
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Chlorine-radical-mediated C–H oxygenation reaction under light irradiation
Yuki
Itabashi
 a,
Haruyasu
Asahara
*bc and
Kei
Ohkubo
*ac
a,
Haruyasu
Asahara
*bc and
Kei
Ohkubo
*ac
aInstitute for Open and Transdisciplinary Research Initiatives, Osaka University, 1-6 Yamadaoka, Suita, Osaka 565-0871, Japan. E-mail: ohkubo@irdd.osaka-u.ac.jp
bGraduate School of Pharmaceutical Sciences, Osaka University, 1-6 Yamadaoka, Suita, Osaka 565-0871, Japan. E-mail: asahara@phs.osaka-u.ac.jp
cInstitute for Advanced Co-Creation Studies, Osaka University, 1-6 Yamadaoka, Suita, Osaka 565-0871, Japan
First published on 5th May 2023
Abstract
The oxygenation of C(sp3)–H bonds is a key chemical reaction in the production of fine chemicals and pharmaceuticals. Hydrogen atom transfer has recently gained significant attention for its ability to functionalise C–H bonds. The C–H bond can be activated by transferring the H radical to a hydrogen acceptor such as the chlorine radical (Cl˙). Thus, the Cl˙ generated by light irradiation can be used to initiate C–H oxygenation reactions, in which molecular oxygen is used as the oxidant. In this review article, we have summarised the recent advances in the field of Cl˙-mediated C–H oxygenation reactions. Reactions with catalysts such as metal chlorides, organophotoredox catalysts (acridinium ions), and chlorine dioxide radicals under light irradiation have been discussed. We conclude the review by providing suggestions for future research studies in the field. We expect that this review article will provide valuable information for the development of Cl˙-mediated C–H oxygenation reactions, contribute to the understanding of the reactivity of Cl˙, and inspire other useful synthetic chemical applications for C–H oxygenation reactions.
 Yuki Itabashi | Yuki Itabashi received his PhD from the University of Tokyo. He worked as a postdoctoral researcher at the University of Tokyo and Nagoya University in the national museum of emerging science and innovation. He moved to Osaka University as a postdoctoral fellow (Prof. Ohkubo group). He joined as a specially appointed assistant professor in Institute for Open and Transdisciplinary Research Initiatives, Osaka University (Prof. Ohkubo group). |
 Haruyasu Asahara | Haruyasu Asahara received his PhD degree in 2010 from Osaka University. After completion of his degree he was appointed as a Postdoc with Prof. Minakata (Osaka University). From 2010, he worked as a Humboldt Fellow in Prof. Herbert Mayr group (Ludwig-Maximilians-Universität München, Germany). He then moved to a postdoctoral position with Prof. Akashi group (Osaka University). He worked in Prof. Nishiwaki group (Kochi University of Technology, 2013–2016). In 2017, he returned to Osaka University (the group of Prof. Inoue and Prof. Ohkubo). In 2019, he joined the graduate school of pharmaceutical sciences at Osaka University as associate professor. |
 Kei Ohkubo | Dr Kei Ohkubo, Chemistry Doctor (PhD-engineering), now is a Professor of Chemistry of Institute for Advanced Co-Creation Studies and Open and Transdisciplinary Research Initiatives, Osaka University, Japan. He earned his PhD degree from Graduate School of Engineering, Osaka University in 2001. He was working as a JSPS fellow and a JST research fellow at Osaka University (2001–2005), a designated associate professor in Osaka University (2005–2014) and a specially appointed professor at Osaka University (2014–2017). He has been a full professor at Osaka University since 2017. His research fields are photochemistry, organic photocatalysis and oxygenation chemistry. |
1. Introduction
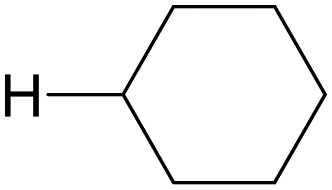
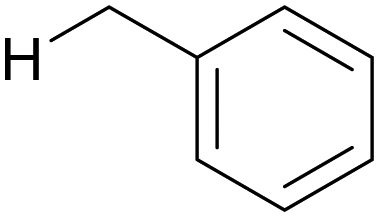
The oxygenation of C(sp3)–H bonds is a key chemical reaction in the production of fine chemicals and pharmaceuticals.1–4 As shown in Table 1, these reactions typically require harsh conditions such as a high temperature and/or high pressure and strong oxidants owing to their high bond dissociation energies (BDEs, >90 kcal mol−1).5 The use of molecular oxygen (O2) as an oxidant is favourable as it is eco-friendly and sustainable;6 however, O2 cannot activate C–H bonds under mild conditions.| C–H bond | H–X bond | ||||
|---|---|---|---|---|---|
| H–CH3 | 105 |
|
100 | H–Cl | 103 |
| H–CH2CH3 | 101 | H–Br | 88 | ||
| H–CH(CH3)2 | 98 |
|
90 | H–l | 71 |
| H–C(CH3)3 | 96 | ||||
| H–CCl3 | 96 | ||||
Hydrogen atom transfer (HAT) is one of the most effective tools to cleave the C–H bond and activate it.7 HAT has recently garnered significant attention owing to its ability to functionalise complex molecules via C–H bond activation. As a fundamental radical reaction, HAT involves the transfer of a hydrogen atom from a hydrogen donor (C–H) to a hydrogen acceptor (X˙), which is also known as the radical reagent. The progress of HAT reactions can be estimated by comparing the BDEs of the bonds that would be cleaved (C–H) and those that would be formed (H–X), as shown in Scheme 1. Therefore, a radical reagent that forms more stable bonds than those that would be cleaved can enable direct functionalisation through HAT. Several radical species (X˙), including O-, N-, S-, B-, and halogen-centred radicals, are known to be suitable HAT reagents.8–11 Halogen radicals have attracted much attention owing to their easy accessibility and high reactivity.
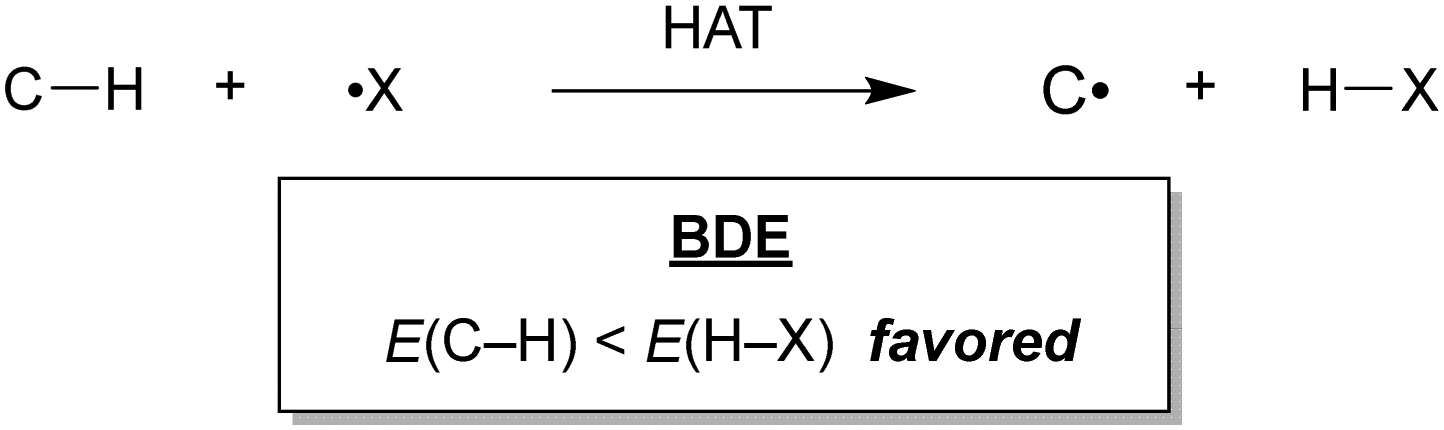 | ||
| Scheme 1 Hydrogen atom transfer (HAT). | ||
In particular, a chlorine radical (Cl˙) is a powerful hydrogen acceptor and valuable HAT agent. HAT from hydrocarbons to Cl˙ affords HCl with a H–Cl bond stable to homolytic cleavage. The C–H BDEs of the hydrocarbons listed in Table 1 are generally lower than the bond formation energy of H–Cl (103 kcal mol−1). Thus, Cl˙ can easily cleave the C–H bonds of alkanes. However, Br˙ and I˙ have little or no ability to induce HAT from alkanes owing to the low bond formation energies of H–Br and H–I (88 and 71 kcal mol−1, respectively).12 The formation of two Cl˙ radicals from Cl2 upon UV irradiation or heating is a well-established synthetic organic radical reaction for the chlorination of hydrocarbons.13,14 However, Cl2 gas is not suitable for the synthesis of fine chemicals because of its poor site selectivity and potential hazards. In this context, different methods for the generation of Cl˙ have been developed for the photocatalytic oxidation of Cl− and photo-cleavage of the M–Cl bond in the past decade, which has increased the synthetic utility of Cl˙. Carbon radicals produced by the action of Cl˙ readily react with molecular oxygen under O2 atmosphere to produce oxygenated products (Scheme 2). Cl˙ is a strong oxidant that enables the transfer of one electron from an electron-rich substrate. Substrates containing benzyl groups can undergo single-electron transfer (SET) to form a radical cation, which subsequently deprotonates to form a stable benzyl radical (Scheme 3). The likelihood of SET can be estimated by comparing the redox potentials of the substrate and chlorine radical (E[Cl˙/Cl−] = +2.03 V vs. SCE (saturated calomel electrode)).15 The addition of oxygen to these benzyl radicals yields oxygenated products, as shown in Scheme 2. Therefore, the oxygenation of benzyl radicals may involve the SET mechanism.
