
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis and structural evaluation of closed-shell folded and open-shell twisted hexabenzo[5.6.7]quinarene†‡
Tomohiko
Nishiuchi
 *ab,
Kazuyuki
Uchida
a and
Takashi
Kubo
*ab
*ab,
Kazuyuki
Uchida
a and
Takashi
Kubo
*ab
aDepartment of Chemistry, Graduate School of Science, Osaka University, Toyonaka, Osaka 560-0043, Japan. E-mail: nishiuchit13@chem.sci.osaka-u.ac.jp; kubo@chem.sci.osaka-u.ac.jp
bInnovative Catalysis Science Division, Institute for Open and Transdisciplinary Research Initiatives, (ICS-OTRI), Osaka University, Suita, Osaka 565-0871, Japan
First published on 26th May 2023
Abstract
We describe the synthesis and characterization of hexabenzo[5.6.7]quinarene, which is composed of an anthraquinodimethane (AQD) central core that is end-capped with fluorenyl and dibenzosuberenyl groups. Due to strong intramolecular spin–spin interaction through the central AQD unit, this compound is obtained as a stable folded form. Isolation of the stable twisted dication by oxidation and reduction of the dication yields a twisted triplet species, which thermally reverts to the folded form. This is a spin-switching system based on a combination of chemical oxidation/reduction and thermal stimulation.
Thiele's hydrocarbon (TH)1 is composed of two triphenyl methyl radical units and is sometimes considered as a pre-biradical molecule due to the quinoidal π-skeleton. However, the strong intramolecular spin–spin interaction of its two unpaired electrons results in a stable closed-shell species with a large singlet-triplet energy gap (ΔES–T, Fig. 1a). With the recent research interest in biradicaloid2 and multiradicaloid π-systems3 for the development of molecule-based spintronic materials, there is growing attention towards spin-state controllable molecules by external stimuli such as heating,4 photoirradiation,5 or mechanical stress.6 To introduce an open-shell nature to the TH skeleton, an anthraquinodimethane (AQD) unit has been employed as its central core to create steric repulsion between the terminal aromatic rings and the central AQD unit. This facilitates transformation to a biradical form by twisting the ethylene units (Fig. 1b). In fact, several AQD-extended TH derivatives end-capped with tricyclic aromatic units such as fluorenyl (FLU),7 9,10-dihydro-10,10-dimethyl-9-anthryl(DMA),8 or dibenzosuberenyl (DBS)9 units have been synthesized as systems that can be switched from a stable closed-shell ground state to an open-shell biradical state via heating or photoirradiation. Typically, AQD-extended TH derivatives have symmetric terminal π-skeletons, which leads to homolytic cleavage of the π-bond orbitals on the C
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bonds, resulting in a biradical state.10 On the other hand, it is also interesting to investigate whether heterolytic π-bond cleavage of CC bonds, leading to a zwitterionic state, is possible or not in a biradical system. Several zwitterionic biradicaloids with heteroatom(s) embedded in their skeletons have been synthesized.11 Due to the difference in electronegativity between carbon and heteroatoms, electron-transfer between them can occur, changing the electronic state from biradical to zwitterion and vice versa. Moreover, several neutral planar hydrocarbon π-systems including five- and seven-membered rings have been suggested to exhibit a resonance structure between a biradical state and an electron-polarized state.12
