
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceElectrogenerated chemiluminescence at boron-doped diamond electrodes
Andrea
Fiorani
 *a,
Giovanni
Valenti
b,
Francesco
Paolucci
b and
Yasuaki
Einaga
a
*a,
Giovanni
Valenti
b,
Francesco
Paolucci
b and
Yasuaki
Einaga
a
aDepartment of Chemistry, Keio University, 3-14-1 Hiyoshi, Yokohama 223-8522, Japan. E-mail: andrea.fiorani@keio.jp
bDepartment of Chemistry “G. Ciamician”, University of Bologna, Via Selmi 2, 40126 Bologna, Italy
First published on 17th May 2023
Abstract
Electrogenerated chemiluminescence (ECL) refers to the phenomenon of light emission from molecular species which is triggered by an electrochemical reaction. Therefore, like most electrochemical systems, the electrode material plays a pivotal role and much effort has been made in order to find the best material for ECL, in terms of light signal intensity and long-term stability, especially after the development of ECL for analytical applications. In this article, we will introduce and highlight the distinctive features of boron-doped diamond (BDD) as an electrode material for ECL which has complementary properties compared to the most common metals (e.g., Au or Pt) and carbon materials (e.g., glassy carbon, carbon nanotubes and graphene). Boron-doped diamond electrodes emerged as novel electrodes, gaining more and more interest from the electrochemical community for their peculiar characteristics such as a wide solvent window, low capacitance, resistance to fouling and mechanical robustness. Furthermore, compared to metal electrodes, BDD does not form an oxide layer in aqueous solutions, and the sp3 carbon hybridization gives BDD the ability to enable peculiar electrochemical reactions that are not possible on sp2 carbon materials. Electrogenerated chemiluminescence investigations with boron-doped diamond electrodes have been reported for common ECL systems (luminophores and co-reactants), and special ECL that is only possible on BDD which includes the in situ electrochemical generation of the co-reactant.
 Andrea Fiorani | Andrea Fiorani graduated in chemistry from the University of Bologna, where he also obtained his PhD in chemistry. After working as a postdoctoral fellow in the group of Prof Francesco Paolucci, and shortly in the lab of Prof Wolfgang Schuhmann, he joined the laboratory of Prof Yasuaki Einaga at Keio University from 2018, where he is currently a postdoc fellow (Tokunin Jokyō). From 2019 to 2021, Dr Fiorani has been fellow of the Japan Society for the Promotion of Science. His research includes electrochemiluminescence, electrode materials and chemical conversion by electrochemical reactions. |
 Giovanni Valenti | Giovanni Valenti obtained his PhD degree in chemistry from the University of Bologna in 2010 under the guidance of Prof. Paolucci. He was appointed as an assistant professor in 2013 and he has been a visiting fellow at the Dublin City University (Prof Förster) and at the University of Texas (Prof Bard). In 2021 he was selected as an emerging investigator from ChemCommun and he became an associate professor of chemistry at Department “Ciamician” (University of Bologna). He applies electrochemical tools to study molecules and nanomaterials, for light generation (electrochemiluminescence), for early diagnosis (biosensors) and for energy application (CO2RR, HER and OER). |
 Francesco Paolucci | Francesco Paolucci is a professor of chemistry at the University of Bologna, and since 2001 he has been leading the electrochemistry group (EMFM) at the Department of Chemistry. He spent sabbatical leaves at the University of Southampton (1990–1991), Ecole Normale Supérieure in Paris (1995), University of Bourdeaux (2007, 2010, and 2012), and Keio University (2015). His current research includes fundamental themes of molecular and material electrochemistries in energy-related applications and in the development of detection tools of biomarkers and toxins. He spends a great part of his spare time on cooking (enthusiastically but with a great deal of irreproducibility). |
 Yasuaki Einaga | Yasuaki Einaga received his BS (1994), MS (1996), and PhD (1999) degrees from the University of Tokyo. After 2 years as a research associate at the University of Tokyo, he started his faculty career as an assistant professor at Keio University in2001, where he was promoted to full professor in 2011. He has been also a research director of JST-CREST (2011–2014), and JST-ACCEL (2014–2020). He was awarded “The Chemical Society of Japan Award for Creative Work” in 2016 for his pioneering work in diamond electrodes. His research interests include functional materials science, photochemistry, and electrochemistry. |
Introduction
Electrogenerated chemiluminescence, also known as electrochemiluminescence (ECL), is the emission of photons from a molecular species following a homogeneous high-energy (2–3 eV) electron transfer process in solution, which is triggered by a heterogeneous electrochemical reaction. The first experiments on luminescence induced by electrolysis date back to the 1920s1,2 and 1954.3 The development of ECL as it is known nowadays began in the mid-1960s with the pioneering works of Hercules,4 Chandross5 and Bard.6 Nowadays, the research on ECL is mainly focused on analytical applications, mostly concerning important biological targets found in serum, plasma, whole blood, and cells.7,8 The advantages peculiar to ECL are (1) high sensitivity achieved from amplification of the signal, (2) a broad dynamic range, (3) a low background as a result of the decoupling of electrochemical stimulus from the light signal, and (4) fast measurement. However, the molecular electrochemistry approach is still very important.9 The latter comprises the development of new luminophores such as inorganic complexes10,11 or organic molecules,12 either freely diffusing in solution or embedded within nanosystems,13–17 and ECL imaging with spatial and temporal resolution for the investigation of reaction mechanisms,18–26 and biological applications.27–34 Because ECL is primarily triggered by a heterogeneous electrochemical reaction, electrode materials are crucial in the signal development and intensity, and this subject was already reviewed for common electrode materials.35 Here, we focus our attention on doped diamonds, as special electrode materials with their own peculiarities, for application in electrochemiluminescence.Doped diamonds are semiconducting materials, and have received increasing attention in the electrochemical field over the last few decades, in electroanalytical chemistry,36–38 organic and inorganic electrosynthesis,39,40 and water treatment.41 Nitrogen, phosphorus and boron are the typical elements used for diamond doping, although boron is the most used element. Boron-doped diamond (BDD) is a p-type semiconductor material, and has several notable features when used as an electrode, such as a wide potential window, low capacitive currents, high chemical and physical stability, the ability to endure very high potential, and better resistance to fouling compared to other conventional electrodes.38 In particular, due to the wide potential window provided by BDD, both oxidation and reduction ECL reactions are accessible without interference from water oxidation or proton reduction, in addition to new pathways for the in situ co-reactant generation directly. Furthermore, depending on the growth of the diamond, the amount of boron concentration, and the pretreatment before the electrochemical measurement, the electrochemical properties of BDD can be adjusted at will, making it an even more interesting electrode material for electrochemists.
Because the research on ECL at BDD electrodes investigates mainly the co-reactant mechanisms,42 a useful classification is the distinction among the luminophores/co-reactants used, i.e., the reaction mechanism involved in the light generation. These include tris(bipyridine)ruthenium(II), and luminol as luminophores, and tri-n-propylamine, peroxydisulphate, and hydrogen peroxide as co-reactants, in particular for the in situ generation of the co-reactant, as developed by our research groups.