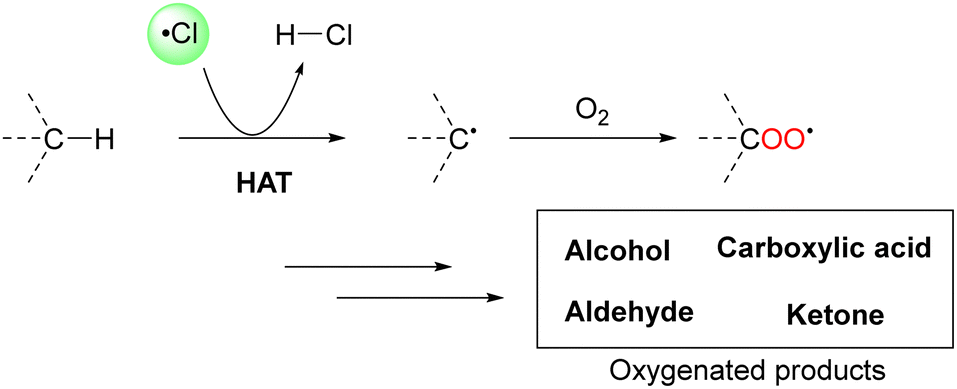 | ||
| Scheme 2 C–H oxygenation via HAT by the action of Cl˙. | ||
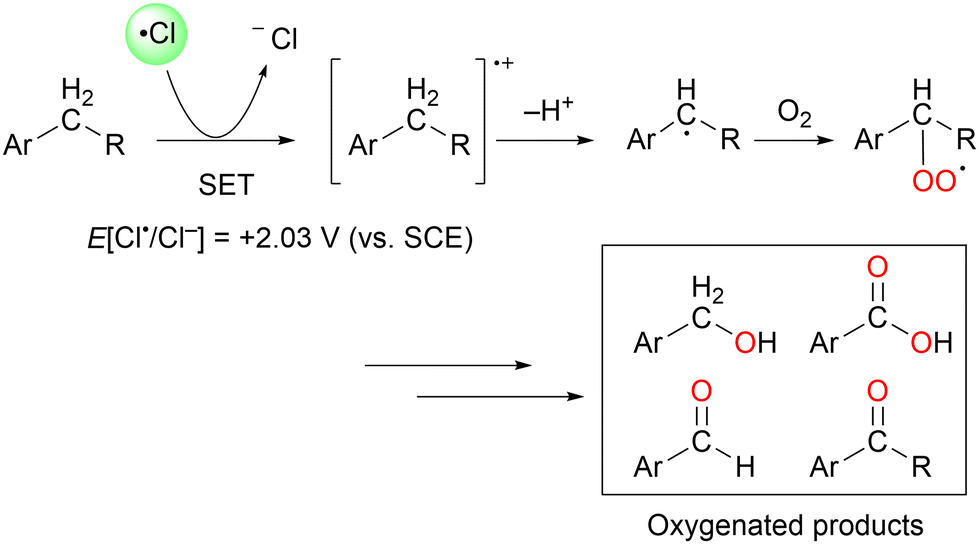 | ||
| Scheme 3 C–H oxygenation via SET with Cl˙. | ||
Molecular oxygen (O2) is an ideal reagent for economical and environmentally benign oxygenation reactions because of its abundant availability and nontoxicity.16–20 O2 does not react with hydrocarbons because the reaction of triplet oxygen (3O23Σg−) with a singlet organic substrate is a spin-forbidden process. Nevertheless, O2 can be converted to reactive oxygen species (ROS), such as singlet oxygen (1O2) by energy transfer or superoxide anion (O2˙−) by electron-transfer reduction, to facilitate a spin-allowed process for the oxygenation of hydrocarbons. However, the direct activation of O2 to produce ROS without a transition metal catalyst is challenging21–24 because it requires a photosensitiser under light irradiation. In addition, ROS do not react with hydrocarbons such as alkanes and aromatic substrates.
In this review article, we summarise the recent progress in the field of Cl˙-mediated C–H oxygenation reactions under light irradiation. However, reactions initiated by the addition of Cl˙ to alkenes are not discussed. As previous review articles have provided an overview of C–C bond-formation reactions,25 this article focuses only on C–H oxygenation reactions. We discuss C–H oxygenation reactions initiated by metal chlorides (iron, copper, and cerium), organophotoredox catalysts (acridinium ion), and chlorine dioxide radicals. Finally, we suggest future directions for research in the field.
2. Chlorine-radical-mediated C–H oxygenation reactions
2.1 Metal chlorides
Transition metal chlorides can be excited by UV- or visible-light irradiation, resulting in ligand-to-metal charge transfer (LMCT) electronic transitions. These transitions involve the transfer of an electron from an occupied orbital of a chlorine ligand to a vacant orbital of the metal,24–27 which must be on a lower energy level; high-valent metals such as Fe(III), Cu(II), and Ce(IV) are suitable for these transitions. The transfer of an electron from a M–Cl bonding molecular orbital to a vacant metal orbital and from a Cl lone pair to a M–Cl antibonding molecular orbital ultimately lead to homolytic cleavage and subsequent formation of a one-electron-reduced metal (Mn−1) and Cl˙ (Scheme 4). Kochi first applied this process to organic synthesis in 1962. CuCl2 undergoes photolysis to produce Cl˙, which oxidises isopropanol and chlorinates the benzyl and allyl positions.28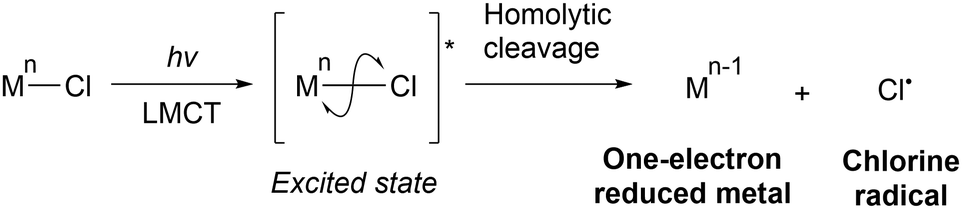 | ||
| Scheme 4 Cl˙ generation via photoinduced ligand-to-metal charge transfer (LMCT). | ||
HAT from aliphatic C–H bonds to Cl˙ produces HCl and carbon radical species. Under O2 atmosphere, carbon radicals react with molecular oxygen to afford oxygenated products because the reaction is thermodynamically favourable. Finally, the catalyst is regenerated via the oxidation of molecular oxygen by HCl (Scheme 5). In recent years, several metal-catalysed reactions other than oxygenation that are initiated by HAT from C–H bonds to chlorine radicals have been reported.29–43
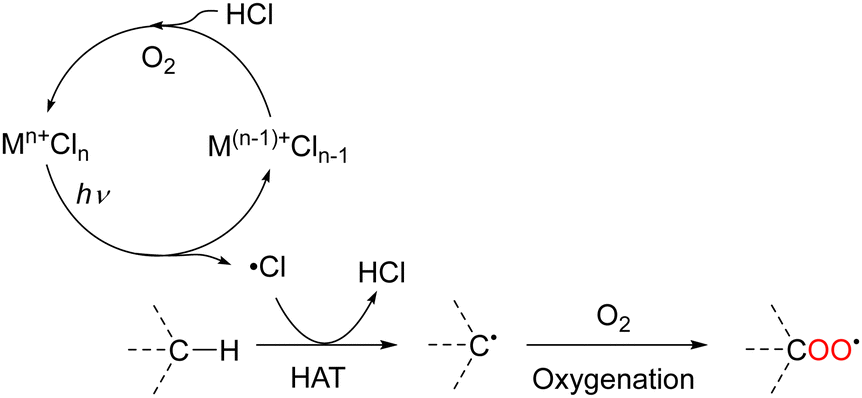 | ||
| Scheme 5 Mechanism of transition–metal–chloride-catalysed oxygenation via HAT. | ||
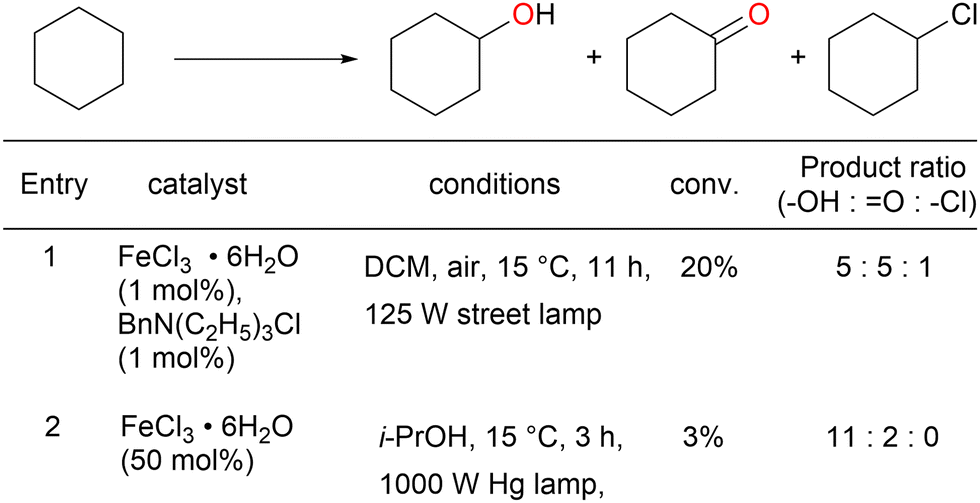 | ||
| Scheme 6 Oxygenation of cyclohexane with iron chloride. | ||
|
The kinetic isotope effect estimated using C6H12 and C6D12 was 1.6 ± 0.3, which indicates the occurrence of C–H bond cleavage.
A reaction mechanism for the oxidation reaction was reported in a subsequent study: Cl˙ and FeCl2 are formed from FeCl3 under light irradiation, and hydrogen is abstracted from the alkane by Cl˙.51 The C–H bond at the benzyl position can be more easily cleaved than those at the other alkane positions, thus contributing to efficient oxygenation at this position. A primary C–H bond at the benzyl position is typically oxidised to the corresponding alcohol or aldehyde and even carboxylic acid, whereas secondary and tertiary C–H bonds are oxidised to the corresponding alcohols or ketones. Further oxidation to carboxylic acid in the latter case occurs via C–C bond cleavage only under certain conditions. Barbier et al. reported an early example of benzylic C–H oxygenation, where a secondary C–H bond afforded a ketone. Tetralin afforded 1-tetralone in high yield in the presence of 10 equivalents of FeCl3 and 30 equivalents of water under UV irradiation (Scheme 7).52 Indan and diphenylmethane also tolerated the oxidation reaction well, but the oxidation of toluene and ethylbenzenes afforded <10% product yields. Therefore, the scope of this reaction was narrow. Jiang et al. developed the secondary benzylic C–H oxygenation to ketones. The oxidation of substrates such as ethylbenzene, fluorene, and xanthene at the benzyl position in the presence of FeCl3·6H2O and LiBr under blue-light irradiation afforded moderate to high yields of the desired products (Scheme 8).53 The addition of an equal amount of LiBr to the that of the iron catalyst improved the reaction to afford the desired oxygenated products in higher yields than when only FeCl3 was used. However, the authors explained that the reaction mechanism did not involve HAT by the Cl˙ generated by the photoexcitation of FeCl3; instead, the reaction occurred via SET from the substrate to the photoexcited iron. This was confirmed by the same oxygenation reactions proceeding well in the presence of Fe(NO3)2 and FeSO4.54 However, since Cl˙ can be generated in oxidation reactions using FeCl3 as the photocatalyst, it cannot be ruled out that both HAT and SET mechanisms are involved in this reaction simultaneously.
