
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Concise total synthesis and structure revision of metacridamides A and B†
Masahito
Yoshida
 *ab,
Yuhi
Okoshi
a and
Hideo
Kigoshi
abc
*ab,
Yuhi
Okoshi
a and
Hideo
Kigoshi
abc
aDegree Programs in Pure and Applied Sciences, Graduate School of Science and Technology, University of Tsukuba, 1-1-1 Tennodai, Tsukuba, Ibaraki 305-8571, Japan. E-mail: masahito@chem.tsukuba.ac.jp
bDepartment of Chemistry, Faculty of Pure and Applied Sciences, University of Tsukuba, 1-1-1 Tennodai, Tsukuba, Ibaraki 305-8571, Japan
cAlliance for the Research on the Mediterranean and North Africa (ARENA), University of Tsukuba, 1-1-1 Tennodai, Tsukuba, Ibaraki 305-8571, Japan
First published on 26th July 2023
Abstract
Concise total synthesis of metacridamides A and B was accomplished through repetitive vinylogous Mukaiyama aldol reactions and ynamide-mediated macrolactonization. Spectral data of both synthetic products were identical to those of the natural products, resulting in the revision of the absolute configuration of the C-9 position to be S.
Metacridamides A (1a) and B (1b) were isolated from the conidia of Metarhizium acridum by Krasnoff et al. in 2012 as cytotoxic natural products that form a 17-membered macrocycle consisting of a nonaketide chain and a Phe residue1 (Fig. 1). Structure elucidation of 1a has been performed by mass spectrometric and NMR spectroscopic analyses, and the absolute configuration has also been determined by X-ray single crystal analysis to be 3S, 8S, 9R, 12S, 13S, 16S, 17R. Notably, 1a exhibits moderate cytotoxicity against Caco-2, MCF-7, and HepG2/C3A (IC50 = 6.2, 11.0, and 10.8 μM, respectively), while 1b shows cytotoxicity only against HepG2/C3A (IC50 = 18.2 μM) of the three cancer cells. Thus, the acetyl group at the C-13 position in 1a should be important for inducing the cytotoxic effect. However, the role of the acetyl group in the mechanism of action has been unclear, for example, an improvement of cell permeability or contribution to the binding affinity. Therefore, synthetic studies of metacridamides A (1a) and B (1b) should be an important and interesting route to achieve elucidation of the mechanism of action. We now report the first total synthesis and structure revision of metacridamides A (1a) and B (1b) by the repetitive vinylogous Mukaiyama aldol reaction and ynamide-mediated macrolactonization.
 | ||
| Fig. 1 Reported structures of metacridamides A (1a) and B (1b). | ||
During our synthetic efforts for the metacridamides, Ghosh, S. et al. reported the total synthesis of the proposed structure of metacridamide B (1b), in which the absolute configuration of the C-9 carbon was suggested to be revised as the S configuration.2 Additionally, we carefully validated the X-ray structure of metacridamide A (1a) (CCDC 840273†) at the beginning of our synthetic study of 1a, resulting in the absolute configuration of the C-9 carbon that was found to be S. Based on these considerations, the structure of metacridamide A (1a) should be revised to 1a′. Therefore, we designed our synthetic strategy for 1a′ as illustrated in Scheme 1. The desired structure of 1a′ can be synthesized by macrolactonization of the cyclization precursor 2, which can be simply prepared by coupling the acid 4 with a phenylalanine derivative 3. Notably, the acid 4 possesses the repeated structure as highlighted, therefore, we planned the synthesis of 4 using the repetitive vinylogous Mukaiyama aldol reaction (VMAR)3,4 from the aldehyde 8. Repetitive VMAR has been previously attempted for the synthesis of (+)-TMC-151C reported by Kobayashi et al.5 Elongation of the polyketide chain by VMAR using an α,β-unsaturated aldehyde was problematic due to the low reactivity of the α,β-unsaturated aldehyde,6 therefore, the reaction was investigated in the presence of excess amount of a Lewis acid. However, the decomposition of the polyketide structure was observed due to the removal of protecting groups under strong Lewis acidic conditions. According to the previous report, we designed the intermediate 5 possessing Lewis acid-tolerant protecting groups. The intermediate 5 can be prepared using the α,β-unsaturated aldehyde 6 by two repetitive VMARs with the N,O-ketene acetal 7 from the known aldehyde 8. Based on the total synthesis of 1a′, 1b′ could be directly converted by chemoselective hydrolysis of the acetyl group without cleavage of the macrolactone skeleton.
