
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRemote template effect in the synthesis of bipyridine-strapped porphyrins†
Mathilde
Berthe
a,
Yoshiyuki
Kagawa
b,
Axel
Riquet‡
 a,
Takashi
Hayashi
b,
Jean
Weiss
*a and
Jennifer A.
Wytko
*a
a,
Takashi
Hayashi
b,
Jean
Weiss
*a and
Jennifer A.
Wytko
*a
aInstitut de Chimie de Strasbourg, UMR 7177 CNRS-Université de Strasbourg, 4 rue Blaise Pascal, Strasbourg 67000, France. E-mail: jwytko@unistra.fr
bDepartment of Applied Chemistry, #C4-621 Graduate School of Engineering, Osaka University, 2-1 Yamadaoka, Suita, Osaka 565-0871, Japan
First published on 9th May 2023
Abstract
A bipyridine-strapped porphyrin was prepared using a remote template effect of alkali or transition metal cations in the bipyridine subunit to enhance the yield 10-fold. The flexibility of the bipyridine-strap also allowed the synthesis of a doubly strapped porphyrin.
Strapped porphyrins have long been considered essential in the design of hemoprotein models. With a few exceptions, the introduction of straps and caps on one side of a porphyrin ring requires long and tedious syntheses. Among the few efficient syntheses of strapped porphyrins, the malonate-based cyclisation of picket-fence derivatives developed by the group of Boitrel allows the introduction of various functional straps on the gram scale.1 To date, the most efficient strapped-porphyrin synthesis (60–70% yield) is of a 1,10-phenanthroline-strapped porphyrin21 (Fig. 1) for which the final step is the condensation of a dialdehyde derivative of 2,9-diphenyl-phenanthroline with two dipyrrylmethane3 moieties. The presence of the 2,9-diphenylphenanthroline motif has been particularly important in the development of [1(FeII–CuI)]+ complexes4 that mimic the four-electron reduction of oxygen during the final stage of the respiratory process of cytochrome c oxidase (CcO).5 The strong stabilisation of the CuI in the phenanthroline strap together with the effects of basicity of auxiliary axial ligands on the ferrous porphyrin core are the major parameters controlling the electrocatalytic properties of the [1(FeII–CuI)]+ complex. However, the lack of adaptability of the CuI coordination sphere in these rigid CcO models was never addressed. To this end, synthetic approaches to obtain a flexible 2,2′-bipyridine-strapped porphyrin 2 (Fig. 1) have been developed and led to the original findings reported hereafter.
 | ||
| Fig. 1 Phenanthroline- and bipyridine-strapped porphyrins. | ||
In the solid state, in basic medium or in organic solvents, 2,2′-bipyridine (bipy) adopts an anti conformation,6 whereas the syn conformation is observed in acidic medium7 or upon coordination to a metal. In the latter case, the bipy acts as a bidentate ligand and a wide variety of stable metal complexes can be formed. Metal-induced switching of the bipy configuration has been used advantageously to spatially arrange remote substituents to turn on/off or enhance desired properties such as allosteric binding,8 chirality,9 energy transfer,10 peptide folding11 or catalytic activity12 among others. A parallel can be drawn between this metal-induced spatial orientation and that observed in the metal template effect in which the coordination geometry of a metal centre correctly orients functional groups for subsequent reactions, often a macrocyclisation. An elegant example is the copper(I)-templated synthesis of catenanes13 for which the tetrahedral coordination geometry of the metal centre orients phenol substituents of two phenanthroline ligands for intramolecular cyclisations that generate two interlocked rings. The same copper(I) template strategy was later used to synthesise catenanes containing a 2,2′-bipyridine moiety.14 We have now applied this principle to the synthesis of a bipyridine-strapped porphyrin 2 (Fig. 1) using a remote metal template effect to increase the yield 10-fold for the porphyrin condensation step.
