
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEngineering ketoreductases for the enantioselective synthesis of chiral alcohols†
Li
Qiao‡
a,
Zhiyuan
Luo‡
a,
Haomin
Chen
a,
Pengfei
Zhang
 a,
Anming
Wang
*a and
Roger A.
Sheldon
*bc
a,
Anming
Wang
*a and
Roger A.
Sheldon
*bc
aCollege of Materials, Chemistry and Chemical Engineering; Key Laboratory of Organosilicon Chemistry and Material Technology, Ministry of Education; Hangzhou Normal University, Zhejiang Province, Hangzhou, 311121, Zhejiang, P. R. China. E-mail: waming@hznu.edu.cn
bMolecular Sciences Institute, School of Chemistry, University of the Witwatersrand, PO Wits. 2050, Johannesburg, South Africa. E-mail: roger.sheldon@wits.ac.za
cDepartment of Biotechnology, Section BOC, Delft University of Technology, van der Maasweg 9, 2629 HZ Delft, The Netherlands
First published on 5th May 2023
Abstract
The use of engineered ketoreductases (KREDS), both as whole microbial cells and isolated enzymes, in the highly enantiospecific reduction of prochiral ketones is reviewed. The homochiral alcohol products are key intermediates in, for example, pharmaceuticals synthesis. The application of sophisticated protein engineering and enzyme immobilisation techniques to increase industrial viability are discussed.
 Li Qiao | Li Qiao earned her MS degree (2018) from Hangzhou Normal University under the supervision of Prof. Pengfei Zhang and completed her PhD degree (2022) in chemistry at Zhejiang University under the supervision of Prof. Yanguang Wang and Prof. Ping Lu. Now she is a postdoctoral researcher in the Chemical Synthetic Biology lab at Hangzhou Normal University. Her current research focuses on organic photosynthesis and building photobiocatalysis systems based on biological semiconductors. |
 Zhiyuan Luo | Zhiyuan Luo received his BSc degree in 2019 and is currently a master degree candidate in the Chemical Synthetic Biology lab under supervision of Prof. Anming Wang at the College of Material, Chemistry and Chemical Engineering, Hangzhou Normal University, China (HZNU). His research focuses on the covalent immobilization of proteins through Tag/Catcher system and artificial amino acids for biocatalytic applications. |
 Haomin Chen | Haomin Chen received his BSc degree in 2021. He is currently studying in the Chemical Synthetic Biology lab under supervision of Prof. Anming Wang at the College of Materials, Chemistry and Chemical Engineering, Hangzhou Normal University, China (HZNU). His research focuses on biocatalysis, especially multi-enzymatic catalysis of fine chemicals. |
 Pengfei Zhang | Pengfei Zhang was born in Anhui, China. He obtained his Bachelor's degree (1986) from Huaibei Normal University, China, and completed his PhD degree (2001) at Zhejiang University under the supervision of Prof. Zhenchu Chen. Currently, he is a professor in the College of Materials, Chemistry and Chemical Engineering at Hangzhou Normal University. His current research effort focuses on developing green and efficient synthetic methodologies for coupling reaction, carbohydrate chemistry, enzymatic catalytic reaction, and chemical industry. |
 Anming Wang | Anming Wang obtained his PhD in biotechnology and pharmaceutical engineering at Nanjing Tech University. From 2012 to 2016 he was appointed as postdoctoral researcher at Zhejiang University of Technology under the supervision of Prof. Yuguo Zheng and Prof. Yinchu Shen. Now he is a professor in the college of Materials, Chemistry and Chemical Engineering at Hangzhou Normal University. His research interests cover the fields of biocatalysis and its kinetic studies, including the design, reconstruction and engineering of enzyme using genetic-encoded non-standard amino acid. |
 Roger A. Sheldon | Roger Sheldon, a recognised authority on Green Chemistry, is widely known for developing the E factor for assessing environmental impacts of chemical processes. He is currently Distinguished Professor of Biocatalysis Engineering at the University of the Witwatersrand (SA). He received the RSC Green Chemistry Award and the Biocat Lifetime Achievement Award for important and lasting contributions to biocatalysis and was elected a Fellow of the Royal Society in 2015 and an honorary Fellow of the RSC in 2019. He has a PhD from Leicester University (UK) and was at Shell Research Amsterdam (1969–1980), DSM-Andeno (1980–1990), Delft University of Technology (1991–2007) and CEO of CLEA Technologies (2006–2015). |
1. Introduction
Ketoreductases (KREDs) are omnipresent in nature. They are key catalysts in metabolic pathways in all living organisms. The use of KREDs for the asymmetric reduction of ketones in organic synthesis dates back to the use of Baker's yeast (Saccharomyces cerevisiae) or similar whole cell biocatalysts in the 1960s.1 Such whole cell biocatalysts have the advantage of accommodating a broad range of substrates. However, this is because they produce a mixture of KRED isoenzymes with differing substrate preferences. On the one hand, broad substrate scope is a boon but, on the other hand it can lead to a compromised stereoselectivity owing to competition with other KREDs, perhaps having the opposite stereopreference.Moreover, the slow growth and low substrate and product concentrations observed with wild-type strains of yeasts translate to low catalyst and volumetric productivities (g L−1 h−1) resulting in complex downstream processing and the requirement for specialized know-how and equipment that is not generally present in the chemical industry. Organic chemists in general have a strong preference for isolated enzymes over whole cell biocatalysts because the former do not require any special treatment or equipment. However, it is worth noting, in this context, that Baker's yeast can be immobilised and used in continuous processing.2
The advent of recombinant DNA technology and the development of efficient cofactor regeneration concepts in the early 1980s tipped the scales in favour of the use of isolated KREDs. This led, in the first decade of this century, to the availability of a rapidly growing number of isolated KREDs that had been optimised using directed evolution technologies, thus rendering yeast-based reductions largely obsolete. The exquisite enantioselectivities of these enzymes repeatedly outperformed those obtained with other methods and KREDs replaced platinum-group metal catalysed asymmetric hydrogenation as the method of choice for the synthesis of chiral alcohols.3–5 Indeed, two decades ago an ee of say 95% would have been referred to as excellent. Nowadays, an ee of 99% is quite pedestrian and an ee of >99.9% or >99.99% would be called excellent. This remarkable achievement of efficient, green and sustainable production of single enantiomers of chiral alcohols on an industrial scale can be largely attributed to advances in genome mining, computer aided design, directed evolution and high-throughput screening in the last two decades.4,6,7 The products are key intermediates in the synthesis of, inter alia, pharmaceuticals, flavours and fragrances and pesticides,
The introduction of beneficial mutations by directed evolution can increase the thermal stability and activity of enzymes and increase their substrate range, including even a reversal in enantiomeric preference. For industrial viability it is essential to regenerate the reduced state of the expensive cofactors (NADH or NADPH). This is achieved using a large excess of a co-substrate, usually isopropanol, which is oxidised with concomitant reduction of the KRED. However, not all (oxidised forms of) KREDs accept isopropanol as a substrate. In that case, a second enzyme is added that catalyses the reduction by a co-substrate. Combinations that are typically used for this purpose are glucose/glucose dehydrogenase (GDH) and formate/formate dehydrogenase (FDH). The overall efficiency can be further improved by co-immobilisation of the enzymes or by constructing fusion proteins of the two enzymes. (see Section 3.2).
2. Classification and characteristics
We are concerned here with a broad class of oxidoreductases that when operating in one direction are all referred to as alcohol dehydrogenases (ADHs). When operating in the opposite direction they are all carbonyl reductases (CRs). The difference is that when operating in this direction they are sub-divided into ketoreductases (KREDs) and aldehyde reductases. The latter are abbreviated to AKRs which stands for Aldo Keto Reductases.New members of this superfamily of enzymes are regularly identified in the growing number of genome sequences that have become available.8 Within these two main classes there are various sub-classes. For example, the short-chain dehydrogenases/reductases (SDRs), bearing a name that refers to the reaction in both directions. In this review, if the substrate is a ketone we prefer to call the enzyme a KRED. The acronym CR for carbonyl reductase has become largely redundant.
Short-chain dehydrogenase/reductases (SDRs) were first characterised in Drosophila melanogaster.9 They are also a member of the NAD(P)H-dependent oxidoreductase superfamily10,11 and have similar catalytic properties but with a shorter chain length. This old superfamily of metabolic enzymes is present in all organisms12 and currently has more than 47![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 primary structures available in sequence databases and over 300 crystal structures deposited in the Protein Data Bank. They are divided into seven major classes namely: classical, extended, intermediate, complex, atypical, divergent and unknown. Classical and extended SDRs include almost 30000 members across 150 subfamilies and they are ubiquitous in plants, fungi, and bacteria.9
000 primary structures available in sequence databases and over 300 crystal structures deposited in the Protein Data Bank. They are divided into seven major classes namely: classical, extended, intermediate, complex, atypical, divergent and unknown. Classical and extended SDRs include almost 30000 members across 150 subfamilies and they are ubiquitous in plants, fungi, and bacteria.9
Although SDRs exhibit diversity in structure and function, they generally have three features in common: (1) a cofactor binding site (TGxxxGxG), (2) a catalytic residue motif YxxxK, and (3) a conserved three-dimensional structure made up of a “Rossmann fold” and a β-sheet sandwiched between three α-helices on each side.13 The active site is formed by a triad/tetrad with highly conserved Tyr, Lys, Ser (and Asn) residues. The pairwise sequence identity between different enzymes is low, typically 10–30%, but all available three-dimensional structures (∼20) display a highly similar α/β folding pattern.14,15
Most SDRs have a 250–350-residue core structure, frequently with additional N- or C-terminal transmembrane domains or signal peptides. Conserved sequence regions cover a variable N-terminal TGxxxGxG motif as part of the nucleotide binding region and the active site with a triad of catalytically important Ser, Tyr, and Lys residues, of which Tyr is the most conserved residue within the whole family. The functions of the residues at these particular sites have been elucidated by a combination of chemical modification, sequence comparisons, structure analyses, and site-directed mutagenesis.16
Aldoketoreductases (AKRs) exist in nearly all phyla and form a superfamily of proteins characterised by their common three-dimensional structure and reaction mechanism for catalysing the NAD(P)H-dependent reduction of carbonyl groups. They are mainly monomeric soluble proteins (34–37 kDa) with similar three-dimensional structures involving a parallel β-8/α-8-barrel fold.17 AKRs accept a wide range of substrates and are potential drug targets.18,19 Over 190 members have been identified in species ranging from prokaryotes to plants, fungi, and animals. They are grouped into 16 families having unique structural features that influence their substrates and kinetics. They are mostly cytosolic and are active as monomers. The single amino acid chain of, on average, 320 residues folds into a (α/β)8-barrel, an evolutionarily highly conserved structure originally observed in triose-phosphate isomerase (TIM) and therefore also known as a TIM barrel structure. In contrast to another 32 superfamilies adopting this motif, AKRs do not require metal ions for functionality, and bind NAD(P)H cofactor in an extended anti-conformation so that they can catalyse 4-pro-R hydride transfer and thus belong to the large group of A-face oxidoreductases.18
The reactions catalysed by AKRs involve an ordered bi–bi kinetic mechanism and general acid–base catalysis.20 The cofactor binds first, followed by tethering of the substrate. In the reduction direction, the 4-pro-R hydride from NAD(P)H is transferred to the substrate carbonyl group. Simultaneously, the carbonyl oxygen is protonated by a conserved tyrosine acting as a general acid. Oxidation proceeds in the reverse sequence with the tyrosine acting as a general base. Asp 50, Tyr 55, Lys 84 and His 117 form the catalytic tetrad (based on residue numbering in AKR1C9),17,21 where the Tyr is the general acid–base. Conservation of these residues is strong and underlines their role in catalysis. Asp 50 is present in 99 per cent, Tyr 55 in 97 per cent, Lys 84 in 97 per cent and His 117 in 88 per cent of all annotated AKRs on the AKR website. Consistent substitution of these key amino acids, however, especially of the histidine, is a characteristic for some AKR subfamilies. In AKR1D isoforms, which catalyse the 5β-reduction of Δ4-3-ketosteroids, glutamate replaces His 117.17 Interestingly, mutational analysis in AKR1C9 demonstrates that this single amino acid substitution is sufficient to convert this 3-ketosteroid reductase to a steroid 5β double-bond reductase.22 Although these four conserved amino acids seem to play a crucial role in the catalytic mechanism, targeted mutation studies (e.g. in AKR1B123 and AKR1C924) did not indicate a complete loss of enzymatic activity and suggested that, in some instances, proximity of the hydride donor to the acceptor is sufficient to catalyse a reaction.
3. Enzyme engineering
3.1. Improving reductases by directed evolution
Virtually all KRED variants that are currently used on an industrial scale have been optimised using directed evolution. Gong and co-workers,25 for example, used directed evolution to evolve the native KRED, lactobacillus brevis ketoreductase (LbCR), in order to increase its thermal stability and activity. Some variants had increased thermal stability while others displayed increased activity. Subsequently, variants with both improved thermostability and activity were developed by combining additivity and cooperative mutational effects. Analysis of variant structures demonstrated that increased thermostability was largely due to rigidification of flexible loops around the active site through the formation of additional hydrogen bonds and hydrophobic interactions. The best variant, LbCRM8, displayed a 1944-fold increase in half-life at 40 °C and a 3.2-fold improvement in catalytic efficiency compared with the wide-type enzyme.Using only 1 g L−1 of lyophilized E. coli cells, co-expressing LbCRM8 and glucose dehydrogenase BmGDH, with 300 g L−1 substrate loading, t-butyl 6-cyano-(5R)-hydroxy-3-oxo-hexanoate 1 was completely reduced, in 6 h at 40 °C, to the atorvastatin 3 intermediate, t-butyl 6-cyano-(3R,5R)-dihydroxy hexanoate 2 with >99.5% de and a STY of 1.05 kg L−1 d−1 (Scheme 1).25
 | ||
| Scheme 1 Asymmetric reduction of 1 by E. coli cells coexpressing LbCRM8 and BmGDH to synthesise atorvastatin intermediate 2. Reproduced from ref. 25 with permission from American Chemical Society, copyright 2019. | ||
(S)-2-Chloro-1-(3,4-difluorophenyl) ethanol ((S)-CFPL) is an intermediate for the drug ticagrelor, and is currently manufactured via a chemical route. To develop a biocatalytic process to (S)-CFPL, an inventory of KREDs from the Chryseobacterium sp. CA49 was rescreened, and ChKRED20 was found to catalyse the stereoselective reduction of the ketone precursor in 99% ee. Screening of an error-prone PCR library of the wild-type ChKRED20, led to the identification of two variants, each bearing a single amino acid substitution (H145L and L205M), with significantly increased activity. Subsequently, the two critical positions were randomised, using saturation mutagenesis, to afford several variants with enhanced activity. Among them, the variant L205A was the best performer with a specific activity ten times that of the wild-type. Its kcat/Km and half-life at 50 °C were increased 15 times and 70%, respectively. The variant catalysed the complete conversion of 150 and 200 g L−1 substrate within 6 and 20 h, respectively, to yield enantiopure (S)-CFPL with an isolated yield of 95%.30
Similarly, Ou-Yang and coworkers32 reported that mutations at residues Phe92, Tyr208, Met144, Phe145 of CgKR1, which are important for the binding pocket, afforded engineered CgKR1s with high activity and stereoselectivity towards benzo-fused cyclic ketones. Particularly, the whole-cell biocatalysts prepared by the co-expression of CgKR1 variants and GDH displayed excellent stereoselectivity (>99.9% ee) in the reduction of N-ethyl-N-methyl-2-(3-oxo-2,3-dihydro-1H-inden-5-yl) acetamide, the key intermediate of the anti-Alzheimer drug ladostigil (TV3326).