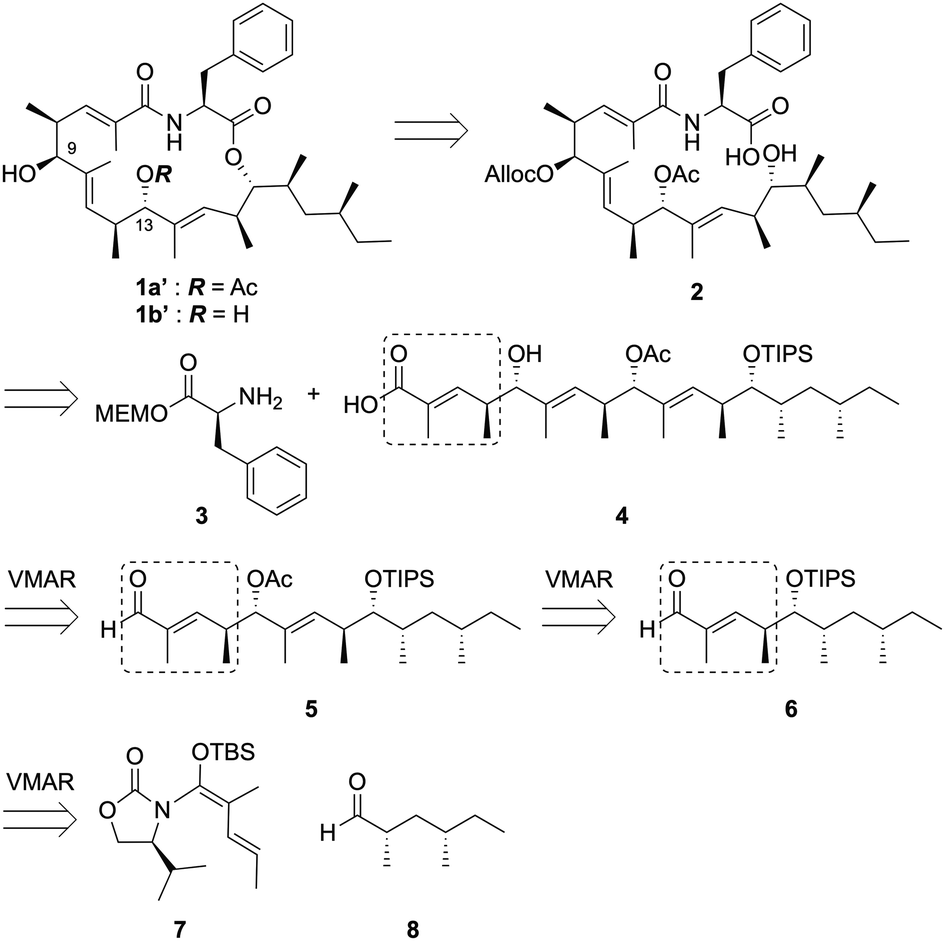 | ||
| Scheme 1 Retrosynthesis of 1a′ and 1b′. | ||
We initially synthesized the α,β-unsaturated aldehyde 6 from the commercially-available (S)-2-methyl-1-butanol (9) through the first VMAR as shown in Scheme 2. The acid 10 was prepared from the alcohol 9 according to the procedure reported by Rezanka et al.,7 then the coupling of 10 with a chiral oxazolidinone 11 was performed using a mixed anhydride method (PivCl/Et3N/LiCl) to afford the acylated compound 12 in 80% yield. After diastereoselective alkylation of 12 leading to 13, partial reduction of 13 using DIBAL-H provided the desired aldehyde 8. Next, the first VMAR with the aldehyde 8 was attempted using TiCl4 at −40 °C, and the desired 14 was successfully obtained in quantitative yield as a single diastereomer.8 The resulting hydroxy group in 14 was protected by a TIPS group to provide the silyl ether 15, which was subjected to partial reduction using DIBAL-H at −78 °C to afford the desired α,β-unsaturated aldehyde 6 in 79% yield.