The bipy dialdehyde strap 3 (Scheme 1) was synthesised by coupling of the pinacol-borane 415 with 6,6′-dibromo-2,2′-bipyridine under standard Suzuki–Miyuara conditions. Single crystals of 3 suitable for X-ray diffraction were obtained by slow diffusion of cyclohexane in CH2Cl2 solutions. In the solid state, bipyridine 3 adopts an anti conformation (Fig. 2) with the aldehyde groups separated by a C–C distance of 17 Å.16 This orientation and separation should obviously disfavour the reaction involving the two aldehyde functions to form the porphyrin macrocycle in the final step. However, the strongly acidic medium used in porphyrin synthesis was expected to favour the syn conformation necessary for cyclisation.
 | ||
| Scheme 1 Synthesis of the bipyridine strap 3. | ||
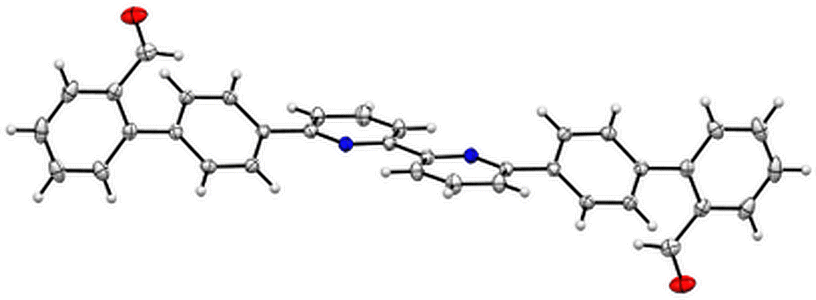 | ||
| Fig. 2 X-ray structure of the dialdehyde strap of the bipy 3. | ||
For direct comparison with the efficient method used to prepare the phenanthroline-strapped porphyrin 1,2 the synthesis of the bipy-strapped porphyrin 2 was first attempted by reaction of 3 with two equivalents of dipyrrylmethane3 under highly diluted conditions (CH2Cl2, 10−4 M)17 in the presence of excess trifluoroacetic acid. Subsequent oxidation with 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (DDQ), basic treatment, and purification by both column chromatography and recrystallisation gave porphyrin 2 in a 4% yield. The effect of metal templation on the efficiency of porphyrin formation was then explored using a template that does not directly act on the porphyrin ring but which preorganises a remote part of the targeted product.
A lithium cation was first chosen as a metal template because this spherical, monovalent alkali cation is known to bind to bipy, albeit weakly,18 but would not interfere with the porphyrin condensation reaction. Addition of five equivalents of the LiOAc had only a small effect on the formation of 2 and enhanced the yield from 4% to 9%; nevertheless, this increase was encouraging. Due to the tendency of Cu(I) to form entwined bis-bipyridine complexes, the use of a Cu(I) template (0.5 equivalents) was also explored with the additional hope of forming a catenated14 bis-porphyrin. After oxidation, neutralisation with NEt3 and treatment with EDTA and KCN to remove any copper, mass spectrometry analysis of the crude product showed traces of a catenane as well as more intense peaks for unreacted dialdehyde 3, porphyrin 2, a pseudo-rotaxane comprised of 2 and 3, and other unidentified products (see ESI†). After purification, the main fraction still contained numerous porphyrins with close Rf values, which dissuaded us from continuing the purification.
Among the other transition metal cations that form complexes with 2,2′-bipyridine, Zn(II) seemed well-adapted for our purpose for two reasons. First, this metal cation can form stable complexes in which only one bipyridine ligand is bound to the metal.19 Second, this metal centre can be easily removed if bound within the porphyrin macrocycle during the synthesis. The addition of one equivalent of Zn(OAc)2·2H2O to the porphyrin synthesis (Scheme 2) increased the yield of porphyrin 2 to 41%. A derivative of compound 2 with zinc(II) coordinated to the bipy was never isolated probably because excess triethylamine was used during the work-up of the reaction mixture. The zinc(II) porphyrin was also not isolated but could be obtained by metallation of porphyrin 2 with Zn(OAc)2·2H2O in CHCl3/MeOH to afford [2Zn] in 85% yield.