C bonds, resulting in a biradical state.10 On the other hand, it is also interesting to investigate whether heterolytic π-bond cleavage of CC bonds, leading to a zwitterionic state, is possible or not in a biradical system. Several zwitterionic biradicaloids with heteroatom(s) embedded in their skeletons have been synthesized.11 Due to the difference in electronegativity between carbon and heteroatoms, electron-transfer between them can occur, changing the electronic state from biradical to zwitterion and vice versa. Moreover, several neutral planar hydrocarbon π-systems including five- and seven-membered rings have been suggested to exhibit a resonance structure between a biradical state and an electron-polarized state.12
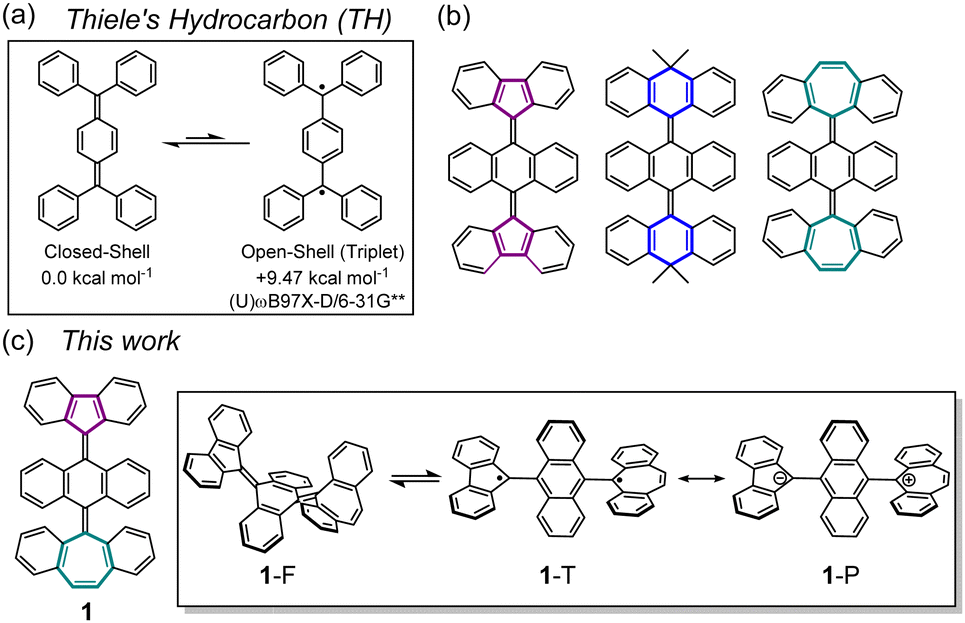 | ||
| Fig. 1 (a) The structure of Thiele's hydrocarbon. (b) Typical examples of reported π-extended THs, having fluorenyl, 9,10-dihydro-10,10-dimethyl-9-anthryl, or dibenzosuberenyl end-caps. (c) The structure of hexabenzo[5.6.7]quinarene 1 and its structural isomers 1-F (closed-sell) and 1-T (open-shell) as well as a formal resonance structure of 1-P (polarized form). | ||
π-extended [5.6.7]quinarene131, composed of an AQD central core end-capped with FLU and DBS groups, is a good candidate for understanding the nature of biradical and polarized character and for investigating whether these states can coexist in a pure hydrocarbon skeleton (Fig. 1c). Previously, we investigated persistent FLU and DBS radicals protected by a 9-anthryl ring and a redox potential difference (ΔEredox) of the DBS radical (Eox) and the FLU radical (Ere) was determined to be 0.80 V, which is expected to be a moderate ΔEredox to construct a neutral charge-transfer (CT) complex (Fig. S1, ESI‡).14 Herein, we report the synthesis of compound 1 and elucidate the structure and properties of folded closed-shell 1-F and twisted open-shell 1-T that can be switched by a combination of chemical oxidation/reduction and thermal stimulation. We also discuss whether twisted polarized species 1-P can exist.
The synthesis of 1 is shown in Scheme 1. To introduce a non-symmetric terminal π-skeleton into the AQM core, key intermediate 3, possessing an anthrone and DBS unit, was synthesized in two steps from 9,10-dibromoanthracene and dibenzosuberenone. Compound 2 was converted to 3 by treatment with an excess amount of trifluoroacetic acid and water. Although the Barton–Kellogg reaction is a well-known method to construct an overcrowded ethylene15 to introduce a FLU unit into carbonyl compound 3, we employed the Peterson-olefination reaction16 because the stable FLU anion and 9-(trimethylsilyl)fluorenyl anion 4 are readily prepared by n-butyllithium in a one-pot reaction. The reaction of 3 and 4 afforded target hexabenzo[5.6.7]quinarene 1 as a pale yellow solid in 74% yield.
 | ||
| Scheme 1 Synthetic route to 1 and its dication 12+. Reagents and conditions: (a) (i) 1.0 eq. of n-BuLi, −78 °C, THF, and 30 min; (ii) 0.8 eq. of dibenzosuberenone, −78 °C to RT.; (b) TFA/H2O, 1,2-dichloroethane; and (c) 4.0 eq. of antimony pentachloride, dichloromethane. | ||
To evaluate the structure of 1, single crystal X-ray diffraction and 1H NMR measurements were conducted. These measurements revealed that compound 1 possesses a folded structure (1-F) rather than a twisted one. 1-F can exhibit exo- and endo-forms of the DBS unit, and the exo-form was identified by X-ray crystallography in the solid state (Fig. 2a and Fig. S3, ESI‡). In addition, the 2D NOESY 1H NMR spectrum showed that only the exo-form is present even in solution, as confirmed by cross peaks between a-f and i–j protons (Fig. S5, ESI‡). The DFT calculations showed that the endo-form is 2.27 kcal mol−1 higher in energy than the exo-form, probably because the endo-form of 1-F has larger steric repulsion between the DBS unit and central AQD core than that of the exo-form. From the X-ray analysis, the CC bond lengths a, b, and c are 1.357 Å, 1.348 Å, and 1.339 Å, respectively. This indicates that, despite the anthracene bridge, the intramolecular spin–spin interaction between the FLU and DBS units is strong, resulting in a folded closed-shell structure.
