
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceWater splitting and CO2 reduction over an AgSr2Ta5O15 photocatalyst developed by a valence band control strategy†
Tomoaki
Takayama‡
 a,
Akihide
Iwase§
a and
Akihiko
Kudo
*ab
a,
Akihide
Iwase§
a and
Akihiko
Kudo
*ab
aDepartment of Applied Chemistry, Faculty of Science, Tokyo University of Science, 1-3 Kagurazaka, Shinjuku-ku, Tokyo 162-8601, Japan. E-mail: a-kudo@rs.tus.ac.jp
bTokyo University of Science, Research Institute of Science and Technology, Carbon Value Research Center, Japan
First published on 22nd May 2023
Abstract
Ag+ substitution was applied to a tungsten-bronze-type metal oxide. An AgSr2Ta5O15 photocatalyst has emerged for water splitting and CO2 reduction. DFT calculation and diffuse reflection spectra revealed that the Ag d-orbital formed a new valence band, leading to a narrow band gap (3.91 eV) compared to that of NaSr2Ta5O15 (4.11 eV).
Photocatalytic water splitting and CO2 reduction using water as an electron donor are attractive from the viewpoint of artificial photosynthesis.1,2 Metal oxides have been widely studied as photocatalysts.1 This is because most metal oxides are stable under photocatalytic reaction conditions. However, these metal oxide photocatalysts generally possess wide band gaps, resulting in limitation of their responses to irradiation lights with shorter wavelengths. In this context, many efforts have been made to narrow their band gaps.
Band engineering,3 including valence band control, doping, and solid solution formation, is an effective strategy for narrowing the band gap. Among them, the valence band control technique has indeed developed metal oxide photocatalysts with narrower band gaps. In this strategy, Cu+(3d10),4–9 Ag+(4d10),10–16 Sn2+(5s2),17–20 Pb2+(6s2),13,21 and Bi3+(6s2)13,22–25 make new valence bands at shallower levels than O2p-based valence bands. Among the photocatalysts developed by this strategy, AgTaO310 and Na0.5Bi0.5TiO324 photocatalysts with perovskite structures efficiently split water to H2 and O2 in stoichiometric amounts. However, the numbers of valence-band-controlled metal oxide photocatalysts for water splitting in a one-photon excitation mechanism are still limited to mainly the typical perovskites, resulting in a few crystal structures being useful for the valence band control strategy.
A tungsten-bronze-type crystal structure is one of the families of perovskite materials which are generally denoted as the chemical formula ABO3. A notable feature in the crystal structure of tungsten bronze is that it is easy to replace A and A′ site cations of its chemical formula (AA′2M5O15) with various cations. For example, the A site can contain Na+ and K+ cations, and the A′ sites can contain Ca2+, Sr2+, and Ba2+ cations, whereas the M sites can contain Nb5+ and Ta5+ cations. To date, it has been reported that KCaSrTa5O1526,27 and KSr2Ta5O1528 show photocatalytic activities for water splitting and CO2 reduction using water as an electron donor. A series of AA′2Ta5O15 (A = K, Na; A′ = Sr, Ba)29,30 and K2RETa5O15 (RE = rare earth metal)31 are also tungsten-bronze-type photocatalysts. Therefore, these materials are expected to contribute to expanding the crystal structures for a valence band controlled photocatalyst.
In this study, we successfully prepared AgSr2Ta5O15 with a tungsten-bronze-type structure and compared its band structure to that of NaSr2Ta5O15 based on diffuse reflectance spectra and density functional theory (DFT) calculation. Moreover, the photocatalytic property of AgSr2Ta5O15 for water splitting and CO2 reduction was evaluated.
NaSr2Ta5O15 was prepared by a polymerized complex method (PC method) that usually gives high photocatalytic performances compared with a conventional solid-state reaction.27 AgSr2Ta5O15 was prepared by a solid-state reaction because it would not be easy to prepare it by the PC method due to the isolation of metallic Ag. The obtained AgSr2Ta5O15 was treated with an aqueous HNO3 solution, if necessary. Their crystal phases were identified by X-ray diffraction. Their diffuse reflectance spectra were obtained using the Kubelka–Munk method. Particle shapes of AgSr2Ta5O15 and NaSr2Ta5O15 were observed using a scanning electron microscope. Photocatalytic CO2 reduction and water splitting were conducted using a gas-flow system equipped with inner irradiation cells made of quartz and Pyrex, and a 400 W high-pressure mercury lamp. Density functional theory (DFT) calculations were performed using the CASTEP code to obtain band structures of AgSr2Ta5O15 and NaSr2Ta5O15. The detailed information of these experiments is summarized in the supporting information (including Fig. S1–S3 and Table S1, ESI†).