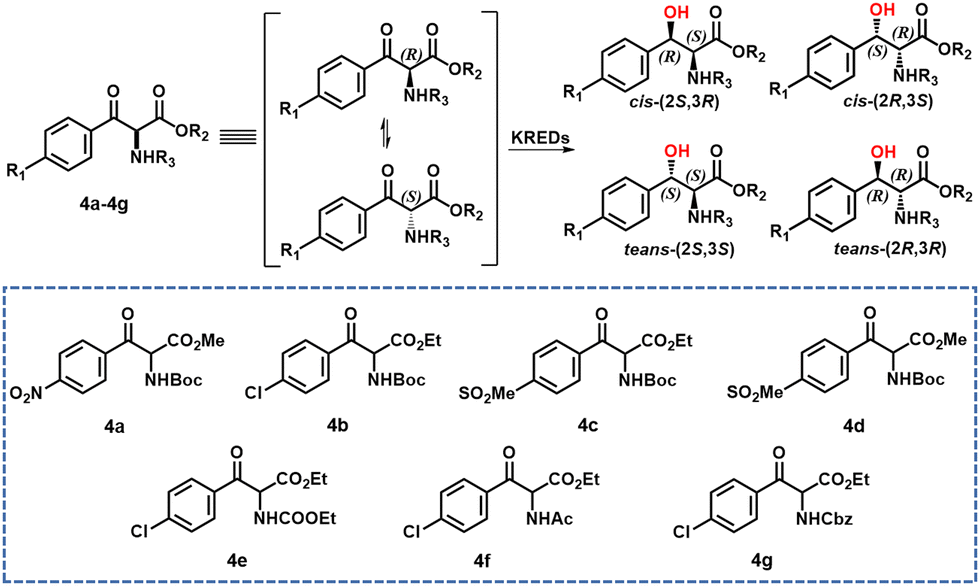
Tang and co-workers33 used structure-guided rational directed evolution to improve the dynamic reductive kinetic resolution (DYRKR) of bulky α-amino-β-keto esters catalysed by a wild-type KRED from Exiguobacterium sp. F42. A number of variants were identified with remarkably improved activity and excellent stereoselectivity. In particular, a variant, M30, comprising 9 mutatations exhibited excellent stereoselectivity (>99% dr, >99% de) and high conversion (>99%) for six α-amino β-keto esters.
Similarly, novel and practical chemoenzymatic routes were developed for the synthesis of chloramphenicol and florfenicol through the application of enzymatic DYRKR to establish the two stereocentres of the amino alcohols in >99% dr, >99% de and >99% conversion from 100 g L−1 α-amino β-keto esters 4a and 4c, respectively (Scheme 2). Crystal structure and molecular dynamics studies revealed the potential molecular basis for activity improvement and the mechanism of stereoselectivity control at the atomic level.33
 | ||
| Scheme 2 Efficient and stereoselective bioreduction of bulky α-amino β-keto esters. | ||
KRED-catalysed stereoselective reduction of methyl 8-chloro-6-oxooctanoate constitutes an efficient approach to the synthesis of methyl (R)-8-chloro-6-hydroxyoctanoate, a key intermediate in the synthesis of α-lipoic acid. Chen and coworkers34 employed structure-guided protein engineering of SsCR from Sporobolomyces salmonicolor, assisted by comparative analysis of enzyme/substrate binding modes in pre-reaction-state and free-state molecular dynamics (MD) simulations, to develop a versatile KRED. The variant displayed a 3-fold increased kcat (17 s−1) towards methyl 8-chloro-6-oxooctanoate relative to the wild-type KRED in addition to improved stereoselectivity (98.0% ee), and no substrate inhibition. Moreover, insights were gained into the origins of the enhanced catalytic activity and stereoselectivity by conducting complete deconvolution experiments and MD simulations. Using 60 g L−1 of wet E. coli cells co-expressing the SsCR variant and glucose dehydrogenase (GDH), and 0.05 mM NADP, complete reduction of 260 g L−1 of 1a was achieved, furnishing methyl (R)-8-chloro-6-hydroxyoctanoate with 98.0% ee and a space-time yield of 391 g L−1 d−1.
3.2. Fusion proteins
KRED/ADH fusion proteins are produced by connecting these sequentially acting enzymes end to end through a linker peptide. The linker peptide sequence and length can influence the folding and limit the stability, flexibility, oligomeric state and solubility of the fusion protein and, hence, affect its function or lead to expression failure. To improve cofactor regeneration and catalytic efficiency, Xu and coworkers35 constructed a KRED/GDH fusion protein of a (CR2-L-GDH) combination with a (GGGGSGGGGSGGGGS) sequence as the peptide linker. As a result, (S)-(−)-N,N-dimethyl-3-hydroxy-3-(2-thienyl)-1-propanamine, (S)-DHTP, the key intermediate in the synthesis of duloxetine, was obtained in 98% yield with >99.9% ee at 20 g L−1 substrate loading by asymmetric reduction of DKTP (N,N-dimethyl-3-keto-3-(2-thienyl)-1-propanamine).Sührer and coworkers36 constructed a MycFDH(7M)-SG-KR fusion protein, from a 3-ketoacyl-[acyl-carrier-protein]-reductase (KR) from Synechococcus PCC 7942 and a variant formate dehydrogenase from Mycobacterium vaccae N10 (MycFDH). The fusion protein was used in the asymmetric reduction of ethylbenzoyl acetate. The chiral alcohol product is an alternative precursor of fluoxetine. Cells containing the fusion protein exhibited a 2-fold higher initial reaction rate than cells containing a mixture of the free enzymes. The improved performance of the fusion protein was attributed to an improved Km,EBA of the KRED subunit resulting from changes in its overall structure caused by its N-terminal fusion rather than to substrate channeling.37,38
Mass transfer limitations are often observed when enzymes are immobilised on porous carriers. In a recent study,39 it was shown that co-immobilisation of a dehydrogenase and a cofactor-regenerating enzyme resulted in more efficient catalysis compared to the use of two different carriers – especially at low cofactor concentrations. The immobilisation of fusion proteins can lead to a uniform distribution of the two biocatalysts on a solid support and a high catalyst density as the fusion protein occupies less area on the carrier than the two individual enzymes. Therefore, the generation of fusion proteins such as MycFDH(7M)-SG-KR could also be beneficial for multi-step enzymatic conversions involving immobilised enzymes.
4. Immobilisation of reductases
Reductase catalysis requires a reliable cofactor regeneration system supported by a dehydrogenase. Unfortunately, these enzymes often suffer from low stability and difficult recovery, resulting in high production costs and environmental pollution from single use enzymes. In contrast, co-immobilisation of enzymes improves their stability by hindering subunit dissociation, thus decreasing aggregation, autolysis and proteolysis. and enhancing enzyme rigidification, and the formation of favorable microenvironments. We also note that immobilisation avoids problems observed with emulsions using soluble enzymes and facilitates product isolation.Co-immobilised enzymes can be roughly divided into on-carrier and carrier-free. Cross-linked enzyme aggregates (CLEAs) are a well-known example of the latter.40,41 A variety of reactive functional groups inherent to enzymes, such as NH2, SH and OH, can be used to affect cross-coupling. Moreover, the introduction of non-canonical amino acids (ncAAs), bearing highly reactive groups such as N3, C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CH and NC, through genetic coding, significantly expands the number of functional groups that can be introduced. Traditionally, carrier-free immobilisation of enzymes42 or whole cells43,44 involves the use of glutaraldehyde as a cross-linker. This results in higher stability but is often accompanied by a substantial loss of activity owing to random formation of covalent bonds with reactive groups in or close to the active site.
CH and NC, through genetic coding, significantly expands the number of functional groups that can be introduced. Traditionally, carrier-free immobilisation of enzymes42 or whole cells43,44 involves the use of glutaraldehyde as a cross-linker. This results in higher stability but is often accompanied by a substantial loss of activity owing to random formation of covalent bonds with reactive groups in or close to the active site.
We have reported the use of ncAAs to enable the cross-linking of KREDs. The ncAAs can be inserted into proteins at preselected sites, for subsequent cross-linking, using genetic code expansion. Thus, KRED variants were obtained by five-point insertion, at preselected sites, of the ncAA, p-azido-L-phenylalanine (pAzF). The variants were subsequently cross-linked, directly from cell lysate, using a Cu-free bio-orthogonal click reaction with sym-dibenzo-1,5-cyclooctadiene-3,7-diyne 5 (Scheme 3).45,46 The resulting CLEs exhibited improved catalytic efficiencies and thermal stabilities.
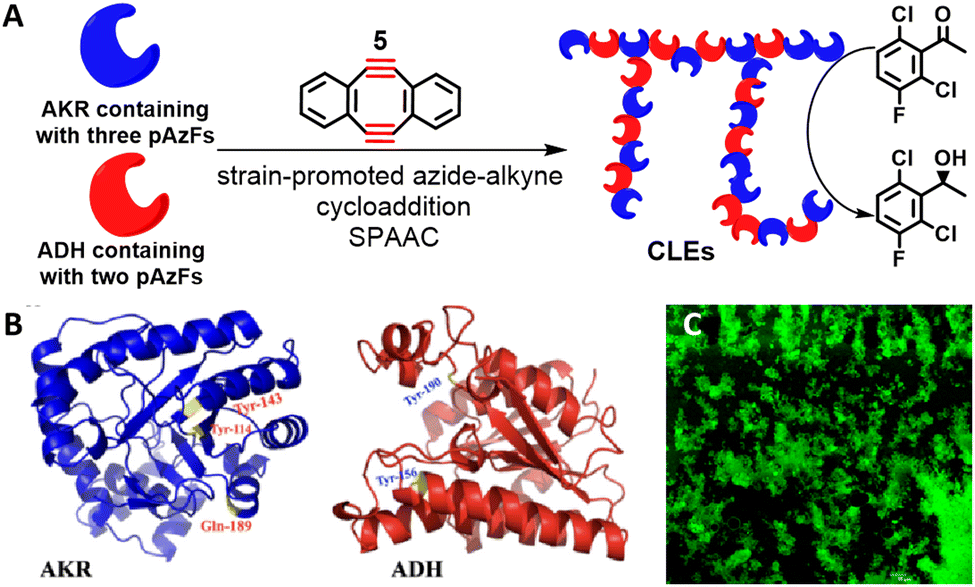 | ||
| Scheme 3 SPAAC reaction mediated co-immobilisation of dual-enzymes (A); the mutation sites of AKR and ADH (B); the confocal laser scanning microscope image of CLEs (C). Reproduced from ref. 44 with permission from Elsevier, copyright 2022. | ||
Combi-CLEs of the KRED and GDH were prepared from mixed cell lysates of pAzF substituted variants of both the KRED and GDH which were bioothogonally cross-linked with the diyne. The resulting combi-CLEs of the KRED and GDH exhibited an enhanced activity in the synthesis of (R)-1-[3,5-bis (trifluoromethyl)phenyl] ethanol by reduction of the corresponding prochiral ketone.45,46 Similarly, (S)-1-(2,6-dichloro-3-fluorophenyl) ethanol was prepared in 76% yield and 99.99% ee, and the reusability of the combi-CLEs demonstrated.48 Cross-linking, via cycloaddition of the azide functionalities with the cyclooctadiyne, is promoted by microwave irradiation and visible-light and affords an immobilised biocatalyst that is conveniently separated by centrifugation. More examples of bio-orthogonal immobilisation strategies can be found in our recently published review.47
Immobilisation on aldehyde agarose relies on the reaction between deprotonated ε-amino groups of surface lysines and aldehyde groups on the carrier, under alkaline conditions. For example, KRED1-Pglu (from Pichia glucozyma) and BmGDH (from Bacillus megaterium) were immobilised on aldehyde agarose and used in the asymmetric reduction of ketones under continuous flow conditions in a packed bed (Scheme 4).48 Their stability towards organic solvent (DMSO) was far superior to that of the free enzymes. No significant loss of activity was observed over a period of 15 days continuous conversion of 4-nitroacetophenone to afford (S)-1-(4-nitrophenyl) ethanol 6b in 97–98% yield and >98% ee. The flow conditions were similarly applied to the asymmetric reduction of 1-phenylpropane-1,2-dione, 1-phenylbutane-1,3-dione and ethyl secodione.
 | ||
| Scheme 4 Continuous stereoselective reduction of diketones with immobilised KRED1-Pglu/BmGDH in a flow reactor. Reproduced from ref. 48 with permission from Elsevier, copyright 2017. | ||
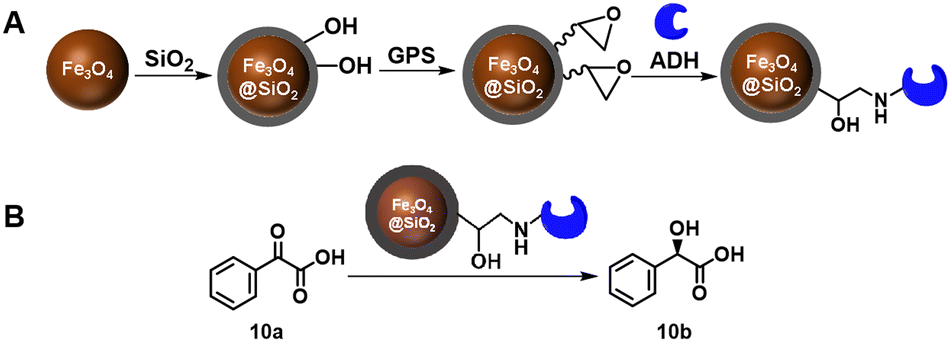
Epoxy resins are widely used as carrier materials for enzymes. Covalent immobilisation of an alcohol dehydrogenase (ADH) from Saccharomyces cerevisiae using epoxy functionalised magnetic Fe3O4@SiO2 nanoparticles, for example, endowed the ADH with broad pH tolerance, high thermostability and 84% activity recovery after five cycles in the reduction of phenylglyoxylic acid to afford (R)-mandelic acid 10b, of undisclosed optical purity, in 64% yield (Scheme 5).49
 | ||
| Scheme 5 ADH immobilised on the Fe3O4@SiO2-epoxy nanoparticles. | ||
Mesoporous ZnO/carbon composite,50 and organic–inorganic hybrid nanoflowers51 have also been applied in co-immobilisation of enzymes with excellent activity retention and improved stability towards heat, pH changes and organic solvents. Similarly, advanced 3D printing technology is a promising low-cost method for immobilising enzymes based on the ability to create countless high-resolution structures from digital models.52,53 Pei and coworkers54 used 3D printing technology, for example, to load the reductase AKR-IA, containing two amino acid mutations (Ile189 + Asn256), uniformly in calcium alginate, with significant improvements in reusability.
Enzymes and whole cell biocatalysts are miniature dispersions and their rates and productivities can be enhanced under continuous flow conditions. Potential advantages of flow-based devices are: increased mixing efficiency, controlled scaling factors, improved safety ratings, in-line control and product recovery (Scheme 4). For example, the engineered ketoreductase, apKRED-9, derived from Acetobacter pasteurianus 386B, was immobilised on glutaraldehyde-activated amino polymer beads or by poly(ethylenimine) (CEP) mediated coaggregation followed by glutaraldehyde cross-linking.55 Both immobilisates exhibited an excellent performance in the continuous flow reduction of 3-ketotetrahydrothiophene. With the apKRED-9-CEP, for example, (R)-tetrahydrothiophene-3-ol was obtained in 76% yield, 99.9% ee and an STY of 729 g L−1 d−1 after 64h of continuous operation.
