 1c) with an opening at the origin that can be modulated by playing with isotopes.
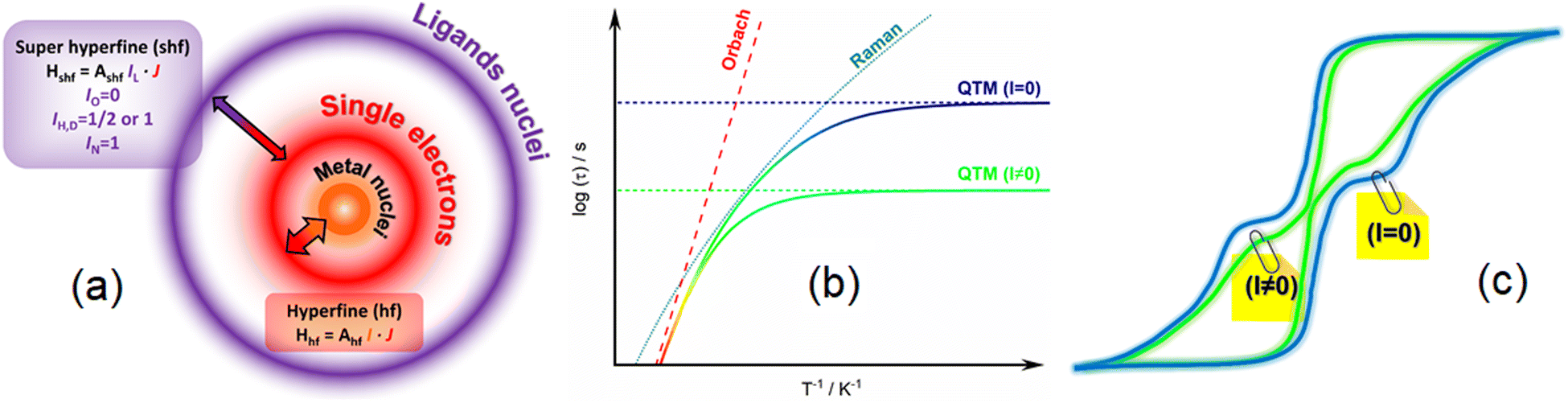 | ||
| Scheme 1 (a) Scheme of the hyperfine interaction (hf) between the metal nuclei (I, orange) and the single electrons and super-hyperfine interaction (shf) between the single electrons and the ligands nuclei (IL, purple). (b) Thermal variation of the relaxation time with representation of the three main relaxation mechanisms: Orbach (red), Raman (turquoise blue) and QTM for I = 0 (blue) and I ≠ 0 (green). (c) Scheme of hysteresis loops for two isotopologues with I = 0 (blue) and I ≠ 0 (green). | ||
3. Hyperfine interactions influence slow magnetic relaxation
3.1. Naturally pure isotopic non-Kramers lanthanide SMM
The first magnetic investigation of the nuclear spin effect was performed in 2005 by W. Wernsdorfer and colleagues54 on the bis(phthalocyaninato)terbium anion [Pc2Tb]− (1) (Pc = dianion of phthalocyanine) deeply explored for its remarkable SMM properties some years before.55–57 The latter coordination complex displayed magnetic relaxation occurring through the thermally activated Orbach process above 25 K.58,59 Clear evidence of QTM was highlighted on a diluted sample of 1 in an Y(III) diamagnetic isomorphous matrix (1@Y) to cancel the intermolecular dipolar interactions.60 This investigation was possible thanks to magnetic hysteresis measurements at very low temperature (0.04 K) using the micro-SQUID technique (SQUID = Superconducting QUantum Interference Device)61,62 with the magnetic field aligned parallel to the easy axis of magnetization by the transverse field method.63 The QTM in 1@Y resulted in a staircase-like structure of the hysteresis loop and was attributed to the interaction between the electronic spin (4f8, J = 6) and the nuclear spin (I = 3/2). Thus, the Zeeman diagram for the eight ∣Jz〉∣Iz〉 states created from the combination of the Jz = ±6 doublets and I = 3/2 quartets gave level intersections at 13 different magnetic-field positions which are all observed in the experimental hysteresis loop. The 13 step positions were reproduced by using Ahf = 0.0173 cm−1 and P = 0.010 cm−1 where Ahf is the constant of the hyperfine interaction (AhfJI) and P is the constant of the nuclear quadrupole interaction term (P{Iz2 − 1/3I(I + 1)}).
 | ||
| Fig. 1 (a) Representation of the molecule of 3. Iodine anion, free oxophosphine and solvent molecules of crystallization are omitted for clarity. Ho: light green, P: orange, O: red, C: grey, H: white. (b) Zeeman energy diagram (green lines) and field dependence of the relaxation time determined by AC measurements at 8 K for 3 (blue) and 3@Y (red). Grey circles represent the avoided level crossing due to the hyperfine interactions. Inset: Zoom view at zero field for the lowest doublet with (green) and without (purple) hyperfine interactions. (c) Normalized magnetic hysteresis loops for a single crystal of 3. (d) Derivatives of the hysteresis loops with predicted magnetic field positions of the avoided level crossings (black dashed lines). Adapted with permission from ref. 39. Copyright 2017, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim. | ||
In 2021, M.-L. Tong and colleagues reported a 3d4f heterobimetallic SMM of formula HoNi5(quinha)5F2(dfpy)10](ClO4)·2EtOH (4) where H2quinha = quinaldichydroxamic acid and dfpy = 3,5-difluoropyridine.65 The Ho(III) is surrounded by the metallacrown and two axial fluoride ions leading to a similar D5h pentagonal-bipyramidal geometry than in 3. 4 displayed slow magnetic relaxation with a record energy barrier of 577 cm−1 for a Ho(III) based SMM. The introduction of the [15-MCNi-5] metallacrown induced incoherent QTM with field dependent oscillation of the barrier height and of the relaxation time. This field dependence was attributed to the joint effect of the hyperfine interactions at the Ho(III) ion and the exchange coupling with surrounding Ni(II) ions. This phenomenon is promising for the manipulation of memory spin qubits.
Hyperfine interactions have also been studied for single atom magnets (SAMs). Especially Holmium SAMs have been deposited on the MgO(100) surface.66 When adsorbed on this surface, holmium atoms have a J = 8 ground state manifold which is split by the crystal field created by the on-top oxygen adsorption site.67 X-ray magnetic circular dichroism (XMCD) allowed the measurement of long-lived magnetization lifetimes. Nevertheless, the zero-field stability of Ho on the MgO surface required the use of antiferromagnetic spin-polarized scanning tunnelling microscopy (SP-STM).67 This stability allowed identification of the ground state Jz = ±8 and the existence of avoided level crossings that couple the positive and negative spin manifolds and allow transitions via Landau–Zener tunnelling.
The magnetic investigations at very low temperature of single atom and single ion magnets involving Tb(III) and Ho(III) non-Kramers ions, demonstrated that the non-zero nuclear spin provides significant hyperfine interaction. In the case of non-Kramer ions, the hyperfine interaction participates to cancel the QTM at zero field while it provokes avoided level crossings leading to field-induced QTM. Finally, the Zeeman diagram can be established if the following three components are considered: crystal field, hyperfine interaction and nuclear quadrupole interaction.