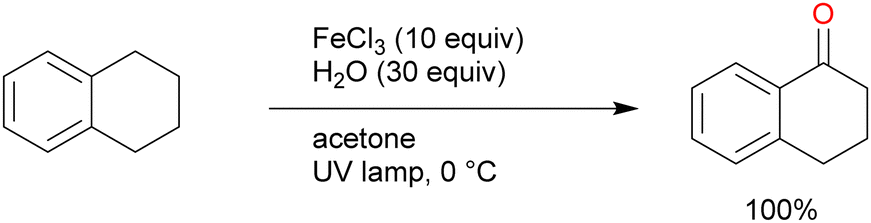 | ||
| Scheme 7 Benzylic C–H oxygenation in the presence of the FeCl3 catalyst. | ||
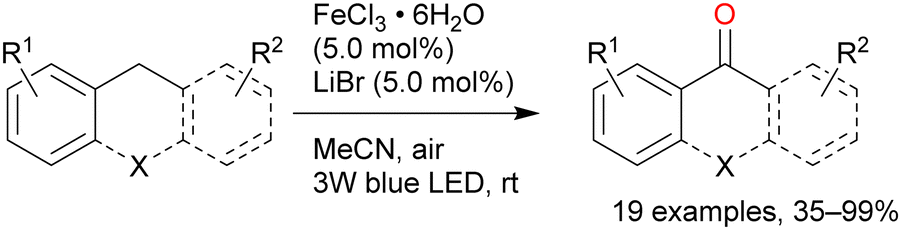 | ||
| Scheme 8 Iron-catalysed oxygenation of benzyl C–H to a ketone. | ||
Oxygenation reactions of primary, secondary, and tertiary C–H bonds directly afford carboxylic acids in the presence of catalytic amounts of FeCl3, tetrabutylammonium chloride, and 2,2,2-trichloroethanol (Scheme 9). Many toluene derivatives possessing electron-withdrawing or electron-donating groups and heterocyclic aromatics such as thiophene and pyridine have been oxidised at the benzyl position; even substrates with secondary and tertiary benzyl C–H bonds afforded carboxylic acids via C–C bond cleavage.55
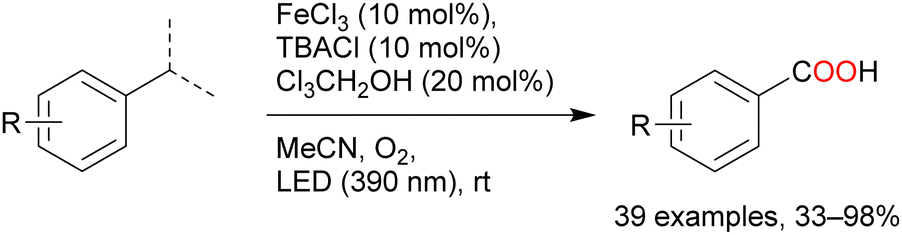 | ||
| Scheme 9 Iron-catalysed oxygenation of benzyl C–H to carboxylic acid. | ||
The oxidation of 1,3-diphenylpropane by FeCl2 under O2 atmosphere afforded benzoic acid in a high yield (87%) with CO2 as a by-product via C–C bond cleavage (Scheme 10).56 In addition, polystyrene and its derivatives [styrene-allyl alcohol copolymer, poly(styrene-co-acrylonitrile), ABS (acrylonitrile-butadiene-styrene), and styrene-maleic anhydride copolymer] afforded benzoic acid in moderate yields via oxidative degradation of the substrate with 100% conversion. Stache et al. further developed methods to degrade the high-molecular-weight polystyrene (MW > 90![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000) to a molecular weight of less than 1000 and obtained up to 23 mol% benzoyl products using 10 wt% FeCl3 as the catalyst and acetone as the solvent under white-light irradiation. Commercially available polystyrenes were efficiently degraded by this method and have been used in a photoflow process.57
000) to a molecular weight of less than 1000 and obtained up to 23 mol% benzoyl products using 10 wt% FeCl3 as the catalyst and acetone as the solvent under white-light irradiation. Commercially available polystyrenes were efficiently degraded by this method and have been used in a photoflow process.57
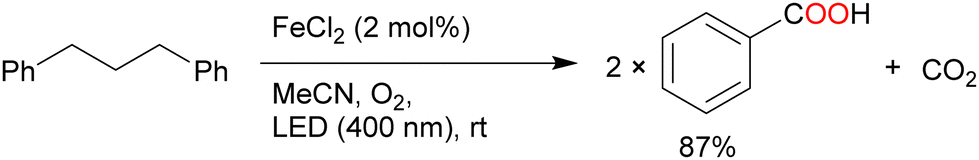 | ||
| Scheme 10 Oxygenation of 1,3-diphenylpropane by FeCl2 for polymer degradation. | ||
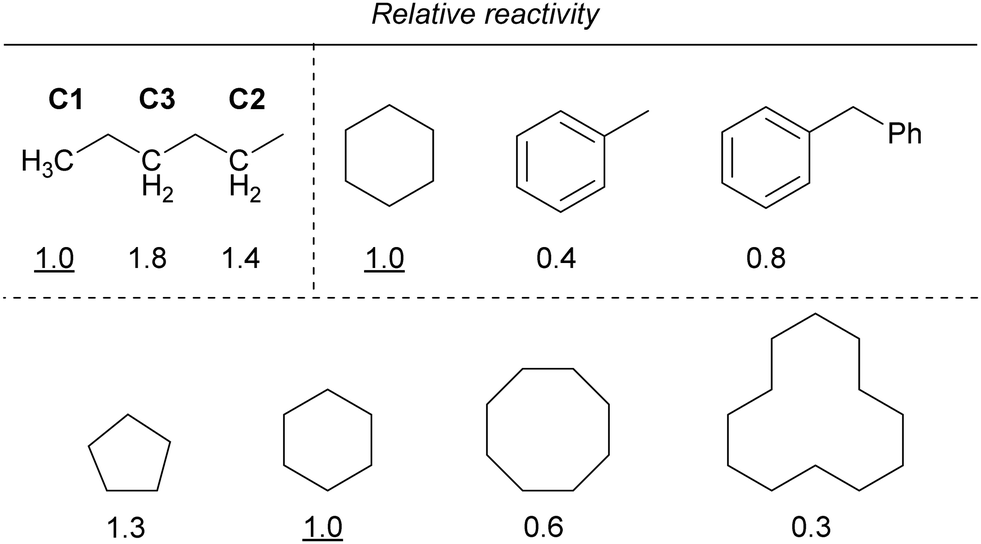
|
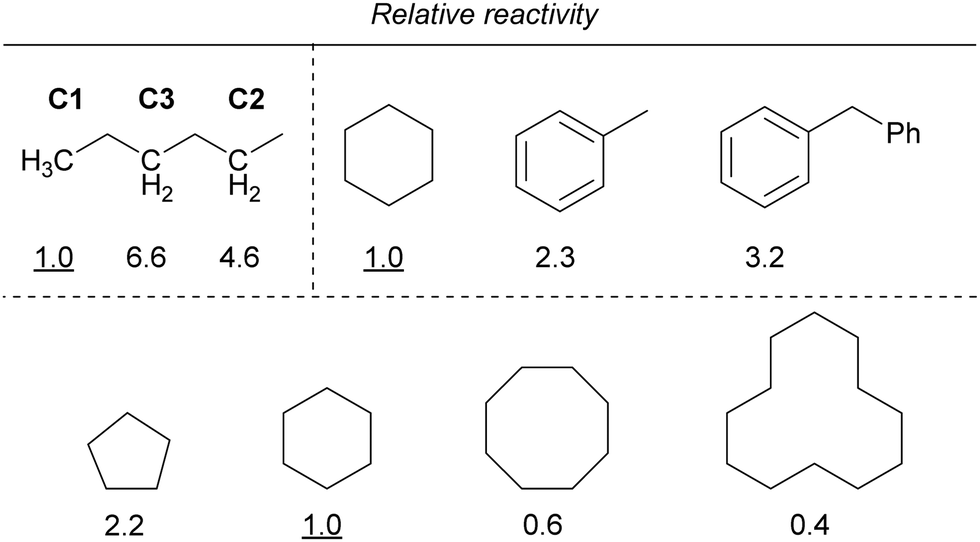
Furthermore, the reactivity of the C–H bond at the benzyl position was lower than that of the C–H bond at the alkane position. In addition, the reactivity decreased as the ring size of the cycloalkanes increased, similar to that observed for iron. If the reaction proceeds via C–H bond cleavage by the Cl˙ generated by the light irradiation of copper, the relative reactivities should not be different from those in the case of iron. Therefore, other mechanisms are also probably involved in addition to the Cl˙-mediated mechanism.