4.1. Covalent immobilisation
Chen and coworkers56 reported the preparation of a magnetic combi-CLEA of 3-quinuclidinone reductase (QNR) and glucose dehydrogenase from E. coli BL21 on amino-functionalized Fe3O4 nanoparticles using ammonium sulfate as precipitant and glutaraldehyde as the cross-linker, directly from cell lysates. The m-combi-CLEA was used in the enantioselective synthesis of (R)-3-quinuclidinol. The m-combi-CLEAs exhibited improved operational and thermal stability and enhanced catalytic performance in the asymmetric reduction of 3-quinuclidinone (33 g L−1) to (R)-3-quinuclidinol in 100% yield and 100% ee after 3 h. The biocatalyst retained 80% activity after 70 days storage and more than 40% of its initial activity after ten cycles.Orthogonal cell surface tags have been used to produce whole-cell biocatalysts. Peschke57 for example, used three different orthogonal labeling systems for whole cell immobilisation. The 39 amino acid streptavidin-binding peptide (SBP) tag, for example, binds to the protein streptavidin (STV) with high affinity and is commonly used in the chromatographic purification of recombinant proteins. The SpyTag/SpyCatcher system consists of the 113 amino acid SpyCatcher (SC) protein, which generates a covalent isopeptide bond between one of its lysine residues and an aspartate residue of the 13 amino acid SpyTag peptide. The third example involves the self-labeling Halo-based oligonucleotide binder (HOB) tag protein (293 amino acids) that forms a covalent bond with chloroalkyl functionalities in a similar fashion to the HaloTag protein, which is commonly used for imaging in cell biology. HOB was genetically engineered to bind to chloroalkyl groups attached to DNA oligonucleotides and DNA nanostructures with a significantly higher efficiency than the HaloTag system.
4.2. Noncovalent fixation
An improved sol–gel process involving the use of hollow silica microspheres as a carrier was applied for the co-immobilisation of whole cells of Escherichia coli with Chromobacterium violaceum ω-transaminase activity and Lodderomyces elongisporus with ketoreductase activity. The sol–gel entrapment protocol required sol formation by acid-catalysed hydrolysis of tetraethyl orthosilicate (TEOS). A suspension of whole cells and hollow silica particles in phosphate buffer was added to silica sol to entrap the cells adsorbed onto the supporting silica particles during gelation. The co-immobilised cells with two different biocatalytic activities could perform a cascade of reactions to convert racemic 2-amino-4-phenylbutane or 2-aminoheptane into a mixture of the corresponding enantiomerically pure R amine and S alcohol. The co-immobilised whole cells had better activity up to 24 h of continuous-flow mode operation, and the final conversion was higher for the co-immobilised whole cells (47%) than for the single-cell column system (42%).58The histidine groups in his-tagged proteins coordinate to Zr4+ ions59 and Zhang and co-workers59 used a coordination polymer derived from Zr4+ and 2-methylimidazole as a carrier for co-immobilisation of a His-tagged KRED and glucose-6-phosphate dehydrogenase (G6PD). The resulting immobilised KRED/G6PD was employed as a nanoreactor for the reduction of chalcone to dihydrochalcone, with cofactor regeneration, in aqueous media. The biocatalyst exhibited good storage stability and tolerance towards elevated temperatures and pH changes. More than 95% chalcone conversion to dihydrochalcone was observed in 15 min and 80% of its initial activity was retained after 4 cycles.59
Basso and co-workers reported60 the immobilisation of a NADPH-dependent, His-tagged KRED and the co-immobilisation of a KRED and GDH on a methacrylic acid/iminodiacetic acid functionalised resin. The adsorption method provided up to 4 times higher specific activity compared with covalent, ionic, and hydrophobic methods. Recycling in batch showed excellent stability over 10 cycles in aqueous and 5 cycles in organic media. NADPH cofactor recycling was studied in batch and continuous flow using isopropanol as co-substrate with separately immobilised and co-immobilised KRED and GDH, in the presence of glucose. The best results were obtained in the presence of the co-immobilised KRED and GDH, while the lowest conversion was obtained by using the two enzymes separately immobilised. Using cofactor regeneration systems, an NADPH concentration 1000 times lower than the substrate concentration can be used while maintaining the enzyme productivity.
Rebros and coworkers61 studied the co-immobilisation of a KRED and glucose dehydrogenase (GDH) on highly cross-linked agarose (Sepharose) via affinity interaction between His-tagged enzymes (six histidine residues on the N-terminus of the protein) and the agarose matrix charged with Ni2+ ions. The immobilised enzymes were applied to the asymmetric reduction of 1-ethyl-2-methyl acetoacetate in a semi-continuous flow reactor at substrate conversions of >95%. Immobilisation allowed the use of higher substrate concentrations in comparison with free enzymes. The immobilised system was also tested on bulky ketones and significant enhancements were observed compared with the free enzymes.
5. Scope in the enantioselective synthesis of chiral alcohols
5.1. Hydroxy esters
Optically pure hydroxy-substituted carboxylic acids are key building blocks in the synthesis of bioactive compounds. (R)-α-Lipoic acid [(R)-14], for example, is a natural antioxidant, an enzyme cofactor and an over-the-counter nutritional supplement. Xu and Zheng and coworkers62,63 used an ε-keto ester reductase (CpAR2) isolated from Candida parapsilosis for the enantioselective reduction of 8-chloro-6-oxooctanoate 11a, a key step in the synthesis of (R)-14 (Scheme 6). Ethyl (R)-8-chloro-6-hydroxy octanoate 11ba was obtained with a space-time yield of 530 g L−1 d−1 and >99% ee.62 Genome mining and protein engineering were used to obtain a variant (CpAR2S131Y/Q252I) that exhibited identical enantioselectivity and a space-time yield of 566 g L−1 d−1 using one fifth of the wild-type reductase loading.63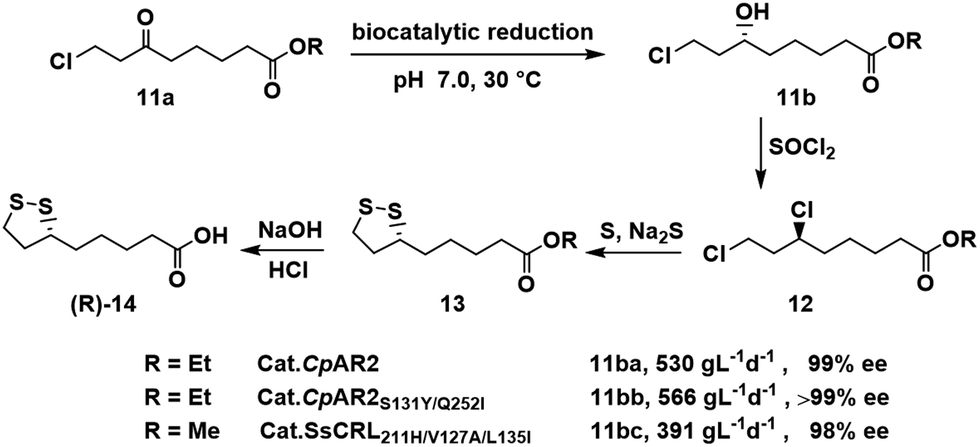 | ||
| Scheme 6 Synthesis of (R)-α-lipoic acid [(R)-14]. | ||
Similarly, three rounds of structure-guided protein engineering of an ε-keto ester reductase (CBS 6045) from Scheffersomyces stipites (SsCR), afforded a triple mutant SsCRL211H/V127A/L135I, with good catalytic activity (kcat = 16.8 s−1) and no substrate inhibition, in the synthesis of methyl (R)-8-chloro-6-hydroxyoctanoate 11bc with 98.0% ee and a space time yield of 391 g L−1 d−1.34
Chiral hydroxy esters are key intermediates in the industrial synthesis of various statins, cholesterol lowering agents that are used to treat cardiovascular diseases. The most well-known statin is atorvastatin, the block-buster drug Lipitor. A key intermediate in the industrial synthesis of atorvastatin is the R-enantiomer of ethyl 4-cyano-3-hydroxybutanoate 15c. In the industrial process, developed by Codexis, it is produced by the KRED/GDH-catalysed enantioselective reduction of ethyl 4-chloroacetoacetate 15a to ethyl (S)-4-chloro-3-hydroxybutyrate 15b, in 96% isolated yield and >99.9% ee, using glucose as the stoichiometric reductant. The chloro ester is subsequently converted to 15c, in >99.5% ee, by halohydrin dehalogenase catalysed reaction with NaCN (Scheme 7). For industrial viability the activities of all three enzymes were dramatically improved by directed evolution using DNA shuffling.64 KRED-catalysed reduction of the second carbonyl group with cofactor regeneration with GDH/glucose affords t-butyl 6-cyano-(3R,5R)-dihydroxyhexanoate 2 which is further converted to atorvastatin.
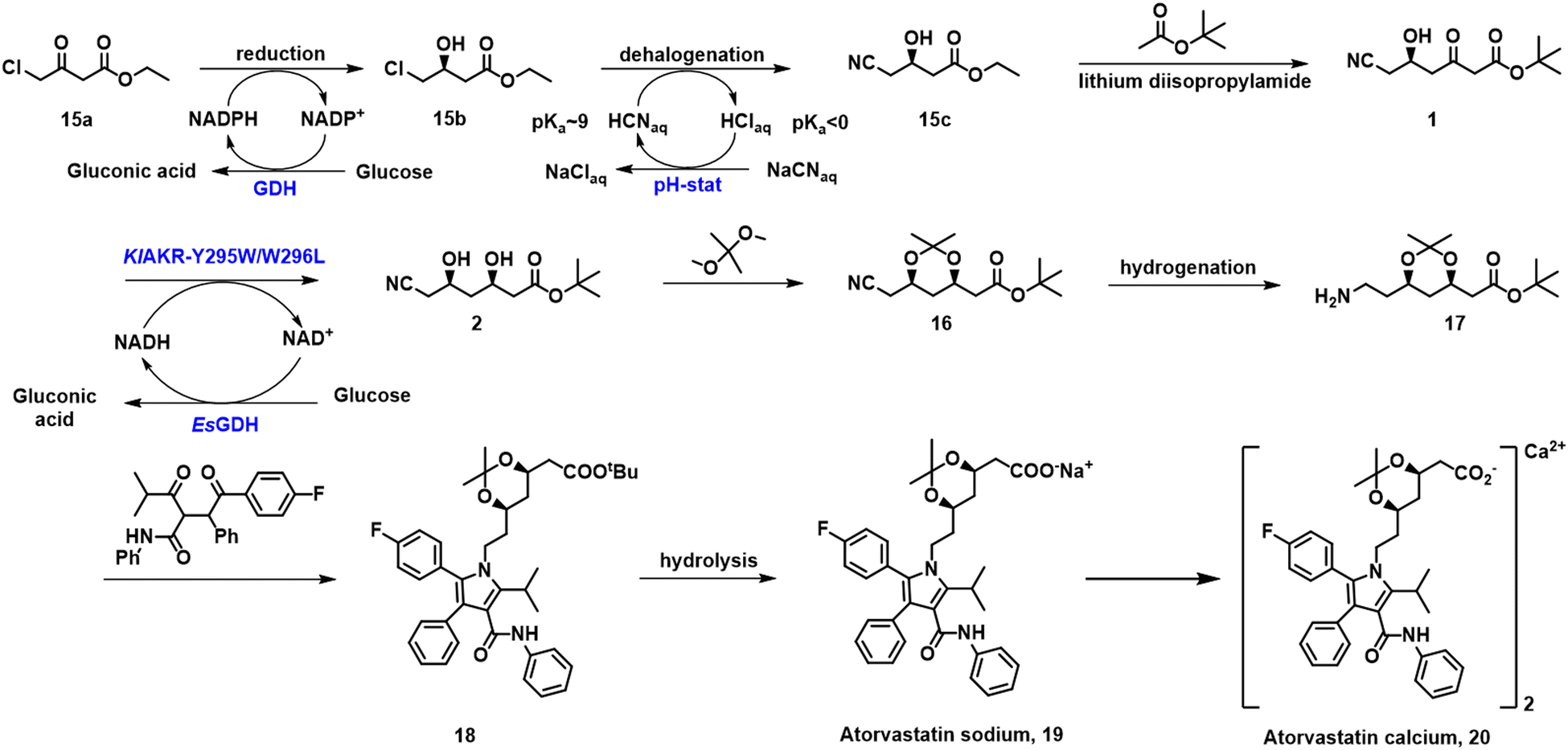 | ||
| Scheme 7 Synthesis of atorvastatins. | ||
Reductase KlAKR from Kluyveromyces lactis exhibited perfect diastereoselectivity but poor activity toward 1.65 In contrast, co-expression of the KlAKR-Y295W/W296L variant, derived from site-saturation mutagenesis, in E. coli BL21 (DE3) together with Exiguobacterium sibiricum glucose dehydrogenase (EsGDH), enabled the formation of 2 in 243 kg m−3.66 Immobilisation of reconstructed E. coli BL21 cells in Celite-polyethyleneimine (PEI)-glutaraldehyde improved the stability of the biocatalyst which could be reused 7 times giving a total of 17.5 g 2 per gram in dep > 99.5%.43 Zheng and Wang and coworkers used a variant of the KmAKR from Kluyveromyces marxianus, obtained following multiple rounds of directed evolution, for the efficient asymmetric reduction of 1.44,67–71 The variant exhibited a longer half-life, an enhanced thermostability and activity, and afforded 2 with a space-time yield of 1.8 kg L−1 d−1 and a dep >99.5%.70 Similarly, LbCR protein from Lactobacillus brevis catalysed the reduction of 1 to the 3R, 5R-diol. Co-expression of a variant LbCR and BmGDH in E. coli improved the STY of 2 from 351 g L−1 d−1 to 1.5 kg L−1 d−1 with over 99.5% de.25,72
Various KREDs, e.g. the stereoselective carbonyl reductase (RtSCR9) from Rhodosporidium toruloides73 and the alcohol dehydrogenase, KleADH, from Klebsiella oxytoca74 have been successfully used for the synthesis of tert-butyl (3R,5S)-6-chloro-3,5-dihydroxyhexanoate by asymmetric reduction of the keto ester precursor. Industrial scale reactions were performed in aqueous or aqueous biphasic media (water–octanol) using E. coli harboring the SCR gene.73,75RtSCR9 was evolved, through random and site-saturation mutagenesis, to produce positive variants that gave tert-butyl (3R,5S)-6-chloro-3,5-dihydroxyhexanoate in 753 mmol L−1 h−1 g−1 wet cell weight.76
(R)- or (S)-Ethyl-3-hydroxybutyrate bearing H or methyl at the C2 position61 and halogen (Cl, Br) or trifluoromethyl77 at the C4 position can also be prepared by KRED catalysed reduction of the corresponding keto ester. Co-immobilisation of the KRED from Hansenula polymorpha78 and GDH (25:1) using Lentikats PVA entrapment technology retained approximately 80% of the original activity in the asymmetric reduction of ethyl-2-methylacetoacetate 21a to ethyl 3-hydroxy-2-methylbutanoate 21b (Scheme 8).79 The KRED used here was obtained by Hanson et al.78 who first sequenced and cloned the KRED gene in H. polymorpha.