3.2. Isotopically enriched Kramers lanthanide SMM
 | ||
| Fig. 2 (a) Representation of the molecule of 5, n-hexane molecule of crystallization is omitted for clarity. (b) Thermal variation of the relaxation time of the magnetization for 5 (black dots), 1615 (blue dots), 1625 (orange dots), 1635 (green dots), 1645 (red dots), 1615@Y (blue circles), 1625@Y (orange circles), 1635@Y (green circles) and 1645@Y (red circles) in a zero applied magnetic field in the temperature range of 2–15 K. Full lines are a guide for the eyes. (c) The normalized magnetic hysteresis loop at 0.46 K and a sweep rate of 16 Oe s−1 for 1625 (orange line) and 1635 (green line). In the inset is a zoomed view of the origin. Adapted from ref. 69–72. (d) Thermal variation of the relaxation times extracted from ac susceptibility (full symbols) and μSR (empty symbols) for 1615 (red) and 1645 (blue). | ||
This study showed that the trend seen using standard magnetometry is confirmed via a local probe technique that captures fine details of the spin dynamics.
A step forward in the hyperfine interaction effect on SMM magnetic dynamics was achieved with the highlighting of the hyperfine constant (Ahf) effect. Indeed, the study of the two pure isotopes 1615@Y and 1635@Y both with I = 5/2 but with different Ahf values demonstrated that 1615@Y relaxed four times slower than 1635@Y because Ahf for 1615 < Ahf1635.72 The consequences of hyperfine interactions on the magnetic relaxation are visible on the hysteresis loops measured at 0.46 K with a magnetic bistability in zero-magnetic field for the nuclear spin free 1625@Y compound while the hysteresis loop is closed for 1635@Y (Fig. 2c). One could also observe a staircase-like structure of the hysteresis loop for 1635@Y because the hyperfine interactions provoke avoided level crossings leading to field-induced QTM.
This hyperfine interaction effect on dynamics of SMM was also confirmed on a different molecular system involving the same Dy(tta)3 unit associated with a different TTF-based ligand (4,5-bis(methylthio)-tetrathiafulvalene-2-(benzothiazole-2pyridine)).73
Finally, the two nuclear spin isomers of the magnetically diluted Dy(III) analogue of 1 were studied in case of the tetraethyl ammonium salt.74 While both Et4N[163Dy@YPc2] (1636@Y) and Et4N[163Dy@YPc2] (1646@Y) displayed slow magnetic relaxation, the nuclear spin free 1646@Y is designed for SMM applications whereas 1636@Y could be used as a multilevel nuclear spin qubit (called qudit with d = 6) for quantum information processing (see part 4 of this review).
In 2019, the isotopic enrichment of lanthanide SMMs allowed us to suggest the existence of another phenomenon at the origin of fast relaxation of the magnetization at zero applied magnetic field.75 The three SMMs of formula [Dy(tBuO)Cl(THF)5][BPh4] (7),76 [K(18-crown-6-ether)(THF)2][Dy(BIPM)2] (8)77 (BIPM = C{PPh2NSiMe3}2) and [Dy(Cpttt)2][B(C6F5)4] (9) (Cpttt = C5H2tBu3-1,2,4)37 were selected because of their high energy barrier. The hysteresis loops of the natural isotope samples, magnetically diluted samples and 164Dy isotope and diluted samples were measured and compared showing that neither dipolar interactions nor hyperfine interaction are solely responsible for the QTM at zero field. The authors demonstrated that the vibrational modes that most impact the first Dy(III) coordination sphere, are active at energies which follow the trend 7 < 8 < 9. These energies can be correlated with the coercive field values of the complexes. Therefore, the authors suggested that molecular vibrations could be at the origin of the fast relaxation of the magnetization at zero field through a so-called “vibrational QTM”.
The same year, the possibility of synthesizing pure isotope lanthanide SMM opens the way to the use of spectroscopies such as Synchrotron Mössbauer Spectroscopy (SMS). Local moments and spin-relaxation dynamics were already determined in iron-containing SMMs using 57Fe Mössbauer spectroscopy78–81 but this technique has not yet been used to investigate the magnetic properties of lanthanide SMMs. V. Schünemann, A. K. Powell and colleagues carried out 161Dy time-domain SMS using 161Dy nucleus and hyperfine interaction as a probe for the dysprosium magnetization.82 Thus the authors were able to determine the magnetic hyperfine field of the isotopologue [161Dy(Cy3PO)2(H2O)5]Br3·2(Cy3PO)·2H2O·2EtOH (10) (Cy3PO = tricyclohexylphosphine oxide)83 with a value of 582.3(5) T which is significantly larger than that of the free-ion Dy(III) with MJ = ±15/2 as the ground state.84–86 The difference was attributed to the Fermi contact between the s and 4f electrons of the Dy(III) ion which is influenced by the coordinating Cy3PO and H2O ligands.
The previously described work of N. Chilton and colleagues75 suggested that the SMM performances limitation could be due to the fast relaxation at zero applied magnetic field due to molecular vibrations. Thus, a probe of such vibrations is required to investigate their role in the magnetic relaxation. In this context, the incorporation of a Mössbauer active nucleus such as 161Dy allowed the detection of vibrational properties of a material using the Nuclear Resonance Vibrational Spectroscopy (NRVS) also referring to Nuclear Inelastic Scattering (NIS) or Nuclear Resonant Inelastic X-ray Scattering (NRIXS).87–89 NRVS detects all the vibrational modes that include a Dy displacement90 and it was applied to compound 10.91 Extraction of the vibration modes gave some vibration with significant intensity at the energies corresponding to the electronic transitions leading to the conclusion that no relaxation process occurred involving electronic transitions in resonance with molecular vibrations with dysprosium displacements.
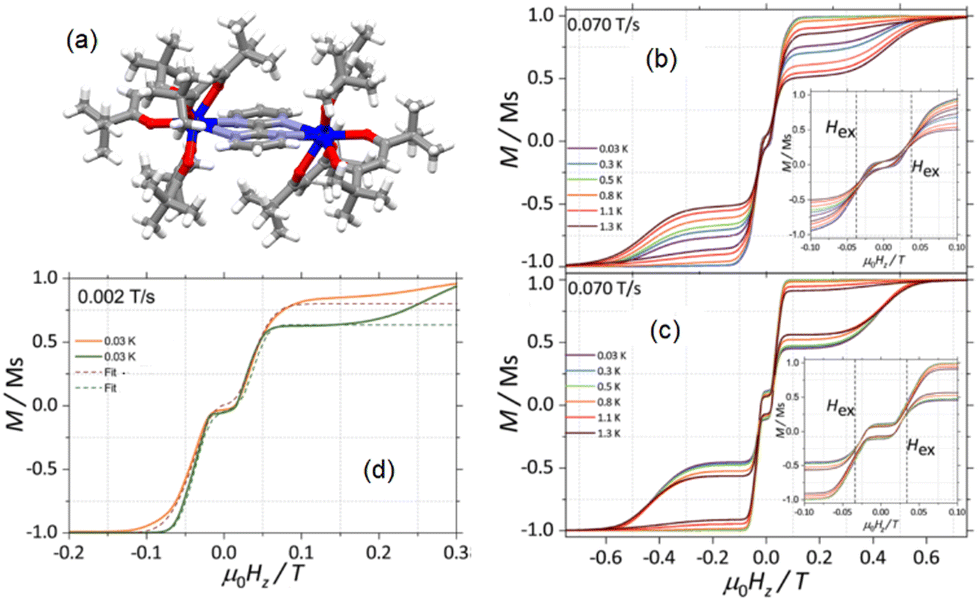 | ||
| Fig. 3 (a) X-ray structure on the single crystal for 12. Field dependence of the magnetization for 16312 (b) and 16412 (c) in the temperature range of 0.03–1.3 K at a field sweep rate of 0.070 T s−1. In the insets: zoomed-in view of the origin region. (d) Fits of the magnetization curves of 16312 (orange) and 16412 (green). Adapted from ref. 94. | ||
These observations are in contradiction with what was observed with the dinuclear compound 11. Nevertheless, it is worth noticing that the AF interaction in 12 is about four times weaker than in 11. A similar tunnelling rate was determined in both isotopologues by fitting the hysteresis loops (Fig. 3d) confirming that the QTM is not solely responsible for the magnetic relaxation of the isotopologues. For the first time in the study of the hyperfine coupling effect on the magnetic relaxation of a SMM, the authors proposed an explanation for the difference of opening of the hysteresis loop in an applied magnetic field. They proposed that the splitting of the larger spectrum of hyperfine state for 16312 due to the splitting of I = 5/2 in eight states under the hyperfine coupling94 allows a better coupling to acoustic phonons. In other words, the efficiency of the under-barrier relaxation process such as the direct relaxation process is increased due to the modulation of the electric field of the magnetic ion through phonons22,95,96 leading to a reduced hysteresis loop for 16312 than for 16412 (Fig. 3d).