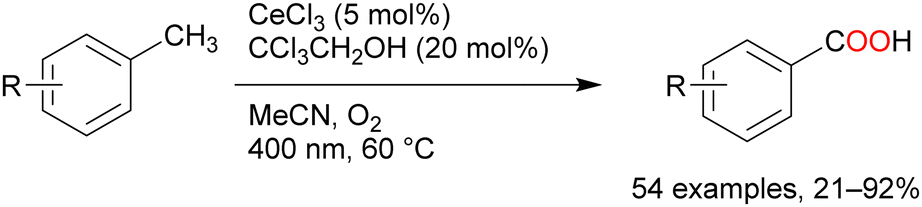 | ||
| Scheme 11 Cerium-catalysed benzyl C–H oxygenation. | ||
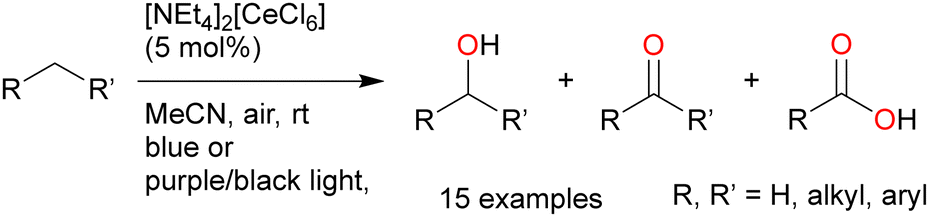 | ||
| Scheme 12 Cerium-catalysed aliphatic C–H oxygenation. | ||
2.2 Organophotoredox catalysts
Over the past half century, organic dyes have been used for strong-electron-transfer oxidation or reduction, where the dyes are irradiated with UV or visible light to generate an excited state. Organophotoredox catalysts have received considerable attention as a ‘green tool’ for conducting various organic transformations such as oxidation, C–C coupling, and C–hetero atom bond formation.61,62 The metal-free functionalisation of alkanes is challenging because the activation of chemically stable alkanes requires a powerful C(sp3)–H abstraction reagent. Cl˙ can cleave C–H bonds to yield alkyl radicals under mild conditions (vide supra). Cl˙ can be photochemically and electrochemically generated from Cl− as an inexpensive and safe HAT-active source via one-electron oxidation.63,64 The one-electron oxidation potential of Cl− is +1.15 V vs. SCE in MeCN.65–67 An appropriate photocatalyst will have a more positive one-electron reduction potential than that of Cl− in the photoexcited state to facilitate the negative free energy change of electron transfer (ΔGet <0). Consequently, the alkyl radical generated by the electron transfer oxidation of Cl− reacts with molecular oxygen to produce alkyl peroxyl radicals.Visible-light irradiation of the absorption band of Acr+–Mes in an O2-saturated MeCN solution containing toluene using a xenon lamp afforded benzaldehyde in 2% yield (Scheme 13).77 The one-electron oxidation potential of toluene (2.20 V vs. SCE) is more positive than that of Acr+–Mes. Hence, the electron transfer oxidation of toluene did not occur in this case. However, in the presence of HCl under the same irradiation conditions, toluene underwent oxidation to concomitantly produce benzaldehyde (60%), benzoic acid (20%), and H2O2 (60%) after 7 h of light irradiation (Scheme 13). The photocatalytic mechanism of the oxygenation of toluene catalysed by Acr+–Mes and HCl is shown in Scheme 14. The photoexcitation of Acr+–Mes affords the long-lived electron-transfer state Acr˙–Mes˙+via intramolecular electron transfer from the strongly reducing mesitylene moiety to the strongly oxidising acridinium moiety (Ered = 2.06 V and Eox = –0.57 V vs. SCE, respectively). The Acr˙ and Mes˙+ moieties reduce and oxidise O2 and Cl− to produce O2˙− and Cl˙, respectively. The hydrogen of the methyl group of toluene is abstracted by Cl˙ to afford the benzyl radical, which is followed by rapid O2 addition to afford the peroxyl radical. This peroxyl radical produces benzaldehyde and benzyl alcohol via disproportionation. Benzyl alcohol is readily oxygenated by Acr˙–Mes˙+ to yield benzaldehyde, while O2˙− disproportionates in the presence of protons to yield H2O2 and O2. Further oxygenation of benzaldehyde by Acr+–Mes and HCl yields benzoic acid via the abstraction of hydrogen from the formyl group of benzaldehyde by Cl˙.
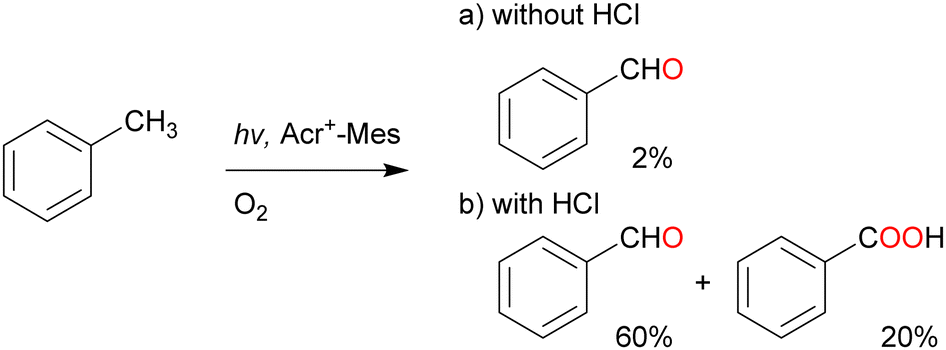 | ||
| Scheme 13 Acr+–Mes catalysed photo-oxygenation of toluene with or without HCl. | ||
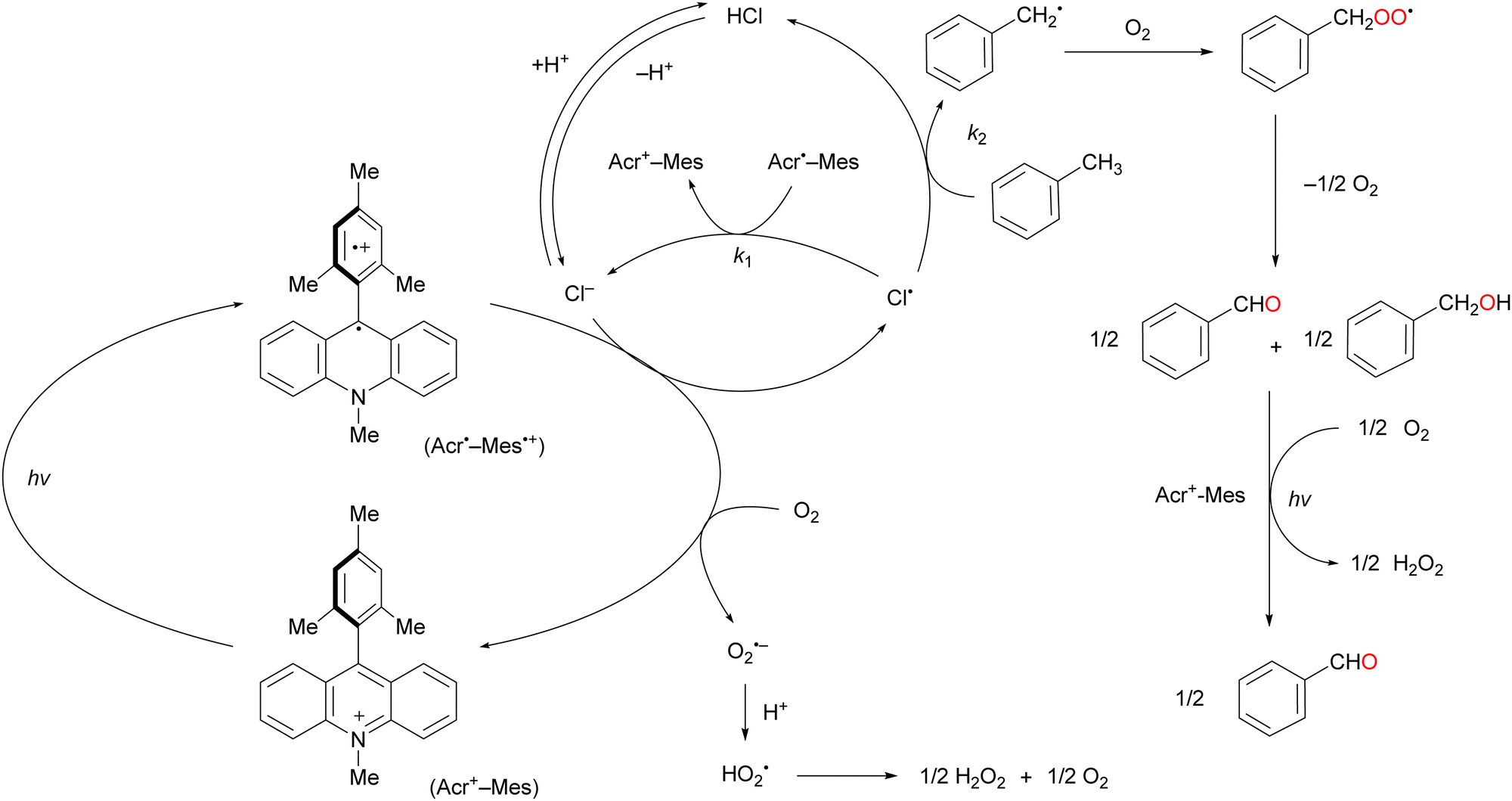 | ||
| Scheme 14 Reaction mechanism of the photooxygenation of toluene catalysed by Acr+–Mes and HCl. | ||
The visible-light-mediated oxidative lactonisation of 2-methyl-1,1′-biaryls produces benzocoumarins (Scheme 15).78 Multiple C–H functionalisation processes occur in this reaction in the presence of oxygen. Other 1,1′-biaryl derivatives such as 2-aldehydes, alcohols, and carboxylic acids also produce good results in this reaction.
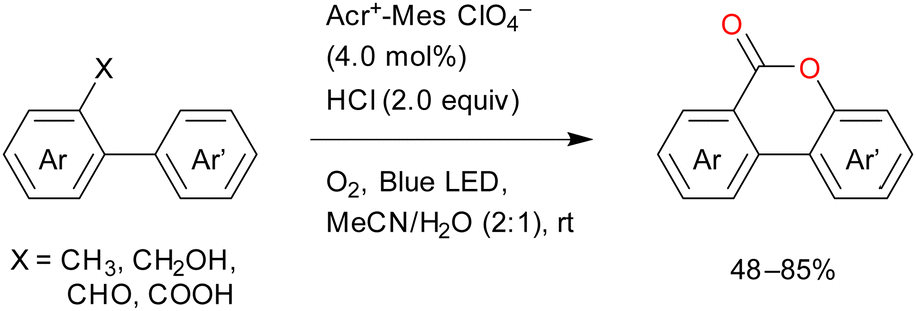 | ||
| Scheme 15 Reaction mechanism for the photooxygenation of toluene catalysed by Acr+–Mes and HCl. | ||
Unlike the one-electron oxidation potentials of aromatic substrates, those of C1–C6 alkanes are too positive (Eox >3.0 V vs. SCE) because of the large ionisation potential. As Cl˙ can cleave strong C–H bonds, it has been used to activate the C–H bond of alkanes such as cyclohexane to afford C–C or C–Cl bonds in a photochemical reaction.79,80 The combination of the acridinium photoredox catalyst and HCl produces an HAT-active Cl˙ for the C−H oxygenation of cyclohexane.81 The photooxygenation of cyclohexane by O2 occurs efficiently in an O2-saturated MeCN solution containing Acr+–Mes and HCl under visible-light irradiation to yield cyclohexanone, cyclohexanol, and H2O2.
The photocatalytic mechanism of Acr+–Mes-catalysed cyclohexane oxygenation is shown in Scheme 16. The reaction is initiated by intramolecular photoinduced electron transfer from the mesitylene moiety to the singlet excited state of the acridinium ion moiety of Acr+–Mes to form an electron transfer state (Acr˙–Mes˙+).68,69 The Mes˙+ moiety oxidises Cl− to Cl˙, whereas the Acr˙ moiety reduces O2 to O2˙−. The produced Cl˙ reacts with cyclohexane to form a cyclohexyl radical via C–H bond dissociation.82 The cyclohexyl radical undergoes O2 addition to afford the peroxyl radical, which disproportionates to the final oxygenated products, cyclohexanol and cyclohexanone. Although the cleavage of the C–H bonds of cyclohexane is difficult because of the high BDE (97 kcal mol−1), the Cl˙ generated by the light irradiation of Cl2 in the gas phase can readily cleave them. In addition, O2˙− disproportionates in the presence of protons to yield H2O2 and O2.