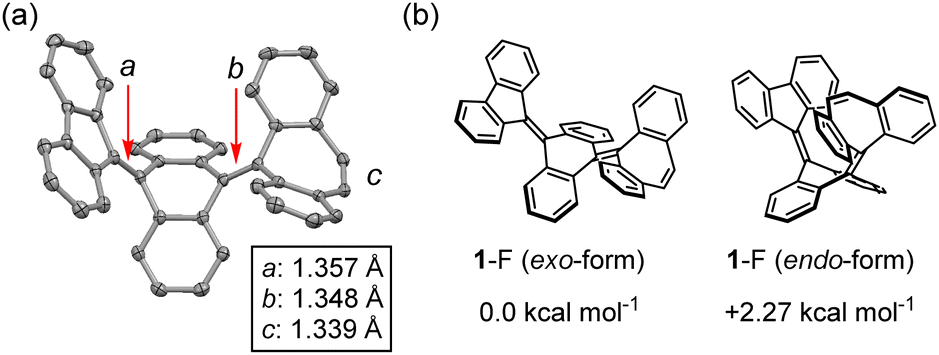 | ||
| Fig. 2 (a) X-ray crystallography of 1-F. Hydrogen atoms are omitted for clarity. (b) Comparison of the energy difference ΔE between exo- and endo-forms of 1-F (ωB97X-D/6-31G**). | ||
Next, the structural transformation of 1-F into the twisted structure 1-T was investigated. UV-vis absorption measurements of 1-F in various solvents such as dichloromethane (DCM), toluene, and DMF were attempted to evaluate solvent-induced charge transfer, but no significant difference in absorption maximum (λabs = 354–356 nm) or absorption coefficient (ε = ca. 23![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cm−1 M−1) was observed among them (Fig. S6, ESI‡). Mechanical grinding of 1-F to generate 1-T was also attempted,6e but no color change in the solid state was observed. This observation is in accordance with DFT calculations, which revealed that the twisted biradical structure 1-T is 20 kcal mol−1 higher in energy than 1-F (Fig. S7, ESI‡). Photoirradiation at 365 nm to 1-F did not induce isomerization to 1-T. Despite these results, the cyclic voltammogram of 1-F showed unique irreversible waves (Fig. 3b). There are three anodic peaks at E = −1.06, +0.24, and +0.92 V and three cathodic peaks at E = +0.41, −0.22, and −2.05 V. This observation indicates that the quinoidal folded form of 1-F undergoes a one-step two-electron oxidation/reduction process, producing the twisted dication 12+ at E = +0.92 V or dianion 12− at E = −2.05 V, respectively (Fig. 3c). Due to the different redox potentials of charged FLU (EOx1/2 = +0.28 V, E1/2Re = −1.11 V) and DBS (E1/2Ox = −0.30 V, Epc = −1.78 V) units (Fig. S1, ESI‡),14 the twisted dication 12+ or dianion 12− showed a stepwise reduction or oxidation, respectively, with redox potentials corresponding to those of FLU and DBS radicals attached with a 9-anthryl unit, affording the twisted biradical form 1-T. It is considered that the generated 1-T thermally reverted to 1-F due to the irreversible nature of the waves. Thus, it is expected that the twisted biradical form 1-T can be generated by reduction of dication 12+ at low temperatures.