 | ||
| Scheme 2 Synthesis of α,β-unsaturated aldehyde 6 through the 1st VMAR. | ||
The second VMAR with the α,β-unsaturated aldehyde 6 was investigated, and the details of the investigation are shown in Table 1. We initially attempted the same reaction conditions as used for the 1st VMAR with the saturated aldehyde 8, however, the desired product 168 was obtained in 14% yield along with the recovery of the aldehyde 6 (entry 1). On the other hand, the N,O-ketene acetal 7 was completely consumed under the stated conditions, indicating that the decomposition of 7 could be faster than the desired coupling due to the low reactivity of the α,β-unsaturated aldehyde 6. Therefore, the reaction using an excess amount of 7 was next attempted, resulting in a slight improvement of the yield of the desired product 16 up to 25% (entry 2). In addition, an increase in the reaction temperature did not significantly improve the yield and selectivity (entry 3). To promote the first VMAR using the low reactive 6, we next investigated the reaction in the presence of an excess amount of TiCl4 and the N,O-ketene acetal 7. As expected, VMAR of the α,β-unsaturated aldehyde 6 proceeded, but the stereoselectivity of VMAR was completely inverted to provide undesired 17 in 58% yield (entry 4). Hosokawa et al. previously reported that the VMAR of saturated aldehydes promoted by an excess Lewis acid selectively provided the syn-aldol product,9 therefore, an equivalent of the Lewis acid should be strictly controlled to achieve the anti-selective VMAR with an α,β-unsaturated aldehyde. Based on the above results and further investigations,10 we attempted the reaction using 0.5 eq. of TiCl4 at −40 °C, resulting in an improvement of the yield of the desired 16 up to 36% (brsm 67%) along with the recovery of substrates 6 and 7 (entry 5).
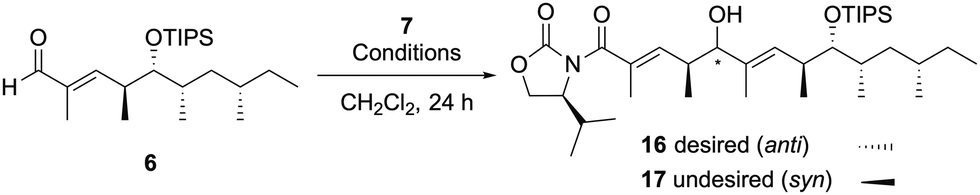
We next investigated the preparation of the allyl ester 20 through the third VMAR (Scheme 3).