Fig. 1(A) shows X-ray diffraction patterns (XRDs) of AgSr2Ta5O15 (not treated with an aqueous HNO3 solution; denoted with “w/o HNO3”) and NaSr2Ta5O15. The diffraction peaks of both samples were well consistent with those of a powder diffraction file (PDF) of NaSr2Ta5O15. This is because of the resembled ionic radii of Ag+ (115 pm) and Na+ (102 pm) in the 6 coordination number. For AgSr2Ta5O15 (w/o HNO3), AgTaO3 and metallic Ag were not observed in the XRD pattern (Fig. S4, ESI†). Scanning electron microscopy (SEM) was performed to observe AgSr2Ta5O15 and NaSr2Ta5O15 particles (Fig. 1(B) and Fig. S5, ESI†). Sintered particles with a size of several hundred nm were observed. Diffuse reflectance spectra (DRS) of AgSr2Ta5O15 and NaSr2Ta5O are shown in Fig. 1(C). The absorption spectrum red-shifted by replacing Na+ with Ag+, resulting in their band gaps of 3.91 eV for AgSr2Ta5O15 and 4.11 eV for NaSr2Ta5O15. The reason why the band gap was narrowed by containing Ag+ is due to a new valence band formed by the hybridization of O2p and Ag4d orbitals. These physicochemical analyses indicate that AgSr2Ta5O15 of a tungsten-bronze-type crystal structure was successfully formed, and that its band gap was narrowed by Ag+ substitution as seen for NaTaO3 and AgTaO3.10 Density functional theory (DFT) calculation was performed to discuss the band structures of AgSr2Ta5O15 and NaSr2Ta5O15, as shown in Fig. 2. Conduction band minima of both samples were mainly formed by Ta d-orbital and O p-orbital, whereas both valence band maxima contained O p-orbital. Most importantly, the Ag d-orbital took part in making the valence band of AgSr2Ta5O15, whereas the Na s-orbital did not make such a valence band. This is well consistent with the band structure of the AgTaO3 photocatalyst.10 Thus, Ag+ substitution is effective to narrow the band gap of the tungsten-bronze-type metal oxide by forming the new valence band. The calculated band gaps of AgSr2Ta5O15 and NaSr2Ta5O15 were 3.25 and 3.28 eV, respectively. These values are small as compared to those experimentally obtained from DRS, even though HSE06 was used. This might be due to deviation of the optimized structures in the DFT calculation from the experimentally obtained samples; e.g., structural isomerism (Fig. S1 and S2, ESI†).32–34
 | ||
| Fig. 1 (A) XRD patterns of (a) AgSr2Ta5O15 (w/o HNO3) and (b) NaSr2Ta5O15, and PDF of (c) NaSr2Ta5O15 (PDF No.: 39-1439). (B) SEM image of AgSr2Ta5O15 (w/o HNO3). (C) DRS of (d) AgSr2Ta5O15 (w/o HNO3) and (e) NaSr2Ta5O15. | ||
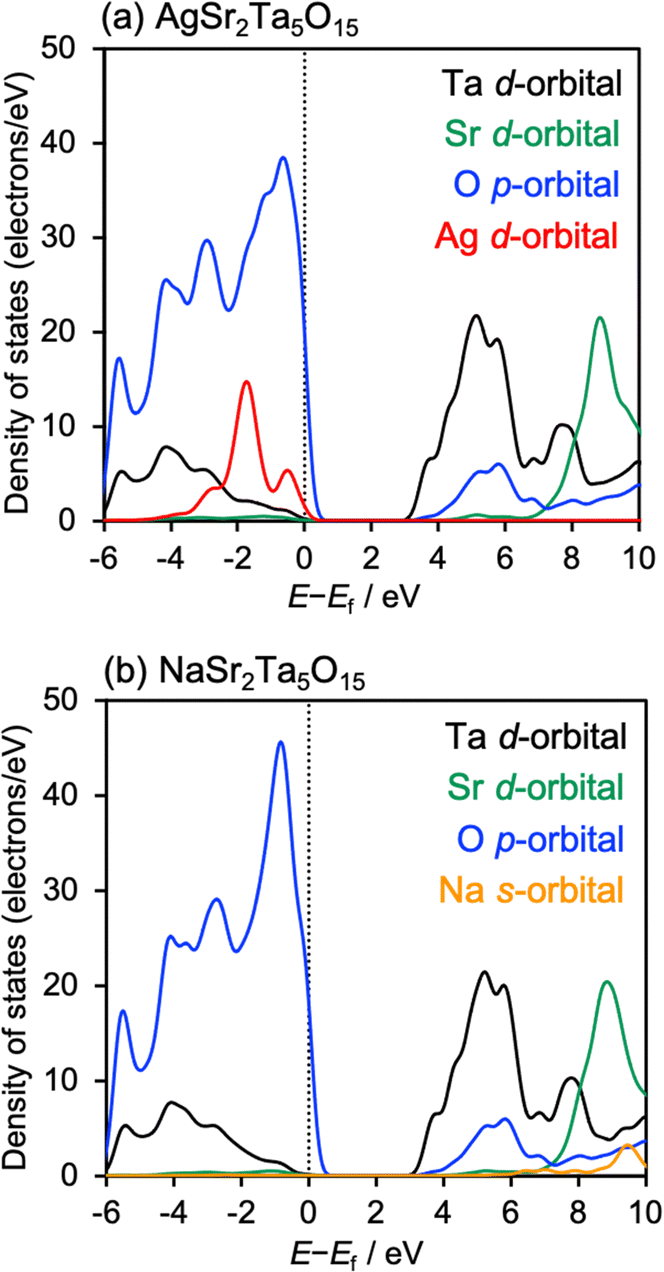 | ||
| Fig. 2 DOS of (a) AgSr2Ta5O15 and (b) NaSr2Ta5O15. The dotted lines indicate their Fermi levels. | ||