Previously, the nuclear spin driven QTM phenomena was studied using a theoretical model for both terbium and dysprosium dinuclear complexes i.e. quadruple-decker phthalocyanine complexes without isotopic enrichment.97 The authors demonstrated that under zero applied magnetic field, the f–f dipolar interactions supressed the QTMs while at 1000 Oe they enhanced the nuclear spin driven QTM leading to faster magnetic relaxation for dinuclear rather than mononuclear complexes.
The last Dy(III)-based system investigated by isotopic enrichment is a 3d4f heterobimetallic complex in which the Dy(III) centre is associated with a diamagnetic Zn(II) both coordinated to a Schiff base ligand (L2 = N,N′-bis(3-methoxysalicylidene)phenylene-1,2-diamine). This compound of formula [Zn2(L2)2DyCl3]·2H2O (13) highlighted good SMM performances thanks to the polarization of the phenoxido oxygen atoms and favouring axial distribution of the electron distribution.98–101
The magnetic dynamics of the isotopically enriched compounds 16313, 13 and 16213 were studied (Fig. 4a) as well as the magnetic hysteresis loops for magnetically diluted analogues 16313@Y, 13@Y and 16213@Y (Fig. 4b).102 The investigation of the thermal dependence of the relaxation time of the magnetization under zero applied magnetic field demonstrated that the nuclear spin free isotopologue (16213) relaxed slightly slower than 16313 with the natural compound relaxing with a relaxation time at 2 K in between those of the two pure isotopes as already observed for isotopic enrichment of pure mononuclear lanthanide compounds.69,72 The isotopic enrichment effect was also visible under an applied dc field on the opening of the hysteresis loops for magnetically diluted samples (Fig. 4b). Indeed, the opening of the hysteresis loop for 16213@Y is wider than for the natural element which is itself wider than for 16313@Y. This behaviour was attributed to the possible interaction between the MJ sublevels and the matrix as proposed by M. Ruben and colleagues93 in the previously related example of this review. It is worth noticing that this explanation could be applied to compound 5 for which the effect of the isotopic enrichment on the opening of the hysteresis loop under applied magnetic field was also observed.
 | ||
| Fig. 4 (a) Temperature dependence of the magnetic relaxation time for 16213 (red), 13 (black) and 16313 (blue). Full and dashed lines are the best fitted curves and separated Raman (purple), Orbach (green) and QTM (colour of the compound) contributions, respectively. (b) Normalized magnetic hysteresis loops at 0.5 K at a sweep rate of 16 Oe s−1 for 16213@Y (orange), 13@Y (grey) and 16313@Y (light blue). Insets: Zoomed-in view of the origin region. Adapted from ref. 102. | ||
 | ||
| Fig. 5 Thermal variation of the magnetic relaxation time for the three isotopologues 17314@Y (blue), 14@Y (black) and 17414@Y (red) in a zero applied magnetic field and temperature range 2–6 K. Full and dashed lines are the best-fitted curves and separated Orbach (green), Raman (orange) and QTM (colour of the compounds) contributions, respectively. Reprinted with permission from ref. 104. Copyright 2021, American Chemical Society. | ||
 | ||
| Fig. 6 (a) The molecular structure of 15·yH2O, sodium and water inorganic network was omitted for clarity. (b) Thermal dependence of the relaxation time of the magnetization for Na9[ErxY1−x(W5O18)2]·yH2O in a zero applied magnetic field. Adapted from ref. 112. | ||
The authors hypothesized that the collective emission of a single phonon by many spins could play a decisive role in the explanation of the observed trend.117 These observations were confirmed when some of us studied the solvato-modulation of the magnetic bistability in 15·yH2O (y ranges from 35 to 6).118 The nuclear spin-free 16615·35H2O was also studied showing that it relaxes faster than the nuclear spin active 16715·35H2O following the opposite trend observed by F. Luis and colleagues. In both papers, a considerable broadening of the out of phase component of the magnetic susceptibility in the quantum tunnelling regime was observed, a sign of the coexistence of multiple spin–lattice relaxation processes associated to the splitting of the ground state doublet MJ = ±13/2 into 2I + 1 states (I = 7/2) due to the hyperfine interaction. It is worth noticing that an isotopic enrichment investigation was realized only on this system and nowadays no definitive explanations have been given.