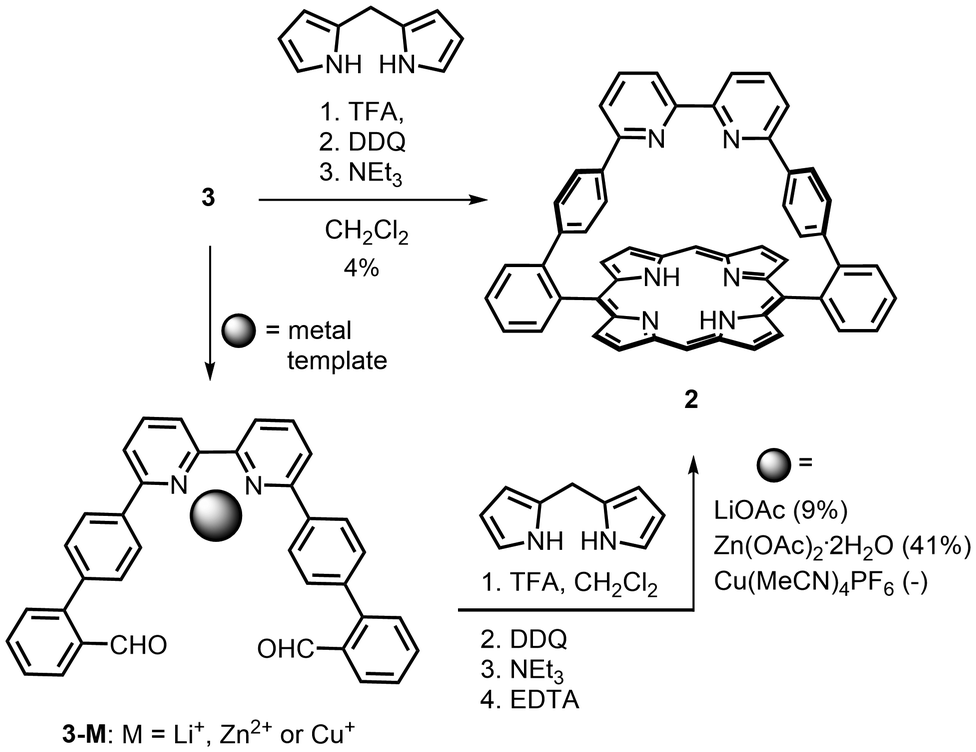 | ||
| Scheme 2 Synthesis of porphyrin 2 in the absence and in the presence of templates. | ||
UV-visible spectroscopy provided evidence of zinc(II) binding to the bipy 3 (see ESI†). Spectral changes in the UV region were observed upon addition of zinc(II) acetate to bipy 3 under reaction conditions (excess CF3CO2H, CH2Cl2, 10−4 M). No spectral changes were observed when the zinc salt was added to bipy 3 in the absence of CF3CO2H. Trifluoroacetic acid protonates the acetate ligands of the metal salt, thus generating a zinc(II) salt available for bipy coordination and weakly-coordinating trifluoroacetate anions.
The 1H NMR spectrum of 2 in CDCl3 displayed signals for both the bipyridine strap and the porphyrin, including a singlet at 10.14 ppm for the porphyrin meso protons and a shielded singlet at −3.15 ppm for the two NH protons of the porphyrin core. Based on the number of signals, the molecule appears to have a C2 symmetry axis perpendicular to the pyridine–pyridine bond. MALDI-TOF MS confirmed the mass expected for the strapped structure of 2. The UV-visible spectrum of 2 is identical to that of the phenanthroline-strapped porphyrin 12 and displays a Soret band (413 nm), four Q bands (506, 539, 579 and 635 nm) and a shoulder with an ill-defined maximum near 586 nm (see ESI†). The emission spectrum of 2 is identical to that of 1 with emission bands at 636 and 702 nm (see ESI†).