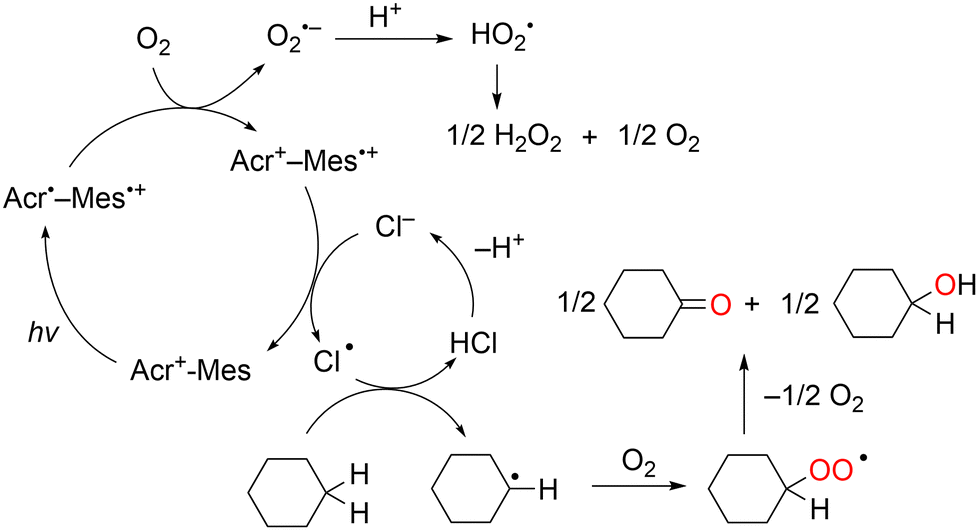 | ||
| Scheme 16 Photocatalytic mechanism of Acr+–Mes-catalysed cyclohexane oxygenation. | ||
2.3 Chlorine-dioxide-mediated C–H oxygenation reactions
The chlorine dioxide radical (ClO2˙), which is composed of chlorine and oxygen, is prepared by the oxidation of sodium chlorite (NaClO2) with HCl (Eq. 1). It is a photochemically reactive yellow gas that absorbs UV-visible light (λmax = 358 nm), as shown in Fig. 1.83
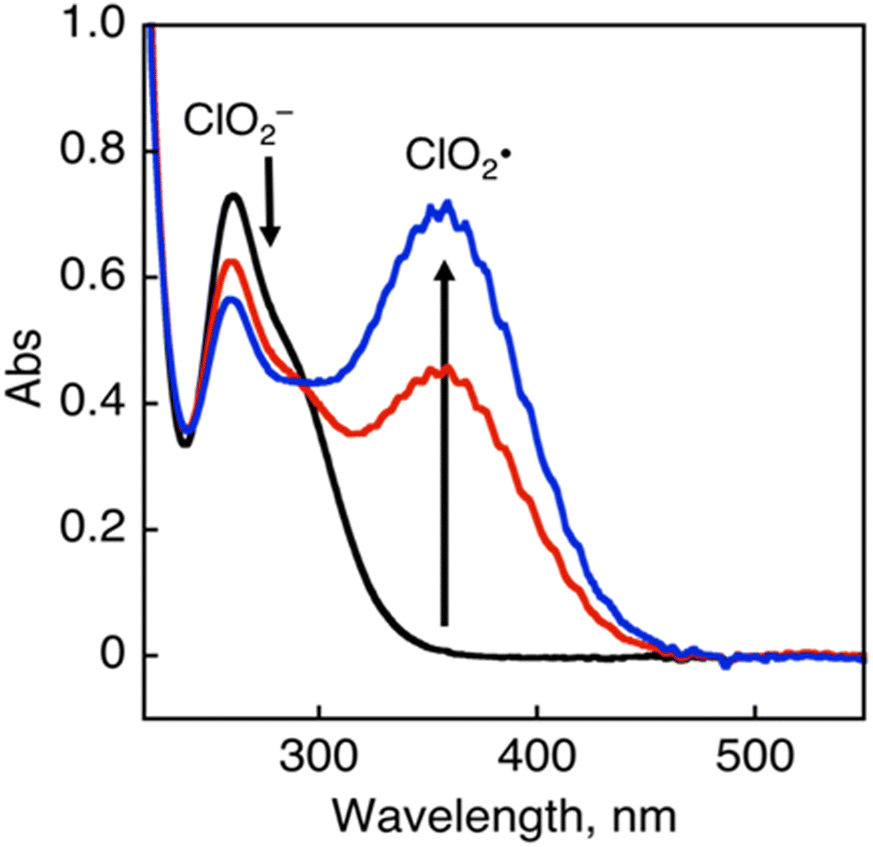 | ||
| Fig. 1 UV-Vis absorption spectral change of ClO2˙ generation in the reaction of NaClO2 with HCl. | ||
The photoexcitation of ClO2˙ affords one molecule each of Cl˙ and singlet oxygen (1O2).84 Singlet oxygen has a higher reactivity than ground state triplet oxygen. Cl˙ then reacts with CH4 to form CH3˙ and HCl via C–H bond dissociation. The addition of singlet oxygen to CH3˙ occurs rapidly, affording the methyl peroxyl radical (CH3OO˙). Thus, ClO2˙ is an effective initiator of CH4 oxygenation without chlorination.
The photochemical oxygenation of CH4 occurs in a two-phase system comprising perfluorohexane (PFH) and water under ambient conditions (298 K, 1 atm). PFH is an effective solvent for the oxygenation of CH4 as only the substrates and intermediates such as CH4, O2, and ClO2˙ dissolve in PFH but not the products methanol and formic acid, thus facilitating product isolation. The yields of methanol and formic acid were 14% and 85%, respectively. The CH4 conversion was 99% and the selectivity for CH3OH and HCOOH formation was >99% based on the initial concentration of CH4. The formation of further oxygenated products, such as CO and CO2, was not observed under the reported reaction conditions.84 The reaction mechanism for the oxygenation of CH4 with ClO2˙ is shown in Scheme 17. ClO2˙ photochemically converts Cl˙ and singlet O2via bond rearrangement from Cl–O–Cl to Cl–O–O.85 The bimolecular reaction of CH3OO˙, generated by the addition of singlet oxygen to CH3˙, affords the methoxy radical (CH3O˙). CH3O˙ reacts with CH4 to yield CH3OH and regenerate CH3˙. The rearrangement from CH3O˙ to CH2(OH)˙ also competes with the reaction with CH4. The second radical chain reaction (B) with O2 and CH4via CH2(OH)OO˙ produces methyl hydroperoxide (CH3OOH) and regenerates CH2(OH)˙. Finally, CH3OOH decomposes to HCOOH. The chain reactions are terminated by the disproportionation of CH3OO˙ to yield CH3OH and HCHO. However, HCHO is not observed as a product because it is immediately oxidised to HCOOH by autoxidation or Pinnick oxidation.86
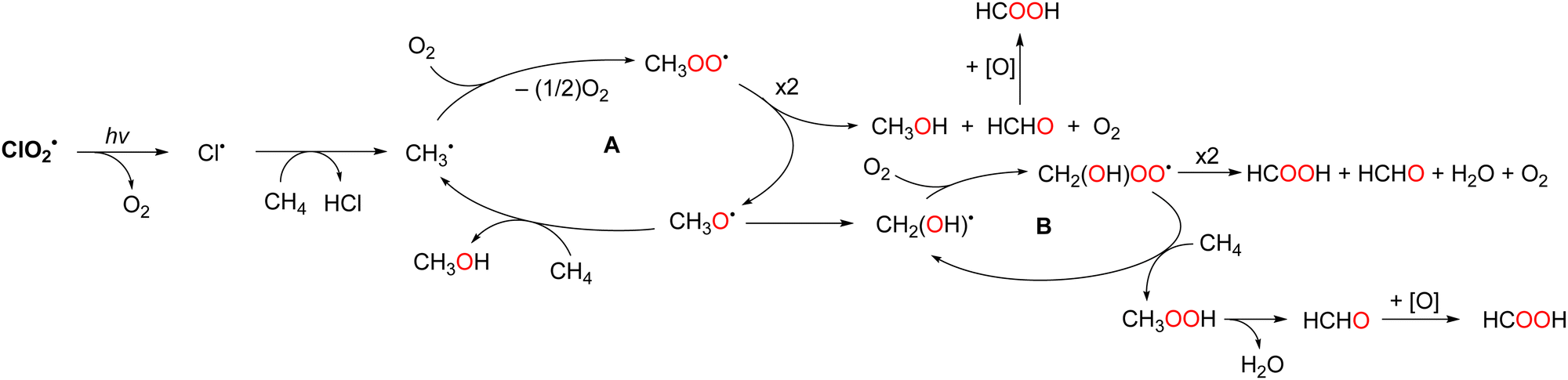 | ||
| Scheme 17 Radical chain reactions during the ClO2˙ oxygenation of CH4 to CH3OH and HCOOH. | ||
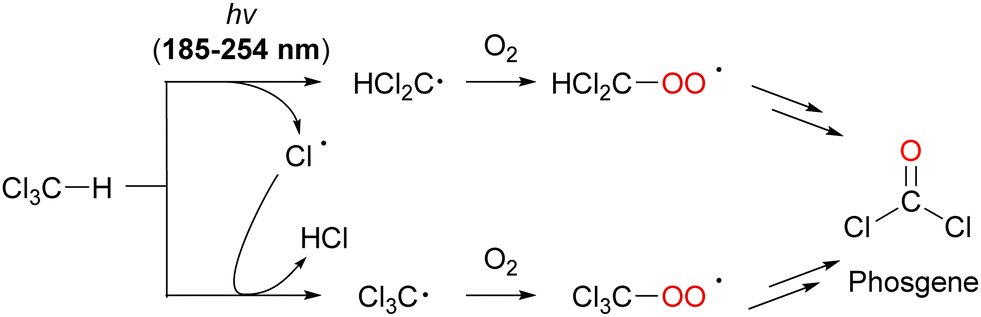 | ||
| Scheme 18 Generation of phosgene (COCl2) by the oxidative degradation of CHCl3 under UV-light irradiation. | ||
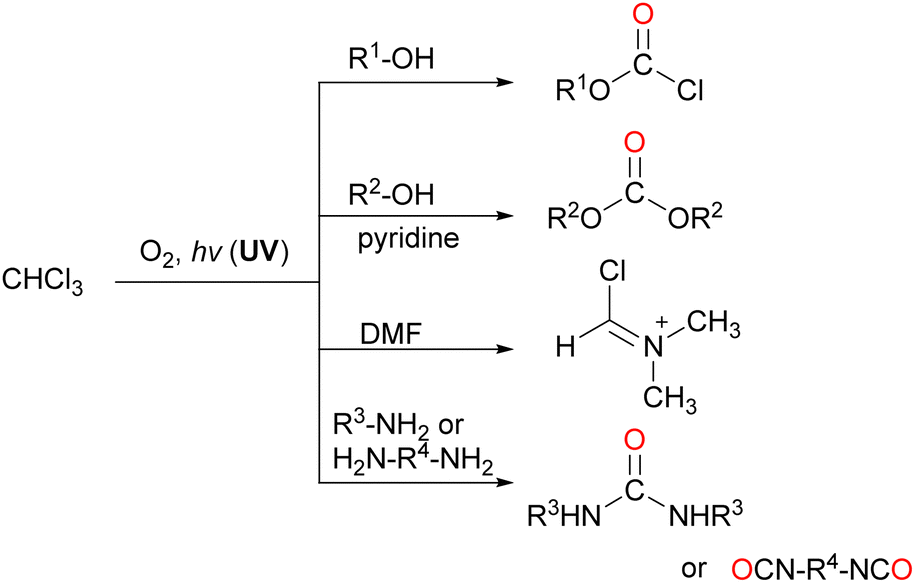 | ||
| Scheme 19 Synthetic organic reactions using COCl2 generated by the oxidative decomposition of CHCl3. | ||
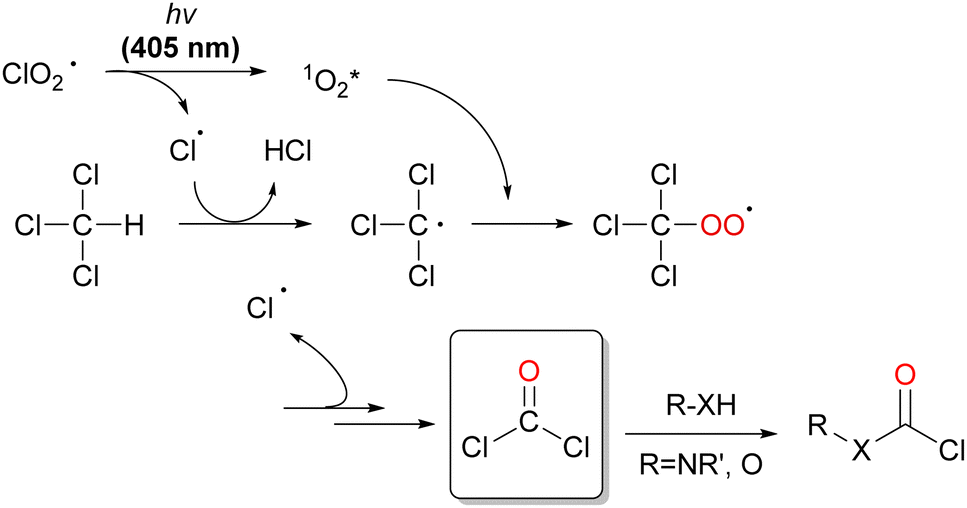 | ||
| Scheme 20 COCl2 generation via the formation of ClO2˙ under visible-light irradiation and its synthetic applications. | ||
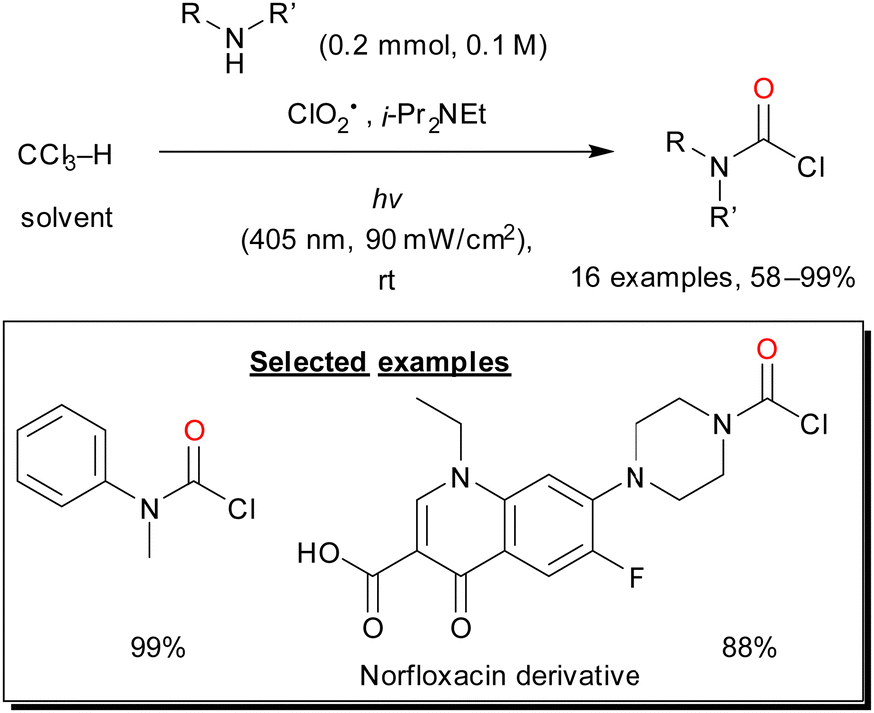 | ||
| Scheme 21 In situ preparation of COCl2 through CHCl3 oxygenation using visible-light-activated ClO2˙ and further reaction with amines to afford carbamoyl chlorides. | ||
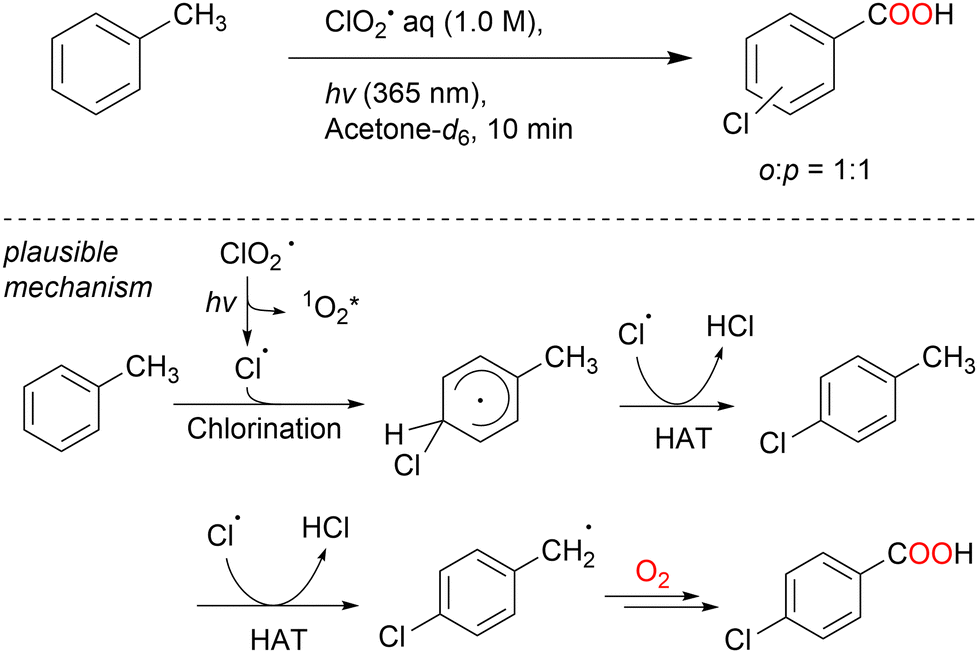 | ||
| Scheme 22 Chlorocarboxylation of toluene using ClO2˙ under LED (365 nm) irradiation. | ||
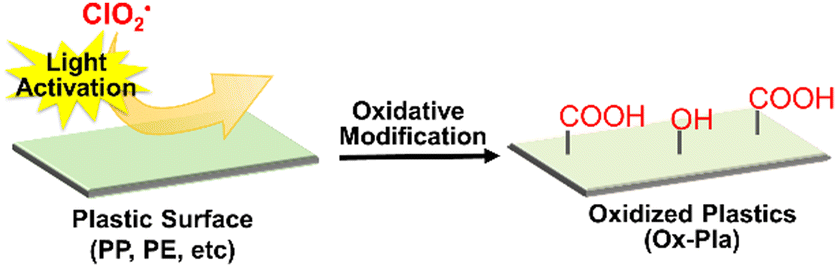 | ||
| Fig. 2 Polymer surface oxygenation by photoactivated ClO2˙. | ||