3.3. “Super-hyperfine interactions” influence on slow magnetic relaxation
We have discussed the hyperfine coupling between the electronic and nuclear spins of the lanthanide centre has been discussed in isotopically enriched SMMs as well as its effect on the slow magnetic relaxation in zero applied magnetic field (QTM) and applied magnetic field (direct processes) for mono-, bi- and heterobi–nuclear complexes involving oblate Dy(III) ion and prolate Yb(III) and Er(III) ions. Nevertheless, the interaction between the electronic spins of the metal and the nuclear spins of the first coordination sphere (super-hyperfine interaction) could have also a significant influence on the magnetic dynamic of the SMM. This effect was demonstrated in 2000 on the well-known Fe8 cluster by performing a partial deuteration (DFe8) and by comparing the relaxation rates.48 It is only very recently that similar investigation was carried out for a lanthanide SMM.119 The mononuclear Ho(III) SMM 3 was partially deuterated by replacing the five equatorial H2O coordinated molecules with five D2O (D3). The authors demonstrated that the difference in the nuclear spin value for protium (I = 1/2) and deuterium (I = 1) and the presence of significant super-hyperfine interaction allowed the formation of super-hyperfine electronic energy spectra which differ for 3 and D3 leading to sizeable differences in their magnetic relaxation rates (Fig. 7).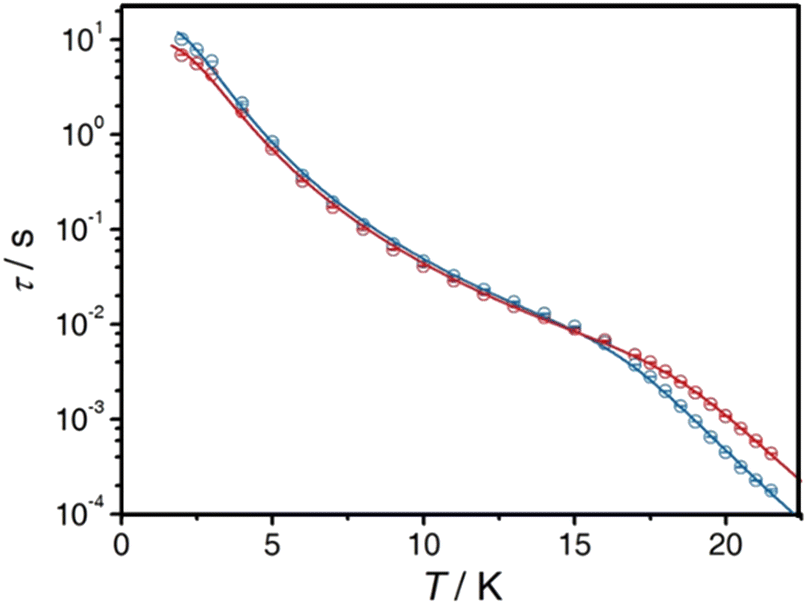 | ||
| Fig. 7 Thermal dependence of the magnetic relaxation time for 3 (blue) and D3 (red). Reprinted with permission from ref. 119. Copyright 2021, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim. | ||
4. Future development of isotopologues
In the last few years, the role of the hyperfine interaction was investigated in spin Qubits and Qudits. In this context, M. Ruben, W. Wernsdorfer and colleagues reviewed how the first mononuclear lanthanide SMM ((TbPc2)−, 1) can be a molecular nuclear spin qudit (d = 4) which displays all the requirements to perform quantum operations i.e. isolation, initialization, read out, long coherence times and manipulations.120Another mononuclear field-induced SMM namely Yb(trensal) (Fig. 8a) (H3trensal = 2,2′,2′′-tris(salicylideneimino)trimethylamine) (16) was studied for its potential applications in quantum information processing.121 The dilution of 16 in a diamagnetic isomorphous Lu(III) matrix (16@Lu) allowed the observation of a Hahn echo by oriented single-crystal echo-detected field-swept (EDFS) X-band pulsed EPR experiment.122 The phase memory time and the spin–lattice relaxation time were found to be independent from the isotopes because of the applied magnetic field used for the EDFS pulsed EPR experiment leading to magnetic relaxation through Raman-like processes and not through the electron nuclear spins coupling. Transient nutation experiments highlighted more than 70 Rabi oscillations of the spin echo (Fig. 8b) proving that this molecular system could be used as an electronic Qubit (Seff = 1/2). Soon after, 16@Lu was isotopically enriched in a nuclear spin active 173Yb isotope (I = 5/2) (17316@Lu).123 NMR measurements concluded that 17316@Lu molecules behave as a nuclear qudit (I = 5/2, d = 6) coupled to an electronic qubit (Seff = 1/2). Strong hyperfine interaction and long coherence times are key parameters to rapidly and coherently manipulate the nuclear qudit. 17316@Lu can encode a qubit thanks to its nuclear qudit with embedded basic quantum error correction (Fig. 8c).
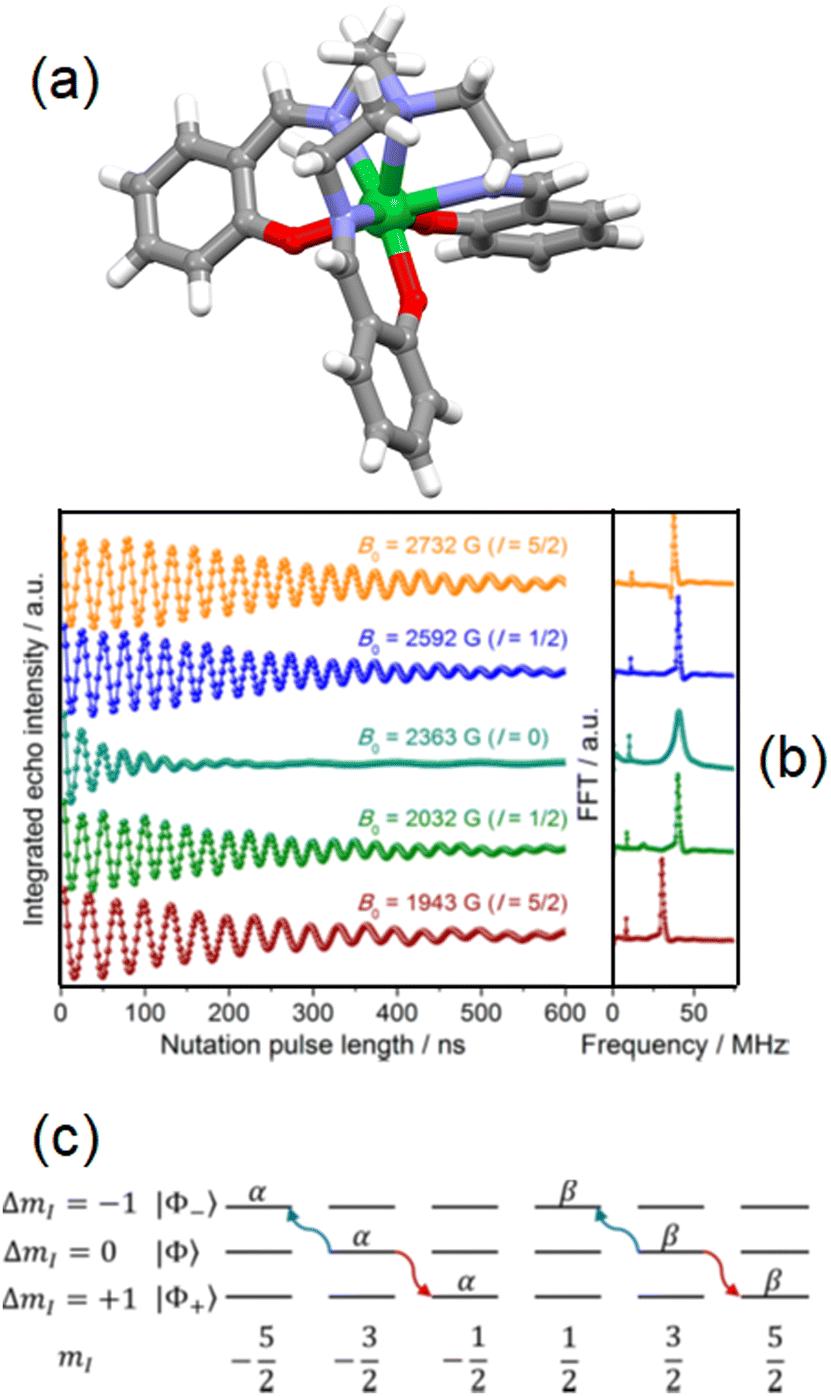 | ||
| Fig. 8 (a) Molecular structure of 16. (b) Echo intensity at selected field positions (left) and its corresponding Fourier transforms (right). (c) Encoding of a qubit protected from amplitude shift errors in the nuclear levels of 17316@Lu. Reprinted with permission from ref. 121. Copyright 2016, American Chemical Society and from ref. 122. Copyright 2018, American Chemical Society. | ||
Finally, W. Wernsdorfer and M. Ruben124 proposed the elaboration of multinuclear lanthanide metal complexes to reach the aim of scalability of the available Hilbert space which is expressed by the qudit-dimension. Indeed, the indirect coupling between the multiple nuclear spin units via their electron spins and bridging ligands will exponentially extend the Hilbert space available for Quantum Information Processing.