Photocatalytic performances of AgSr2Ta5O15 and NaSr2Ta5O15 were evaluated, as shown in Table 1. NaSr2Ta5O15 with NiO and Ag cocatalysts showed higher photocatalytic activities for water splitting and CO2 reduction than AgSr2Ta5O15 (entries 1, 2, 5, 7). This is probably due to the enhancement of recombination between photogenerated e− and h+ at a silver site, more or less. Next, let us see AgSr2Ta5O15 newly obtained in the present study.
| Entry | Photocatalyst | HNO3 treatment | Cocatalyst (wt%) | Gas | NaHCO3 addition | Activity/μmol h−1 | SelCO % | ||
|---|---|---|---|---|---|---|---|---|---|
| H2 | O2 | CO | |||||||
| Photocatalyst: 0.5 g, Reactant solution: water (350 mL), Reactor: a gas-flow system with an inner irradiation cell made of quartz, Light source: a 400 W high-pressure mercury lamp, Concentration of the aqueous NaHCO3 solution: 0.1 mol L−1. Selectivity of CO formation (SelCO) was estimated as follows; SelCO (%) = (the CO formation rate)/[(the H2 formation rate) + (the CO formation rate)] × 100. | |||||||||
| 1 | NaSr2Ta5O15 | No | NiO(0.2) | Ar | No | 1365 | 714 | — | — |
| 2 | NaSr2Ta5O15 | No | Ag(0.5) | CO2 | Yes | 25 | 29 | 29 | 54 |
| 3 | AgSr2Ta5O15 | No | — | Ar | No | 80 | 34 | 0 | 0 |
| 4 | AgSr2Ta5O15 | No | NiO(0.2) | Ar | No | 197 | 76 | — | — |
| 5 | AgSr2Ta5O15 | Yes | NiO(0.2) | Ar | No | 457 | 204 | — | — |
| 6 | AgSr2Ta5O15 | Yes | Ag(0.5) | CO2 | No | 25 | 11 | 2.5 | 9.1 |
| 7 | AgSr2Ta5O15 | Yes | Ag(0.5) | CO2 | Yes | 17 | 12 | 13 | 43 |
| 8 | AgSr2Ta5O15 | Yes | Ag(3) | CO2 | No | 15 | 5.2 | 6.2 | 29 |
| 9 | AgSr2Ta5O15 | Yes | Ag(3) | CO2 | Yes | 6.2 | 9.3 | 21 | 77 |
Bare AgSr2Ta5O15 split water into H2 and O2 (entry 3). Its performance was improved by loading NiO, which is a typical cocatalyst for water splitting (entry 4).3 Considering the literature about AgTaO3,10 there is a possibility that Ag nanoparticles are segregated on the surface. Therefore, AgSr2Ta5O15 was treated with an aqueous HNO3 solution to remove the Ag nanoparticles, resulting in an improvement in the water splitting performance (entry 5). The treated AgSr2Ta5O15 photocatalyst was applied to CO2 reduction using water as an electron donor (entries 6–9). We have reported that an Ag cocatalyst loaded by impregnation works as an active site for CO2 reduction to form CO.2,35–37 The Ag-impregnated AgSr2Ta5O15 produced not only H2 and O2 but also CO under CO2 gas conditions. Because bare AgSr2Ta5O15 did not give CO under Ar (entry 3), we can conclude that the evolved CO was generated from not contamination of carbonous species over the photocatalyst but CO2. When the amount of Ag cocatalyst was increased, the CO formation rate and its selectivity were improved. The addition of NaHCO3 was effective for CO2 reduction.