Reductive-oxidation co-reactant
Peroxydisulfate is a well-known co-reactant for reductive-oxidation ECL (Scheme 1).43–45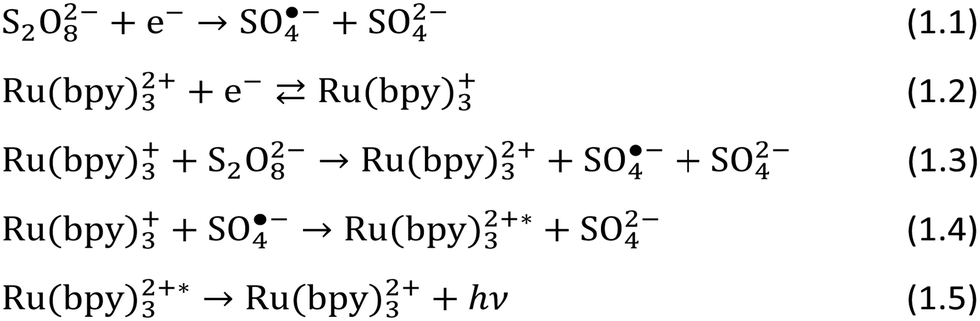 | ||
| Scheme 1 Reductive-oxidation ECL mechanism for Ru(bpy)32+/S2O82−. | ||
The ECL of tris(bipyridine)ruthenium(II), [Ru(bpy)3]2+/peroxydisulfate (S2O82−) at a BDD electrode was investigated through cyclic voltammetry and the results were compared with those of Pt and GC electrodes (Fig. 1).46
 | ||
| Fig. 1 ECL (a) and corresponding cyclic voltammograms (b) with BDD (black), GC (blue), and Pt (red) electrodes (0.635 cm2) for 10 μM Ru(bpy)32+ and 100 μM S2O82− in 200 mM phosphate buffer (pH 6.8). Scan rate: 100 mV s−1. In a, the curves are shifted for clarity. ECL and peak potential as a function of pH (c). ECL as a function of peroxydisulfate concentration (d). Reproduced from ref. 46 with permission of the American Chemical Society. | ||
No ECL signals were observed with Pt and GC electrodes, although there was a detectable ECL emission with the GC electrode when the Ru(bpy)32+ concentration was increased by 5 times. In contrast, the ECL signal from the BDD electrode was clearly evident. The rate of hydrogen evolution, which is related to the cathodic current (BDD < GC ≪ Pt), adversely affects the ECL emission, preventing light detection, and hindering the reduction of Ru(bpy)32+ and peroxydisulfate in favour of proton reduction. The higher overpotential for proton reduction at BDD, compared to GC and Pt, makes diamond electrodes far superior for reductive–oxidation ECL in aqueous solutions.
A pH variation, from 9 to 4, resulted in two distinctive effects: an increase in the ECL emission which was ascribed to lower quenching by hydroxyl ions of sulphate radical anions, and a shift to negative potentials of the ECL peak. Although the cathodic current for hydrogen evolution increased, this was not a limiting factor that hindered the ECL emission, at least until pH 4, while at pH 3, the ECL decreased and it completely turned off at pH 2. However, this might be responsible for the potential shift of the ECL peak to more negative values as the pH decreased.
The ECL response as a function of peroxydisulfate concentration was linear at low peroxydisulfate concentrations (1–100 μM), and the maximum emission occurred at 10 mM, while at 100 mM, the emission decreased due to the oxidative quenching of the excited state of Ru(bpy)32+ by peroxydisulfate ions. Interestingly, we found new evidence of ECL emission for solutions with a Ru(bpy)32+/peroxydisulfate ratio down to 1/1000, while previous data from White et al.43 reported a value of 1/20 and Yamazaki-Nishida et al.47 reported a ratio of 1/200. This increases the range of available persulfate concentrations that can be used profitably without interference from oxidative quenching, which will enable a wider range of Ru(bpy)32+ detection.
Oxidative–reduction co-reactant
Tri-n-propylamine is the most used and studied tertiary amine co-reactant in ECL for inorganic complexes, mainly of ruthenium and iridium.48–50 The [Ru(bpy)3]2+/tri-n-propylamine (TPrA) couple is the benchmark for academic research19 and the only available system in commercial applications of ECL for clinical analysis,7,8 with previous attempts to investigate this system also on BDD.51–55 The oxidative–reduction mechanism leading to ECL emission has been long established (Scheme 2), and this mechanism applies when Ru(bpy)32+ is freely diffusing in solution, i.e., the direct oxidation at the electrode is possible (eqn (2.4)).48 | ||
| Scheme 2 Oxidative-reduction ECL mechanism for Ru(bpy)32+ and TPrA. P1 is the degradation product of TPrA˙ by oxidation and following hydrolysis. | ||
The ECL from this mechanism is reported in Fig. 2 for the anodic oxidized (AO) and cathodic reduced (CR) surface states of BDD.56 CR-BDD performed better compared to AO-BDD, with higher intensity and lower onset potential of emission. This is a direct consequence of different kinetics for TPrA oxidation, where a higher current for CR-BDD leads to higher ECL intensity. This was further confirmed by measuring the rate of electron transfer (kET, Fig. 2b). CR-BDD has higher hydrophobicity than AO-BDD as a consequence of the hydrogen terminated surface, facilitating the adsorption of the hydrophobic TPrA molecule before oxidation. The lower onset potential for ECL emission, especially before Ru(bpy)32+ oxidation, depends on TPrA oxidation as well, although it is more affected by a different mechanism which requires only TPrA oxidation (Scheme 3).
 | ||
| Fig. 2 ECL intensity and cyclic voltammograms (a) at 100 mV s−1 for 10 μM Ru(bpy)32+ and 100 mM TPrA in 0.2 M phosphate buffer (pH 8.0). Logarithmic plot of kET for TPrA oxidation at anodic oxidised (AO, red), and cathodic reduced (CR, black) BDD electrodes (b). Potential referred to Ag/AgCl (KCl sat’d). Reproduced from ref. 56 with permission of the American Chemical Society. | ||
 | ||
| Scheme 3 Mechanism involved in ECL emission before Ru(bpy)32+ oxidation, triggered only TPrA oxidation. P1 is the degradation product of TPrA˙ from oxidation and following hydrolysis. | ||
Furthermore, this mechanism is of outmost importance because it is the only active mechanism leading to ECL emission in bead-based immunoassay analysis.
Interestingly, the mechanism swapping from Ru(bpy)32+ free diffusing to labelled on beads is evident from the pH effect on the TPrA oxidation and follow up reactions (Fig. 3).
 | ||
| Fig. 3 Normalised ECL as a function of pH at the 1% BDD electrode: Ru(bpy)32+ free diffusing in solution (black), and Ru(bpy)32+ labelled on 2.8 μm beads deposited on the electrode (red). Author's own elaboration based on data available from ref. 56. | ||
The ECL relationship with pH is dictated by two opposite effects: (i) a pH increase will bring a higher amount of free TPrA available for oxidation (i.e., TPrAH+ is not electroactive, eqn (3.1) and (3.2)); meanwhile, (ii) higher concentrations of hydroxyl ions promote the TPrA˙+ deprotonation by proton scavenging which leads to lower ECL (eqn (3.3)–(3.5)).
The ECL generated by BDD has been investigated by imaging single Ru(bpy)32+ labelled beads (Fig. 4) which mimics exactly an ECL immunoassay. According to the mechanism proposed by Miao et al.,49 which was re-examined by Zanut et al.,19 the emission is described using Scheme 3. ECL from Ru(bpy)32+ labelled beads is clearly evident from BDD, although AO-BDD resulted in higher emission compared to CR-BDD, as opposite to Ru(bpy)32+ in solution.
 | ||
| Fig. 4 ECL imaging from 2.8 μm Ru(bpy)32+ labelled beads for AO-BDD (a) and CR-BDD (b). The images were obtained by applying a constant potential of 1.7 V (vs. Ag/AgCl, 3 M KCl) for 4 s. Scale bar: 10 μm. Comparison of the bead profile lines (c) and integrated ECL (d) for AO-BDD (red) and CR-BDD (black). ECL intensity obtained from single Ru(bpy)32+ labelled beads at 100 mV s−1 through CV (e). Solution: 100 mM TPrA in 0.2 M phosphate buffer (pH 7.0). Reproduced from ref. 56 with permission of the American Chemical Society. | ||
To assess the full potential of BDD in analytical applications, the ECL has been benchmarked against Pt electrodes which is used in analytical instrumentation with bead conjugates for clinical analysis.57,58 In this case, the electrodes are covered with a layer of Ru(bpy)32+ labelled beads, and the ECL is the sum of all single bead emissions.