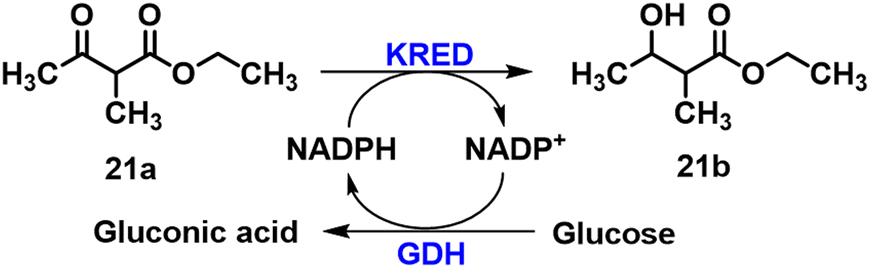 | ||
| Scheme 8 Biosynthesis of ethyl 3-hydroxy-2-methylbutanoate 22b. | ||
Besides, the (R)-enantiomer of ethyl 4-chloro-3-hydroxybutanoate obtained by biocatalytic reduction of 15a with a variety of KREDS,80,81 is a useful building block for the synthesis of L-carnitine.
Similarly, a crude dual-enzyme system composed of a KRED and Lactobacillus kefir alcohol dehydrogenase (LkADH) afforded tert-butyl (R)-3-hydroxy-5-hexenoate 22b in 96% yield and >99.9% ee on a pilot plant scale (Scheme 9).82 Subsequently, the KRED and LkADH were co-immobilised, by entrapment in a polyvinyl alcohol (PVA) gel, and used in continuous operation in a fixed bed reactor, affording 22b, in 93% isolated yield and a STY of 22 g L−1 h−1, with >99.9% ee.83
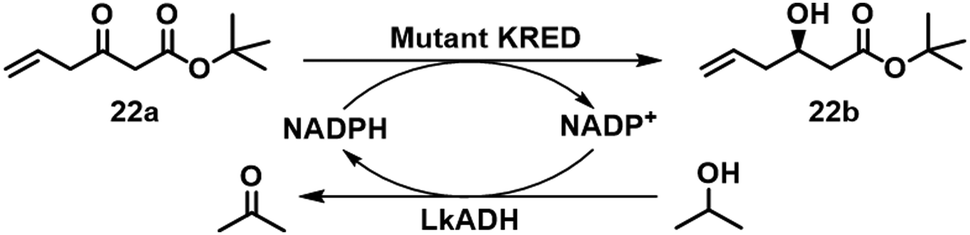 | ||
| Scheme 9 Biosynthesis of tert-butyl (R)-3-hydroxyl-5-hexenoate 22b. | ||
Biocatalytic asymmetric reduction of prochiral aromatic keto esters affords the corresponding chiral α, β, and γ-hydroxy esters, which are valuable synthons used in organic synthesis.84–86 Ethyl (R)-2-hydroxy-4-phenylbutyrate (R)-23b, for example, is a key intermediate in the synthesis of angiotensin converting enzyme (ACE) inhibitors, such as enalapril. Xu and coworkers87 used genome mining to discover new enzymes for the synthesis of (R)-23b (Scheme 10A). An enzyme, CgKR2, obtained from Candida glabrata and expressed in E. coli, produced (R)-23b with >99% ee and a space-time yield of 700 g L−1 d−1 when the lyophilised E. coli cells were used together with lyophilised GDH and glucose for cofactor regeneration.
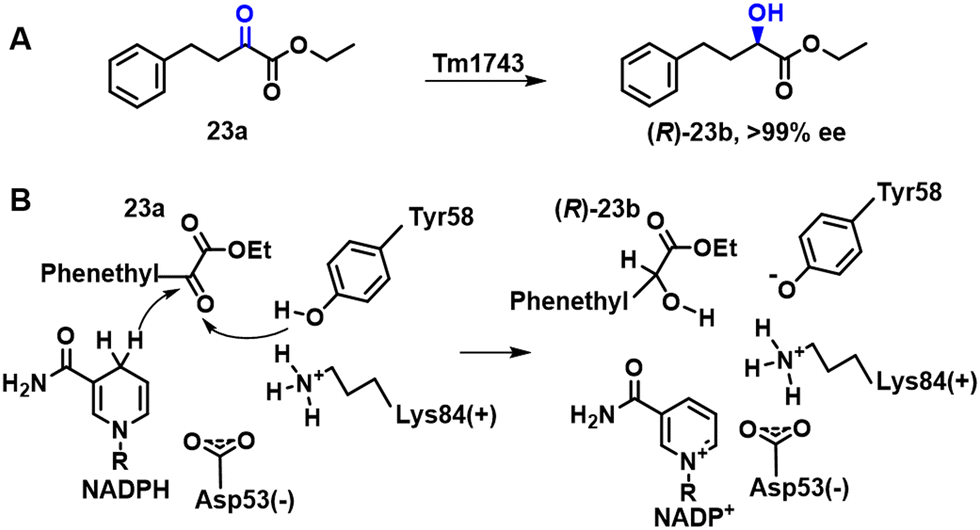 | ||
| Scheme 10 The possible catalytic mechanism of Tm1743 reduction of 23a to 23b (A); the biosynthesis of 23b from 23a (B). | ||
A KRED, Tm1743, from Thermotoga maritima, exhibited good activity with the keto ester precursor, ethyl-2-oxo-4-phenyl butyrate 23a, but was S-selective. Semi-rational protein engineering involving saturation mutagenesis was used to manipulate substrate-binding and invert the enantioselectivity of Tm1743 (Scheme 10B).88 The best Tm1743 variant afforded the R enantiomer in 99.4% ee.
5.2. Hydroxy ketones
Chiral hydroxy alkyl ketones are important building blocks for pharmaceuticals that are obtained by enzymatic reduction of the corresponding prochiral diketone. For example, the (R,R)-butane-2,3-diol dehydrogenase, BcBDH, from Bacillus clausii DSM 8716T catalyses the stereoselective reduction of α-diketones to the corresponding (R)-hydroxyketone.89 Similarly, fungal and human KREDs catalysed the enantioselective production of α-hydroxy ketones from symmetrical diketone precursors (Scheme 11A) albeit with opposite stereopreferences.90 The fungal enzyme, AKR5G was dominantly R-selective while the human AKR1B1 and yeast derived AKR3C1 gave mainly the S-product. Reactions were performed with pure enzymes combined with an NADPH-regenerating system consisting of glucose-6-phosphate and GDH. Further reduction of the α-hydroxyketone products affords the corresponding α-diols. In particular, the acetoin reductase ReAR from R. erythropolis WZ010, cloned and expressed in E.coli, produced (2S,3S)-2,3-butanediol 26 with absolute stereospecificity in alcoholic fermentation when using phenethyl alcohol (±)-27 as hydrogen source (Scheme 11B).91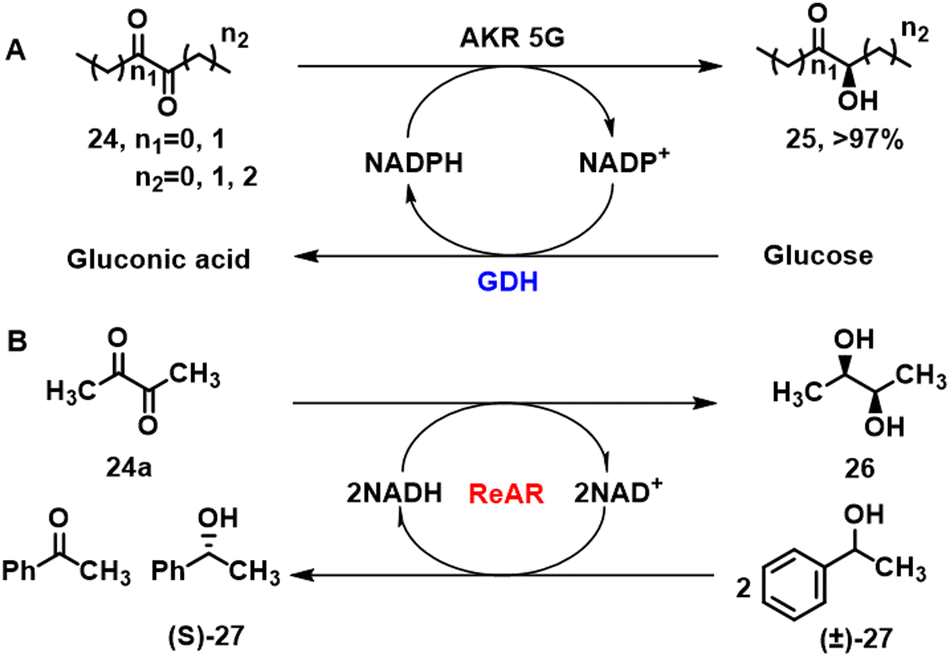 | ||
| Scheme 11 AKR 5G catalysed asymmetric reduction of diketones (A);90 reductase ReAR enabled asymmetric reduction of diacetyl (B).91 | ||
Erythritol is a four-carbon sugar alcohol that is used as a sweetener and flavour enhancer in food and beverages. Engineering NADPH cofactor metabolism by overexpression of genes ZWF1 and GND1 encoding glucose-6-phosphate dehydrogenase and 6-phosphogluconate dehydrogenase enabled the production of erythritol with a titer of 190 g L−1 using glucose as the main carbon source.92
5.3. Benzylic alcohols by reduction of aryl alkyl ketones
In the biocatalytic asymmetric reduction of aryl alkyl ketones93 the reductases have to cope with a wide variety of substituents in the aromatic ring, both electron-donating (e.g. alkyl, alkoxy, amine), and electron-withdrawing (e.g. nitro, cyano, halogen, trifluoromethyl, carboxylate ester),48,94,95 and bulky aryl,96 not to mention other aromatic ring systems.97 For example, Lactobacillus composti reductase SDR, heterologously expressed in E. coli, catalysed the enantioselective reduction of acetophenone to R-1-phenylethanol in >99% ee and 562.8 g L−1 d−1 STY.98Similarly, the KRED from Bacillus aryabhatti, BaSDR1 catalyses the enantioselective reduction of o-haloacetophenones. The catalytic efficiency, which is far below what is necessary for industrial viability, was improved through fine tuning of the substrate binding mode. Variants, Q139S, D253Y and Q139S/D253Y exhibited significantly enhanced activities without any loss in the high stereoselectivities (>99% ee). E. coli whole cells expressing the BaSDR1 variant were successfully applied in the stereoselective synthesis of chiral pharmaceutical intermediates such as (S)-1-(2-chlorophenyl)ethanol, (S)-1-(2,4-difluorophenyl)ethanol and (S)-1-(2,6-difluorophenyl) ethanol in 99% ee, at 95% conversion in a relatively short time.99
Similarly, a carbonyl reductase from Gluconobacter oxydans (GoCR), overexpressed in E. coli BL21 cells together with a cofactor regeneration system employing isopropanol as co-substrate, catalysed the stereoselective reduction of a series of fluoro-, chloro-, and bromo-substituted acetophenones. The corresponding S-alcohols, of interest as pharmaceutical intermediates, were obtained in excellent enantioselectivity (>99% ee) at high substrate loadings (Scheme 12).94 For example, 450 mM (S)-1-(2-chlorophenyl)-ethanol (>99% ee) was produced in 18 h from 500 mM o-chloroacetophenone. Due to the different electronegativities and van der Waals radii of F, Cl, and Br the various substrates react at different rates but the high stereoselectivities are retained in all cases.
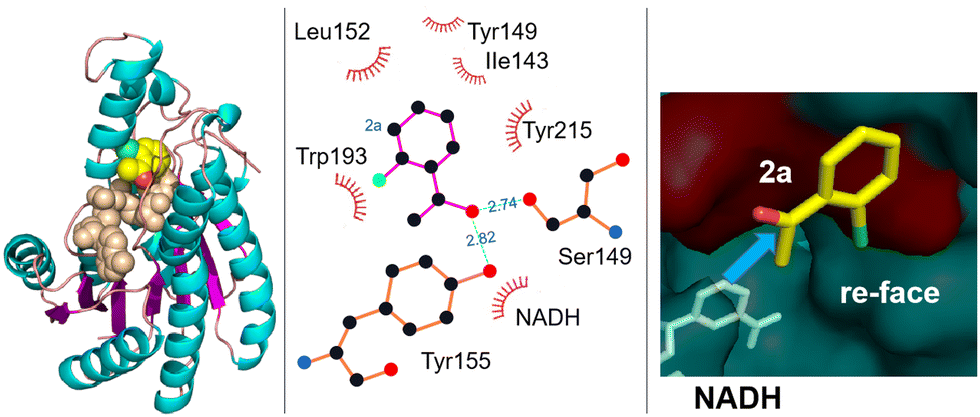 | ||
| Scheme 12 Substrate-binding model in GoCR. Reproduced from ref. 94 with permission from Elsevier, copyright 2015. | ||
Similarly, a variant, Val182Ala, of carbonyl reductase YueD from Bacillus subtilis was designed to expand its catalytic pocket and alter substrate orientation by replacing Val181 with Ala181. In conjunction with a GDH cofactor regeneration system and xylose as co-substrate, the whole cells of YueD catalysed the reduction of 500 mM halogenated acetophenones, to afford the corresponding S-alcohols in high enantioselectivity (99% ee) at conversions of 85–99%.100
(S)-1-(2,6-Dichloro-3-fluorophenyl) ethanol is an intermediate for anticancer drugs. It was obtained in up to 91% yield and 99.98% ee using a KRED, cloned from Thermotoga maritima MSB8, that contained the ncAA, p-azido-L-phenylalanine (pAzF) at preselected sites.101 The variant was immobilised, carrier free and directly from cell lysate, by cross-linking using light-promoted alkyne–azide cycloaddition with sym-dibenzo-1,5-cyclooctadiene-3,7-diyne 5 (see earlier).