Single crystals of 2 were obtained by slow vapour diffusion of cyclohexane into a dichloromethane solution of 2.20 Single crystals of metallated porphyrin [2Zn] were obtained by slow vapour diffusion of cyclohexane into a dichloromethane solution of [2Zn] containing 5% acetone.21 Both porphyrins crystallise with solvent molecules, but only the apical acetone bound to the zinc(II) atoms of [2Zn] is shown (Fig. 3d). A comparison of the crystal structures of 2 and [2Zn] (Fig. 3) with that of the phenanthroline analogue 122 shows significant differences that are a consequence of rotational freedom of the bipy strap. Whereas the free base structure of 1 shows a strong tilt (68°) of the rigid phenanthroline strap over the porphyrin plane along the (meso-Ph-C) axis,22 the straps of 2 and [2Zn] are upright with respect to the porphyrin plane. Despite the classical 65° dihedral angle between the meso-phenyls and the porphyrin plane, a centroid placed between the two bipy N atoms is normal to the mean plane passing through the centroid of the N4 porphyrin macrocycle. Possible constraints in the strap are relieved through the 31° and 33° rotation of the pyridine–pyridine bond of the bipy in 2 and [2Zn], respectively.
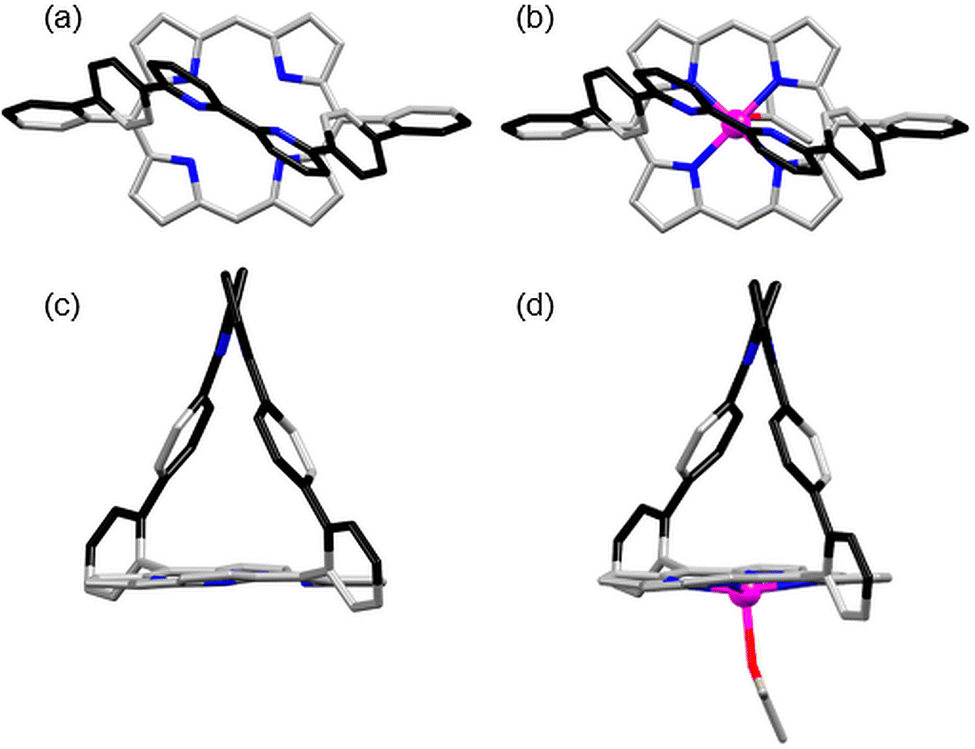 | ||
| Fig. 3 (a) and (c) Top and side views of the X-ray structure of the free base porphyrin 2. (b) Top view of the X-ray structure of the zinc porphyrin [2Zn]. (d) Side view of the crystal structure of [2Zn] that shows the apical acetone bound to the zinc(II) atom. Other solvent molecules and hydrogen atoms are omitted for clarity. | ||
Over the last several decades, several unsuccessful attempts were made to form a double phenanthroline-strapped porphyrin. The reduced constraint of the bipy strap in porphyrin 2 prompted us to explore the synthesis of a double bipy-strapped species by taking advantage of the remote zinc(II) template effect. The cyclisation of the dialdehyde strap 3 with 2 equiv. of pyrrole in the presence of 1 equiv. of Zn(OAc)2·2H2O (Scheme 3) gave the doubly strapped porphyrin 5 in 7% yield after tedious chromatographic purification. This yield is surprisingly high for the condensation of 16 functions to form eight new C–C bonds. The success of this reaction marks a significant difference between the bipy strap and the more rigid phenanthroline strap.