As shown in Fig. 5, AO-BDD has a superior ability to produce ECL as the emission reached a value 70% higher compared to Pt electrodes.
 | ||
| Fig. 5 Integrated ECL from Ru(bpy)32+ labelled beads for AO-BDD at 2.4 V (red), CR-BDD at 1.6 V (black), and Pt at 1.5 V (green). Solutions: 100 mM TPrA in 0.2 M phosphate buffer (pH 7) for both AO-BDD and CR-BDD electrodes; 180 mM TPrA and 0.1 wt % C12E9 surfactant in 0.2 M phosphate buffer (pH 6.9) for Pt electrode. Reproduced from ref. 56 with permission of the American Chemical Society. | ||
Metal electrodes, such as Pt, are known to develop a layer of metal oxide during potential application that suppresses the ECL signal by hindering TPrA oxidation. For this reason, they benefit from the addition of surfactants to prevent the formation of the surface oxides and favour the TPrA adsorption, therefore enhancing the ECL signal.35,59,60 However, BDD could achieve a higher ECL signal without surfactant addition; it showed a signal-to-noise ratio for AO-BDD that was 2 times higher compared to Pt electrodes, and noteworthily, the amount of TPrA needed was lowered from 180 to 100 mM with a significant savings of this toxic and relatively expensive chemical.56 These results can be a step forward for its practical application beyond fundamental studies to expand the frontiers of ECL immunoassays and BDD electrodes.
As a concluding remark, we would like to draw attention to the opposite ECL outcome of the CR-BDD and AO-BDD when used in homogeneous (Fig. 2) or heterogeneous (Fig. 4 and 5) systems. Although the better performance of CR-BDD compared to AO-BDD in homogeneous systems has been straightforwardly attributed to the faster oxidation rate of TPrA (Fig. 2b), the better performance of AO-BDD compared to CR-BDD in heterogeneous systems does not have a suitable explanation. This subject is still a matter of investigation; however we want to point out that these opposing results are not necessarily in contradiction as the ECL systems are homogeneous vs heterogeneous, and the optimal potentials applied are different. In addition, the same results on beads were confirmed independently by two methods: (1) single bead emission detected using a CCD camera (Fig. 4), and (2) collective bead signals measured using a PMT (Fig. 5).
Co-reactant in situ generation
Besides analytical applications,37 BDD is well-known for electrochemical advanced oxidation processes, in particular the production of oxidants from hydroxyl radical mediated processes.61–63The reaction sequence initiated by hydroxyl radicals can generate radicals active as co-reactants if particular species are present in solution. We proved this concept by the generation in situ of peroxydisulfate and hydrogen peroxide, as co-reactants for Ru(bpy)32+ and luminol, by oxidation of sulfate and carbonate, respectively. The procedure is rather straightforward, not requiring any particular electrode geometry, with the reactive co-reactant generated on the electrode surface for a short time.
Peroxydisulphate co-reactant from sulphate oxidation
The reaction mechanism for the generation of the peroxydisulphate co-reactant from sulphate oxidation and following ECL reaction with Ru(bpy)32+ is depicted in Fig. 6.64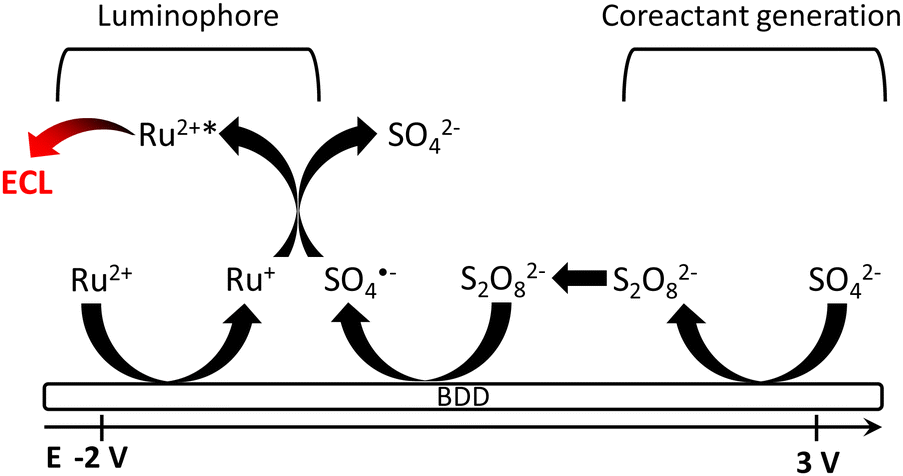 | ||
| Fig. 6 Reaction mechanism of electrochemiluminescence from Ru(bpy)32+ on BDD electrode with sulphate ions. Ru = Ru(bpy)3. Reproduced from ref. 64 with permission of the American Chemical Society. | ||
The ability of BDD (i) to promote peroxydisulphate generation with high efficiency was coupled with (ii) the high overpotential for the hydrogen evolution reaction to allow, in the end, the efficient ECL generation in a Ru(bpy)32+/SO42− aqueous solution. The mechanism begins with sulphate oxidation at BDD (Scheme 4) during the first positive scan of cyclic voltammetry.
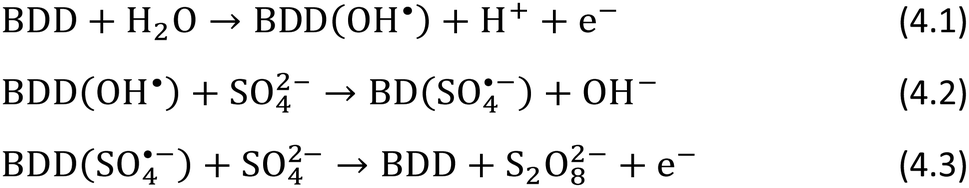 | ||
| Scheme 4 Electrogeneration of peroxydisulphate at BDD by sulphate oxidation. | ||
Then, it proceeds with the ECL mechanism of Ru(bpy)32+/peroxydisulphate as reported previously in Scheme 1 during the negative potential scan and emission started at −1.5 V, in conjunction with Ru(bpy)32+ reduction to Ru(bpy)3+ (Fig. 7).
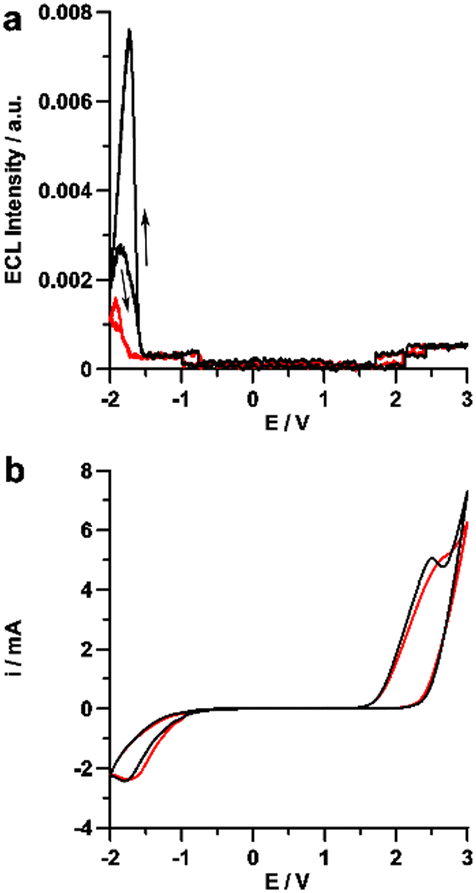 | ||
| Fig. 7 ECL (a) and corresponding cyclic voltammograms (b) comparison between 0.1 M Na2SO4 (black) and 0.1 M KClO4 (red) with 5 μM Ru(bpy)3Cl2 in water solution. Scan rate 100 mV s−1, potential referred to Ag/AgCl (KCl sat’d). Reproduced from ref. 64 with permission of the American Chemical Society. | ||
On the other hand, the cyclic voltammetry curves obtained in the absence of sulphate ions, that is, in aqueous KClO4 (Fig. 7a, red line) showed only a much lower ECL emission which has been ascribed to annihilation mechanism (eqn (1)) from Ru(bpy)32+ oxidation and reduction during cyclic voltammetry. Such a mechanism, which involves a couple of fluorophores in either their oxidized and reduced form, is generally unobserved in aqueous media where the prevailing hydrogen evolution reaction prevents the formation of the reduced species Ru(bpy)3+, while in the present case it would be made possible by the high overpotential for hydrogen evolution on BDD.