This is the first convenient chemical method for the surface C–H oxygenation of olefinic plastics such as PP and PE without the use of heavy metal oxides. Oxygen functional groups such as carboxyl groups were introduced onto the polymer surface via this surface oxidative modification, and cationic dyes were immobilised. In addition, the polar oxygen functional groups introduced on the polymer surface can facilitate metal adhesion; for instance, the oxidised PP film exhibits good adhesion of an aluminium plate without any adhesives (Fig. 3(a)).110 In addition, this modification method has been used to successfully develop electroless metal plating without exfoliation (Fig. 3(b)). The functional groups on the oxygenated polymer surface allow for further modification with various organic compounds, which could have significant implications in synthetic polymer chemistry. For instance, our research group successfully immobilised cyclodextrins on PP nonwoven fabrics via the ester coupling reaction of carboxyl groups that are introduced by photooxidation.111 This highly functional PP nonwoven fabric can sustainably load and release drugs. This surface modification has also been applied to polycarbonate (an engineering plastic),112 ABS resin,113 and polyhydroxyalkanoic acid (a biomass plastic).114
 | ||
| Fig. 3 Photographs of (a) the oxidized polypropylene (PP) films before and after the thermocompression bonding of Al/PP/Al without any adhesive and (b) Cu and Ni electroless plated PP films. | ||
2.4 Others
Recently, C–H oxygenation reactions using the abundant and inexpensive HCl as the Cl˙ source without metal or organocatalysts have also been developed. Wang et al. reported the benzyl oxidation of alkylarenes using HCl as the catalyst and molecular oxygen as the oxidant (Scheme 23).115 They attributed this reaction to the generation of singlet oxygen from the photoexcitation of the substrate and the consequent formation of Cl˙ from Cl−. However, direct excitation of the substrates is unlikely under visible-light irradiation (395–400 nm) as the substrates do not absorb visible light. In addition, energy transfer from the excited substrate to oxygen has not been sufficiently investigated using techniques such as cyclic voltammetry. Further follow-up studies are expected. | ||
| Scheme 23 HCl-catalysed aerobic oxidation of alkylarenes. | ||
3. Conclusion and outlook
The direct oxygenation of C(sp3)–H bonds is an important chemical conversion process in industrial and fine chemical synthesis. It can be a step-, time-, and energy-saving method in the construction of complex organic compounds compared with the classical synthetic approach. Oxygenation using molecular oxygen is an optimal method for economic and environmental reasons because O2 is abundant and non-toxic. However, O2 cannot react directly with hydrocarbons and should be converted to singlet oxygen to facilitate C–H bond cleavage. In this article, we reviewed C–H oxygenation reactions initiated by the Cl˙ generated by light irradiation and molecular oxygen. Cl˙ is an attractive HAT reagent and electron acceptor (SET source) as it has strong oxidative properties, and the Cl atoms form strong bonds with hydrogen atoms. In addition, the generation of Cl˙ by light irradiation is effective in terms of the energy required and ease of controlling the reaction. These features meet the increasing demand for sustainable and environment-friendly chemical processes and Cl˙-mediated oxygenation reactions are expected to be further developed in the future.Further development is expected to complement the current electrochemical techniques.