 | ||
| Scheme 3 Synthesis of a doubly strapped free base porphyrin 5. Only the outer frame of the porphyrin is drawn for sake of clarity. | ||
In the 1H NMR spectrum of 5 in CDCl3, the most notable feature is the absence of a signal near 10 ppm for the meso protons of a porphyrin. The observation of a singlet for the eight beta pyrrolic protons at 8.57 ppm and a singlet at −2.65 ppm for the NH protons confirms the presence of the porphyrin macrocycle. Each doublet for the ortho and meta protons (labelled o and m in Scheme 3) of the phenyl spacer at 6.92 and 6.73 ppm integrates for eight protons, thus indicating the presence of two straps. Despite drying this compound under vacuum for one week, CH2Cl2 from the recrystallisation was still visible in the NMR spectra. The ESI HRMS spectrum showed a molecular peak at 1223.4525 corresponding to the expected mass ([M+H+] of the protonated double-strapped porphyrin 5. The most notable features of the UV-visible spectrum of 5 (see ESI†) are the presence of five Q bands and a 17–20 nm red-shift of all absorption bands compared to those of 12 and 2 (see ESI†). Both emission bands of 5 (658 and 728 nm) display a bathochromic shift compared to those of 123 and 2 (see ESI†).
The X-ray structure of single crystals, obtained by diffusion of MeOH into a CH2Cl2 solution of 5, confirmed the atomic connectivity and the double-strapped nature of 5, as shown in Fig. 4.24 The pyridine–pyridine rings display torsions of 25° and 47°, and the straps adopt positions similar to that in 2. The ruffling of the porphyrin macrocycle is clearly more pronounced in 5 than in 2, probably to accommodate the strain of the second strap. One cavity between a strap (in green) and the porphyrin contains one molecule of methanol and one molecule of CH2Cl2 whereas the opposite cavity (in red) contains only a molecule of methanol. The hydrogen atoms of the alcohol groups are 2.28–2.59 Å from the nitrogen atoms of the bipyridines (see ESI†). The hydrogen atoms of the CH2Cl2 are 2.69 Å and 2.86 Å from the closest bipy nitrogen atoms. One Cl atom points towards an aromatic C(meso) and C(α-pyrrolic) bond with Cl⋯C distances of 3.59 Å and 3.69 Å, respectively and respective C–Cl⋯C(aromatic) bond angles of 173.9° and 163.3° (see ESI†). These observations suggest that the CH2Cl2 molecule is stabilised by a combination of weak hydrogen bonding and weak halogen-pi interactions. The presence of both solvents accentuates the uprightness of the strap shown in green.
 | ||
| Fig. 4 Side (a) and top (b) views of the X-ray structure of porphyrin 5. The three solvent molecules (two MeOH and CH2Cl2) are shown only in view (a). Hydrogen atoms are omitted for clarity. | ||
In-depth studies are in progress to determine if the flexibility of the bipyridine affects the properties of an iron(II)–copper(I) derivative of porphyrin 2 as a cytochrome c oxidase mimic. In addition, both porphyrins 2 and 5 are structurally suitable to bind two or three distinct metal centres, respectively. Such multi-metallic structures are of interest for EPR studies of multi-spin scaffolds or as a basis for double threaded rotaxanes with synergestic functionality.