5. Conclusion and outlook
This feature article summarized the studies dealing with the isotopic enrichments in lanthanide single-molecule magnets and their effects on physical properties such as slow magnetic relaxation, magnetic memories or spin qudits behaviour. This overview covered Kramers and non-Kramers lanthanide SMMs with oblate and prolate electronic distributions for both mononuclear and polynuclear complexes.Some key points can be taken away considering the advances reviewed herein:
1. Pure nuclear spin active (I ≠ 0) Tb(III) and Ho(III) based SMMs allowed the observation of interactions between electronic and nuclear spins (hyperfine coupling) and determination of the hyperfine interaction constant (Ahf).
2. Dynamics study of two 161Dy and 164Dy SMM isotopologues demonstrated the effect of hyperfine coupling on QTM and magnetic bistability at 0 Oe while the thermally activated relaxation processes (Raman and Orbach) were not affected. The magnetic bistability at H ≠ 0 was also altered due to the coupling between I and the acoustic phonons. Not only has the nuclear spin value had an influence on the QTM but also the hyperfine coupling constant.
3. The isotopic enrichment combined with magnetic dilution made it possible to highlight the existence of another phenomenon at the origin of fast magnetic relaxation at 0 Oe called vibrational QTM induced by molecular vibrations.
4. Extrapolations of isotopic enrichment to polynuclear complexes demonstrated that an effect on slow magnetic relaxation can be observed in the case of magnetic interaction of a magnitude weaker than the hyperfine interactions.
5. Isotopic enrichment for prolate Yb(III)-based SMMs led to a similar trend on slow magnetic relaxation time than for Dy(III)-based SMMs while studies on Er(III)-based SMM concluded an opposite trend.
6. Isotopic enrichment of the coordinated ligands (for example H2O vs. D2O) could have an influence on the slow magnetic relaxation. The origin could be attributed to the effect of the changing mass on molecular vibrations or/and to superhyperfine interactions. However the process is not fully understood at this time.
7. Two SMMs isotopologues can find potential applications for magnetic memory or multi-level nuclear spin qudits depending if they are nuclear spin active or not, respectively.
The field of isotopic enrichment in lanthanide SMMs has been growing for less than 10 years, however, it already demonstrated that pure isotopes of SMMs could have potential applications in high-density storage with nuclear spin-free SMMs. Indeed, such SMMs have reduced QTM efficiency and they permitted to highlight current limitations of SMM performances because of the fast magnetic relaxation through molecular vibrations. The latter might be managed by designing specific molecular systems. Additionally, the isotopic enrichment of SMMs, especially pure nuclear spin active isotopologues, opened the route to potential applications in Quantum Information Processing because the electronic qubit can be coupled to nuclear qudit. The topic of isotopic enrichment of lanthanide SMMs is a perfect example of the need for both chemists and physicists to work hand in hand.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Université de Rennes, CNRS, and the European Commission through the ERC-CoG 725184 MULTIPROSMM (project n. 725184).Notes and references
- E. Maxwell, Phys. Rev., 1950, 78, 477 CrossRef CAS.
- R. Blinc, J. Phys. Chem. Solids, 1960, 13, 204–211 CrossRef CAS.
- N. W. Ashcroft and M. Cyrot, Europhys. Lett., 1993, 23, 605–608 CrossRef CAS.
- A. Greco and R. Zeyher, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 60, 1296–1302 CrossRef CAS.
- C. C. Tong and K. C. Hwang, J. Phys. Chem. C, 2007, 111, 3490–3494 CrossRef CAS.
- T. Kosone, C. Kachi-Terajima, C. Kanadani, T. Saito and T. Kitazawa, Chem. Lett., 2008, 37, 754–755 CrossRef CAS.
- K. Hosoya, T. Kitazawa, M. Takahashi, M. Takeda, J. F. Meunier, G. Molnar and A. Bousseksou, Phys. Chem. Chem. Phys., 2003, 5, 1682–1688 RSC.
- T. W. Ebbesen, J. S. Tsai, K. Tanigaki, J. Tabuchi, Y. Shimakawa, Y. Kubo, I. Hirosawa and J. Mizuki, Nature, 1992, 355, 620–622 CrossRef CAS.
- J. P. Franck, I. Isaac, W. Chen, J. Chrzanowski and J. C. Irwin, Phys. Rev. B: Condens. Matter Mater. Phys., 1998, 58, 5189 CrossRef CAS.
- M. S. Fuhrer, K. Cherrey, A. Zettl, M. L. Cohen and V. H. Crespi, Phys. Rev. Lett., 1999, 83, 404 CrossRef CAS.
- G.-M. Zhao, K. Conder, H. Keller and K. A. Müller, Nature, 1996, 381, 676–678 CrossRef CAS.
- S. Cantekin, D. W. R. Balkenende, M. M. J. Smulders, A. R. A. Palmans and E. W. Meijer, Nat. Chem., 2011, 3, 42–46 CrossRef CAS PubMed.
- A. Shengelaya, G. M. Zhao, C. M. Aegerter, K. Conder, I. M. Savic and H. Keller, Phys. Rev. Lett., 1999, 83, 5142–5145 CrossRef CAS.
- P. W. Fowler and W. T. Raynes, Mol. Phys., 1981, 43, 65–82 CrossRef CAS.
- M. H. Rummeli, M. Loffler, C. Kramberger, F. Simon, F. Fulop, O. Jost, R. Schonfelder, A. Gruneis, T. Gemming, W. Pompe, B. Buchner and T. Pichler, J. Phys. Chem. C, 2007, 111, 4094–4098 CrossRef.
- R. Watanabe, H. Ishiyama, G. Maruta and S. Takeda, Polyhedron, 2005, 24, 2599–2606 CrossRef CAS.
- R. H. Liu, T. Wu, G. Wu, H. Chen, X. F. Wang, Y. L. Xie, J. J. Ying, Y. J. Yan, Q. J. Li, B. C. Shi, W. S. Chu, Z. Y. Wu and X. H. Chen, Nature, 2009, 459, 64–67 CrossRef CAS PubMed.
- K. A. Muller, Z. Phys. B: Condens. Matter, 1990, 80, 193–201 CrossRef.
- D. D. Lawrie, J. P. Franck and C. T. Lin, Phys. C, 1998, 297, 59–63 CrossRef CAS.
- R. Ofer, G. Bazalitsky, A. Kanigel, A. Keren, A. Auerbach, J. S. Lord and A. Amato, Phys. Rev. B: Condens. Matter Mater. Phys., 2006, 74, 220508R CrossRef.
- R. Giraud, W. Wernsdorfer, A. M. Tkachuk, D. Mailly and B. Barbara, Phys. Rev. Lett., 2001, 87, 5 CrossRef PubMed.
- A. Abragam and B. Bleaney, Electron Paramagnetic Resonance of transition Ions, Clarendon Press, Oxford, 1970 Search PubMed.
- J. A. Weil and J. R. Bolton, Electron Paramagnetic Resonance: Elementary Theory and Practical Applications, John Wiley & Sons, Inc, 2006 Search PubMed.
- T. Kubo, T. Goto, T. Koshiba, K. Takeda and K. Awaga, Phys. Rev. B: Condens. Matter Mater. Phys., 2002, 65, 224425 CrossRef.