(R)-[3,5-bis(Trifluoromethyl)phenyl] ethanol (R)-28b is a valuable chiral intermediate for the synthesis of the anti-emetic drugs Aprepitant and Fosaprepitant. Among diverse reductases102 tested for (R)-28b synthesis, the S154Y variant of carbonyl reductase LxCAR from Leifsonia xyli functions with either NADPH or NADH as cofactor.103 The catalytic triad of LxCAR is composed of Ser144, Tyr157, and Lys161 residues, in which Tyr157 is responsible for positioning of the substrate, 28a, in the required orientation by π–π stacking (Scheme 13A). S154Y-LxCAR, produced by replacing Ser154 by Tyr154 afforded 28a in 83% yield and 99.4% ee at 256 g L−1 substrate loading by establishing extra noncovalent contacts with trifluoromethyl groups.104 The asymmetric synthesis of (R)-28b from 28a also proceeded smoothly in whole cells expressing a fusion protein of LkCR (carbonyl reductase from Lactobacillus kefir), discovered by genome data mining, and BsGDH (GDH from Bacillus subtilis) by using 10 nm rigid linker peptides with helical ER/K motifs (Scheme 13B).105
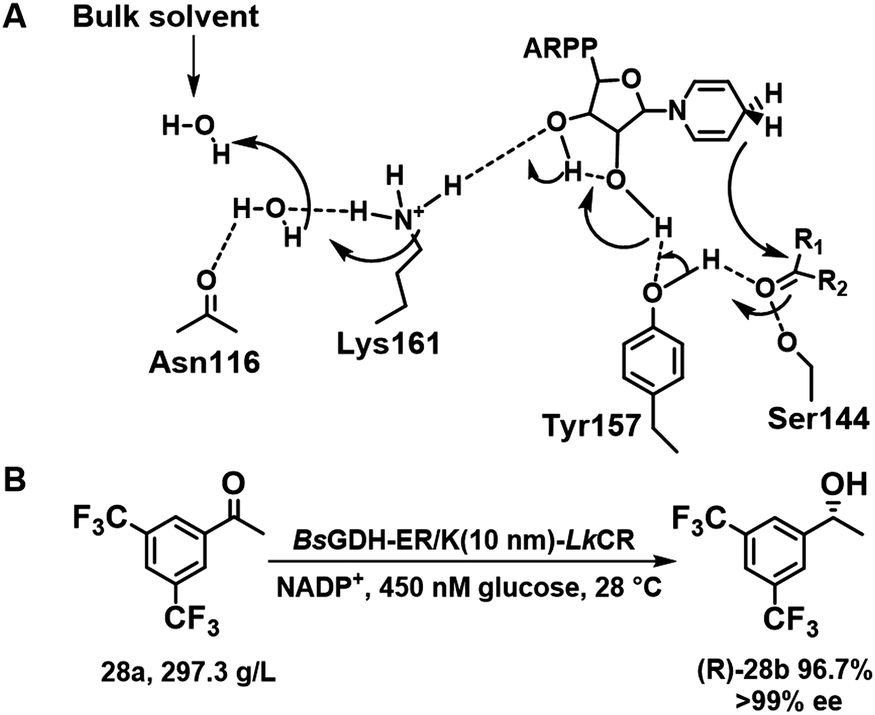 | ||
| Scheme 13 Schematic relay mechanism of LxCAR in (R)-28b synthesis (A);104 the synthesis of (R)-28b in whole cell (B).105 Reproduced from ref. 104 with permission from Elsevier, copyright 2022. | ||
Rivastigmine [(S)-3-[1-(dimethylamino)-ethyl]phenyl ethyl-(methyl)carbamate] 31 is an oral acetylcholinesterase inhibitor used for treatment of Alzheimer disease. Conventional chemical and enzymatic routes to rivastigmine involve the use of catalytic asymmetric hydrogenation or a lipase or transaminase.106 Sethi and Bhandya and co-workers,107 in contrast, used a KRED selected from a commercially available enzyme test kit of 80 KREDs for the enantioselective reduction of 3-N-ethyl-N-methylcarbomoyloxy acetophenone 29a in aqueous DMSO. The N-ethyl-methyl-carbamic acid-3-[(1S)-hydroxy-ethyl]-phenyl ester 29b was obtained in >99% ee (Scheme 14). The same ester was obtained in 99.99% ee using whole cells of bacillus subtilis containing a KRED and GDH.108
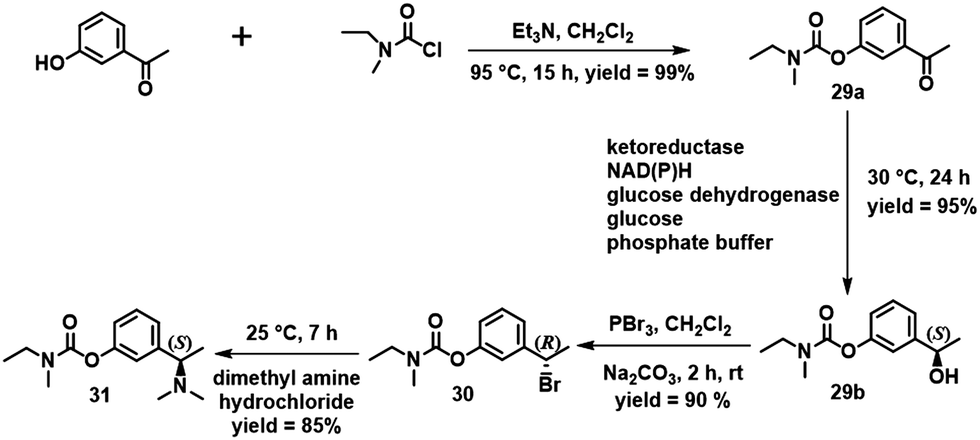 | ||
| Scheme 14 Synthesis of (S)-rivastigmine 31 from 3-hydroxy acetopheneone. Reproduced from ref. 107 with permission from Elsevier, copyright 2013. | ||
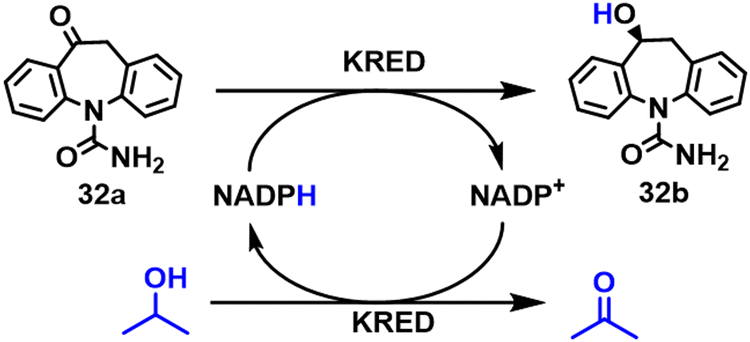
(S)-Licarbazepine 32b is used in the treatment of epilepsy. Codexis workers developed a process for the production of 32b by KRED catalysed reduction of oxcarbazepine.109 Using a variant of Lactobacillus kefir KRED, and an excess of isopropanol for cofactor regeneration, they obtained 32b in >95% isolated yield at >99% conversion with >98% chemical purity and >99% ee from >100 g L−1 (0.39 M) substrate and an enzyme loading of 1% w/w. (Scheme 15).
 | ||
| Scheme 15 Enzymatic synthesis of (S)-licarbazepine. | ||
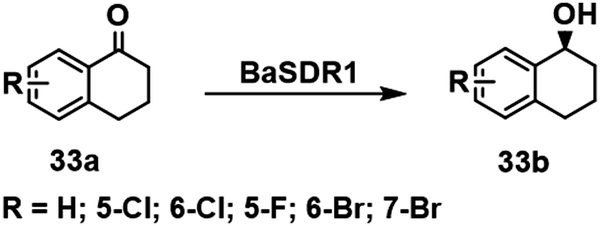
α-Tetralols and their halogenated derivatives are important chiral intermediates in the synthesis of pharmaceuticals such as sertraline. The activity of a KRED, BaSDR1, from Bacillus Aryabhattai towards bulky tetralones was improved using rational design to adjust the steric bulk and hydrophobicity of residues affecting the approach of α tetralone to the catalytic residues in the active site.110 The resulting variants exhibited enhanced activities and stereoselectivities in the asymmetric reduction of prochiral halogenated tetralones (Scheme 16). For example, the activity of variant Q237V/I291F towards 7-fluoro-α-tetralone was 16-fold that of the wild-type enzyme.
 | ||
| Scheme 16 The BaSDR1 protein catalyzed synthesis of chiral α-tetralinols. | ||
5.4. Aromatic amino alcohols
R-Phenylephrine 33b is an α1-adrenoceptor agonist that is used as a nasal decongestant. It is produced by asymmetric reduction of 1-(3-hydroxyphenyl)-2-(methylamino) ethanone 33a catalysed by a short-chain dehydrogenase/reductase from Serratia quinivorans BCRC 14811 (SmSDR).111 The catalytic activity of SmSDR depends on a conserved tetrad formed by Tyr158, Lys162, Ser144 and Asn118 residues (Scheme 17A). The activity of SmSDR is increased by replacing Phe98 and Phe202 with residues that stabilise its flexible region. Scheme 17 shows the possible mechanism of SmSDR converting 34a into 34b (the hydrogen atom of the nicotinamide is close to C3 atom of 34a, which initiates hydride transfer to this atom. Subsequent protonation of TYR158 by a water molecule then reduces the carbonyl group to hydroxyl).111 Hsu and co-workers112 constructed recombinant E. coli cells expressing an SDR cloned from Serratia quinivorans BCRC 14811) and used it to produce S-phenylephrine in 99% ee. The latter can be, in principle, converted into 34bvia a Walden inversion.112 Similarly, a carbonyl reductase from Sporobolomyces salmonicolor AKU4429 produced with the aid of semi−rational engineering catalysed the enantioselective reduction of β-amino ketones to the corresponding chiral γ-amino alcohols.113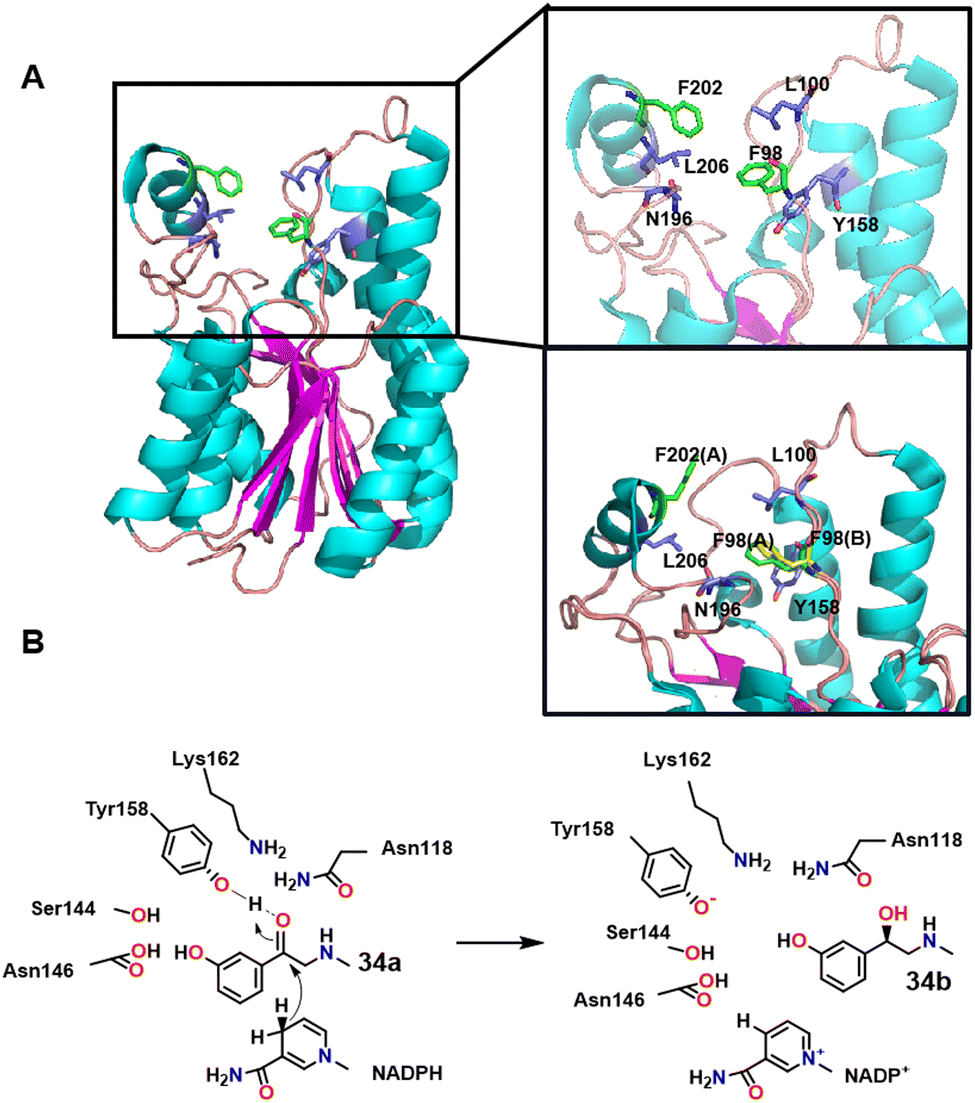 | ||
| Scheme 17 Structure of SmSDR (A); possible mechanism of SmSDR for the conversion of 34a into 34b (B). | ||
5.5. β-Hydroxy sulfones
Apremilast 36 is a chiral β-hydroxylsulfone used in the treatment of plaque psoriasis and psoriatic arthritis. The active stereoisomer has the R configuration. Two KREDs have been identified with complementary stereoselectivity for the reduction of the prochiral ketone precursor (Scheme 18).114 Thanks to the stronger binding strength of sulfone groups and proton donor Tyr175-OH, the LfSDR1-V186A/E141I variant of Lactobacillus fermenturn short-chain dehydrogenase/reductase 1 (LfSDR1)) protein exhibits R selectivity. The S-preference of CgKR1-F92I (variant of Candida glabrata ketoreductase 1 (CgKR1)) protein towards aromatic β-keto sulfones, in contrast, is attributed to π–π interactions of phenyl groups with Tyr208 and NADPH, and hydrogen bond formation between the sulfone group and Ala136.114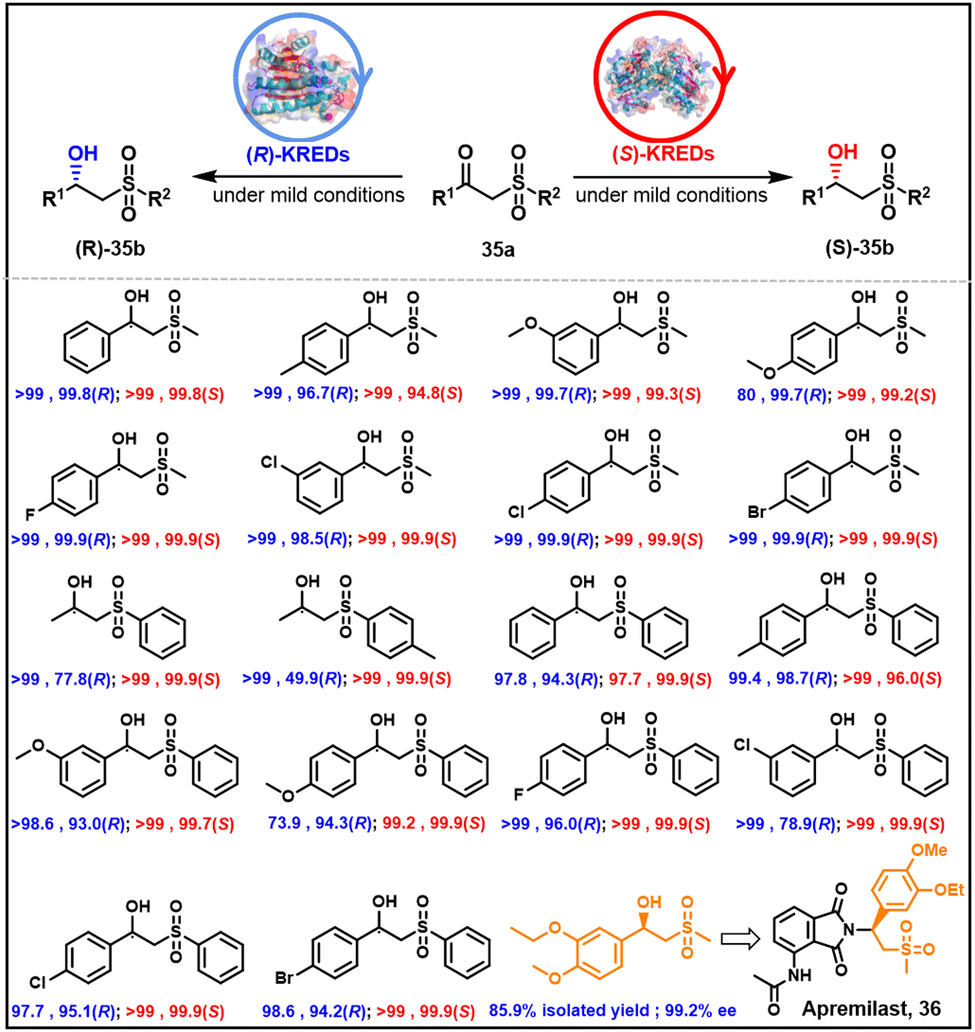 | ||
| Scheme 18 KREDs controlled enantioselective reduction of β-keto sulfones. | ||
5.6. Aryl halohydrins
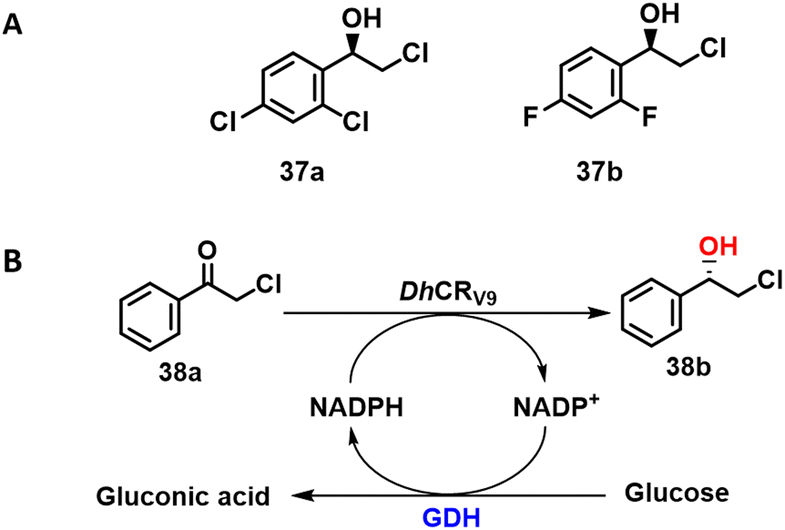
KREDs were successfully applied in the synthesis of a variety of chiral halohydrins that are important intermediates in the synthesis of various pharmaceuticals.115–117 For example, the KRED cloned from Scheffersomyces stipitis CBS 6045 SsCR exhibited a specific activity of 65 U mg−1 protein and excellent enantioselectivity (99.9% ee) in the conversion of the prochiral ketone precursor to (R)-2-chloro-1-(2,4-dichlorophenyl) ethanol) 37a with 268 g L−1 d−1 STY.118 The product is an intermediate in the synthesis of various antifungal agents, such as miconazole and sertaconazole.The structurally related (S)-2-chloro-1-(3,4-difluorophenyl) ethanol 37b is an intermediate for the drug ticagrelor. A mutant, L205M, obtained from an error prone PCR library of Chryseobacterium sp reductase (ChKRED20) combined excellent enantioselectivity (>99% ee) and 95% isolated yield with a specific activity ten times that of the wild-type enzyme (Scheme 19A).119 Similarly, Xu and co-workers116 used combinatorial mutation of conserved residues to identify key residues in a Debaryomyces hansenii KRED for the highly selective synthesis of S-halohydrins from α-chloroacetophenone (Scheme 19B).