| Ru(bpy)3+ + Ru(bpy)33+ ⇄ Ru(bpy)32+* + Ru(bpy)32+ | (1) |
Oxidation charge for sulphate oxidation increased linearly with oxidation time, thus suggesting that the formation of surface reactive species (eqn (4.1)), rather than diffusion of the sulphate, controls the oxidation process. In contrast, the ECL signals decreased linearly with the square root of oxidation time for sulphate, indicating that the efficiency of the overall ECL generation process is limited by diffusion of the sulphate precursor to the electrode and of electrogenerated peroxydisulfate toward the bulk of solution (Fig. 8a).
 | ||
| Fig. 8 ECL intensity transients measured by chronoamperometry with 5 μM Ru(bpy)3Cl2: (a) in 0.1 M Na2SO4 aqueous solution (inset: integrated ECL intensity vs. square root of sulphate oxidation time step, tox). (b) ECL at various Na2SO4 concentrations (Inset: integrated ECL intensity as a function of concentration). Reproduced from ref. 64 with permission of the American Chemical Society. | ||
The ECL generation was investigated at various sulphate concentrations. There was a linear range from 10−3 M to 0.1 M, followed by a slower increase of ECL emission which reached a plateau at [SO42−] ≈ 0.5 M, stable until 1 M. However, the charge for oxidation was linear until ≈0.6 M and kept increasing until 1 M. The observed trend of ECL intensity vs. sulphate precursor concentration can be ascribed to the known ability of peroxydisulfate ions to effectively quench the excited state Ru(bpy)32+*, as demonstrated previously (Fig. 1d).
Hydrogen peroxide co-reactant from carbonate oxidation
If carbonate is used instead of sulphate, it is possible to obtain a co-reactant for Ru(bpy)32+ through the same procedure already presented, although not carbonate radicals, but hydrogen peroxide will be involved in the ECL generation.65 The sequence of chemical reactions involved in this Ru(bpy)32+/carbonate ECL system is reported in Scheme 5 for hydrogen peroxide production. | ||
| Scheme 5 Hydrogen peroxide production from carbonate oxidation at BDD. | ||
The ECL generation involves reactions reported in Scheme 6, in agreement with the “reductive–oxidation” mechanism for H2O2.66
 | ||
| Scheme 6 Reductive–oxidation ECL mechanism for Ru(bpy)32+/H2O2. | ||
ECL can be obtained by cyclic voltammetry of Ru(bpy)32+ in carbonate aqueous solution (Fig. 9), first scanning the potential from 0 V to 2.5 V to generate the hydrogen peroxide (Scheme 5), followed by scanning to a negative potential up to −2.5 V to generate the ECL emission (Scheme 6) concurrently to Ru(bpy)32+ reduction at −1.5 V.
 | ||
| Fig. 9 ECL (a) and corresponding cyclic voltammograms (b) comparison between 0.1 M NaClO4 (black) and 0.1 M Na2CO3 (red) with of 10 μM Ru(bpy)3Cl2 in water solution (pH 11.5). Scan rate 100 mV s−1, potential referred to Ag/AgCl (KCl sat’d). Reproduced from ref. 65 with permission of the American Chemical Society. | ||
The ECL emission was found to increase linearly from 1.8 to 2.5 V, which indicates an increase in hydrogen peroxide production from the electrogenerated peroxydicarbonate (Fig. 10a).
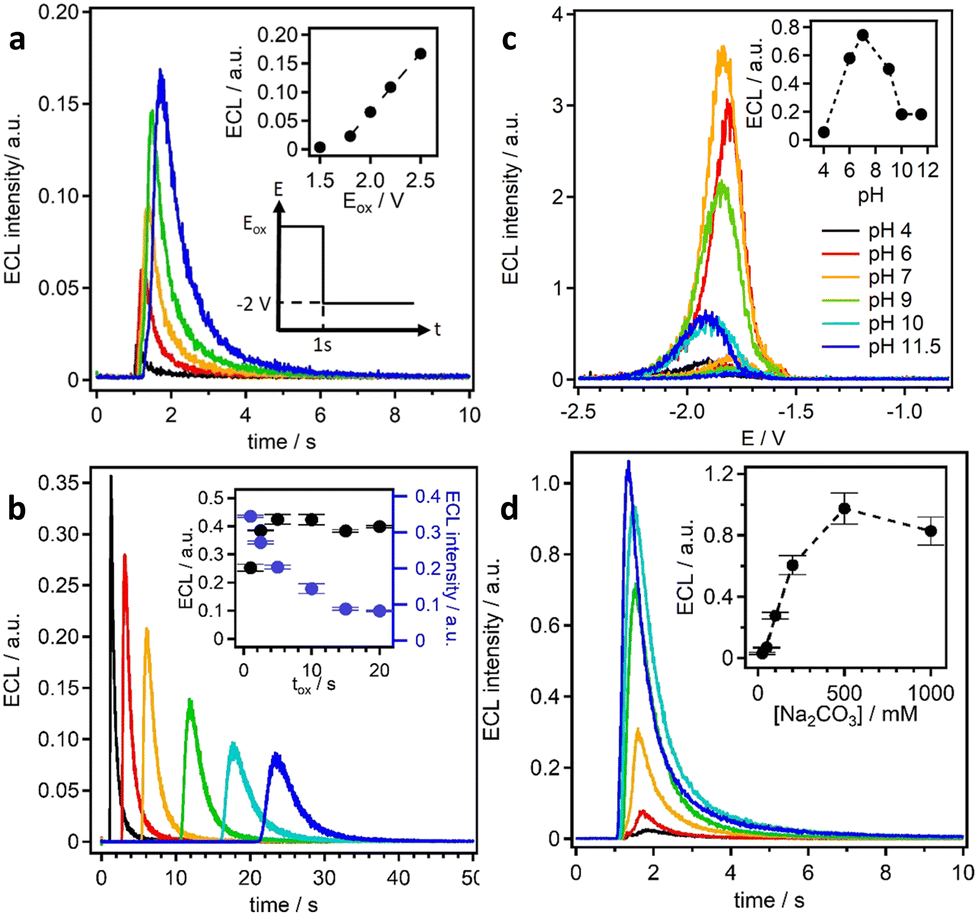 | ||
| Fig. 10 ECL emission as function of: oxidation potential, Eox (a); oxidation time, tox (b); pH (c); carbonate concentration (d). Reproduced from ref. 65 with permission of the American Chemical Society. | ||
If the oxidation time was increased, the ECL emission showed an increase up to 5 s and then nearly a stable emission up to 20 s (Fig. 10b). The ECL is highly affected by the gradient of the co-reactant in the diffusion layer. In fact, the diffusion layer thickness for carbonate within 1–20 s oxidation times ranges roughly from 100 to 600 μm. This results in a lower peak intensity, in broader ECL emission, and an increased delay time to reach the maximum intensity.
Because carbonate is a weak base, the effect of pH on the ECL signal was investigated to highlight the effect of the carbonate/bicarbonate couple. The oxidation current was found to decrease with decreasing pH, and because in the neutral region the concentration of carbonate is less dominant compared to that of the bicarbonate, the current measured could be ascribed the carbonate ions rather than bicarbonate. However, the ECL signal increased as the pH decreased from 12 to 7, while a further decrease in pH to 4 also resulted in a decrease in the ECL emission, with the maximum ECL emission being observed at pH 7. In this case, the stability of the hydrogen peroxide generated in solution might be responsible for the ECL emission, as hydrogen peroxide is more stable at neutral or acidic pH. Moreover, carbonate can promote the decomposition of hydrogen peroxide. In conclusion, the ECL emission as the pH decreases is influenced by two trends: (i) the decreasing carbonate oxidation current therefore, low hydrogen peroxide production, and (ii) the increasing hydrogen peroxide stability.