Future studies can focus on the following topics:
(1) Selective oxygenation reactions of alkyls can be developed by controlling steric hindrance using reactive ligands, which exhibit selectivity even when the C–H bonds have similar bond strengths (radical stability).116 On the other hand, the following points should be noted in Cl˙-mediated oxygenation using metal chlorides as the Cl source: reactions where metal chlorides are used as the Cl˙ source can proceed via SET from the substrate to the metal. This should be confirmed by conducting the reaction in the presence of metal species other than chlorides (e.g., sulfates and nitrates) and Cl˙ trapping experiments. Both HAT and SET are possible mechanisms for the benzylic C–H oxygenation. Even though elucidating the reaction mechanism is challenging owing to competing reactions, the presence or absence of SET can be validated by determining the redox potentials, and the presence of radical cations can be detected by electron spin resonance (ESR). In addition, density-functional-theory-based mechanistic studies have been demonstrated to be useful in most of the original papers in this review.
(2) The use of acridinium and other organic photocatalysts can potentially improve the reaction efficiency and selectivity. However, these photocatalysts need to have the appropriate oxidation ability and chemical stability.
(3) Reactions of chlorine dioxide, such as methane oxidation and plastic surface modification, should be developed as industrial processes. Scale-up and safety assessment are topics to be studied in the future.
We hope that this review will encourage interest in C–H oxygenation reactions with Cl radicals and contribute to the understanding of the reactivity of Cl radicals, thereby facilitating their use in further useful and wide-range synthetic chemical applications.
Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This work was supported by JSPS KAKENHI Grants (JP23K13709 to Y. I., JP20K05606 to H. A., JP20H02779, and JP20H04819 to K. O.); the NEDO Grant 17101509-0 to H. A.; and the JST OPERA Grant JPMJOP1861.References
- M. S. Chen and M. C. White, Science, 2010, 327, 566–571 CrossRef CAS PubMed.
- M. A. Bigi, S. A. Reed and M. C. White, Nat. Chem., 2011, 3, 216–222 CrossRef CAS PubMed.
- P. E. Gormisky and M. C. White, J. Am. Chem. Soc., 2011, 133, 12584–12589 CrossRef CAS PubMed.
- M. C. White, Science, 2012, 335, 807–809 CrossRef CAS PubMed.
- Y.-R. Luo, Comprehensive Handbook of Chemical Bond Energies, CRC Press, Boca Raton, FL, 2007 Search PubMed.
- C. Tang, X. Qiu, Z. Cheng and N. Jiao, Chem. Soc. Rev., 2021, 50, 8067–8101 RSC.
- L. Capaldo, D. Ravelli and M. Fagnoni, Chem. Rev., 2022, 122, 1875–1924 CrossRef CAS PubMed.
- L. Capaldo and D. Ravelli, Eur. J. Org. Chem., 2017, 2056–2071 CrossRef CAS PubMed.
- H. Cao, X. Tang, H. Tang, Y. Yuan and J. Wu, Chem Catal., 2021, 1, 523–598 CrossRef CAS.
- G. Lei, M. Xu, R. Chang, I. Funes-Ardoiz and J. Ye, J. Am. Chem. Soc., 2021, 143, 11251–11261 CrossRef CAS PubMed.
- L. Capaldo, T. Noël and D. Ravelli, Chem. Catal., 2022, 2, 957–966 CrossRef CAS.
- S. J. Blanksby and G. B. Ellison, Acc. Chem. Res., 2003, 36, 255–263 CrossRef CAS PubMed.
- E. T. McBee and H. B. Hass, Ind. Eng. Chem., 1941, 33, 137–142 CrossRef CAS.
- E. T. McBee, H. B. Hass, C. M. Neher and M. Strickland, Ind. Eng. Chem., 1942, 34, 296–300 CrossRef CAS.
- A. A. Isse, C. Y. Lin, M. L. Coote and A. Gennaro, J. Phys. Chem. B, 2011, 115, 678–684 CrossRef CAS PubMed.
- R. A. Sheldon, J. Chem. Technol. Biotechnol., 1997, 68, 381–388 CrossRef CAS.
- R. A. Sheldon, Chem. Ind., 1997, 1, 12–15 Search PubMed.
- Oxygenases and Model Systems, ed. T. Funabiki, Kluwer, Dordrecht, The Netherlands, 1997 Search PubMed.
- D. H. R. Barton, A. E. Martell and D. T. Sawyer, The Activation of Dioxygen and Homogeneous Catalytic Oxidation, Plenum Press, New York, 1993 Search PubMed.
- L. L. Simandi, Dioxygen Activation and Homogeneous Catalytic Oxidation, Elsevier, Amsterdam, 1991 Search PubMed.
- T. Mukaiyama, Aldrichimica Acta, 1996, 29, 59–76 CAS.
- R. A. Sheldon, I. W. C. E. Arends and A. Dijksman, Catal. Today, 2000, 57, 157–166 CrossRef CAS.
- J. Tsuji, Synthesis, 1984, 369–384 CrossRef CAS.
- D. H. R. Barton, Tetrahedron, 1998, 54, 5805–5817 CrossRef CAS.
- S. Bonciolini, T. Noël and L. Capaldo, Eur. J. Org. Chem., 2022, e202200417 CAS.
- Y. Abderrazak, A. Bhattacharyya and O. Reiser, Angew. Chem., Int. Ed., 2021, 60, 21100–21115 CrossRef CAS PubMed.
- F. Julia, ChemCatChem, 2022, 14, e202200916 CrossRef CAS.
- J. K. Kochi, J. Am. Chem. Soc., 1962, 84, 2121–2127 CrossRef CAS.
- Y. C. Kang, S. M. Treacy and T. Rovis, ACS Catal., 2021, 11, 7442–7449 CrossRef CAS PubMed.
- Q. Zhang, S. Liu, J. Lei, Y. Zhang, C. Meng, C. Duan and Y. Jin, Org. Lett., 2022, 24, 1901–1906 CrossRef CAS PubMed.
- A. Chinchole, M. A. Henriquez, D. Cortes-Arriagada, A. R. Cabrera and O. Reiser, ACS Catal., 2022, 12, 13549–13554 CrossRef CAS.
- S. M. Treacy and T. Rovis, J. Am. Chem. Soc., 2021, 143, 2729–2735 CrossRef CAS PubMed.
- A. Hu, J. J. Guo, H. Pan and Z. Zuo, Science, 2018, 361, 668–672 CrossRef CAS PubMed.
- J. Xu, H. Cai, J. Shen, C. Shen, J. Wu, P. Zhang and X. Liu, J. Org. Chem., 2021, 86, 17816–17832 CrossRef CAS PubMed.
- B. J. Shields and A. G. Doyle, J. Am. Chem. Soc., 2016, 138, 12719–12722 CrossRef CAS PubMed.
- H. P. Deng, X. Z. Fan, Z. H. Chen, Q. H. Xu and J. Wu, J. Am. Chem. Soc., 2017, 139, 13579–13584 CrossRef CAS PubMed.
- S. K. Kariofillis, B. J. Shields, M. A. Tekle-Smith, M. J. Zacuto and A. G. Doyle, J. Am. Chem. Soc., 2020, 142, 7683–7689 CrossRef CAS PubMed.
- T. Sato, S. Yoshiie, T. Imamura, K. Hasegawa, M. Miyahara, S. Yamamura and O. Ito, Bull. Chem. Soc. Jpn., 1977, 50, 2714–2730 CrossRef CAS.
- T. Sato, S. Yamaguchi and H. Kaneko, Tetrahedron Lett., 1979, 20, 1863–1864 CrossRef.
- T. Sato, H. Kaneko and T. Takahashi, Chem. Lett., 1981, 1469–1472 CrossRef CAS.
- M. Yamane, Y. Kanzaki, H. Mitsunuma and M. Kanai, Org. Lett., 2022, 24, 1486–1490 CrossRef CAS PubMed.
- G. B. Panetti, Q. Yang, M. R. Gau, P. J. Carroll, P. J. Walsh and E. J. Schelter, Chem Catal., 2022, 2, 853–866 CrossRef CAS.
- D. Birnthaler, R. Narobe, E. Lopez-Berguno, C. Haag and B. König, ACS Catal., 2023, 13, 1125–1132 CrossRef CAS.
- H. Inoue, K. Tamaki, N. Komakine and E. Imoto, Bull. Chem. Soc. Jpn., 1966, 39, 1577–1582 CrossRef CAS.
- H. Inoue, K. Tamaki, N. Komakine and E. Imoto, Bull. Chem. Soc. Jpn., 1967, 40, 875–880 CrossRef CAS.
- H. Inoue, N. Komakine and E. Imoto, Bull. Chem. Soc. Jpn., 1968, 41, 2726–2733 CrossRef CAS.
- H. Inoue, M. Izumi and E. Imoto, Chem. Lett., 1973, 571–572 CrossRef CAS.
- G. B. Shul’pin and M. M. Kats, React. Kinet. Catal. Lett., 1990, 41, 239–243 CrossRef.
- G. B. Shul'pin and A. N. Druzhinina, Mendeleev Commun., 1992, 2, 36–37 CrossRef.
- K. Takaki, J. Yamamoto, K. Komeyama, T. Kawabata and K. Takehira, Bull. Chem. Soc. Jpn., 2004, 77, 2251–2255 CrossRef CAS.
- G. B. Shul’pin, G. V. Nizova and Y. N. Kozlov, New J. Chem., 1996, 20, 1243–1256 Search PubMed.
- M. Barbier, Helv. Chim. Acta, 1984, 67, 866–869 CrossRef CAS.
- S. Li, B. Zhu, R. Lee, B. Qiao and Z. Jiang, Org. Chem. Front., 2018, 5, 380–385 RSC.
- J. Gu, Y. Wan, H. Ma, H. Zhu, H. Bu, Y. Zhou, W. Zhang, Z.-G. Wu and Y. Li, Tetrahedron, 2021, 93, 132298 CrossRef CAS.