 | ||
| Scheme 19 The structure of 37a and 37b (A);119 the biosynthesis of 38b (B). | ||
(S)-3-Chloro-1-phenyl-1-propanol is an intermediate in the production of the anti-depressant, (S)-fluoxetine. The NADH-dependent KRED from Novosphingobium aromaticivorans catalysed its synthesis in 99.6% ee by asymmetric reduction of 100 g L−1 3-chloro-1-phenyl-1-propanone.120,121
5.7. Phenyl-1,2-ethanediol
Yarrowia lipolytica is one of the most extensively studied non-conventional yeasts. Two Yarrowia lipolytica KREDs, YaCRI and YaCRII, identified by homologous sequence amplification, were expressed in rec. E.coli and used to reduce a variety of 2-hydroxyacetophenones 39a–41a to the corresponding (S)-1-phenyl-1,2-ethanediols 39b–41b (Scheme 20).122 YaCRII exhibited a broader substrate spectrum owing to a slightly narrower substrate entrance site. Both showed excellent enantioselectivity, good heat and acid stability and resistance to organic solvents, albeit with rather low activity.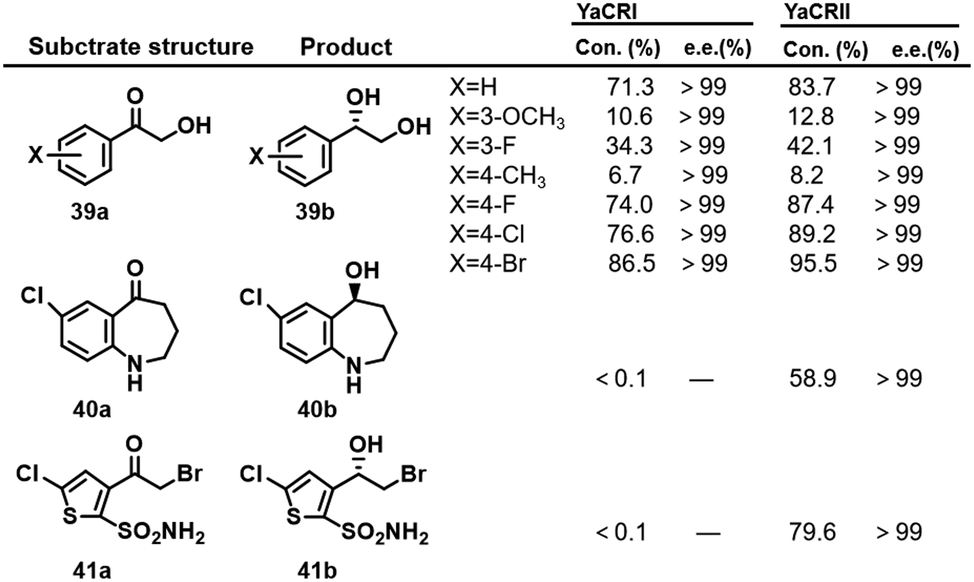 | ||
| Scheme 20 The biotransformation of ketols catalysed by YaCRII.122 Con. represents conversion. | ||
Similarly, the R-selective KRED from Candida papasilosis catalysed the reduction of 2-hydroxyacetophenone to (R)-1-phenyl-1, 2-ethanediol but with low rate and yield. Xiao and coworkers123 addressed this issue by constructing a fusion co-expression system, containing the R-KRED and GDH from Bacillus sp. YX-1, in an E.coli host. This optimised system produced R-PED in 99.9% ee and 99.9% yield after 24 h at 35 °C and pH 7.5.
5.8. Aromatic β-hydroxy nitriles
Chiral β-hydroxy nitriles are the precursors of (R)-β-hydroxy carboxylic acids 42 and intermediates in the synthesis of many biologically active compounds, such as serotonin/norepinephrine reuptake inhibitors.124 Romano and coworkers125 reported the asymmetric reduction of aromatic β-ketonitriles 42a to the (R)-β-hydroxy nitriles 42b using a NADPH-dependent KRED (1-Pglu) from Pichia glucozyma CBS 5766 with cofactor regeneration using glucose/GDH (Scheme 21).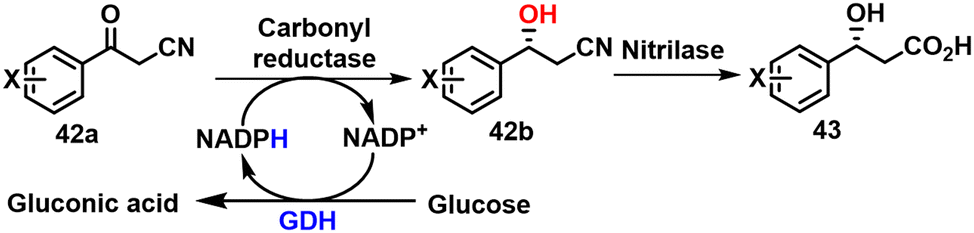 | ||
| Scheme 21 The biosynthesis route to (R)-β-hydroxy carboxylic acids. | ||
5.9. Other aromatic alcohols
A key chiral intermediate for (S)-duloxetine 50, a versatile anti-depressant is produced by asymmetric reduction of N,N-dimethyl-3-keto-3-(2-thienyl)-1-propanamine 44a catalysed by KREDs derived from various bacteria and yeasts.126–128 Zheng and co-workers,128 for example, used a KRED, mined from Rhodosporidium toruloides (RtSCR9) and co-expressed with GDH in E. coli. (S)-3-(dimethylamino)-1-(2-thienyl)-1-propanol 44b was produced in 92% yield and 99.9% ee at high substrate loading (1000 mM) with an STY of 23 mmol L−1 h−1 g DCW−1 on a 200 g scale (Scheme 22).128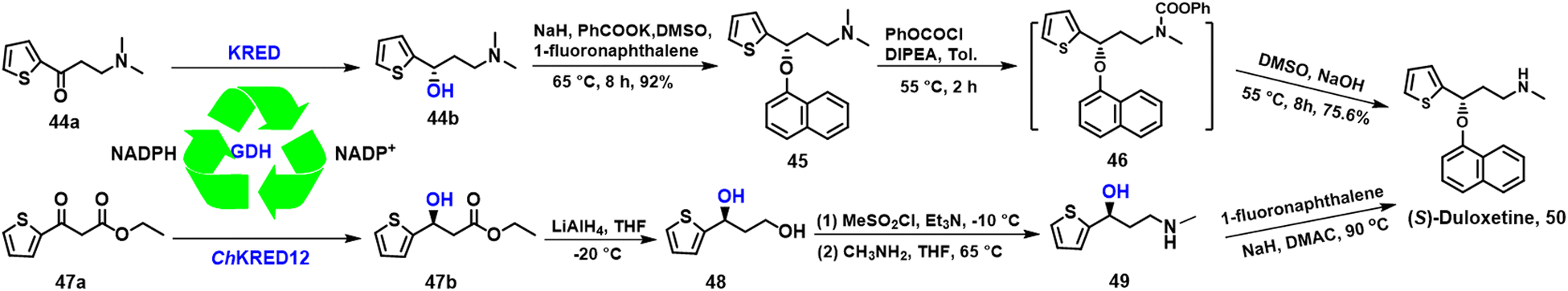 | ||
| Scheme 22 Chemical synthesis of (S)-duloxetine. | ||
Alternatively, (S)-duloxetine 50 can be produced via asymmetric reduction of ethyl 3-oxo-3-(2-thienyl) propanoate 47a catalysed by ChKRED15 from the genome of Chryseobacterium sp. CA49 and expressed in E. coli, The catalytic activity and stability of ChKRED15 was significantly improved by a single mutation of S12G. (S)-3-hydroxy-3-(2-thienyl) propanoate 47b was produced in >99% ee, with a space-time yield of 289 g L−1 d−1 at 100 g L−1 substrate loading (Scheme 22).129
5.10. Chiral heterocyclic alcohols
Many intermediates of drugs, vitamins and food additives are chiral heterocyclic alcohols that are amenable to synthesis by KRED catalysed asymmetric reduction of the corresponding ketones. (R)-pantolactone 51b, for example, is the precursor of calcium (R)-pantothenate (vitamin B5) and R-panthenol used as food additives. We showed that it can be prepared by KRED catalysed asymmetric reduction of ketopantoyl lactone ester precursor 51a, thereby avoiding the use of HCN in the chemical route which involves a racemate resolution.130 Thus, a putative conjugated polyketone reductase (CPR) gene from Candida orthopsilosis (CorCPR) was discovered by genome mining and expressed in E. coli. The rec-CorCPR catalysed the reduction of 50 mM KPL to 51b in >99% ee at 99% conversion within 5 h.131 Subsequently, the CPR and GDH genes were co-expressed in E. coli in order to achieve efficient NADPH regeneration from glucose and R-PL was obtained in whole-cell catalysis at a rate of 2.1 g h−1 g−1 DCW in a fed-batch biphasic system (Scheme 23).132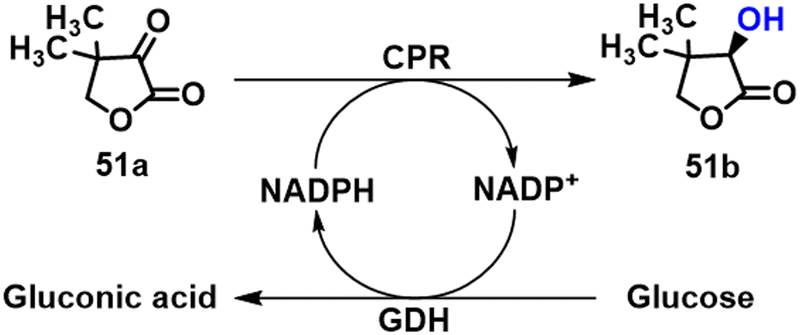 | ||
| Scheme 23 The biosynthesis of 51b. | ||
(S)-N-Boc-3-hydroxypiperidine 52b is the key chiral intermediate in the synthesis of the anti-cancer drug, ibrutinib.133–136 It is produced by KRED catalysed reduction of the prochiral ketone N-Boc-3-piperidone 52a (Scheme 24). For example, the KRED from Candida glabrata, co-expressed in E. coli with GDH from a Bacillus sp., afforded 52b in >99% ee at >99% conversion.136
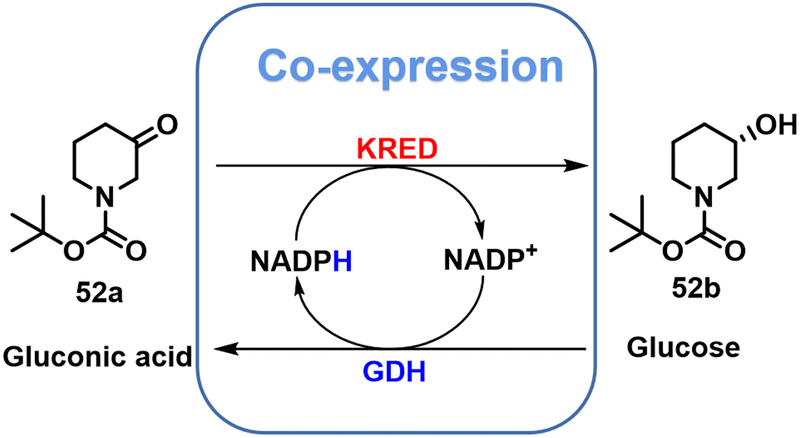 | ||
| Scheme 24 Asymmetric synthesis of 52b by co-expressing KRED and GDH. | ||
Similarly, a KRED, CprCR, from Candida parapsilosis, co-expressed with GDH from Bacillus megaterium in E. coli with added glucose, at 100 g L−1 substrate and 10% whole cell loading, afforded 52b in 99.8% ee at 98% conversion.137 In yet another variation, ChKRED03 from Chryseobacterium sp. catalysed the complete conversion (99%) of 200 g L−1 substrate within 3 h to afford 52b in >99% ee.138
Other examples of chiral heterocyclic alcohols, generally key building blocks for drugs and vitamins, that have been prepared by KRED catalysed reduction of the prochiral ketones include (R)-3-quinuclidinol 53,139tert-butyl ((2R,4R)-2-methyltetrahydro-2H-pyran-4-yl)carbamate 54140 and (R)-tetrahydrothiophene-3-ol 55 (Scheme 25).55
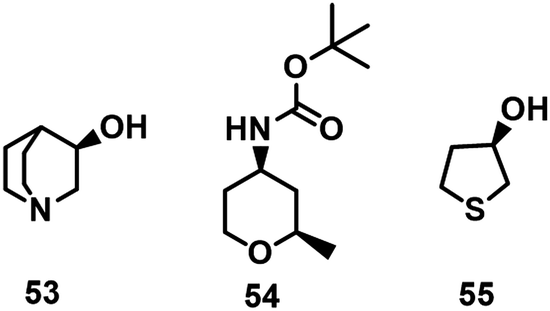 | ||
| Scheme 25 The structure of 53, 54, 55. | ||
6. Conclusions and future perspectives
Hopefully, we have shown that KRED catalysed reduction of prochiral ketones is a versatile, industrially viable technology for the synthesis of homochiral alcohols, in near perfect enantioselectiities, as key intermediates for a broad palette of pharmaceuticals and other high value added products. These developments, over the last two decades, were enabled by the application of (meta)genome mining in conjunction with bioinformatics for enzyme discovery and modern protein and enzyme engineering techniques for their optimisation. In the future they will be augmented by extensive use of machine-learning assisted directed evolution.141,142 These developments provide solid theoretical foundations for future improvement of existing KREDs and the development of new ones.The use of computational de novo protein design is an emerging field that enables the creation of new enzymes with desired activities. It enables the construction of specific functional proteins when the structure and function of natural proteins do not meet the needs of industrial or medical applications.51
A limiting factor in many KRED catalysed reductions is the efficiency of in situ regeneration of the NAD(P)H cofactor. During enzymatic regeneration, the reaction systems have to tolerate an overabundance of by-product that could result in enzyme deactivation. The most widely used GDH demonstrates the highest activity (up to 550 U g−1) and stability, however, it generates stoichiometric amounts of the water-soluble co-product gluconic acid which can also require extra downstream treatment costs.143 In the ongoing general trend towards the use of electrons and photons instead of reagents, in a sustainable low carbon economy,144 electrochemical,145 and photolytic146 cofactor regeneration are emerging methodologies. Despite being dependent on electron mediators for effective 1,4-NAD(P)H regeneration efficiency, both types of regenerative systems are robust, easy-to-assemble components that, if they are recyclable, give them potential for large-scale integrated processes including continuous flow.