The ECL emission as a function of carbonate concentration was found to be linear within the range of 0.025–0.2 M and reached a plateau at 0.5 M. The decrease at 1 M carbonate can be explained by the possibility of quenching the Ru(bpy)32+ excited state by hydrogen peroxide, similar to peroxydisulphate. The carbonate oxidation increased with carbonate concentration, suggesting that hydrogen peroxide production is also increasing. The ECL efficiency compared to that of hydrogen peroxide was 20%, and it reached the maximum at 200 mM of carbonate.
Attenuated total reflectance–IR spectroscopy revealed a difference in absorption when the carbonate oxidation was performed in H2O or D2O (Fig. 11).
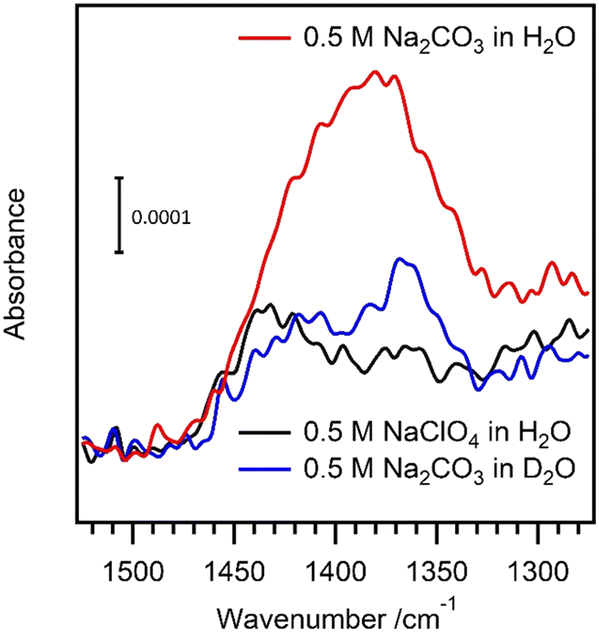 | ||
| Fig. 11 Attenuated total reflectance–IR spectra on a BDD electrode: 0.5 M Na2CO3 in H2O (red), in D2O (blue); 0.5 M NaClO4 in H2O (black). Oxidation at 2.5 V (vs. Ag/AgCl, KCl sat’d). Reproduced from ref. 65 with permission of the American Chemical Society. | ||
Only oxidation of carbonate in H2O, as opposed to carbonate in D2O or perchlorate in H2O showed a signal at around 1380 cm−1 which can be interpreted by slow or even no peroxydicarbonate hydrolysis to generate hydrogen peroxide in D2O. This also implies that hydrogen peroxide was not generated during oxidation without carbonate, which is consistent with the lack of relevant ECL emission in NaClO4.
In this particular process for hydrogen peroxide production, Ru(bpy)32+ can be replaced with luminol, a classical compound for hydrogen peroxide detection in chemiluminescence,67 and electrochemiluminescence.23,67–70 The classical ECL of luminol and hydrogen peroxide by using BDD was reported previously, for example, our group investigated a particular application for the detection of hypochlorite,71 in addition to investigations of reaction mechanisms.72–74
ECL from the luminol/carbonate system can be obtained through an oxidation step only (Fig. 12).75
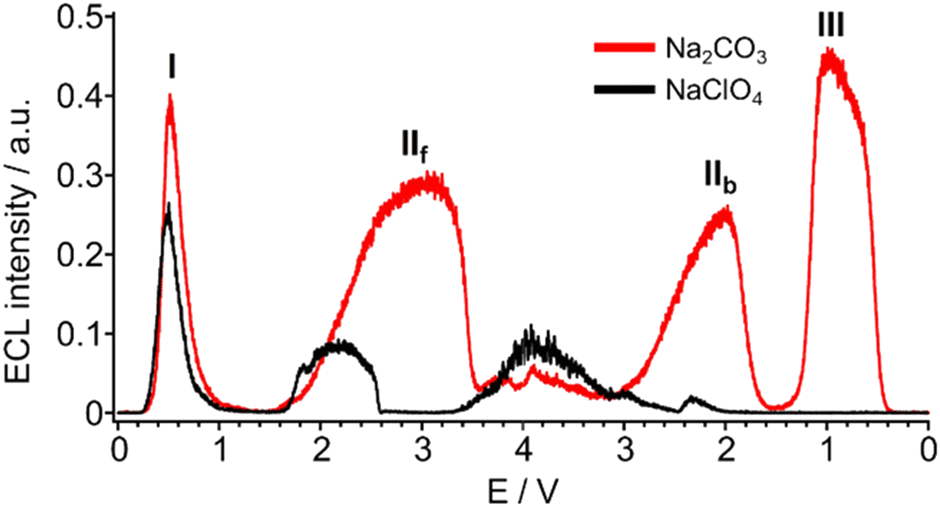 | ||
| Fig. 12 ECL intensity by cyclic voltammetry of 100 μM luminol in 100 mM Na2CO3 (red) and in 100 mM NaClO4 (black) at pH 12. Scan rate, 100 mV s−1. Reference electrode: Ag/AgCl (KCl Sat’d). For clarity, the potentials of forward and backward scans have been plotted consecutively, and peaks are named using roman numerals. Subscripts “f” and “b” stand for forward and backward, respectively. Reproduced from ref. 75 with permission of the American Chemical Society. | ||
The ECL emission was obtained in the whole potential range from 0 V to 4 V, both in the forward and backward scan, for a total of 4 peaks.
The peak I is the ECL from direct luminol oxidation, while peak II is peculiar of BDD where hydrogen peroxide is generated by carbonate oxidation (Scheme 5). Peak III was associated to accumulation of hydrogen peroxide in bulk solution.
The reaction of luminol and hydrogen peroxide is reported in Scheme 7.
 | ||
| Scheme 7 Simplified ECL reaction mechanism of luminol and hydrogen peroxide. L = luminol, 3AP = 3-aminophthalate dianion. | ||
Direct correlation with carbonate concentration was obtained for peak IIf, validating the direct involvement of carbonate oxidation in the ECL development (Fig. 13a); therefore we focused our attention on this peak for the following pH investigation.
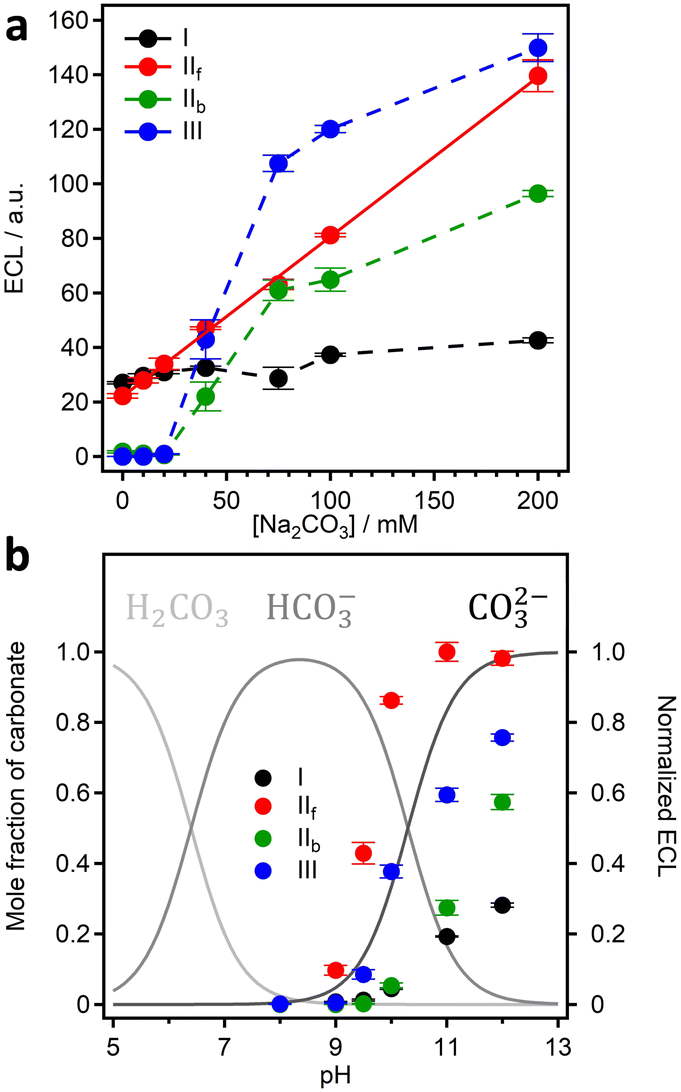 | ||
| Fig. 13 (a) ECL emission as a function of carbonate concentration and (b) mole fraction of carbonate (left y-axis), and ECL emission (right y-axis) as a function of pH (ECL signals are normalized to the maximum of peak IIf). Reproduced from ref. 75 with permission of the American Chemical Society. | ||
In fact, it was possible to shed light on the species between carbonate and bicarbonate which underwent oxidation at the BDD electrode. The ECL emission was not detected when bicarbonate was the prevalent species in solution (pH 8), while the ECL emission increased promptly with the molar fraction of carbonate (Fig. 13b). This clearly confirms that the electrochemical oxidation in carbonated solutions on the BDD electrode is specifically on carbonate rather than bicarbonate or both anions. We reported this evidence for the first time,75 which was later suggested by other researchers.76
In conclusion, the in situ production of the co-reactant from inactive species permitted to realise new ECL pathways which could not be obtained using common electrode materials, highlighting the outstanding ability of BDD. As a highly promising transduction method in analytical chemistry, ECL by in situ production of the co-reactant might hypothetically fulfil this task. However, the emission intensities are generally low (vide infra) which might preclude real analytical applications. In contrast, it could find application in mechanistic investigation, for example, as we showed in the discrimination of carbonate/bicarbonate oxidation for the production of hydrogen peroxide.