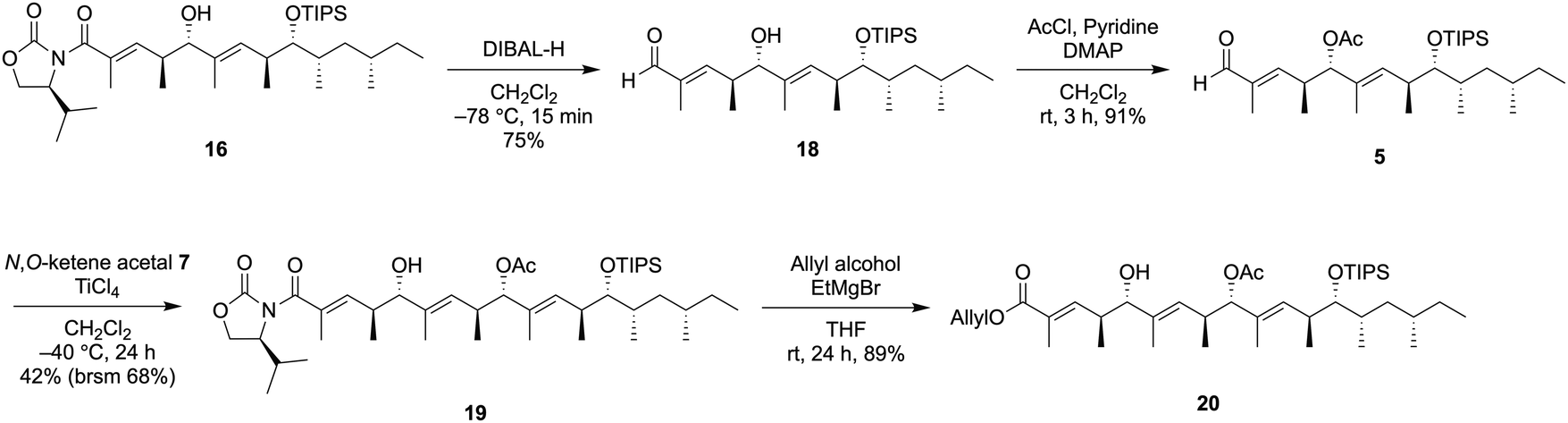 | ||
| Scheme 3 Synthesis of allyl ester 20. | ||
Reductive removal of the chiral auxiliary in 16 using DIBAL-H provided the aldehyde 18, and the hydroxy group in the resulting 18 was protected by an acetyl group to provide 5 in 90% yield. The third VMAR with the aldehyde 5 was carried out in the same manner as the second VMAR, leading to the desired 198 in 42% yield along with the recovery of aldehyde 5 (brsm 68%). Finally, the removal of the chiral auxiliary in 19 using AllyloxyMgBr provided the allyl ester 20 without losing the acetyl group.
For the construction of a macrolactone skeleton, the cyclization precursor 2 was prepared as shown in Scheme 4. Removal of the allyl group in 20 using Pd(PPh3)4/morpholine led to the acid 4, which was amidated with a phenylalanine derivative 3 to afford the amide 21 in quantitative yield. The secondary hydroxy group in 21 was protected with an Alloc group to provide the amide 22, and the removal of the MEM and TIPS groups in 22 was investigated under acidic conditions. However, simultaneous removal of both protecting groups failed using various acidic conditions due to the decomposition of 22, therefore, stepwise deprotection was next investigated. The MEM group was readily removed using 2 M HCl/dioxane at room temperature, and the remaining TIPS ether was successfully cleaved by treatment with 30% aq. HF/acetonitrile to provide the desired cyclization precursor 2 in 74% yield.
 | ||
| Scheme 4 Synthesis of the cyclization precursor 2. | ||
Macrolactonization of the resulting cyclization precursor 2 was next investigated, and the results are summarized in Table 2.
|
|
|||
|---|---|---|---|
| Entry | Conditions | Yield (%) | |
| 23 | 24 | ||
| 1 | MNBA, DMAPO, toluene | 0 | 42 |
| 2 | TCBC, Et3N, DMAP, toluene | 0 | 56 |
| 3 |

|
65 | Trace |
| 2) 15 mol% TsOH·H2O, (CH2Cl)2 | |||
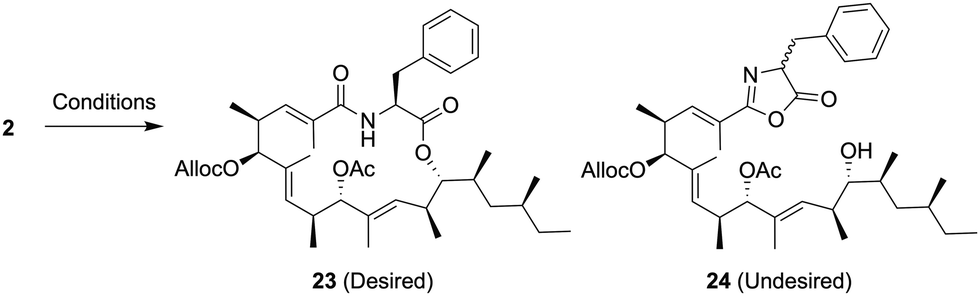
The reaction using 2-methyl-6-nitrobenzoic anhydride (MNBA)/DMAPO11 was initially attempted, however, the desired macrolactone 23 was not obtained (entry 1). In addition, the macrolactonization of 2 using 2,4,6-trichlorobenzoyl chloride (TCBC)/Et3N12 gave the same result as already described (entry 2). Under both conditions, the oxazolone 24 was generated by intramolecular cyclization of the amide group to an activated ester, indicating that the nucleophilicity of a carbonyl moiety in the amide group should be reduced, and protonation of the corresponding carbonyl group would prevent the formation of the undesired oxazolone 24. Recently, Zhao et al. reported the ynamide-mediated macrolactonization in the presence of 5 mol% TsOH·H2O leading to the corresponding macrolactone without epimerization.13 After the formation of a vinyl ester intermediate from 2 and N-methylynetoluenesulfonamide (MYTsA),14 macrolactonization by activation using 15 mol% TsOH·H2O successfully provided the desired macrolactone 23 in 65% yield without forming the undesired products such as the oxazolone 24 (entry 3).