Fig. 3 shows photocatalytic CO2 reduction over Ag(3 wt%)-loaded AgSr2Ta5O15 (Table 1 entries 8, 9). H2, CO, and O2 were formed from water dissolved with CO2 gas (1 atm). The activity was improved by adding NaHCO3. It is reported that the hydrogen carbonate enhances O2 production in water splitting over a Pt/TiO2 photocatalyst.38 The selectivity for CO formation (SelCO) was also drastically improved up to 77%. This is because of the smooth supply of CO2 molecules onto the active site due to a chemical equilibrium of the hydrogen carbonate.2,36 The ratios of H2/O2 and (H2 + CO)/O2 were slightly beyond stoichiometry (i.e., beyond two). According to the literature about AgTaO3,10 a part of the evolved O2 might be adsorbed on the AgTaO3 photocatalyst, resulting in the O2 evolution being slightly less than the stoichiometry in an early period for water splitting. For the present CO2 reduction over Ag/AgSr2Ta5O15, the rates of gas evolution became gradually close to stoichiometry with the reaction time (Fig. S6, ESI†). Therefore, there is a possibility that the evolved O2 was partly adsorbed on AgSr2Ta5O15.
 | ||
| Fig. 3 CO2 reduction using water as an electron donor over the Ag (3 wt%)-loaded AgSr2Ta5O15 photocatalyst. Photocatalyst: 0.5 g, Reactant solution: water (350 mL), Reactor: a gas-flow system with an inner irradiation cell made of quartz, Light source: a 400 W high-pressure mercury lamp, Concentration of the aqueous NaHCO3 solution: 0.1 mol L−1. | ||
Fig. 4 shows the effect of the wavelength of the irradiation light on photocatalytic CO2 reduction over AgSr2Ta5O15 without HNO3 treatment (w/o HNO3). AgSr2Ta5O15 (w/o HNO3) was active for CO2 reduction even without additional Ag cocatalyst loading (1st run). The CO formation rate and its selectivity were improved by adding NaHCO3 (2nd run). This result implies that a small amount of Ag nanoparticles would be segregated on the surface of AgSr2Ta5O15 prepared by a solid-state reaction as seen in AgTaO3, though it was not detected by XRD and DRS. The segregated Ag worked as the active site for CO2 reduction. Additionally, it is noteworthy that the AgSr2Ta5O15 (w/o HNO3) produced H2, CO, and O2 under irradiation of light with wavelength longer than 300 nm. This is a feature of the AgSr2Ta5O15 possessing a red-shifted absorption edge as shown in Fig. 1(C) being different from NaSr2Ta5O15. AgSr2Ta5O15 is superior to NaSr2Ta5O15 in this photoresponse. Thus, AgSr2Ta5O15 (w/o HNO3) is a photocatalyst that is active not only for water splitting but also for CO2 reduction using water as an electron donor.