Effect of boron concentration
The level of boron doping can affect greatly the electrochemical properties of BDD,78i.e., the ECL generation and intensity; therefore we investigated this effect for a range of boron concentrations near the transition of hopping-metallic conduction, as it was observed near 3 × 1020 cm−3 (Fig. 14).79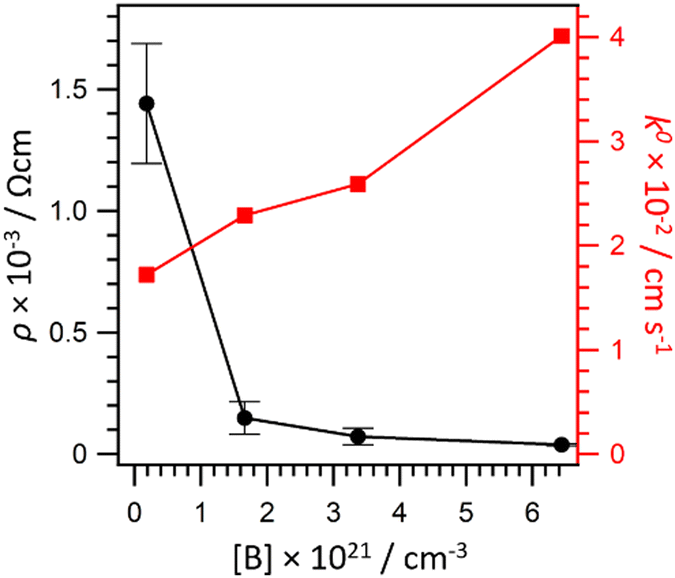 | ||
| Fig. 14 Resistivity of the BDD electrodes (black) and electron transfer constant for the reduction of Ru(NH3)63+ (red) as a function of the boron doping level. Reproduced from ref. 77 with permission of the American Chemical Society. | ||
The ECL emission was evaluated for all the previous systems (Fig. 15). Ru(bpy)32+/TPrA was confirmed to be a superior ECL couple, also on BDD. The tunable proprieties of BDD, as the concentration of boron is changed, have shown that every ECL system has its own optimal boron concentration. Furthermore, this proves that BDD offers a remarkable advantage compared to electrodes where the properties are intrinsic of the material used because the properties can be tuned through boron doping and electrochemical pretreatment, which later show a remarkable effect on the ECL emission.
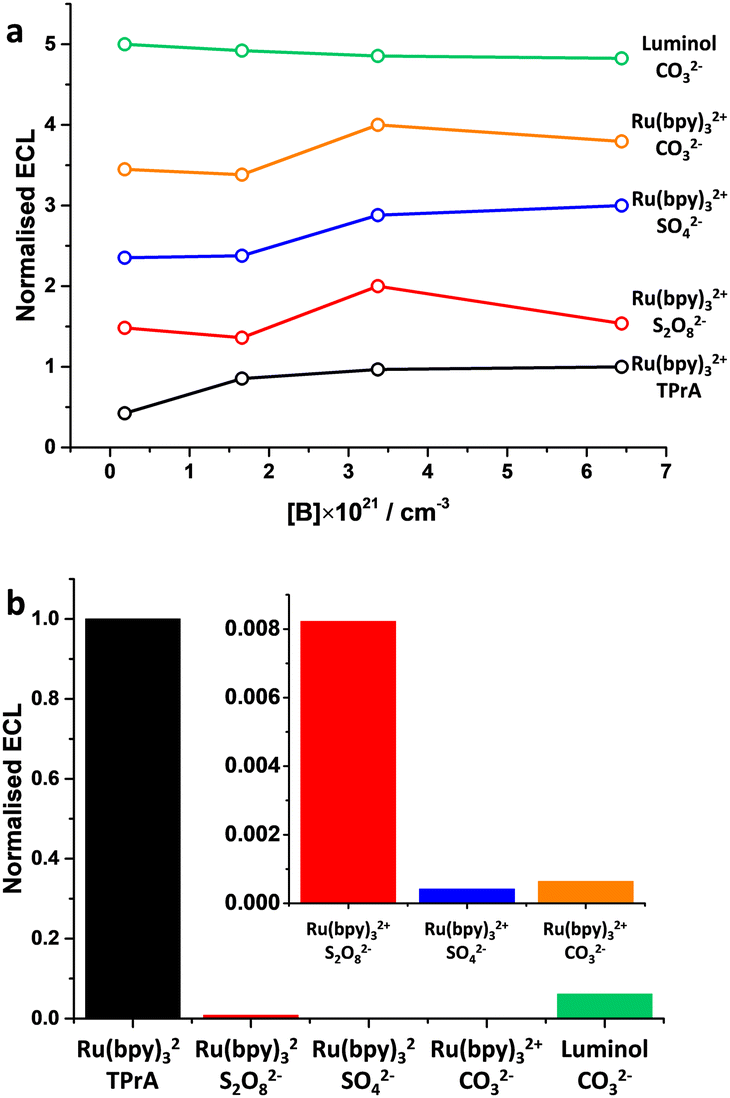 | ||
| Fig. 15 (a) ECL emission as a function of the boron doping level in BDD; (b) relative ECL emission normalised to Ru(bpy)32+/TPrA for the best boron doping level from (a). Author's own elaboration based on data available from ref. 77. | ||
Boron concentrations in the range 3–6 × 1021 cm−3 were found generally the most suitable for ECL applications. While higher boron concentrations can result in the insertion of a non-negligible amount of sp2 carbon during BDD fabrication, lower boron concentrations might exhibit sluggish electron transfer kinetics. This highlights that care must be taken when investigating the electrochemical system of interest to select the most appropriate boron concentration. For this reason, the effect of boron doping should be considered as a general parameter worth the optimization. In addition, we can infer that cathodic reduction of the BDD surface is more suitable for oxidation reactions, while it is the opposite for reduction reactions where anodic oxidation of the BDD surface seems more appropriate.