Having the macrolactone 23 successfully in hand, the total synthesis of 1a′ and 1b′ was attempted as shown in Scheme 5. Removal of the Alloc group using Pd(PPh3)4/N-methylaniline smoothly proceeded to afford the desired 1a′ in quantitative yield.
 | ||
| Scheme 5 Total synthesis of metacridamides A (1a′) and B (1b′) | ||
In addition, direct conversion from 1a′ to 1b′ should be a challenging issue by hydrolysis without degradation of the macrolactone ring. Several investigations of the chemoselective removal of the acetyl moiety in 1a′ led us to find the best conditions (2 M aq LiOH in THF, 60 °C) to afford the desired 1b′ as the sole product. To our delight, the 1H and 13C NMR spectra of the synthetic 1a’ and 1b′ were identical to those of the natural products, resulting in the correction of the absolute configuration at the C-9 position to S as depicted in the ORTEP drawing of metacridamide A (CCDC 840273†). We evaluated the cytotoxicity of the synthetic metacridamides A and B against MCF-7 and HCT-116 cells. However, both compounds exhibited very weak cytotoxicity (>100 μM), while metacridamide A was reported to be cytotoxic at IC50 = 11 μM against MCF-7 cells. These results indicated that impurities contained in the natural metacridamide A, for example compounds that showed signals at around 3.7 ppm observed in the reported 1H NMR spectra, might induce the cytotoxic effect.
In summary, we have succeeded in the total synthesis and structure revision of metacridamides A and B. The plausible revised structures of the metacridamides were initially proposed by validation of the crystal structure of the natural products. The polyketide chain segment was successfully prepared using repetitive vinylogous Mukaiyama aldol reactions with an α,β-unsaturated aldehyde. Notably, strict control of an equivalent of TiCl4 to the aldehyde was required to implement an excellent anti-selectivity, resulting in the successful elongation of the polyketide chain to provide the desired acid segment. Amidation with a phenylalanine derivative, followed by the stepwise removal of the MEM and TIPS groups afforded the cyclization precursor. Although the widely employed methods for macrolactonization (MNBA/DMAPO or TCBC/Et3N/DMAP) were not suitable, the ynamide-mediated macrolactonization smoothly proceeded in the presence of TsOH·H2O leading to the macrolactone in an acceptable yield. The removal of the Alloc group using Pd(PPh3)4/N-methylaniline furnished the desired metacridamide A, which was subjected to a direct conversion by hydrolysis of the acetate moiety at the C-13 position to provide metacridamide B without decomposition of the macrolactone ring. The spectral data of both synthetic products were identical to those of the natural products, resulting in the revision of the absolute configuration of the C-9 position in metacridamides A and B to be S. The further biological studies including an evaluation of the cytotoxicity against other cancer cells will be reported in due course.
This study was supported by JSPS KAKENHI (grant no. 20H02865 for H. K., 21KK0099 and 22H02207 for M. Y.) and partially supported by the Seeding Program from University of Tsukuba.