000 cm−1 M−1) was observed among them (Fig. S6, ESI‡). Mechanical grinding of 1-F to generate 1-T was also attempted,6e but no color change in the solid state was observed. This observation is in accordance with DFT calculations, which revealed that the twisted biradical structure 1-T is 20 kcal mol−1 higher in energy than 1-F (Fig. S7, ESI‡). Photoirradiation at 365 nm to 1-F did not induce isomerization to 1-T. Despite these results, the cyclic voltammogram of 1-F showed unique irreversible waves (Fig. 3b). There are three anodic peaks at E = −1.06, +0.24, and +0.92 V and three cathodic peaks at E = +0.41, −0.22, and −2.05 V. This observation indicates that the quinoidal folded form of 1-F undergoes a one-step two-electron oxidation/reduction process, producing the twisted dication 12+ at E = +0.92 V or dianion 12− at E = −2.05 V, respectively (Fig. 3c). Due to the different redox potentials of charged FLU (EOx1/2 = +0.28 V, E1/2Re = −1.11 V) and DBS (E1/2Ox = −0.30 V, Epc = −1.78 V) units (Fig. S1, ESI‡),14 the twisted dication 12+ or dianion 12− showed a stepwise reduction or oxidation, respectively, with redox potentials corresponding to those of FLU and DBS radicals attached with a 9-anthryl unit, affording the twisted biradical form 1-T. It is considered that the generated 1-T thermally reverted to 1-F due to the irreversible nature of the waves. Thus, it is expected that the twisted biradical form 1-T can be generated by reduction of dication 12+ at low temperatures.
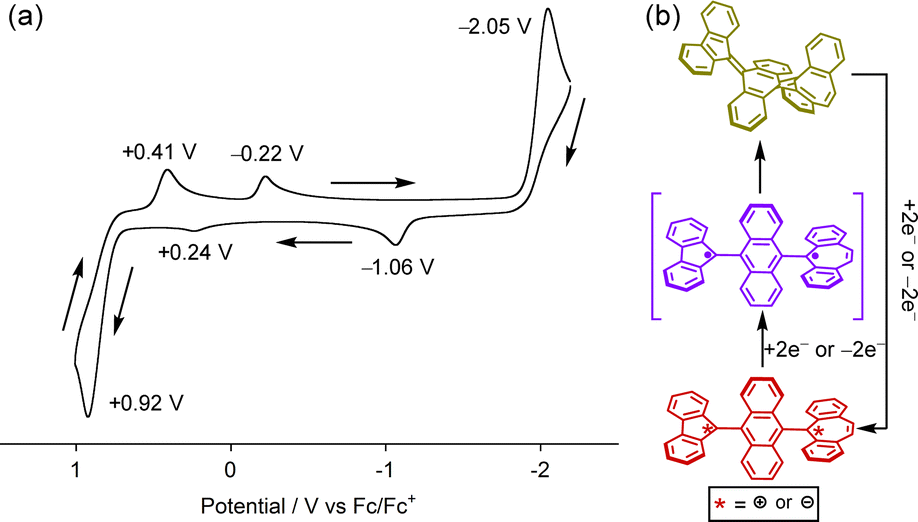 | ||
| Fig. 3 (a) Cyclic voltammogram of 1-F. (0.1 M nBu4NPF6 in CH2Cl2, scan rate = 0.1 V s−1). Black arrows indicate the scan direction. (b) Conformational change of 1 during the oxidation/reduction processes. | ||
The key compound of dication 12+ was prepared using antimony pentachloride as the oxidative reagent in DCM, and it was obtained in a good yield (96%, Scheme 1). Single crystals of 12+were obtained, which were suitable for X-ray crystallography and stable under ambient conditions even though it has an antiaromatic fluorenyl cation unit with large positive NICS(0) = +25.7 ppm (Fig. S8, ESI‡). The X-ray structure of 12+ is shown in Fig. 4 and Fig. S4 (ESI‡). Unlike the structure of 1-F, the central core of 12+ changed from AQD form to anthryl (Ant) form, which is confirmed by the C–C bond length of the central Ant core (Fig. S3 and S4, ESI‡) and C–C bond length between FLU and the Ant unit (a = 1.436 Å) as well as DBS and the Ant unit (b = 1.516 Å). In addition, the twist angles between FLU and Ant (∠FLU-Ant) and DBS and Ant (∠DBS-Ant) are 57.5° and 82.9°, respectively. The smaller twist angle of ∠FLU-Ant is due to lesser steric hindrance of the FLU unit than that of the DBS unit. Therefore, the positive charges are mainly localized on both the DBS and FLU units, but slight charge delocalization on the central Ant unit from the FLU unit is considered, judging from the C–C bond length and the twist angle. This was also suggested by natural population analysis (Fig. S9, ESI‡). The UV-vis spectrum of 12+ exhibited a weak broad band at 1040 nm (Fig. S10, ESI‡). The TD-DFT calculations revealed that the broad band is derived from intramolecular CT from the central Ant core to the electron-deficient FLU unit (Fig. S11 and Table S1, ESI‡).