- G. Zhang, Z. Zhang and R. Zeng, Chin. J. Chem., 2021, 39, 3225–3230 CrossRef CAS.
- M. Wang, J. Wen, Y. Huang and P. Hu, The authors proposed a single-electron transfer process for this reaction based on the fact that it proceeds in the absence of chloride using reagents such as Fe(NO3)3·9H2O, FeOTf3, and FeBr3. However, a Cl˙-mediated HAT process cannot be ruled out for the reaction using FeCl2, ChemSusChem, 2021, 14, 5049–5056 CrossRef CAS PubMed.
- S. Oh and E. E. Stache, J. Am. Chem. Soc., 2022, 144, 5745–5749 CrossRef CAS PubMed.
- C.-C. Wang, G.-X. Zhang, Z.-W. Zuo, R. Zeng, D.-D. Zhai, F. Liu and Z.-J. Shi, Sci. China: Chem., 2021, 64, 1487–1492 CrossRef CAS.
- P. Xie, C. Xue, S. Shi and D. Du, The authors proposed a single-electron transfer mechanism and the formation of a superoxide radical anion based on several control experiments, ChemSusChem, 2021, 14, 2689–2693 CrossRef CAS PubMed.
- Y.-H. Wang, Q. Yang, P. J. Walsh and E. J. Schelter, Org. Chem. Front., 2022, 9, 2612–2620 RSC.
- G. E. M. Crisenza and P. Melchiorre, Nat. Commun., 2020, 11, 803 CrossRef PubMed.
- M. Uygur, J. H. Kuhlmann, M. C. Pérez-Aguilar, D. G. Piekarski and O. G. Mancheño, Green Chem., 2021, 23, 3392–3399 RSC.
- B. E. Conway and G. Ping, J. Chem. Soc., Faraday Trans., 1991, 87, 2705–2714 RSC.
- J. A. Harrison and S. D. Hermijanto, J. Electroanal. Chem. Interfacial Electrochem., 1987, 225, 159–175 CrossRef CAS.
- A. M. Deetz, L. Troian-Gautier, S. A. M. Wehlin, E. J. Piechota and G. J. Meyer, J. Phys. Chem. A, 2021, 125, 9355–9367 CrossRef CAS PubMed.
- I. V. Kadija, J. Electrochem. Soc., 1984, 131, 601–608 CrossRef CAS.
- R. Bevernaegie, S. A. M. Wehlin, E. J. Piechota, M. Abraham, C. Philouze, G. J. Meyer, B. Elias and L. Troian-Gautier, J. Am. Chem. Soc., 2020, 142, 2732–2737 CrossRef CAS PubMed.
- S. Fukuzumi, H. Kotani, K. Ohkubo, S. Ogo, N. V. Tkachenko and H. Lemmetyinen, J. Am. Chem. Soc., 2004, 126, 1600–1601 CrossRef CAS PubMed.
- K. Ohkubo, H. Kotani and S. Fukuzumi, Chem. Commun., 2005, 4520–4522 RSC.
- N. A. Romero and D. A. Nicewicz, Chem. Rev., 2016, 116, 10075–10166 CrossRef CAS PubMed.
- N. Holmberg-Douglas and D. A. Nicewicz, Chem. Rev., 2022, 122, 1925–2016 CrossRef CAS PubMed.
- H. Kotani, K. Ohkubo and S. Fukuzumi, J. Am. Chem. Soc., 2004, 126, 15999–16006 CrossRef CAS PubMed.
- K. Ohkubo, T. Nanjo and S. Fukuzumi, Org. Lett., 2005, 7, 4265–4268 CrossRef CAS PubMed.
- K. Ohkubo, T. Nanjo and S. Fukuzumi, Catal. Today, 2006, 117, 356–361 CrossRef CAS.
- A. G. Griesbeck and M. Cho, Org. Lett., 2007, 9, 611–613 CrossRef CAS PubMed.
- K. Ohkubo, K. Mizushima, R. Iwata, K. Souma, N. Suzuki and S. Fukuzumi, Chem. Commun., 2010, 46, 601–603 RSC.
- K. Ohkubo, K. Mizushima and S. Fukuzumi, Res. Chem. Intermed., 2013, 39, 205–220 CrossRef CAS.
- Z. Luo, Z.-H. Gao, Z.-Y. Song, Y.-F. Han and S. Ye, Org. Biomol. Chem., 2019, 17, 4212–4215 RSC.
- H.-P. Deng, Q. Zhou and J. Wu, Angew. Chem., Int. Ed., 2018, 57, 12661–12665 CrossRef CAS PubMed.
- P. Li, A. M. Deetz, J. Hu, G. J. Meyer and K. Hu, J. Am. Chem. Soc., 2022, 144, 17604–17610 CrossRef CAS PubMed.
- K. Ohkubo, A. Fujimoto and S. Fukuzumi, Chem. Commun., 2011, 47, 8515–8517 RSC.
- B. Mühldorf and R. Wolf, Chem. Commun., 2015, 51, 8425–8428 RSC.
- K. Ohkubo, K. Hirose, T. Shibata, K. Takamori and S. Fukuzumi, J. Phys. Org. Chem., 2017, 30, e3619 CrossRef.
- K. Ohkubo and K. Hirose, Angew. Chem., Int. Ed., 2018, 57, 2126–2129 CrossRef CAS PubMed.
- R. C. Dunn and J. D. Simon, J. Am. Chem. Soc., 1992, 114, 4856–4860 CrossRef CAS.
- E. Dalcanale and F. Montanari, J. Org. Chem., 1986, 51, 567–569 CrossRef CAS.
- T. Alapi and A. Dombi, Chemosphere, 2007, 67, 693–701 CrossRef CAS PubMed.
- A. J. Seidl, L. R. Cohen, L. A. Peña and P. E. Hoggard, Photochem. Photobiol. Sci., 2008, 7, 1373–1377 CrossRef CAS PubMed.
- J. A. Kerr, Chem. Rev., 1966, 66, 465–500 CrossRef CAS.
- Y. Kuwahara, A. Zhang, H. Soma and A. Tsuda, Org. Lett., 2012, 14, 3376–3379 CrossRef CAS PubMed.
- Y. Hashimoto, S. Hosokawa, F. Liang, Y. Suzuki, N. Dai, G. Tana, K. Eda, T. Kakiuchi, T. Okazoe, H. Harada and A. Tsuda, J. Org. Chem., 2021, 86, 9811–9819 CrossRef CAS PubMed.
- Y. Liu, I. Okada and A. Tsuda, Org. Process Res. Dev., 2022, 26, 3336–3344 CrossRef CAS.
- F. Liang, M. Yanai, Y. Suzuki and A. Tsuda, Org. Lett., 2020, 22, 3566–3569 CrossRef CAS PubMed.
- F. Liang, K. Eda, T. Okazoe, A. Wada, N. Mori, K. Konishi and A. Tsuda, J. Org. Chem., 2021, 86, 6504–6517 CrossRef CAS PubMed.
- R. Muranaka, Y. Liu, I. Okada, T. Okazoe and A. Tsuda, ACS Omega, 2022, 7, 5584–5594 CrossRef CAS PubMed.
- T. Sugimoto, T. Kuwahara, F. Liang, H. Wang and A. Tsuda, ACS Omega, 2022, 7, 39250–39257 CrossRef CAS PubMed.
- Y. Suzuki, F. Liang, T. Okazoe, H. Okamoto, Y. Takeuchi and A. Tsuda, Chem. Lett., 2022, 51, 549–551 CrossRef CAS.
- H. Asahara, N. Takao, M. Moriguchi, T. Inoue and K. Ohkubo, Chem. Commun., 2022, 58, 6176–6179 RSC.
- H. Eckert and B. Förster, Angew. Chem., Int. Ed. Engl., 1987, 26, 894–895 CrossRef.
- L. Cotarca, P. Delogu, A. Nardelli and V. Šunjić, Synthesis, 1996, 553–576 CrossRef CAS.
- W. Su, W. Zhong, G. Bian, X. Shi and J. Zhang, Org. Prep. Proced. Int., 2004, 36, 499–547 CrossRef CAS.
- M. O. Ganiu, B. Nepal, J. P. Van Houten and R. Kartika, Tetrahedron, 2020, 76, 131553 CrossRef CAS PubMed.
- L. Cotarca, T. Geller and J. Répási, Org. Process Res. Dev., 2017, 21, 1439–1446 CrossRef CAS.
- S. Ohno, H. Asahara, T. Inoue and K. Ohkubo, RSC Adv., 2022, 12, 31412–31414 RSC.
- S. K. Nemani, R. K. Annavarapu, B. Mohammadian, A. Raiyan, J. Heil, M. A. Haque, A. Abdelaal and H. Sojoudi, Adv. Mater. Interfaces, 2018, 5, 1801247 CrossRef.
- M. Drobota, S. Ursache and M. Aflori, Polymers, 2022, 14, 2307 CrossRef CAS PubMed.
- A. H. Behroozi, V. Vatanpour, L. Meunier, M. Mehrabi and E. H. Koupaie, ACS Appl. Mater. Interfaces, 2023, 15, 13825–13843 CAS.
- A. Vesel, Polymers, 2023, 15, 1109 CrossRef CAS PubMed.
- K. Ohkubo, H. Asahara and T. Inoue, Chem. Commun., 2019, 55, 4723–4726 RSC.
- Y. Jia, J. Chen, H. Asahara, T. Asoh and H. Uyama, ACS Appl. Polym. Mater., 2019, 1, 3452–3458 CrossRef CAS.
- K. Yamamoto, H. Asahara, M. Moriguchi and T. Inoue, Polym. J., 2023, 55, 599–605 CrossRef CAS.
- Y. Jia, H. Asahara, Y. Hsu, T. Asoh and H. Uyama, Appl. Surf. Sci., 2020, 530, 147202 CrossRef CAS.
- Y. Jia, J. Chen, H. Asahara, Y. Hsu, T. Asoh and H. Uyama, Polymer, 2020, 200, 122592 CrossRef CAS.
- Y. Jia, W. Wu, H. Asahara, Y. Hsu, T. Asoh, H. T. Tan, K. Sudesh and H. Uyama, Polym. Degrad. Stab., 2021, 191, 109661 CrossRef CAS.
- K. Niu, X. Shi, L. Ding, Y. Liu, H. Song and Q. Wang, ChemSusChem, 2022, 15, e202102326 CrossRef CAS PubMed.
- M. I. Gonzalez, D. Gygi, Y. Qin, Q. Zhu, E. J. Johnson, Y.-S. Chen and D. G. Nocera, J. Am. Chem. Soc., 2022, 144, 1464–1472 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2023 |