M.B.: synthesis, characterisation, spectroscopy. Y.K. and A.R.: synthesis, characterisation. T.H.: secured funding. J.W.: designed the project, supervised, co-wrote the paper, secured funding. J.A.W.: supervised, co-wrote the paper. All authors reviewed the manuscript.
We thank the CNRS for the IRP SUPRHEME grant and for PhD funding (M. B.) and M. Koepf and T. Storr for fruitful discussions.
Conflicts of interest
There are no conflicts to declare.Notes and references
- A. Didier, L. Michaudet, D. Ricard, V. Baveux-Chambenoît, P. Richard and B. Boitrel, Eur. J. Org. Chem., 2001, 1917 CAS.
- J. A. Wytko, E. Graf and J. Weiss, J. Org. Chem., 1992, 57, 1015 CrossRef CAS.
- (a) P. S. Clezy and G. A. Smythe, Aust. J. Chem., 1969, 22, 239 CrossRef CAS; (b) R. Chong, P. S. Clezy, A. J. Liepa and A. W. Nichol, Aust. J. Chem., 1969, 22, 229 CrossRef CAS.
- (a) F. Melin, C. Boudon, M. Lo, K. J. Schenk, M. Bonin, P. Ochsenbein, M. Gross and J. Weiss, J. Porphyrins Phthalocyanines, 2007, 11, 212 CrossRef CAS; (b) P. Vorburger, M. Lo, S. Choua, M. Bernard, F. Melin, N. Oueslati, C. Boudon, M. Elhabiri, J. A. Wytko, P. Hellwig and J. Weiss, Inorg. Chim. Acta, 2017, 468, 232 CrossRef CAS.
- R. Boulatov, in N4-Macrocyclic Metal Complexes, J. H. Zagal, F. Bedioui and J.-P. Dodelet, Springer, New York, N.Y., 2006, Chapter 1, p. 1–40 Search PubMed.
- (a) P. E. Fielding and R. J. W. LeFevre, J. Chem. Soc., 1951, 1811 RSC; (b) K. J. Nakamoto, Phys. Chem., 1960, 64, 1420 CrossRef CAS.
- S. T. Howard, J. Am. Chem. Soc., 1996, 118, 10269 CrossRef CAS.
- For the first example reported with 2,2′-bipyridine, see: J. Rebek Jr, J. E. Trend, R. V. Wattley and S. Chakravorti, J. Am. Chem. Soc., 1979, 101, 4333 CrossRef.
- T. R. Kelly, M. C. Boxyer, K. V. Bhaskar, F. Bebbington, A. Garcia, F. R. Lang, M. H. Kim and M. P. Jette, J. Am. Chem. Soc., 1994, 116, 3657 CrossRef CAS.
- G. Hungerford, M. Van der Auweraer, J.-C. Chambron, V. Heitz, J.-P. Sauvage, J.-L. Pierre and D. Zurita, Chem. – Eur. J., 1999, 5, 2089 CrossRef CAS.
- J. P. Schneider and J. W. Kelly, J. Am. Chem. Soc., 1995, 117, 2533 CrossRef CAS.
- S. Takebayashi, M. Ikeda, M. Takeuchi and S. Shinkai, Chem. Commun., 2004, 420 RSC.
- C. O. Buchecker-Dietrich, J.-P. Sauvage and J.-P. Kintzinger, Tetrahedron Lett., 1983, 24, 5095 CrossRef.
- J. R. Price, J. K. Clegg, R. R. Fenton, L. F. Lindoy, J. C. McMurtrie, G. V. Meehan, A. Parkin, D. Perkins and P. Turner, Aust. J. Chem., 2009, 62, 1014 CrossRef CAS.