Conclusions
Here we revised the ECL studies conducted in recent years by our research groups, where the most relevant ECL systems have been examined by using boron-doped diamonds as electrode materials. These comprised Ru(bpy)32+/tri-n-propylamine, Ru(bpy)32+/peroxydisulphate, Ru(bpy)32+/sulphate, Ru(bpy)32+/carbonate, and luminol/carbonate. Sulphate and carbonate are not co-reactants for ECL, but thanks to the properties of BDD, it was possible to generate in situ the co-reactants peroxydisulphate and hydrogen peroxide, respectively. From the perspective of in situ co-reactants, we would like to point out that new ECL reactions can be unveiled by using cutting-edge electrode materials, which might advance the ECL toward more robust and sensitive technological applications. As a final remark, although this review focuses on ECL, some of the reaction mechanisms are of general application, and could be helpful in the broad context of electrochemistry with boron-doped diamond electrodes. We have summarised important parameters in Table 1 to show the electrochemiluminescence with boron-doped diamonds at a glance.| BDD B/C | Beads | In situ co-reactant | pH | Rel. ECL | |
|---|---|---|---|---|---|
| Ru(bpy)32+/TPrA | 1% | ✓ | ✗ | 8 | 1 |
| 7 (beads) | |||||
| Ru(bpy)32+/S2O82− | 1% | ✗ | ✗ | 4 | 0.008 |
| Ru(bpy)32+/SO42− | 2% | ✗ | S2O82− | ✗ | 0.0004 |
| Ru(bpy)32+/CO32− | 1% | ✗ | H2O2 | 7 | 0.0006 |
| Luminol/CO32− | 0.1% | ✗ | H2O2 | 11 | 0.06 |
Author contributions
Andrea Fiorani: conceptualization and writing – original draft. All authors: writing – review and editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
AF acknowledges the Japan Society for the Promotion of Science (fellowship ID no. P19333) and grant-in-aid for JSPS fellows (19F19333). GV and FP would like to thanks the Nano-ImmunoEra project. Nano-ImmunoEra project has received funding from the European Union’s MSCA Staff exchange Horizon Europe programme Grant Agreement Number 101086341.References
- T. Dufford, D. Nightingale and L. W. Gaddum, J. Am. Chem. Soc., 1927, 49, 1858–1864 CrossRef.
- N. Harvey, J. Phys. Chem., 1929, 33, 1456–1459 CrossRef CAS.
- V. Vojírõ, Collect. Czech. Chem. Commun., 1954, 19, 868–872 CrossRef.
- D. M. Hercules, Science, 1964, 145, 808–809 CrossRef CAS PubMed.
- R. E. Visco and E. A. Chandross, J. Am. Chem. Soc., 1964, 86, 5350–5351 CrossRef CAS.
- S. V. Santhanam and A. J. Bard, J. Am. Chem. Soc., 1965, 87, 139–140 CrossRef.
- M. M. Richter, Chem. Rev., 2004, 104, 3003–3036 CrossRef CAS PubMed.
- W. Miao, Chem. Rev., 2008, 108, 2506–2553 CrossRef CAS PubMed.
- A. Fiorani, G. Valenti, M. Iurlo, M. Marcaccio and F. Paolucci, Curr. Opin. Electrochem., 2018, 8, 31–38 CrossRef CAS.
- E. Villani, K. Sakanoue, Y. Einaga, S. Inagi and A. Fiorani, J. Electroanal. Chem., 2022, 921, 116677 CrossRef CAS.
- M. A. Haghighatbin, S. E. Laird and C. F. Hogan, Curr. Opin. Electrochem., 2018, 7, 216–223 CrossRef CAS.
- A. Fiorani, M. Difonzo, F. Rizzo and G. Valenti, Curr. Opin. Electrochem., 2022, 34, 100998 CrossRef CAS.
- G. Valenti, E. Rampazzo, S. Kesarkar, D. Genovese, A. Fiorani, A. Zanut, F. Palomba, M. Marcaccio, F. Paolucci and L. Prodi, Coord. Chem. Rev., 2018, 367, 65–81 CrossRef CAS.
- S. Kesarkar, S. Valente, A. Zanut, F. Palomba, A. Fiorani, M. Marcaccio, E. Rampazzo, G. Valenti, F. Paolucci and L. Prodi, J. Phys. Chem. C, 2019, 123, 5686–5691 CrossRef CAS.
- K. Imai, G. Valenti, E. Villani, S. Rapino, E. Rampazzo, M. Marcaccio, L. Prodi and F. Paolucci, J. Phys. Chem. C, 2015, 119, 26111–26118 CrossRef CAS.
- A. Zanut, F. Palomba, M. Rossi Scota, S. Rebeccani, M. Marcaccio, D. Genovese, E. Rampazzo, G. Valenti, F. Paolucci and L. Prodi, Angew. Chem., Int. Ed., 2020, 59, 21858–21863 CrossRef CAS PubMed.
- K. Imai, G. Valenti, E. Villani, S. Rapino, E. Rampazzo, M. Marcaccio, L. Prodi and F. Paolucci, J. Phys. Chem. C, 2015, 119, 26111–26118 CrossRef CAS.
- E. Kerr, E. H. Doeven and P. S. Francis, Curr. Opin. Electrochem., 2022, 35, 101034 CrossRef CAS.
- A. Zanut, A. Fiorani, S. Canola, T. Saito, N. Ziebart, S. Rapino, S. Rebeccani, A. Barbon, T. Irie, H.-P. Josel, F. Negri, M. Marcaccio, M. Windfuhr, K. Imai, G. Valenti and F. Paolucci, Nat. Commun., 2020, 11, 2668 CrossRef CAS PubMed.
- A. Fiorani, D. Han, D. Jiang, D. Fang, F. Paolucci, N. Sojic and G. Valenti, Chem. Sci., 2020, 11, 10496–10500 RSC.
- M. Sentic, M. Milutinovic, F. Kanoufi, D. Manojlovic, S. Arbault and N. Sojic, Chem. Sci., 2014, 5, 2568–2572 RSC.
- W. Guo, P. Zhou, L. Sun, H. Ding and B. Su, Angew. Chem., Int. Ed., 2021, 60, 2089–2093 CrossRef CAS PubMed.
- P. Zhou, S. Hu, W. Guo and B. Su, Fundam. Res., 2022, 2, 682–687 CrossRef CAS.
- P. Zhou, W. Fu, L. Ding, Y. Yan, W. Guo and B. Su, Electrochim. Acta, 2023, 439, 141716 CrossRef CAS.
- C. Ma, W. Wu, L. Li, S. Wu, J. Zhang, Z. Chen and J.-J. Zhu, Chem. Sci., 2018, 9, 6167–6175 RSC.
- E. Villani and S. Inagi, Anal. Chem., 2021, 93, 8152–8160 CrossRef CAS PubMed.
- G. Valenti, S. Scarabino, B. Goudeau, A. Lesch, M. Jović, E. Villani, M. Sentic, S. Rapino, S. Arbault, F. Paolucci and N. Sojic, J. Am. Chem. Soc., 2017, 139, 16830–16837 CrossRef CAS PubMed.
- K. Hiramoto, E. Villani, T. Iwama, K. Komatsu, S. Inagi, K. Y. Inoue, Y. Nashimoto, K. Ino and H. Shiku, Micromachines, 2020, 11, 530 CrossRef PubMed.
- K. Hiramoto, K. Ino, K. Komatsu, Y. Nashimoto and H. Shiku, Biosens. Bioelectron., 2021, 181, 113123 CrossRef CAS PubMed.
- K. Ino, K. Komatsu, K. Hiramoto, Y. Utagawa, Y. Nashimoto and H. Shiku, Electrochim. Acta, 2022, 415, 140240 CrossRef CAS.
- Y. Liu, H. Zhang, B. Li, J. Liu, D. Jiang, B. Liu and N. Sojic, J. Am. Chem. Soc., 2021, 143, 17910–17914 CrossRef CAS PubMed.
- Y. Ma, C. Colin, J. Descamps, S. Arbault and N. Sojic, Angew. Chem., Int. Ed., 2021, 60, 18742–18749 CrossRef CAS PubMed.
- J. Dong, Y. Lu, Y. Xu, F. Chen, J. Yang, Y. Chen and J. Feng, Nature, 2021, 596, 244–249 CrossRef CAS PubMed.
- J. Dong and J. Feng, Anal. Chem., 2023, 95, 374–387 CrossRef CAS PubMed.
- G. Valenti, A. Fiorani, H. Li, N. Sojic and F. Paolucci, ChemElectroChem, 2016, 3, 1990–1997 CrossRef CAS.
- S. Yu, S. Liu, X. Jiang and N. Yang, Carbon, 2022, 200, 517–542 CrossRef CAS.