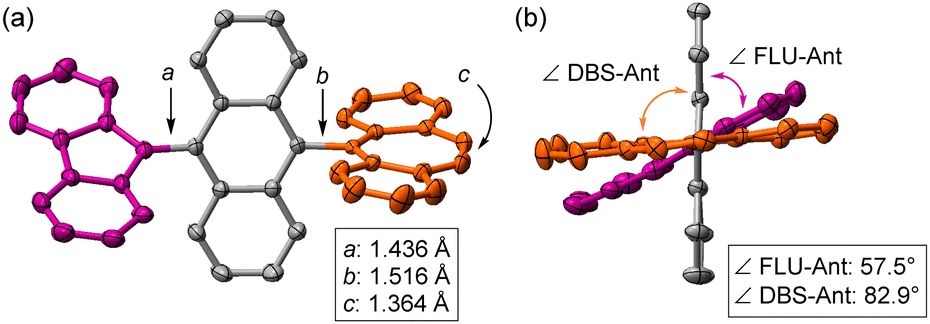 | ||
| Fig. 4 X-ray crystallography of 12+. FLU and DBS units are represented by purple and orange colors, respectively. (a) Front view and C–C bond lengths of a, b, and c. (b) Side view and twist angles between FLU and the central anthryl (Ant) unit (∠FLU-Ant) as well as DBS and the Ant unit (∠DBS-Ant). Protons and counter anions SbCl6− are omitted for clarity. | ||
With the dication 12+ in hand, the reduction to afford the twisted biradical 1-T was conducted by using Zn powder in MeCN solution at −40 °C, affording a dark red powder (Scheme 2). The ESR measurement of biradical 1-T in toluene showed a characteristic pattern for ΔMS = ±1 peaks associated with triplet species (Fig. 5a). Using the point dipole approximation, the average spin–spin distance was estimated to be 6.98 Å, which is the midpoint distance between the ring centers of FLU and DBS units (8.67 Å), and between the 9-position of FLU and 5-position of DBS units (5.82 Å, Fig. 5b). These findings provide unambiguous evidence for the conclusion that the triplet state of twisted 1-T can be generated. In addition, DFT calculations revealed that twisted 1-T has an open-shell ground state singlet with large biradical character y0 = 0.99 estimated from the natural orbital occupation number17 and ΔES–T is only −0.06 kcal mol−1, indicating that the singlet and triplet energy levels are almost degenerated. The recrystallization of 1-T was performed at room temperature, but the obtained crystal structure was 1-F due to the isomerization occurring in solution even though the color of the solution remained slightly dark red. Thus, through a combination of chemical oxidation/reduction and thermal stimulation, compound 1 exhibits spin-switching behavior between 1-F and 1-Tvia12+. Although the experimental determination of the equilibrium kinetics of 1-T to 1-F was attempted, it was not possible to exclude an equilibrium of intermolecular σ-dimerization between DBU units,12c presumably leading to two FLU radical units in the σ-dimer, similar to the equilibrium of the Chichibabin's hydrocarbon (Scheme S1, ESI‡).18
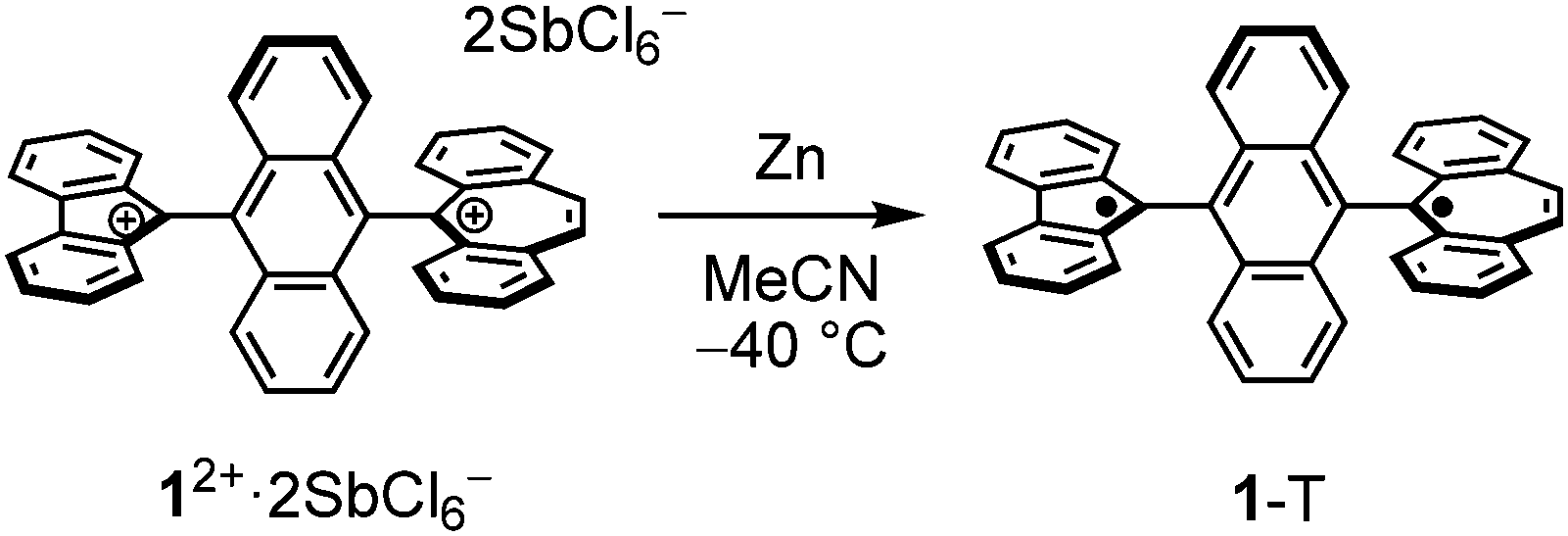 | ||
| Scheme 2 Generation of twisted biradical 1-T from its dication 12+. | ||