 | ||
| Fig. 4 CO2 reduction using water as an electron donor over the AgSr2Ta5O15 (w/o HNO3) photocatalyst without any additional cocatalyst loading. Photocatalyst: 0.5 g, Reactant solution: water (350 mL), Reactor: a gas-flow system with an inner irradiation cell made from quartz (λ > 250 nm) or Pyrex (λ > 300 nm), Light source: a 400 W high-pressure mercury lamp, Concentration of an aqueous NaHCO3 solution: 0.1 mol L−1. | ||
In conclusion, AgSr2Ta5O15 was prepared by a conventional solid-state reaction based on a valence band control strategy using Ag+ substitution. Its crystal structure was almost the same as that of NaSr2Ta5O15 due to the resembled ionic radii of Ag+ and Na+. This Ag+ substitution resulted in red-shift of the absorption spectrum from that of NaSr2Ta5O15. The band gap of AgSr2Ta5O15 was 3.91 eV, while that of NaSr2Ta5O15 was 4.11 eV. DOS estimated by DFT calculations revealed that the Ag d-orbital took part in making the new valence band. Furthermore, AgSr2Ta5O15 was a new photocatalyst for water splitting and CO2 reduction. This study demonstrated the usefulness of the valence band control strategy for the development of new metal oxide photocatalysts, resulting in the AgSr2Ta5O15 photocatalyst with a tungsten-bronze type crystal structure. This finding will contribute to liberating the valence band control strategy from the material design space mainly limited in typical perovskite-type metal oxides for the development of active photocatalysts for water splitting and CO2 reduction.
This work was supported by JSPS KAKENHI, Grants-in-Aid for Scientific Research (A) 23H00248 and Grant Numbers 17H06433 and 17H06440 in Scientific Research on Innovative Areas “Innovations for Light-Energy Conversion (I4LEC)”.
Conflicts of interest
There are no conflicts to declare.Notes and references
- Q. Wang and K. Domen, Chem. Rev., 2020, 120, 919 CrossRef CAS PubMed.
- S. Yoshino, T. Takayama, Y. Yamaguchi, A. Iwase and A. Kudo, Acc. Chem. Res., 2022, 55, 966 CrossRef CAS PubMed.
- A. Kudo and Y. Miseki, Chem. Soc. Rev., 2009, 38, 253 RSC.
- H. Kato, A. Takeda, M. Kobayashi, M. Hara and M. Kakihana, Catal. Sci. Technol., 2013, 3, 3147 RSC.
- K. Iwashina, A. Iwase and A. Kudo, Chem. Sci., 2015, 6, 687 RSC.
- H. Kato, T. Fujisawa, M. Kobayashi and M. Kakihana, Chem. Lett., 2015, 44, 973–975 CrossRef CAS.
- K. Iwashina, A. Iwase, S. Nozawa, S. Adachi and A. Kudo, Chem. Mater., 2016, 28, 4677 CrossRef CAS.
- I. Sullivan, B. Zoellner and P. A. Maggard, Chem. Mater., 2016, 28, 5999 CrossRef CAS.
- B. Zoellner, S. Stuart, C.-C. Chung, D. B. Dougherty, J. Jones and P. A. Maggard, J. Mater. Chem. A, 2016, 4, 3115 RSC.
- H. Kato, H. Kobayashi and A. Kudo, J. Phys. Chem. B, 2002, 106, 12441 CrossRef CAS.
- Y. Hosogi, H. Kato and A. Kudo, J. Mater. Chem., 2008, 18, 647 RSC.
- J. Boltersdorf and P. A. Maggard, ACS Catal., 2013, 3, 2547 CrossRef CAS.
- J. Boltersdorf, T. Wong and P. A. Maggard, ACS Catal., 2013, 3, 2943 CrossRef CAS.
- H. Horie, A. Iwase and A. Kudo, ACS Appl. Mater. Interfaces, 2015, 7, 14638 CrossRef CAS PubMed.
- S. Zong, C. Cheng, J. Shi, Z. Huang, Y. Hu, H. Yang and L. Guo, Chem. – Asian J., 2017, 12, 882 CrossRef CAS PubMed.
- K. Watanabe, A. Iwase, S. Nozawa, S. Adachi and A. Kudo, ACS Sustainable Chem. Eng., 2019, 7, 9881 CrossRef CAS.