In order for processes to be green and sustainable it is of paramount importance that enzymes are recyclable and are not used on a single-use throw-away basis. Improved stability and recyclability are achieved by immobilisation, either carrier-free or on carriers. In order to be cost-effective any loss of activity should be minimal and methods for enzyme immobilisation are becoming increasingly sophisticated in order to achieve this. This is exemplified by the use of ncAAs to enable efficient bio-orthogonal cross-linking of multiple enzymes or attachment to carriers. It is also important for the application of (immobilised) enzymes in continuous processing and the construction of fusion proteins of KREDs for the synthesis of increasingly complex molecules in enzymatic cascade processes.147–149
In short, in the current scenario of increasingly stringent environmental regulations and climate change mitigation, KREDS will play an important role in the sustainable, cost-effective and green production of an increasing number of homochiral secondary alcohols across the pharma, food and fine chemical industries.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (22078079) and the Zhejiang Provincial Ten Thousand Plan for Young Top Talents (2019R51012).References
- R. Csuk and B. I. Glaenzer, Chem. Rev., 1991, 91, 49–97 CrossRef CAS.
- R. Wendhausen, P. J. S. Moran, I. Joekes and J. A. R. Rodrigues, J. Mol. Catal. B: Enzym., 1998, 5, 69–73 CrossRef CAS.
- J. C. Moore, D. J. Pollard, B. Kosjek and P. N. Devine, Acc. Chem. Res., 2007, 40, 1412–1419 CrossRef CAS PubMed.
- Y.-G. Zheng, H.-H. Yin, D.-F. Yu, X. Chen, X.-L. Tang, X.-J. Zhang, Y.-P. Xue, Y.-J. Wang and Z.-Q. Liu, Appl. Microbiol. Biotechnol., 2017, 101, 987–1001 CrossRef CAS PubMed.
- S. M. A. De Wildeman, T. Sonke, H. E. Schoemaker and O. May, Acc. Chem. Res., 2007, 40, 1260–1266 CrossRef CAS PubMed.
- Z. N. Li, H. D. Yang, J. Y. Liu, Z. D. Huang and F. E. Chen, Chem. Rec., 2021, 21, 1611–1630 CrossRef CAS PubMed.
- S. M. Shi and L. Di, Expert Opin. Drug Metab. Toxicol., 2017, 13, 859–870 CrossRef CAS PubMed.
- G. W. Huisman, J. Liang and A. Krebber, Curr. Opin. Chem. Biol., 2010, 14, 122–129 CrossRef CAS PubMed.
- A. Ghatak, N. Bharatham, A. P. Shanbhag, S. Datta and J. Venkatraman, PLoS One, 2017, 12, e0170202 CrossRef PubMed.
- Y. Kallberg, U. Oppermann and B. Persson, FEBS J., 2010, 277, 2375–2386 CrossRef CAS PubMed.
- H. Jörnvall, J. Hedlund, T. Bergman, U. Oppermann and B. Persson, Biochem. Biophys. Res. Commun., 2010, 396, 125–130 CrossRef PubMed.
- C. Filling, K. Berndt, J. Benach, S. Knapp, T. Prozorovski, E. Nordling, R. Ladenstein, H. Jörnvall and U. Oppermann, J. Biol. Chem., 2002, 277, 25677–25684 CrossRef CAS PubMed.
- H. Moummou, Y. Kallberg, L. B. Tonfack, B. Persson and B. van der Rest, BMC Plant Biol., 2012, 12, 219 CrossRef CAS PubMed.
- D. Ghosh and P. Vihko, Chem. – Biol. Interact., 2001, 130–132, 637–650 CrossRef CAS PubMed.
- N. Tanaka, T. Nonaka, M. Nakanishi, Y. Deyashiki, A. Hara and Y. Mitsui, Structure, 1996, 4, 33–45 CrossRef CAS PubMed.
- H. Jornvall, B. Persson, M. Krook, S. Atrian, R. Gonzalez-Duarte, J. Jeffery and D. Ghosh, Biochemistry, 1995, 34, 6003–6013 CrossRef CAS PubMed.
- J. M. Jez, M. J. Bennett, B. P. Schlegel, M. Lewis and T. M. Penning, Biochem. J., 1997, 326, 625–636 CrossRef CAS PubMed.
- R. D. Mindnich and T. M. Penning, Hum. Genomics, 2009, 3, 362 CrossRef CAS PubMed.
- T. M. Penning, Chem. – Biol. Interact., 2015, 234, 236–246 CrossRef CAS PubMed.
- J. W. Trauger, A. Jiang, B. A. Stearns and P. V. LoGrasso, Biochemistry, 2002, 41, 13451–13459 CrossRef CAS PubMed.
- M. J. Bennett, B. P. Schlegel, J. M. Jez, T. M. Penning and M. Lewis, Biochemistry, 1996, 35, 10702–10711 CrossRef CAS PubMed.
- T. M. Penning, H. Ma and J. M. Jez, Chem. – Biol. Interact., 2001, 130–132, 659–671 CrossRef CAS PubMed.
- K. M. Bohren, C. E. Grimshaw, C. J. Lai, D. H. Harrison, D. Ringe, G. A. Petsko and K. H. Gabbay, Biochemistry, 1994, 33, 2021–2032 CrossRef CAS PubMed.
- B. P. Schlegel, K. Ratnam and T. M. Penning, Biochemistry, 1998, 37, 11003–11011 CrossRef CAS PubMed.
- X. M. Gong, Z. Qin, F. L. Li, B. B. Zeng, G. W. Zheng and J. H. Xu, ACS Catal., 2019, 9, 147–153 CrossRef CAS.
- Y. Liu, Z. Y. Li, C. Guo, C. Cui, H. Lin and Z. L. Wu, Process Biochem., 2021, 101, 207–212 CrossRef CAS.
- M. Musil, J. Stourac, J. Bendl, J. Brezovsky, Z. Prokop, J. Zendulka, T. Martinek, D. Bednar and J. Damborsky, Nucleic Acids Res., 2017, 45, W393–W399 CrossRef CAS PubMed.
- D. Bednar, K. Beerens, E. Sebestova, J. Bendl, S. Khare, R. Chaloupkova, Z. Prokop, J. Brezovsky, D. Baker and J. Damborsky, PLoS Comput. Biol., 2015, 11, e1004556 CrossRef PubMed.
- G. W. Zheng, Y. Y. Liu, Q. Chen, L. Huang, H. L. Yu, W. Y. Lou, C. X. Li, Y. P. Bai, A. T. Li and J. H. Xu, ACS Catal., 2017, 7, 7174–7181 CrossRef CAS.
- F. J. Zhao, Y. Liu, X. Q. Pei, C. Guo and Z. L. Wu, Appl. Microbiol. Biotechnol., 2017, 101, 1945–1952 CrossRef CAS PubMed.
- F. Y. Qin, B. Qin, T. Mori, Y. Wang, L. X. Meng, X. Zhang, X. Jia, I. Abe and S. You, ACS Catal., 2016, 6, 6135–6140 CrossRef CAS.
- J. P. Ou-yang, W. H. Zhang, F. Y. Qin, W. G. Zuo, S. Y. Xu, Y. Wang, B. Qin, Y. B. Song and X. Jia, Org. Biomol. Chem., 2017, 15, 7374–7379 RSC.
- J. W. Tang, L. Q. Chen, L. W. Zhang, G. W. Ni, J. Yu, H. Y. Wang, F. L. Zhang, S. G. Yuan, M. Q. Feng and S. X. Chen, Catal. Sci. Technol., 2021, 11, 6755–6769 RSC.
- C. Hu, B. J. Ye, Z. D. Huang and F. Chen, Mol. Catal., 2022, 522, 112208 CrossRef CAS.
- B. L. Taiqiang Sun, Yao Nie, Dong Wang and Yan Xu, Bioresour. Bioprocess., 2017, 4, 21 CrossRef.
- I. Suhrer, M. Haslbeck and K. Castiglione, Process Biochem., 2014, 49, 1527–1532 CrossRef.
- R. J. Conrado, J. D. Varner and M. P. DeLisa, Curr. Opin. Biotechnol, 2008, 19, 492–499 CrossRef CAS PubMed.
- H. Pettersson and G. Pettersson, Biochim. Biophys. Acta, Protein Struct. Mol. Enzymol., 2001, 1549, 155–160 CrossRef CAS PubMed.
- J. Rocha-Martín, B. D. L. Rivas, R. Muñoz, J. M. Guisán and F. López-Gallego, ChemCatChem, 2012, 4, 1279–1288 CrossRef.
- R. A. Sheldon, Catalysts, 2019, 9, 261 CrossRef CAS.
- C. S. Sampaio, J. A. F. Angelotti, R. Fernandez-Lafuente and D. B. Hirata, Int. J. Biol. Macromol., 2022, 215, 434–449 CrossRef CAS PubMed.
- X.-J. Zhang, D. Wu, W.-Z. Wang, M. Cao, Q. Liu, Z.-Q. Liu and Y.-G. Zheng, J. Chem. Technol. Biotechnol., 2021, 96, 3094–3100 CrossRef CAS.
- Y.-J. Wang, X.-P. Chen, W. Shen, Z.-Q. Liu and Y.-G. Zheng, Biochem. Eng. J., 2017, 128, 54–62 CrossRef CAS.
- S. Qiu, Y.-J. Wang, H. Yu, F. Cheng and Y.-G. Zheng, Process Biochem., 2019, 80, 43–51 CrossRef CAS.
- H. Li, R. Wang, A. Wang, J. Zhang, Y. Yin, X. Pei and P. Zhang, ACS Sustainable Chem. Eng., 2020, 8, 6466–6478 CrossRef CAS.
- R. Wang, J. Zhang, Z. Luo, T. Xie, Q. Xiao, X. Pei and A. Wang, Int. J. Biol. Macromol., 2022, 205, 682–691 CrossRef CAS PubMed.
- X. Pei, Z. Luo, L. Qiao, Q. Xiao, P. Zhang, A. Wang and R. A. Sheldon, Chem. Soc. Rev., 2022, 51, 7281–7304 RSC.
- F. Dall'Oglio, M. L. Contente, P. Conti, F. Molinari, D. Monfredi, A. Pinto, D. Romano, D. Ubiali, L. Tamborini and I. Serra, Catal. Commun., 2017, 93, 29–32 CrossRef.
- X.-P. Jiang, T.-T. Lu, C.-H. Liu, X.-M. Ling, M.-Y. Zhuang, J.-X. Zhang and Y.-W. Zhang, Int. J. Biol. Macromol., 2016, 88, 9–17 CrossRef CAS PubMed.
- R. Zhang, J. Jiang, J. Zhou, Y. Xu, R. Xiao, X. Xia and Z. Rao, Adv. Mater., 2018, 30, 1705443 CrossRef PubMed.
- P. Cheng, M. Tang, Z. Chen, W. Liu, X. Jiang, X. Pei and W. Su, React. Chem. Eng., 2020, 5, 1973–1980 RSC.
- J. H. Martin, B. D. Yahata, J. M. Hundley, J. A. Mayer, T. A. Schaedler and T. M. Pollock, Nature, 2017, 549, 365–369 CrossRef CAS PubMed.
- X. Zhou and C.-j Liu, Adv. Funct. Mater., 2017, 27, 1701134 CrossRef.
- R. Pei, W. Jiang, X. Fu, L. Tian and S.-F. Zhou, Chem. Eng. J., 2022, 429, 132293 CrossRef CAS.
- V. Williams, Y. Cui, J. Zhao, H. Fu, X. Jiao, Y. Ma, X. Li, X. Du and N. Zhang, Org. Process Res. Dev., 2022, 26, 1984–1995 CrossRef CAS.
- Y. Chen, Q. Jiang, L. Sun, Q. Li, L. Zhou, Q. Chen, S. Li, M. Yu and W. Li, Catalysts, 2018, 8, 334 CrossRef.
- T. Peschke, K. S. Rabe and C. M. Niemeyer, Angew. Chem., Int. Ed., 2017, 56, 2183–2186 CrossRef CAS PubMed.
- L. Nagy-Gyor, E. Abahazi, V. Bodai, P. Satorhelyi, B. Erdelyi, D. Balogh-Weiser, C. Paizs, G. Hornyanszky and L. Poppe, ChemBioChem, 2018, 19, 1845–1848 CrossRef CAS PubMed.
- S. Q. Zhang, X. Q. Li, Q. P. Yuan, F. Secundo, Y. Li and H. Liang, J. Inorg. Biochem., 2020, 208, 111093 CrossRef CAS PubMed.
- A. Basso, M. S. Brown, A. Cruz-Izquierdo, C. A. Martinez and S. Serban, Org. Process Res. Dev., 2022, 26, 2075–2084 CrossRef CAS.
- M. Plz, T. Petrovicova and M. Rebros, Molecules, 2020, 25, 4278 CrossRef CAS PubMed.
- Y.-J. Zhang, W.-X. Zhang, G.-W. Zheng and J.-H. Xu, Adv. Synth. Catal., 2015, 357, 1697–1702 CrossRef CAS.
- Y. Xu, Q. Chen, Z. J. Zhang, J. H. Xu and G. W. Zheng, ChemBioChem, 2020, 21, 1341–1346 CrossRef CAS PubMed.
- S. K. Ma, J. Gruber, C. Davis, L. Newman, D. Gray, A. Wang, J. Grate, G. W. Huisman and R. A. Sheldon, Green Chem., 2010, 12, 81–86 RSC.
- X. Luo, Y. J. Wang, W. Shen and Y. G. Zheng, J. Biotechnol., 2016, 224, 20–26 CrossRef CAS PubMed.
- Y. J. Wang, W. Shen, X. Luo, Z. Q. Liu and Y. G. Zheng, Biotechnol. Prog., 2017, 33, 1235–1242 CrossRef CAS PubMed.
- H. Yu, S. Qiu, F. Cheng, Y. N. Cheng, Y. J. Wang and Y. G. Zheng, Bioorg. Chem., 2019, 90, 103018 CrossRef CAS PubMed.
- S. Qiu, F. Cheng, L. J. Jin, Y. Chen, S. F. Li, Y. J. Wang and Y. G. Zheng, Bioorg. Chem., 2020, 103, 104228 CrossRef CAS PubMed.