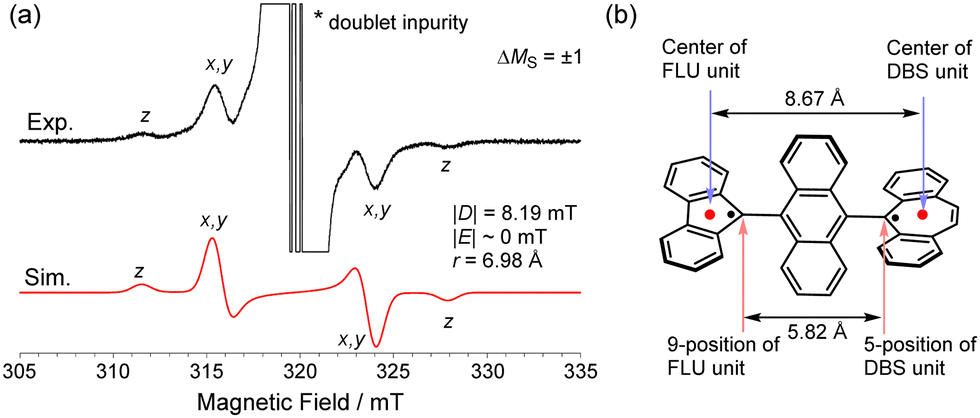 | ||
| Fig. 5 (a) ESR spectrum of 1-T (ΔMS = ±1) in toluene at −113 °C. (b) Calculated structure of 1-T and carbon–carbon distances between the ring centers of the FLU and DBS units and between the 9-position of FLU and 5-position of DBS units (UωB97X-D/6-31G**). | ||
The optical property of 1-T was evaluated to determine whether it can exist in a polarized form 1-P. Unlike dication 12+, 1-T did not show near-infrared absorption bands above 1000 nm and the spectrum is similar to that of the 9-Ant FLU radical (Fig. S2, ESI‡). Although several solvents with different polarity such as DCM, toluene, and DMF were used for the measurement of the UV-vis spectra, only minor differences were observed in the longer-wavelength broad weak band over 700 nm (Fig. S12, ESI‡). TD-DFT calculations revealed that the broad weak band originated from a combination of transitions from the FLU unit to the central Ant core and from the FLU-Ant unit to the DBS unit (Fig. S13 and Table S2, ESI‡). On the other hand, the electrostatic potential surfaces of 1-T in both open-shell singlet and triplet structures showed no significant charge deflection between the FLU and DBS units (Fig. S14, ESI‡). Additionally, negligible difference in absolute spin density of the FLU and DBS units was observed between open-shell singlet and triplet structures (Fig. S15, ESI‡). These results indicate that there is no electron transfer between FLU and DBS in the singlet state and that the polarized form of 1-P cannot be obtained under ambient conditions.
In summary, we synthesized hexabenzo[5.6.7] quinarene 1 and evaluated its properties. Although 1 has a folded form (1-F) as the most stable structure and does not undergo mechanochemical conversion to its twisted form (1-T) in the solid state, dication 12+ could be readily prepared, which has the twisted form, enabling us to obtain twisted 1-T by chemical reduction. The generation of 1-T was unambiguously confirmed from the triplet signals in ESR measurement. Although the polarized form of 1-P could not be observed, a combination of chemical oxidation/reduction and thermal stimulation led to the discovery of spin-switching behaviour between 1-F and 1-Tvia12+.