- Y. Hosogi, H. Kato and A. Kudo, J. Phys. Chem. C, 2008, 112, 17678 CrossRef CAS.
- Y. Hosogi, Y. Shimodaira, H. Kato, H. Kobayashi and A. Kudo, Chem. Mater., 2008, 20, 1299 CrossRef CAS.
- J. Boltersdorf, I. Sullivan, T. L. Shelton, Z. Wu, M. Gray, B. Zoellner, F. E. Osterloh and P. A. Maggard, Chem. Mater., 2016, 28, 8876 CrossRef CAS.
- D. Noureldine and K. Takanabe, Catal. Sci. Technol., 2016, 6, 7656 RSC.
- Y. Shimodaira, H. Kato, H. Kobayashi and A. Kudo, Bull. Chem. Soc. Jpn., 2007, 80, 885 CrossRef CAS.
- A. Kudo, K. Omori and H. Kato, J. Am. Chem. Soc., 1999, 121, 11459 CrossRef CAS.
- H. Fujito, H. Kunioku, D. Kato, H. Suzuki, M. Higashi, H. Kageyama and R. Abe, J. Am. Chem. Soc., 2016, 138, 2082 CrossRef CAS PubMed.
- K. Watanabe, Y. Iikubo, Y. Yamaguchi and A. Kudo, Chem. Commun., 2021, 57, 323 RSC.
- D. Ozaki, H. Suzuki, O. Tomita, Y. Inaguma, K. Nakashima, H. Kageyama and R. Abe, J. Photochem. Photobiol., A, 2021, 408, 113095 CrossRef CAS.
- T. Takayama, K. Tanabe, K. Saito, A. Iwase and A. Kudo, Phys. Chem. Chem. Phys., 2014, 16, 24417 RSC.
- T. Takayama, A. Iwase and A. Kudo, Bull. Chem. Soc. Jpn., 2015, 88, 538 CrossRef CAS.
- Z. Huang, K. Teramura, S. Hosokawa and T. Tanaka, Appl. Catal., B, 2016, 199, 272 CrossRef CAS.
- Z. Huang, S. Yoshizawa, K. Teramura, H. Asakura, S. Hosokawa and T. Tanaka, ACS Omega, 2017, 2, 8187 CrossRef CAS PubMed.
- Z. Huang, S. Yoshizawa, K. Teramura, H. Asakura, S. Hosokawa and T. Tanaka, ACS Sustainable Chem. Eng., 2018, 6, 8247 CrossRef CAS.
- Z. Huang, K. Teramura, H. Asakura, S. Hosokawa and T. Tanaka, Catal. Today, 2018, 300, 173 CrossRef CAS.
- A. Kubo, G. Giorgi and K. Yamashita, J. Phys. Chem. C, 2017, 121, 27813 CrossRef CAS.
- A. Kubo, G. Giorgi and K. Yamashita, Chem. Mater., 2017, 29, 539 CrossRef CAS.
- M. Kaneko, M. Fujii, T. Hisatomi, K. Yamashita and K. Domen, J. Energy Chem., 2019, 36, 7 CrossRef.
- K. Iizuka, T. Wato, Y. Miseki, K. Saito and A. Kudo, J. Am. Chem. Soc., 2011, 133, 20863 CrossRef CAS PubMed.
- H. Nakanishi, K. Iizuka, T. Takayama, A. Iwase and A. Kudo, ChemSusChem, 2017, 10, 112 CrossRef CAS PubMed.
- T. Takayama, H. Nakanishi, M. Matsui, A. Iwase and A. Kudo, J. Photochem. Photobiol., A, 2018, 358, 416 CrossRef CAS.
- K. Sayama and H. Arakawa, J. Chem. Soc., Faraday Trans., 1997, 93, 1647 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3cc01481a |
| ‡ Present address: His current affiliation is Graduate School of Science and Technology, Division of Materials Science, Nara Institute of Science and Technology, 8916-5 Takayama, Ikoma, Nara 630-0192, Japan. |
| § Present address: His current affiliation is Department of Applied Chemistry, School of Science and Technology, Meiji University, Kanagawa 214-8571, Japan. |
| This journal is © The Royal Society of Chemistry 2023 |