- T. Kubo, A. Nagano, T. Goto, K. Takeda and K. Awaga, J. Magn. Magn. Mater., 2004, 272, E727–E729 CrossRef.
- T. Kubo, T. Koshiba, T. Goto, A. Oyamada, Y. Fujii, K. Takeda and K. Awaga, Physica B, 2001, 294, 310–313 CrossRef.
- R. M. Achey, P. L. Kuhns, A. P. Reyes, W. G. Moulton and N. S. Dalal, Phys. Rev. B: Condens. Matter Mater. Phys., 2001, 64, 064420 CrossRef.
- R. Blinc, B. Zalar, A. Gregorovic, D. Arcon, Z. Kutnjak, C. Filipic, A. Levstik, R. M. Achey and N. S. Dalal, Phys. Rev. B: Condens. Matter Mater. Phys., 2003, 67, 094401 CrossRef.
- A. G. Harter, N. E. Chakov, B. Roberts, R. Achey, A. Reyes, P. Kuhns, G. Christou and N. S. Dalal, Inorg. Chem., 2005, 44, 2122–2124 CrossRef CAS PubMed.
- J. Dolinsek, D. Arcon, R. Blinc, P. Vonlanthen, J. L. Gavilano, H. R. Ott, R. M. Achey and N. S. Dalal, Europhys. Lett., 1998, 42, 691–694 CrossRef.
- H. Kohlmann, F. Werner, K. Yvon, G. Hilscher, M. Reissner and G. J. Cuello, Chem. – Eur. J., 2007, 13, 4178–4186 CrossRef CAS PubMed.
- S. Turner, O. Kahn and L. Rabardel, J. Am. Chem. Soc., 1996, 118, 6428–6432 CrossRef CAS.
- J. S. Miller, J. C. Calabrese, A. J. Epstein, R. W. Bigelow, J. H. Zhang and W. M. Reiff, J. Chem. Soc., Chem. Commun., 1986, 1026–1028 RSC.
- R. Sessoli, D. Gatteschi, A. Caneschi and M. A. Novak, Nature, 1993, 365, 141–143 CrossRef CAS.
- D. N. Woodruff, R. E. P. Winpenny and R. A. Layfield, Chem. Rev., 2013, 113, 5110–5148 CrossRef CAS PubMed.
- F.-S. Guo, B. M. Day, Y.-C. Chen, M.-L. Tong, A. Mansikkamäki and R. A. Layfield, Angew. Chem., Int. Ed., 2017, 56, 11445–11449 CrossRef CAS PubMed.
- C. A. P. Goodwin, F. Ortu, D. Reta, N. F. Chilton and D. P. Mills, Nature, 2017, 548, 439–442 CrossRef CAS PubMed.
- C. A. Gould, K. R. McClain, D. Reta, J. G. C. Kragskow, D. A. Marchiori, E. Lachman, E.-S. Choi, J. G. Analytis, R. D. Britt, N. F. Chilton, B. G. Harvey and J. R. Long, Science, 2022, 375, 198–202 CrossRef CAS PubMed.
- Y.-C. Chen, J.-L. Liu, W. Wernsdorfer, D. Liu, L. F. Chibotaru, X.-M. Chen and M.-L. Tong, Angew. Chem., Int. Ed., 2017, 56, 4996–5000 CrossRef CAS PubMed.
- N. Ishikawa, M. Sugita and W. Wernsdorfer, Angew. Chem., Int. Ed., 2005, 44, 2931–2935 CrossRef CAS PubMed.
- J. D. Rinehart and J. R. Long, J. Am. Chem. Soc., 2009, 131, 12558–12559 CrossRef CAS PubMed.
- S. Mossin, B. L. Tran, D. Adhikari, M. Pink, F. W. Heinemann, J. Sutter, R. K. Szilagyi, K. Meyer and D. J. Mindiola, J. Am. Chem. Soc., 2012, 134, 13651–13661 CrossRef CAS PubMed.
- J. M. Zadrozny, D. J. Xiao, M. Atanasov, G. J. Long, F. Grandjean, F. Neese and J. R. Long, Nat. Chem., 2013, 5, 577–581 CrossRef CAS PubMed.
- X.-N. Yao, J.-Z. Du, Y.-Q. Zhang, X.-B. Leng, M.-W. Yang, S.-D. Jiang, Z.-X. Wang, Z.-W. Ouyang, L. Deng, B.-W. Wang and S. Gao, J. Am. Chem. Soc., 2017, 139, 373–380 CrossRef CAS PubMed.
- J. M. Zadrozny and J. R. Long, J. Am. Chem. Soc., 2011, 133, 20732–20734 CrossRef CAS PubMed.
- M. S. Fataftah, J. M. Zadrozny, D. M. Rogers and D. E. Freedman, Inorg. Chem., 2014, 53, 10716–10721 CrossRef CAS PubMed.
- T. M. Ozvat, A. K. Rappé and J. M. Zadrozny, Inorg. Chem., 2022, 61, 778–785 CrossRef CAS PubMed.
- W. Wernsdorfer, A. Caneschi, R. Sessoli, D. Gatteschi, A. Cornia, V. Villar and C. Paulsen, Phys. Rev. Lett., 2000, 84, 2765 CrossRef PubMed.
- M. Evangelisti, F. Luis, F. L. Mettes, R. Sessoli and L. J. de Jongh, Phys. Rev. Lett., 2005, 95, 227206 CrossRef CAS PubMed.
- Y. Furukawa, S. Kawakami, K. Aizawa, K. Kumagai and F. Borsa, Polyhedron, 2003, 22, 2277–2279 CrossRef CAS.
- Y. Furukawa, Y. Hatanaka, K. Kumagai, S. H. Baek and F. Borsa, in Low Temperature Physics, Pts a and B, ed. Y. Takano, S. P. Hershfield, P. J. Hirschfeld and A. M. Goldman, Amer Inst Physics, Melville, 2006, vol. 850, pp. 1147–1148 Search PubMed.
- J. Meija, T. B. Coplen, M. Berglund, W. A. Brand, P. D. Bièvre, M. Gröning, N. E. Holden, J. Irrgeher, R. D. Loss, T. Walczyk and T. Prohaska, Pure Appl. Chem., 2016, 88, 293–306 CrossRef CAS.
- S. T. Liddle and J. van Slageren, Chem. Soc. Rev., 2015, 44, 6655–6669 RSC.
- N. Ishikawa, M. Sugita and W. Wernsdorfer, Angew. Chem., Int. Ed., 2005, 44, 2931–2935 CrossRef CAS PubMed.
- A. De Cian, M. Moussavi, J. Fischer and R. Weiss, Inorg. Chem., 1985, 24, 3162–3167 CrossRef CAS.
- H. Konami, M. Hatano and A. Tajiri, Chem. Phys. Lett., 1989, 160, 163 CrossRef CAS.
- N. Ishikawa, M. Sugita, T. Okubo, N. Tanaka, T. Iino and Y. Kaizu, Inorg. Chem., 2003, 42, 2440 CrossRef CAS PubMed.
- N. Ishikawa, M. Sugita, T. Ishikawa, S. Koshihara and Y. Kaizu, J. Am. Chem. Soc., 2003, 125, 8694–8695 CrossRef CAS PubMed.
- N. Ishikawa, M. Sugita, T. Ishikawa, S. Koshihara and Y. Kaizu, J. Phys. Chem. B, 2004, 108, 11265 CrossRef CAS.