Conflicts of interest
There are no conflicts to declare.Notes and references
- S. B. Krasnoff, U. Englich, P. G. Miller, M. L. Shuler, R. P. Glahn, B. G. G. Donzelli and D. M. Gibson, J. Nat. Prod., 2012, 75, 180 CrossRef PubMed.
- A. Sharma, S. Athe, P. I. Ramesh, K. Vishali and S. Ghosh, Tetrahedron Lett., 2021, 82, 153374 CrossRef CAS.
- S. Shirokawa, M. Kamiyama, T. Nakamura, M. Okada, A. Nakazaki, S. Hosokawa and S. Kobayashi, J. Am. Chem. Soc., 2004, 126, 13604 CrossRef CAS.
- VMAR has been utilized as a powerful tool for the carbon-carbon bond formation. Examples applied in the total synthesis are following references; (a) M. Kalesse, M. Cordes, G. Symkenberg and H. Lu, Nat. Prod. Rep., 2014, 31, 563 RSC; (b) H. Miyatake-Ondozabal, E. Kaufmann and K. Gademann, Angew. Chem., Int. Ed., 2015, 54, 1933 CrossRef CAS; (c) J. Huang and Z. Wang, Org. Lett., 2016, 18, 4702 CrossRef CAS PubMed; (d) Y.-H. Zhang, R. Liu and B. Liu, Chem. Commun., 2017, 53, 5549 RSC; (e) B. Herlé, G. Späth, L. Schreyer and A. Fürstner, Angew. Chem., Int. Ed., 2021, 60, 7893 CrossRef; (f) X. Liang, M. Yoo, T. Schempp, S. Maejima and M. J. Krische, Angew. Chem., Int. Ed., 2022, 61, e202214786 CAS; (g) P. Tian, W. Ye, X. Zhang, Y. Tong, P.-Y. Qian and R. Tong, Chem. Sci., 2022, 13, 12776 RSC.
- R. Matsui, K. Seto, Y. Sato, T. Suzuki, A. Nakazaki and S. Kobayashi, Angew. Chem., Int. Ed., 2010, 50, 683 Search PubMed.
- (a) S. Hosokawa, T. Ogura, H. Togashi and K. Tatsuta, Tetrahedron Lett., 2005, 46, 333 CrossRef CAS; (b) S. Hosokawa, K. Yokota, K. Imamura, Y. Suzuki, M. Kawarasaki and K. Tatsuta, Tetrahedron Lett., 2006, 47, 5415 CrossRef CAS; (c) M. Yamaoka, A. Nakazaki and S. Kobayashi, Tetrahedron Lett., 2010, 51, 287 CrossRef CAS; (d) Y. Iwasaki, R. Matsui, T. Suzuki, A. Nakazaki and S. Kobayashi, Chem. Pharm. Bull., 2011, 59, 522 CrossRef CAS PubMed.
- T. Rezanka and K. Sigler, Eur. J. Org. Chem., 2006, 4284 Search PubMed.
- The absolute configuration of the VMAR product was determined by modified Mosher's method. Relative stereochemistry was determined by correlating to the reported products described in ref. 3 and references therein.
- Y. Mukaeda, T. Kato and S. Hosokawa, Org. Lett., 2012, 14, 5298 CrossRef CAS PubMed.
- The addition of a catalytic amount of water or extension of a reaction time did not improve the yield of desired 16. See ref. 6.
- I. Shiina, R. Ibuka and M. Kubota, Chem. Lett., 2002, 286 CrossRef CAS.
- J. Inanaga, K. Hirata, H. Saeki, T. Katsuki and M. Yamaguchi, Bull. Chem. Soc. Jpn., 1979, 52, 1989 CrossRef CAS.
- M. Yang, X. Wang and J. Zhao, ACS Catal., 2020, 10, 5230 CrossRef CAS.
- L. Hu, S. Xu, Z. Zhao, Y. Yang, Z. Peng, M. Yang, C. Wang and J. Zhao, J. Am. Chem. Soc., 2016, 138, 13135 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3cc01694c |
| This journal is © The Royal Society of Chemistry 2023 |