This study was supported by Grant-in-Aid for Scientific Research (C) (JSPS KAKENHI Grant Number JP20K05475, T. N.) and Transformative Research Areas (A) (Grant Number JP20H05865, T. K.). This work was the result of using research equipment shared in the MEXT Project for promoting public utilization of advanced research infrastructure (Program for supporting construction of core facilities. Grant Number JPMXS0441200023.). Quantum chemical calculations were performed at the Research Center for Computational Science, Okazaki, Japan (Project: 22-IMS-C208 and 23-IMS-C212).
Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) J. Thiele and H. Balhorn, Ber. Dtsch. Chem. Ges., 1904, 37, 1463 CrossRef; (b) L. K. Montgomery, J. C. Huffman, E. A. Jurczak and M. P. Grendze, J. Am. Chem. Soc., 1986, 108, 6004 CrossRef CAS PubMed.
- (a) M. Abe, Chem. Rev., 2013, 113, 7011 CrossRef CAS PubMed; (b) Z. Zeng, X. Shi, C. Chi, J. T. Navarrete, J. Casado and J. Wu, Chem. Soc. Rev., 2015, 44, 6578 RSC; (c) T. Stuyver, B. Chen, T. Zeng, P. Geerlings, F. D. Proft and R. Hoffmann, Chem. Rev., 2019, 119, 11291 CrossRef CAS PubMed; (d) T. Kubo, Bull. Chem. Soc. Jpn., 2021, 94, 2235 CrossRef CAS.
- T. Y. Gopalakrishna, W. Zeng, X. Lu and J. Wu, Chem. Commun., 2018, 54, 2186 RSC.
- (a) C. Wentrup, M. J. Regimbald-Krnel, D. Müller and P. Comba, Angew. Chem., Int. Ed., 2016, 55, 14600 CrossRef CAS PubMed; (b) S. Nishida, Y. Morita, K. Fukui, S. Maki and K. Nakasuji, Angew. Chem., Int. Ed., 2005, 44, 7277 CrossRef CAS PubMed; (c) Y. Hamamoto, Y. Hirao and T. Kubo, J. Phys. Chem. Lett., 2021, 12, 4729 CrossRef CAS PubMed.
- (a) K. Fujita, S. Hatano, D. Kato and J. Abe, Org. Lett., 2008, 10, 3105 CrossRef CAS PubMed; (b) Y. Kishimoto and J. Abe, J. Am. Chem. Soc., 2009, 131, 4227 CrossRef CAS PubMed; (c) K. Uchida, S. Ito, M. Nakano, M. Abe and T. Kubo, J. Am. Chem. Soc., 2016, 138, 2399 CrossRef CAS PubMed.
- (a) T. Nishiuchi, S. Uno, Y. Hirao and T. Kubo, J. Org. Chem., 2016, 81, 2106 CrossRef CAS PubMed; (b) T. Nishiuchi, S. Aibara and T. Kubo, Angew. Chem., Int. Ed., 2018, 57, 16516 CrossRef CAS PubMed; (c) T. Nishiuchi, K. Kisaka and T. Kubo, Angew. Chem., Int. Ed., 2021, 60, 5400 CrossRef CAS PubMed; (d) T. Nishiuchi, S. Aibara, T. Yamakado, R. Kimura, S. Saito, H. Sato and T. Kubo, Chem. – Eur. J., 2022, 28, e202200286 CAS; (e) T. Nishiuchi, S. Aibara, H. Sato and T. Kubo, J. Am. Chem. Soc., 2022, 144, 7479 CrossRef CAS PubMed; (f) T. Nishiuchi, Y. Makihara, R. Kishi, H. Sato and T. Kubo, J. Phys. Org. Chem., 2023, 36, e4451 CrossRef CAS.
- X. Yin, J. Z. Low, K. J. Fallon, D. W. Paley and L. M. Campos, Chem. Sci., 2019, 10, 10733 RSC.
- T. Nishiuchi, R. Ito, E. Stratmann and T. Kubo, J. Org. Chem., 2020, 85, 179 CrossRef CAS PubMed.
- (a) Y. Ishigaki, Y. Hayashi and T. Suzuki, J. Am. Chem. Soc., 2019, 141, 18293 CrossRef CAS PubMed; (b) V. G. Jiménez, P. Mayorga-Burrezo, V. Blanco, V. Lloveras, C. J. Gómez-García, T. Šolomek, J. M. Cuerva, J. Veciana and A. G. Campaña, Chem. Commun., 2020, 56, 12813 RSC; (c) Y. Hayashi, S. Suzuki, T. Suzuki and Y. Ishigaki, J. Am. Chem. Soc., 2023, 145, 2596 CrossRef CAS PubMed.