- F. Habib, P.-H. Lin, J. Long, I. Korobkov, W. Wernsdorfer and M. Murugesu, J. Am. Chem. Soc., 2011, 133, 8830–8833 CrossRef CAS PubMed.
- W. Wernsdorfer, Adv. Chem. Phys., 2001, 118, 99 CAS.
- W. Wernsdorfer, Supercond. Sci. Technol., 2009, 22, 064013 CrossRef.
- W. Wernsdorfer, N. E. Chakov and G. Christou, Phys. Rev. B: Condens. Matter Mater. Phys., 2004, 70, 132413 CrossRef.
- N. Ishikawa, M. Sugita and W. Wernsdorfer, J. Am. Chem. Soc., 2005, 127, 3650–3651 CrossRef CAS PubMed.
- S.-G. Wu, Z.-Y. Ruan, G.-Z. Huang, J.-Y. Zheng, V. Vieru, G. Taran, J. Wang, Y.-C. Chen, J.-L. Liu, L. T. A. Ho, L. F. Chibotaru, W. Wernsdorfer, X.-M. Chen and M.-L. Tong, Chem, 2021, 7, 982–992 CAS.
- F. Donati, S. Rusponi, S. Stepanow, C. Wäckerlin, A. Singha, L. Persichetti, R. Baltic, K. Diller, F. Patthey, E. Fernandes, J. Dreiser, Z. Sljivancanin, K. Kummer, C. Nistor, P. Gambardella and H. Brune, Science, 2016, 352, 318–321 CrossRef CAS PubMed.
- E. Fernandes, F. Donati, F. Patthey, S. Stavric, Z. Sljivancanin and H. Brune, Phys. Rev. B: Condens. Matter Mater. Phys., 2017, 96, 045419 CrossRef.
- P. R. Forrester, F. Patthey, E. Fernandes, D. P. Sblendorio, H. Brune and F. D. Natterer, Phys. Rev. B: Condens. Matter Mater. Phys., 2019, 100, 180405 CrossRef CAS.
- F. Pointillart, K. Bernot, S. Golhen, B. Le Guennic, T. Guizouarn, L. Ouahab and O. Cador, Angew. Chem., Int. Ed., 2015, 54, 1504–1507 CrossRef CAS PubMed.
- T. T. da Cunha, J. Jung, M.-E. Boulon, G. Campo, F. Pointillart, C. L. M. Pereira, B. Le Guennic, O. Cador, K. Bernot, F. Pineider, S. Golhen and L. Ouahab, J. Am. Chem. Soc., 2013, 135, 16332–16335 CrossRef CAS PubMed.
- L. Tesi, Z. Salman, I. Cimatti, F. Pointillart, K. Bernot, M. Mannini and R. Sessoli, Chem. Commun., 2018, 54, 7826 RSC.
- J. Flores Gonzalez, F. Pointillart and O. Cador, Inorg. Chem. Front., 2019, 6, 1081–1085 RSC.
- Y. Kishi, F. Pointillart, B. Lefeuvre, F. Riobé, B. Le Guennic, S. Golhen, O. Cador, O. Maury, H. Fujiwara and L. Ouahab, Chem. Commun., 2017, 53, 3575–3578 RSC.
- E. Moreno-Pineda, M. Damjanovic, O. Fuhr, W. Wernsdorfer and M. Ruben, Angew. Chem., Int. Ed., 2017, 56, 9915–9919 CrossRef CAS PubMed.
- F. Ortu, D. Reta, Y.-S. Ding, C. A. P. Goodwin, M. P. Gregson, E. J. L. McInnes, R. E. P. Winpenny, Y.-Z. Zheng, S. T. Liddle, D. P. Mills and N. F. Chilton, Dalton Trans., 2019, 48, 8541–8545 RSC.
- J. Liu, Y.-C. Chen, J.-L. Liu, V. Vieru, L. Ungur, J.-H. Jia, L. F. Chibotaru, Y. Lan, W. Wernsdorfer, S. Gao, X.-M. Chen and M.-L. Tong, J. Am. Chem. Soc., 2016, 138, 5441–5450 CrossRef CAS PubMed.
- M. Gregson, N. F. Chilton, A.-M. Ariciu, F. Tuna, I. F. Crowe, W. Lewis, A. J. Blake, D. Collison, E. J. L. McInnes, R. E. P. Winpenny and S. T. Liddle, Chem. Sci., 2016, 7, 155–165 RSC.
- S. F. M. Schmidt, C. Koo, V. Mereacre, J. Park, D. W. Heermann, V. Kataev, C. E. Anson, D. Prodius, G. Novitchi, R. Klingeler and A. K. Powell, Inorg. Chem., 2017, 56, 4796 CrossRef CAS PubMed.
- A. Cini, M. Mannini, F. Totti, M. Fittipaldi, G. Spina, A. Chumakov, R. Rgffer, A. Cornia and R. Sessoli, Nat. Commun., 2018, 9, 480 CrossRef PubMed.
- V. Mereacre, A. Baniodeh, C. E. Anson and A. K. Powell, J. Am. Chem. Soc., 2011, 133, 15335–15337 CrossRef CAS PubMed.
- J. M. Zadrozny, D. J. Xiao, J. R. Long, M. Atanasov, F. Neese, F. Grandjean and G. J. Long, Inorg. Chem., 2013, 52, 13123–13131 CrossRef CAS PubMed.
- L. Scherthan, S. F. M. Schmidt, H. Auerbach, T. Hochdörffer, J. A. Wolny, W. Bi, J. Zhao, M. Y. Hu, T. Toellner, E. E. Alp, D. E. Brown, C. E. Anson, A. K. Powell and V. Schünemann, Angew. Chem., Int. Ed., 2019, 58, 3444–3449 CrossRef CAS PubMed.
- Y.-C. Chen, J.-L. Liu, L. Ungur, J. Liu, Q.-W. Li, L.-F. Wang, Z.-P. Ni, L. F. Chibotaru, X.-M. Chen and M.-L. Tong, J. Am. Chem. Soc., 2016, 138, 2829–2837 CrossRef CAS PubMed.
- J. P. Evans, G. A. Stewart, J. M. Cadogan, W. D. Hutchison, E. E. Mitchell and J. E. Downes, Phys. Rev. B: Condens. Matter Mater. Phys., 2017, 95, 54431 CrossRef.
- W. D. Hutchison, G. A. Stewart, J. M. Cadogan, A. Princep, R. Stewart and D. H. Ryan, AIP Adv., 2017, 7, 055702 CrossRef.
- P. C. M. Gubbens, A. M. van der Kraan and K. H. J. Buschow, Hyperfine Interact., 1988, 40, 389 CrossRef CAS.
- M. Seto, Y. Yoda, S. Kikuta, X. W. Zhang and M. Ando, Phys. Rev. Lett., 1995, 74, 3828–3831 CrossRef CAS PubMed.
- W. Sturhahn, T. S. Toellner, E. E. Alp, X. Zhang, M. Ando, Y. Yoda, S. Kikuta, M. Seto, C. W. Kimball and B. Dabrowski, Phys. Rev. Lett., 1995, 74, 3832 CrossRef CAS PubMed.
- E. Gerdau and H. DeWaard, Hyperfine Interact., 1999, 123, 0 CrossRef.
- R. Rchlsberger, Nuclear Condensed Matter Physics with Synchrotron Radiation, Springer STMP 208, Berlin Heidelberg, 2004 Search PubMed.