- S. Qiu, S. Y. Xu, S. F. Li, K. M. Meng, F. Cheng, Y. J. Wang and Y. G. Zheng, Biotechnol. J., 2021, 16, e2100130 CrossRef PubMed.
- S. F. Li, J. Y. Xie, S. Qiu, S. Y. Zhou, Y. J. Wang and Y. G. Zheng, Bioorg. Chem., 2021, 109, 104712 CrossRef CAS PubMed.
- F. Cheng, Y. Chen, S. Qiu, Q.-Y. Zhai, H.-T. Liu, S.-F. Li, C.-Y. Weng, Y.-J. Wang and Y.-G. Zheng, ACS Catal., 2021, 11, 2572–2582 CrossRef CAS.
- X.-M. Gong, G.-W. Zheng, Y.-Y. Liu and J.-H. Xu, Org. Process Res. Dev., 2017, 21, 1349–1354 CrossRef CAS.
- Z. Q. Liu, Z. L. Hu, X. J. Zhang, X. L. Tang, F. Cheng, Y. P. Xue, Y. J. Wang, L. Wu, D. K. Yao, Y. T. Zhou and Y. G. Zheng, Biotechnol. Prog., 2017, 33, 612–620 CrossRef CAS PubMed.
- T. Xu, C. Wang, S. Zhu and G. Zheng, Process Biochem., 2017, 57, 72–79 CrossRef CAS.
- Z. Q. Liu, L. Wu, L. Zheng, W. Z. Wang, X. J. Zhang, L. Q. Jin and Y. G. Zheng, Bioresour. Technol., 2018, 249, 161–167 CrossRef CAS PubMed.
- Z. Q. Liu, H. H. Yin, X. J. Zhang, R. Zhou, Y. M. Wang and Y. G. Zheng, Bioorg. Chem., 2018, 80, 733–740 CrossRef CAS PubMed.
- S. Venkataraman and A. Chadha, J. Fluorine Chem., 2015, 169, 66–71 CrossRef CAS.
- R. L. Hanson, S. Goldberg, A. Goswami, T. P. Tully and R. N. Patel, Adv. Synth. Catal., 2005, 347, 1073–1080 CrossRef CAS.
- T. Petrovicova, K. Markosova, Z. Hegyi, I. Smonou, M. Rosenberg and M. Rebros, Catalysts, 2018, 8, 168 CrossRef CAS.
- S. A. Yoon and H. K. Kim, J. Microbiol. Biotechnol., 2013, 23, 1395–1402 CrossRef CAS PubMed.
- S. Basak, N. G. Sahoo and A. K. Pavanasam, Bioengineered, 2018, 9, 186–195 CrossRef CAS PubMed.
- C. Hu, M. Liu, X. Yue, Z. Huang and F. Chen, Org. Process Res. Dev., 2020, 24, 1700–1706 CrossRef CAS.
- C. Hu, Z. Huang, M. Jiang, Y. Tao, Z. Li, X. Wu, D. Cheng and F. Chen, ACS Sustainable Chem. Eng., 2021, 9, 8990–9000 CrossRef CAS.
- G. A. Applegate, R. W. Cheloha, D. L. Nelson and D. B. Berkowitz, Chem. Commun., 2011, 47, 2420–2422 RSC.
- X. Luo, Y. Zhang, L. Yin, W. Zheng and Y. Fu, 3 Biotech, 2020, 10, 14 CrossRef PubMed.
- I. Sührer, M. Haslbeck and K. Castiglione, Process Biochem., 2014, 49, 1527–1532 CrossRef.
- Y. N. Nai-Dong Shen, H.-M. Ma, L.-J. Wang, C.-X. Li, G.-W. Zheng, J. Zhang and J.-H. Xu, Org. Lett., 2012, 14, 1982–1985 CrossRef PubMed.
- Z. Wang, S. Zhou, S. Zhang, S. Zhang, F. Zhu, X. Jin, Z. Chen and X. Xu, Sci. Rep., 2017, 7, 4007 CrossRef PubMed.
- L. Muschallik, D. Molinnus, M. Jablonski, C. R. Kipp, J. Bongaerts, M. Pohl, T. Wagner, M. J. Schoning, T. Selmer and P. Siegert, RSC Adv., 2020, 10, 12206–12216 RSC.
- E. Calam, S. Porté, M. R. Fernández, J. Farrés, X. Parés and J. A. Biosca, Chem. – Biol. Interact., 2013, 202, 195–203 CrossRef CAS PubMed.
- Z. Wang, Q. Song, M. Yu, Y. Wang, B. Xiong, Y. Zhang, J. Zheng and X. Ying, Appl. Microbiol. Biotechnol., 2014, 98, 641–650 CrossRef CAS PubMed.
- H. Cheng, S. Wang, M. Bilal, X. Ge, C. Zhang, P. Fickers and H. Cheng, Microb. Cell Fact., 2018, 17, 133 CrossRef PubMed.
- J. Zhang, R. Wang, Z. Luo, D. Jia, H. Chen, Q. Xiao, P. Zhang, X. Pei and A. Wang, Mater. Chem. Front., 2022, 6, 182–193 RSC.
- J. Deng, K. Chen, Z. Yao, J. Lin and D. Wei, J. Mol. Catal. B: Enzym., 2015, 118, 1–7 CrossRef CAS.
- T. Schubert, M. R. Kula and M. Muller, Synthesis, 1999, 2045–2048 CrossRef CAS.
- Z. Sun, J. Zhang, D. Yin, G. Xu and Y. Ni, ACS Sustainable Chem. Eng., 2022, 10, 13722–13732 CrossRef CAS.
- R. Preeti, R. Reena, R. Sindhu, M. K. Awasthi, A. Pandey and P. Binod, Environ. Sci. Pollut. Res., 2023, 30, 9036–9047 CrossRef CAS PubMed.
- Y. J. Wang, B. B. Ying, M. Chen, W. Shen, Z. Q. Liu and Y. G. Zheng, World J. Microbiol. Biotechnol., 2017, 33, 144 CrossRef PubMed.
- A. Li, X. Li, W. Pang, Q. Tian, T. Wang and L. Zhang, Catal. Sci. Technol., 2020, 10, 2462–2472 RSC.
- M. Naeem, A. U. Rehman, B. Shen, L. Ye and H. Yu, Biochem. Eng. J., 2018, 137, 62–70 CrossRef CAS.
- H. M. Li, R. Wang, A. M. Wang, J. Zhang, Y. C. Yin, X. L. Pei and P. F. Zhang, ACS Sustainable Chem. Eng., 2020, 8, 6466–6478 CrossRef CAS.
- T.-X. Tang, Y. Liu and Z.-L. Wu, J. Mol. Catal. B: Enzym., 2014, 105, 82–88 CrossRef CAS.
- N. Q. Wang, J. Sun, J. Huang and P. Wang, Appl. Microbiol. Biotechnol., 2014, 98, 8591–8601 CrossRef CAS PubMed.
- J. Li, T. Dinh and R. Phillips, Arch. Biochem. Biophys., 2022, 720, 109158 CrossRef CAS PubMed.
- K. Chen, K. Li, J. Deng, B. Zhang, J. Lin and D. Wei, Microb. Cell Fact., 2016, 15, 191 CrossRef PubMed.
- M. Fuchs, D. Koszelewski, K. Tauber, W. Kroutil and K. Faber, Chem. Commun., 2010, 46, 5500–5502 RSC.
- M. K. Sethi, S. R. Bhandya, N. Maddur, R. Shukla, A. Kumar and V. S. N. Jayalakshmi Mittapalli, Tetrahedron: Asymmetry, 2013, 24, 374–379 CrossRef CAS.
- P. Xie, X. Zhou and L. Zheng, J. Biotechnol., 2019, 289, 64–70 CrossRef CAS PubMed.
- N. K. Modukuru, J. Sukumaran, S. J. Collier, A. S. Chan, A. Gohel, G. W. Huisman, R. Keledjian, K. Narayanaswamy, S. J. Novick, S. M. Palanivel, D. Smith, Z. Wei, B. Wong, W. L. Yeo and D. A. Entwistle, Org. Process Res. Dev., 2014, 18, 810–815 CrossRef CAS.
- A. Li, W. Ting, K. Yang, X. Zhang, D. Yin, Y. Qin and L. Zhang, ChemCatChem, 2021, 13, 4625–4633 CrossRef CAS.
- J. S. Liu, Y. C. Kuan, Y. Tsou, T. Y. Lin, W. H. Hsu, M. T. Yang, J. Y. Lin and W. C. Wang, Sci. Rep., 2018, 8, 2316 CrossRef PubMed.
- G.-J. Peng, Y.-C. Cho, T.-K. Fu, M.-T. Yang and W.-H. Hsu, Process Biochem., 2013, 48, 1509–1515 CrossRef CAS.
- D. Zhang, X. Chen, J. Chi, J. Feng, Q. Wu and D. Zhu, ACS Catal., 2015, 5, 2452–2457 CrossRef CAS.
- J. Guo, X. Gao, D. Qian, H. Wang, X. Jia, W. Zhang, B. Qin and S. You, Org. Biomol. Chem., 2022, 20, 2081–2085 RSC.
- J. Ren, W. Dong, B. Yu, Q. Wu and D. Zhu, Tetrahedron: Asymmetry, 2012, 23, 497–500 CrossRef CAS.
- G. C. Xu, Y. P. Shang, H. L. Yu and J. H. Xu, Chem. Commun., 2015, 51, 15728–15731 RSC.
- F. Qin, B. Qin, T. Mori, Y. Wang, L. Meng, X. Zhang, X. Jia, I. Abe and S. You, ACS Catal., 2016, 6, 6135–6140 CrossRef CAS.
- Y.-P. Shang, Q. Chen, X.-D. Kong, Y.-J. Zhang, J.-H. Xu and H.-L. Yu, Adv. Synth. Catal., 2017, 359, 426–431 CrossRef CAS.
- F. J. Zhao, Y. Liu, X. Q. Pei, C. Guo and Z. L. Wu, Appl. Microbiol. Biotechnol., 2017, 101, 1945–1952 CrossRef CAS PubMed.
- Y. Tian, X. Ma, M. Yang, D. Wei and E. Su, Catal. Commun., 2017, 97, 56–59 CrossRef CAS.
- Y. Tang, G. Zhang, Z. Wang, D. Liu, L. Zhang, Y. Zhou, J. Huang, F. Yu, Z. Yang and G. Ding, Bioresour. Technol., 2018, 250, 457–463 CrossRef CAS PubMed.
- H. L. Zhang, C. Zhang, C. H. Pei, M. N. Han and W. Li, J. Appl. Microbiol., 2019, 126, 127–137 CrossRef CAS PubMed.
- X. Zhou, R. Zhang, Y. Xu, H. Liang, J. Jiang and R. Xiao, Process Biochem., 2015, 50, 1807–1813 CrossRef CAS.
- H. A. Dunming Zhu, C. Mukherjee, Y. Yang, E. R. Biehl and L. Hua, Org. Lett., 2007, 9, 2561–2563 CrossRef PubMed.
- M. L. Contente, I. Serra, F. Molinari, R. Gandolfi, A. Pinto and D. Romano, Tetrahedron, 2016, 72, 3974–3979 CrossRef CAS.
- W. W. Rui Pei, Y. Zhang, L. Tian, W. Jiang and S.-F. Zhou, Catalysts, 2020, 10, 1121 CrossRef.
- R. Pei, X. Fu, L. Tian, S. F. Zhou and W. Jiang, Enzyme Microb. Technol., 2022, 160, 110074 CrossRef CAS PubMed.
- X. Chen, Z. Q. Liu, C. P. Lin and Y. G. Zheng, Bioorg. Chem., 2016, 65, 82–89 CrossRef CAS PubMed.
- Z.-Q. Ren, Y. Liu, X.-Q. Pei, H.-B. Wang and Z.-L. Wu, J. Mol. Catal. B: Enzym., 2015, 113, 76–81 CrossRef CAS.
- P. Cheng, J. Wang, Y. Wu, X. Jiang, X. Pei and W. Su, Enzyme Microb. Technol., 2019, 126, 77–85 CrossRef CAS PubMed.
- J. Wang, P. Cheng, Y. Wu, A. Wang, F. Liu and X. Pei, J. Biotechnol., 2019, 291, 26–34 CrossRef CAS PubMed.
- J. W. Xiaolin Pei, H. Zheng, P. Cheng, Y. Wu, A. Wang and W. Su, React. Chem. Eng., 2020, 5, 531–538 RSC.
- K. Yamada-Onodera, M. Fukui and Y. Tani, J. Biosci. Bioeng., 2007, 103, 174–178 CrossRef CAS PubMed.
- M. He, S. Zhou, M. Cui, X. Jin, D. Lai, S. Zhang, Z. Wang and Z. Chen, Appl. Biochem. Biotechnol., 2017, 181, 1304–1313 CrossRef CAS PubMed.
- X. Ying, J. Zhang, C. Wang, M. Huang, Y. Ji, F. Cheng, M. Yu, Z. Wang and M. Ying, Molecules, 2018, 23, 3117 CrossRef PubMed.
- X. Gao, Q. Pei, N. Zhu, Y. Mou, J. Liang, X. Zhang and S. Feng, Catalysts, 2022, 12, 304 CrossRef CAS.
- J. Chen, M. Yan and L. Xu, World J. Microbiol. Biotechnol., 2017, 33, 61 CrossRef PubMed.
- H.-B. W. Guang-Peng Xua and Z.-L. Wu, Process Biochem., 2016, 51, 881–885 CrossRef.
- A. Uzura, F. Nomoto, A. Sakoda, Y. Nishimoto, M. Kataoka and S. Shimizu, Appl. Microbiol. Biotechnol., 2009, 83, 617–626 CrossRef CAS PubMed.
- M. Burns, W. Bi, H. Kim, M. S. Lall, C. Li and B. T. O’Neill, Org. Process Res. Dev., 2021, 25, 941–946 CrossRef CAS.
- Z. Wu, S. B. J. Kan, R. D. Lewis, B. J. Wittmann and F. H. Arnold, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 8852–8858 CrossRef CAS PubMed.
- F. Cadet, N. Fontaine, G. Li, J. Sanchis, M. Ng Fuk Chong, R. Pandjaitan, I. Vetrivel, B. Offmann and M. T. Reetz, Sci. Rep., 2018, 8, 16757 CrossRef PubMed.
- X. Wang, T. Saba, H. H. P. Yiu, R. F. Howe, J. A. Anderson and J. Shi, Chem, 2017, 2, 621–654 CAS.
- R. A. Sheldon and D. Brady, ChemSusChem, 2022, 15, e202102628 CrossRef CAS PubMed.
- C. Zhang, H. Zhang, J. Pi, L. Zhang and A. Kuhn, Angew. Chem., Int. Ed., 2022, 61, e202111804 CAS.
- Y. Yin, R. Wang, J. Zhang, Z. Luo, Q. Xiao, T. Xie, X. Pei, P. Gao and A. Wang, ACS Appl. Mater. Interfaces, 2021, 13, 41454–41463 CrossRef CAS PubMed.
- K. Rosenthal, U. T. Bornscheuer and S. Lütz, Angew. Chem., Int. Ed., 2022, 61, e202208358 CrossRef CAS PubMed.
- C. An and K. M. Maloney, Curr. Opin. Green Sustainable Chem., 2022, 34, 100591 CrossRef CAS.
- A. I. Benítez-Mateos, D. Roura Padrosa and F. Paradisi, Nat. Chem., 2022, 14, 489–499 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3cc01474f |
| ‡ Li Qiao and Zhiyuan Luo contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2023 |