- L. A. Fontana, M. P. Almeida, A. F. P. Alcântara, V. H. Rigolin, M. A. Ribeiro, W. P. Barros and J. Megiatto, ChemRxiv, 2020, DOI:10.26434/chemrxiv.12625772.v1 preprint.
- Crystal data for 3: C36H24N2O2, M = 516.57, monoclinic, a = 10.7134(5) Å, b = 12.6341(6) Å, c = 10.0230(5) Å, V = 1294.7 Å3, T = 120(2) K, space group P21/c, Z = 2, 43537 reflections measured, 3481 unique (Rint = 0.0248, which were used in all calculations. The R1 [I > 2σ(I)] was 0.0392 with wR2 = 0.1025. CCDC 2240390†.
- J. S. Manka and D. S. Lawrence, Tetrahedron Lett., 1989, 30, 6989 CrossRef CAS.
- (a) J. Ghasemi and M. Shamsipur, J. Coord. Chem., 1992, 26, 337 CrossRef CAS; (b) J. Ghasemi and M. Shamsipur, J. Coord. Chem., 1993, 28, 231 CrossRef CAS; (c) T. Madrakian, A. Afkhami, J. Ghasemi and M. Shamsipur, Polyhedron, 1996, 15, 3647 CrossRef CAS.
- E. E. Benson, A. L. Rheingold and C. P. Kubiak, Inorg. Chem., 2010, 49, 1458 CrossRef CAS.
- Crystal data for 2: C60H46N6, M = 851.03, monoclinic, a = 12.0156(3) Å, b = 15.3535(4) Å, c = 24.3972(6) Å, V = 4493.93 Å3, T = 120(2) K, space group P21/c, Z = 4, 54518 reflections measured, 7944 unique (Rint = 0.0339, which were used in all calculations. The R1 [I > 2σ(I)] was 0.0.578 with wR2 = 0.1551. CCDC 2240391†.
- Crystal data for [2Zn]: C58H40 Cl2N6OZn, M = 973.23, monoclinic, a = 12.0783(3) Å, b = 13.7262(4) Å, c = 27.7976(8) Å, V = 4499.98 Å3, T = 120(2) K, space group P21/n, Z = 4, 108282 reflections measured, 10969 unique (Rint = 0.1329, which were used in all calculations. The R1 [I > 2σ(I)] was 0.0675 with wR2 = 0.1777. CCDC 2240392†.
- P. Ochsenbein, M. Bonin, K. Schenk, J. Froidevaux, J. Wytko, E. Graf and J. Weiss, Eur. J. Inorg. Chem., 1999, 1175 CrossRef CAS.
- D. Paul, J. A. Wytko, M. Koepf and J. Weiss, Inorg. Chem., 2002, 41, 3699 CrossRef CAS PubMed.
- Crystal data for 5: C91H64N8,O2, M = 1372.40, triclinic, a = 13.2810(5) Å, b = 13.2984(5) Å, c = 20.8307(8) Å, V = 3566.4(2) Å3, T = 120(2) K, space group P
![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) , Z = 2, 90821 reflections measured, 12465 unique (Rint = 0.1296, which were used in all calculations. The R1 [I > 2σ(I)] was 0.1310 with wR2 = 0.3423. CCDC 2240393†.
, Z = 2, 90821 reflections measured, 12465 unique (Rint = 0.1296, which were used in all calculations. The R1 [I > 2σ(I)] was 0.1310 with wR2 = 0.3423. CCDC 2240393†.
Footnotes |
| † Electronic supplementary information (ESI) available: Synthesis and spectral characterisation. CCDC 2240390–2240393. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3cc01697h |
| ‡ Present address: UMR CNRS 6226 Institut des Sciences Chimiques de Rennes, Université de Rennes 1, 35042 Rennes Cedex, France. |
| This journal is © The Royal Society of Chemistry 2023 |