- (a) Y. Ishigaki, T. Hashimoto, K. Sugawara, S. Suzuki and T. Suzuki, Angew. Chem., Int. Ed., 2020, 59, 6581 CrossRef CAS PubMed; (b) K. Li, Z. Xu, J. Zu, T. Weng, Z. Chen, S. Sato, J. Wu and Z. Sun, J. Am. Chem. Soc., 2021, 143, 20419 CrossRef CAS PubMed.
- (a) E. Niecke, A. Fuchs, F. Baumeister, M. Nieger and W. W. Schoeller, Angew. Chem., Int. Ed. Engl., 1995, 34, 555 CrossRef CAS; (b) M. Abe, W. Adam and W. M. Nau, J. Am. Chem. Soc., 1998, 120, 11304 CrossRef CAS; (c) Z. Zeng, S. Lee, M. Son, K. Fukuda, P. M. Burrezo, X. Zhu, Q. Qi, R.-W. Li, J. T. L. Navarrete, J. Ding, J. Casado, M. Nakano, D. Kim and J. Wu, J. Am. Chem. Soc., 2015, 137, 8572 CrossRef CAS PubMed; (d) S. Arikawa, A. Shimizu and R. Shintani, Angew. Chem., Int. Ed., 2019, 58, 6415 CrossRef CAS PubMed.
- (a) X. Yang, X. Shi, N. Aratani, T. P. Gonçalves, K.-W. Huang, H. Yamada, C. Chi and Q. Miao, Chem. Sci., 2016, 7, 6176 RSC; (b) X. Fu, H. Han, D. Zhang, H. Yu, Q. Hi and D. Zhao, Chem. Sci., 2020, 11, 5565 RSC; (c) C. Zhen, S. Lu, M. Lin, J. Wu, I. Chao and C. Lin, Chem. – Eur. J., 2021, 27, 16682 CrossRef CAS PubMed; (d) K. Horii, R. Kishi, M. Nakano, D. Shiomi, K. Sato, T. Takui, A. Konishi and M. Yasuda, J. Am. Chem. Soc., 2022, 144, 3370 CrossRef CAS PubMed.
- (a) K. Takahashi, S. Takenaka, Y. Kikuchi, K. Takase and T. Nozoe, Bull. Chem. Soc. Jpn., 1974, 47, 2272 CrossRef; (b) K. Takahashi, I. Oikawa and K. Takase, Chem. Lett., 1974, 1215 CrossRef CAS; (c) K. Takahashi and K. Takase, Tetrahedron Lett., 1975, 15, 245 CrossRef.
- T. Nishiuchi, R. Ito, A. Takada, Y. Yasuda, T. Nagata, E. Stratmann and T. Kubo, Chem. – Asian J., 2019, 14, 1830 CrossRef CAS PubMed.
- (a) H. Staudinger and J. Siegwart, Helv. Chim. Acta, 1920, 3, 833 CrossRef CAS; (b) D. H. R. Barton and B. J. Willis, J. Chem. Soc. D, 1970, 1225 RSC; (c) R. M. Kellogg and S. Wassenaar, Tetrahedron Lett., 1970, 11, 1987 CrossRef; (d) A. M. Schoevaars, W. Kruizinga, R. W. J. Zijlstra, N. Veldman, A. L. Spek and B. L. Feringa, J. Org. Chem., 1997, 62, 4943 CrossRef CAS.
- D. J. Peterson, J. Org. Chem., 1968, 33, 780 CrossRef CAS.
- D. Doehnert and J. Koutecky, J. Am. Chem. Soc., 1980, 102, 1789 CrossRef CAS.
- (a) A. E. Tschitschibabin, Chem. Ber., 1907, 40, 1810 CrossRef CAS; (b) W. J. V. Hart and L. J. Oosterhoff, Mol. Phys., 1970, 18, 281 CrossRef; (c) L. K. Montgomery, J. C. Huffman, E. A. Jurczak and M. P. Grendze, J. Am. Chem. Soc., 1986, 108, 6004 CrossRef CAS PubMed.
Footnotes |
| † This paper is dedicated to the 70th birthday of Prof. Dennis Curran. |
| ‡ Electronic supplementary information (ESI) available. CCDC 2256323, and 2256324. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3cc02157b |
| This journal is © The Royal Society of Chemistry 2023 |