- L. Scherthan, R. F. Pfleger, H. Auerbach, T. Hochdörffer, J. A. Wolny, W. Bi, J. Zhao, M. Y. Hu, E. E. Alp, C. E. Anson, R. Diller, A. K. Powell and V. Schünemann, Angew. Chem., Int. Ed., 2020, 59, 8818–8822 CrossRef CAS PubMed.
- G. Huang, X. Yi, J. Jung, O. Guillou, O. Cador, F. Pointillart, B. Le Guennic and K. Bernot, Eur. J. Inorg. Chem., 2018, 326–332 CrossRef CAS.
- W. Yu, F. Schramm, E. Moreno-Pineda, Y. Lan, O. Fuhr, J. Chen, H. Isshiki, W. Wernsdorfer, W. Wulfhekel and M. Ruben, Beilstein J. Nanotechnol., 2016, 7, 126–137 CrossRef CAS PubMed.
- E. Moreno-Pineda, G. Taran, W. Werndorfer and M. Ruben, Chem. Sci., 2019, 10, 5138–5145 RSC.
- M. Ganzhorn, S. Klyatskaya, M. Ruben and W. Wernsdorfer, Nat. Nanotechnol., 2013, 8, 165–169 CrossRef CAS PubMed.
- M. Ganzhorn, S. Klyatskaya, M. Ruben and W. Wernsdorfer, Nat. Commun., 2016, 7, 11443 CrossRef CAS PubMed.
- T. Fukuda, K. Matsumura and N. Ishikawa, J. Phys. Chem. A, 2013, 117, 10447–10454 CrossRef CAS PubMed.
- J. Long, R. Vallat, R. A. S. Ferreira, L. D. Carlos, F. A. Almeida Paz, Y. Guari and J. Larionova, Chem. Commun., 2012, 48, 9974–9976 RSC.
- A. Upadhyay, S. K. Singh, C. Das, R. Mondol, S. K. Langley, K. S. Murray, G. Rajaraman and M. Shanmugam, Chem. Commun., 2014, 50, 8838–8841 RSC.
- A. Upadhyay, C. Das, S. Vaidya, S. K. Singh, T. Gupta, R. Mondol, S. K. Langley, K. S. Murray, G. Rajaraman and M. Shanmugam, Chem. – Eur. J., 2017, 23, 4903–4916 CrossRef CAS PubMed.
- W.-B. Sun, P.-F. Yan, S.-D. Jiang, B.-W. Wang, Y.-Q. Zhang, H.-F. Li, P. Chen, Z.-M. Wang and S. Gao, Chem. Sci., 2016, 7, 684–691 RSC.
- J. Flores Gonzalez, B. Lefeuvre, B. Degraeve, O. Cador and F. Pointillart, Dalton Trans., 2021, 50, 11466–11471 RSC.
- F. Pointillart, O. Cador, B. Le Guennic and L. Ouahab, Coord. Chem. Rev., 2017, 346, 150–175 CrossRef CAS.
- J. Flores Gonzalez, H. Douib, B. Le Guennic, F. Pointillart and O. Cador, Inorg. Chem., 2021, 60, 540–544 CrossRef CAS PubMed.
- S.-D. Jiang, B.-W. Wang, H.-L. Sun, Z.-M. Wang and S. Gao, J. Am. Chem. Soc., 2011, 133, 4730–4733 CrossRef CAS PubMed.
- K. R. Meihaus and J. R. Long, J. Am. Chem. Soc., 2013, 135, 17952–17957 CrossRef CAS PubMed.
- L. Ungur, J. J. Le Roy, I. Korobkov, M. Murugesu and L. F. Chibotaru, Angew. Chem., 2014, 53, 4413–4417 ( Angew. Chem. , 2014 , 126 , 4502–4506 ) CrossRef CAS PubMed.
- P. Zhang, L. Zhang, C. Wang, S. Xue, S.-Y. Lin and J. Tang, J. Am. Chem. Soc., 2014, 136, 4484–4487 CrossRef CAS PubMed.
- A. J. Brown, D. Pinkowicz, M. R. Saber and K. R. Dunbar, Angew. Chem., Int. Ed., 2015, 54, 5864–5868 ( Angew. Chem. , 2015 , 127 , 5962–5966 ) CrossRef CAS PubMed.
- M. A. AlDamen, S. Cardona-Serra, J. M. Clemente-Juan, E. Coronado, A. Gaita-Ariño, C. Martí-Gastaldo, F. Luis and O. Montero, Inorg. Chem., 2009, 48, 3467–3479 CrossRef CAS PubMed.
- M. A. AlDamen, J. M. Clemente-Juan, E. Coronado, C. Martí-Gastaldo and A. Gaita-Ariño, J. Am. Chem. Soc., 2008, 130, 8874–8875 CrossRef CAS PubMed.
- F. Luis, M. J. Martinez-Perez, O. Montero, E. Coronado, S. Cardonna-Serra, C. Marti-Gastaldo, J. M. Clemente-Juan, J. Sesé, D. Drung and T. Schurig, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 82, 060403 CrossRef.
- R. de and L. Kronig, Physica, 1939, 6, 240 CrossRef.
- J. H. Van Vleck, Phys. Rev., 1940, 57, 426 CrossRef CAS.
- R. Orbach, Proc. R. Soc. London, 1961, A264, 458 Search PubMed.
- G. H. Larson and C. D. Jeffries, Phys. Rev. B: Condens. Matter Mater. Phys., 1966, 145, 311 CrossRef CAS.
- H. N. V. Temperley, Proc. Cambridge Philos. Soc., 1939, 35, 256 CrossRef CAS.
- J. Flores Gonzalez, V. Montigaud, V. Dorcet, K. Bernot, B. Le Guennic, F. Pointillart and O. Cador, Chem. – Eur. J., 2021, 27, 10160–10169 CrossRef CAS PubMed.
- Y. Liu, L. T. A. Ho, G.-Z. Huang, Y.-C. Chen, L. Ungur, J.-L. Liu and M.-L. Tong, Angew. Chem., Int. Ed., 2021, 60, 27282–27287 CrossRef CAS PubMed.
- E. Moreno-Pineda, C. Godfrin, F. Balestro, W. Wernsdorfer and M. Ruben, Chem. Soc. Rev., 2018, 47, 501–513 RSC.
- K. S. Pedersen, J. Dreiser, H. Weihe, R. Sibille, H. V. Johannesen, M. A. Sorensen, B. E. Nielsen, M. Sigrist, H. Mutka, S. Rols, J. Bendix and S. Piligkos, Inorg. Chem., 2015, 54, 7600–7606 CrossRef CAS PubMed.
- K. S. Pedersen, A.-M. Ariciu, S. McAdams, H. Weihe, J. Bendix, F. Tuna and S. Piligkos, J. Am. Chem. Soc., 2016, 138, 5801–5804 CrossRef CAS PubMed.
- R. Hussain, G. Allodi, A. Chiesa, E. Garlatti, D. Mitcov, A. Konstantatos, K. S. Pedersen, R. De Renzi, S. Piligkos and S. Carreta, J. Am. Chem. Soc., 2018, 140, 9814–9818 CrossRef CAS PubMed.
- W. Wernsdorfer and M. Ruben, Adv. Mater., 2019, 31, 1806687 CrossRef PubMed.
| This journal is © The Royal Society of Chemistry 2023 |