- K. Muzyka, J. Sun, T. H. Fereja, Y. Lan, W. Zhang and G. Xu, Anal. Methods, 2019, 11, 397–414 RSC.
- N. Yang, S. Yu, J. V. Macpherson, Y. Einaga, H. Zhao, G. Zhao, G. M. Swain and X. Jiang, Chem. Soc. Rev., 2019, 48, 157–204 RSC.
- S. Lips and S. R. Waldvogel, ChemElectroChem, 2019, 6, 1649–1660 CrossRef CAS.
- J. Sotelo-Gil, E. Cuevas-Yañez and B. A. Frontana-Uribe, Curr. Opin. Electrochem., 2022, 34, 101004 CrossRef CAS.
- O. M. Cornejo, M. F. Murrieta, L. F. Castañeda and J. L. Nava, Curr. Opin. Solid State Mater. Sci., 2021, 25, 100935 CrossRef CAS.
- A. Fiorani, Irkham, G. Valenti, Y. Einaga and F. Paolucci, in Nanocarbon Electrochemistry, ed. N. Yang, G. Zhao and J. Foord, John Wiley & Sons, 2019, pp. 285–321 Search PubMed.
- H. S. White and A. J. Bard, J. Am. Chem. Soc., 1982, 104, 6891–6895 CrossRef CAS.
- E. Villani, G. Valenti, M. Marcaccio, L. Mattarozzi, S. Barison, D. Garoli, S. Cattarin and F. Paolucci, Electrochim. Acta, 2018, 277, 168–175 CrossRef CAS.
- J. Zhang, E. Kerr, K. A. S. Usman, E. H. Doeven, P. S. Francis, L. C. Henderson and J. M. Razal, Chem. Commun., 2020, 56, 10022–10025 RSC.
- A. Fiorani, Irkham, G. Valenti, F. Paolucci and Y. Einaga, Anal. Chem., 2018, 90, 12959–12963 CrossRef CAS PubMed.
- S. Yamazaki-Nishida, Y. Harima and K. Yamashita, J. Electroanal. Chem. Interfacial Electrochem., 1990, 283, 455–458 CrossRef CAS.
- J. K. Leland and M. J. Powell, J. Electrochem. Soc., 1990, 137, 3127–3131 CrossRef CAS.
- W. Miao, J.-P. Choi and A. J. Bard, J. Am. Chem. Soc., 2002, 124, 14478–14485 CrossRef CAS PubMed.
- L. Chen, D. J. Hayne, E. H. Doeven, J. Agugiaro, D. J. D. Wilson, L. C. Henderson, T. U. Connell, Y. H. Nai, R. Alexander, S. Carrara, C. F. Hogan, P. S. Donnelly and P. S. Francis, Chem. Sci., 2019, 10, 8654–8667 RSC.
- K. Honda, M. Yoshimura, T. N. Rao and A. Fujishima, J. Phys. Chem. B, 2003, 107, 1653–1663 CrossRef CAS.
- K. Honda, T. Noda, M. Yoshimura, K. Nakagawa and A. Fujishima, J. Phys. Chem. B, 2004, 108, 16117–16127 CrossRef CAS.
- M. Sentic, F. Virgilio, A. Zanut, D. Manojlovic, S. Arbault, M. Tormen, N. Sojic and P. Ugo, Anal. Bioanal. Chem., 2016, 408, 7085–7094 CrossRef CAS PubMed.
- Y. Yang, J.-W. Oh, Y.-R. Kim, C. Terashima, A. Fujishima, J. S. Kim and H. Kim, Chem. Commun., 2010, 46, 5793–5795 RSC.
- D. V. Snizhkoz, Yu. T. Zholudov, O. M. Bilash, A. V. Kukoba and M. M. Rozhitskii, Russ. J. Electrochem., 2014, 50, 260–266 CrossRef.
- K. Sakanoue, A. Fiorani, C. I. Santo, Irkham, G. Valenti, F. Paolucci and Y. Einaga, ACS Sens., 2022, 7, 1145–1155 CrossRef CAS PubMed.
- K. Imai, S. Watari, T. Sakazume and S. Mitsuyama, Hitachi Rev., 2008, 57, 1–7 Search PubMed.
- K. Erler, Wien. Klin. Wochenschr., Suppl., 1998, 110, 5–10 CAS.
- Y. Zu and A. J. Bard, Anal. Chem., 2000, 72, 3223–3232 CrossRef CAS PubMed.
- Y. Zu and A. J. Bard, Anal. Chem., 2001, 73, 3960–3964 CrossRef CAS PubMed.
- C. Comninellis, A. Kapalka, S. Malato, S. A. Parsons, I. Poulios and D. Mantzavinos, J. Chem. Technol. Biotechnol., 2008, 83, 769–776 CrossRef CAS.
- I. Sirés, E. Brillas, M. A. Oturan, M. A. Rodrigo and M. Panizza, Environ. Sci. Pollut. Res., 2014, 21, 8336–8367 CrossRef PubMed.
- S. O. Ganiyu, C. A. Martínez-Huitle and M. A. Oturan, Curr. Opin. Electrochem., 2021, 27, 100678 CrossRef CAS.
- Irkham, T. Watanabe, A. Fiorani, G. Valenti, F. Paolucci and Y. Einaga, J. Am. Chem. Soc., 2016, 138, 15636–15641 CrossRef CAS PubMed.
- Irkham, A. Fiorani, G. Valenti, N. Kamoshida, F. Paolucci and Y. Einaga, J. Am. Chem. Soc., 2020, 142, 1518–1525 CrossRef CAS PubMed.
- J.-P. Choi and A. J. Bard, Anal. Chim. Acta, 2005, 541, 143–150 CAS.
- P. Khan, D. Idrees, M. A. Moxley, J. A. Corbett, F. Ahmad, G. von Figura, W. S. Sly, A. Waheed and M. I. Hassan, Appl. Biochem. Biotechnol., 2014, 173, 333–355 CrossRef CAS PubMed.
- E. Villani, N. Shida and S. Inagi, Electrochim. Acta, 2021, 389, 138718 CrossRef CAS.
- S. A. Kitte, W. Gao, Y. T. Zholudov, L. Qi, A. Nsabimana, Z. Liu and G. Xu, Anal. Chem., 2017, 89, 9864–9869 CrossRef CAS PubMed.
- S. B. Lee, J. Kwon and J. Kim, Electroanalysis, 2015, 27, 2180–2186 CrossRef CAS.
- I. Rahmawati, E. Saepudin, A. Fiorani, Y. Einaga and T. A. Ivandini, Analyst, 2022, 147, 2696–2702 RSC.
- S. Garcia-Segura, F. Centellas and E. Brillas, J. Phys. Chem. C, 2012, 116, 15500–15504 CrossRef CAS.
- É. Mahé, P. Bornoz, E. Briot, J. Chevalet, C. Comninellis and D. Devilliers, Electrochim. Acta, 2013, 102, 259–273 CrossRef.
- S. Szunerits, M. Manesse, P. Actis, B. Marcus, G. Denuault, C. Jama and R. Boukherroub, Electrochem. Solid-State Lett., 2007, 10, G43–G46 CrossRef CAS.
- Irkham, R. R. Rais, T. A. Ivandini, A. Fiorani and Y. Einaga, Anal. Chem., 2021, 93, 2336–2341 CrossRef CAS PubMed.
- S. Mavrikis, M. Göltz, S. Rosiwal, L. Wang and C. P. de León, ChemSusChem, 2022, 15, e202102137 CrossRef CAS PubMed.
- K. Sakanoue, A. Fiorani, Irkham and Y. Einaga, ACS Appl. Electron. Mater., 2021, 3, 4180–4188 CrossRef CAS.
- J. Xu, K. Natsui, S. Naoi, K. Nakata and Y. Einaga, Diamond Relat. Mater., 2018, 86, 167–172 CrossRef CAS.
- J.-P. Lagrange, A. Deneuville and E. Gheeraert, Diamond Relat. Mater., 1998, 7, 1390–1393 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2023 |